
ABSTRACT
Background
Stargardt disease (STGD1) is an autosomal recessive retinal dystrophy due to mutations in ABCA4, characterized by subretinal deposition of lipofuscin-like substances and bilateral centrifugal vision loss. Despite the tremendous progress made in the understanding of STGD1, there are no approved treatments to date. This review examines the challenges in the development of an effective STGD1 therapy.
Materials and Methods
A literature review was performed through to June 2021 summarizing the spectrum of retinal phenotypes in STGD1, the molecular biology of ABCA4 protein, the in vivo and in vitro models used to investigate the mechanisms of ABCA4 mutations and current clinical trials.
Results
STGD1 phenotypic variability remains an challenge for clinical trial design and patient selection. Pre-clinical development of therapeutic options has been limited by the lack of animal models reflecting the diverse phenotypic spectrum of STDG1. Patient-derived cell lines have facilitated the characterization of splice mutations but the clinical presentation is not always predicted by the effect of specific mutations on retinoid metabolism in cellular models. Current therapies primarily aim to delay vision loss whilst strategies to restore vision are less well developed.
Conclusions
STGD1 therapy development can be accelerated by a deeper understanding of genotype-phenotype correlations.
Introduction
Stargardt disease (OMIM 248200), also known as STGD1, is an autosomal recessive inherited retinal disease caused by biallelic mutations in the ATP-binding cassette transporter subfamily A4 gene (ABCA4, OMIM *601691) (Citation1–3). STGD1 is one of the most common genetic inherited retinal diseases (IRDs) accounting for 12% of IRD-related blindness (Citation4) and was originally described as a macular dystrophy; however, it is now well recognized that the spectrum of the disease can vary from childhood-onset cone-rod dystrophy with rapidly progressive central and peripheral vision loss, to late-onset macular pattern dystrophy-like disease that tends to spare the fovea (Citation5–7). In this era of translational personalized medicine, a deeper understanding of the genotype-phenotype correlation is essential for appropriate patient selection for the emerging gene- and cell-based therapies. Herein, we review the spectrum of STGD1 in humans and the model systems currently available for investigating pathogenesis and treatment. We then discuss the pathophysiology of gene expression, protein function and consequences of ABCA4 mutations and conclude with current therapeutic options.
Human STGD1 phenotype
Clinical assessment of STGD1
The retinal phenotype in STGD1 depends on the combined severity of the two ABCA4 mutations, the duration of the disease and other environmental or modifier gene effects. The first attempt to classify disease stages was based on clinical examination (Citation8) (). As fundus autofluorescence (FAF) imaging enabled better visualization of fleck lesions before they become visible on color photography and delineation of the boundaries of retinal pigment epithelial (RPE) atrophy (Citation9,Citation10) several grading systems based on FAF have also been developed using the 30° or 55° lens of the Heidelberg device (Citation11) and the ultra-widefield Optos camera (Citation12). The relationship between atrophy area on FAF and genotype severity has also been examined (Citation13). Functional classification based on electroretinography (ERG) responses of cone and rod pathways in addition to macular dysfunction has also been developed (Citation14). The value in classifying disease severity based on function is further supported by the demonstration of clinically significant progression in only 22% of the patients from group 1 as compared to 100% from group 3 (Citation14).
Table 1. Classification and staging in ABCA4-associated retinopathy according to outcomes of fundus examination, fundus photography, autofluorescence and electrophysiology
Multimodal imaging and functional assessment are required in the baseline assessment of a patient with suspected STGD1. If possible, it is highly recommended that all immediate family members are also examined to identify late-onset STGD1 without symptoms or masquerading as age-related macular degeneration, and pedigrees with pseudodominant inheritance (Citation16). While the purpose of clinical assessment in STGD1 is to guide genetic analysis and variant interpretation, it is also important to document biomarkers of disease progression and prognosis. Whether STGD1 presents as a pure macular dystrophy, cone dystrophy or cone-rod dystrophy, this condition is uniquely characterised by the excessive deposition of lipofuscin in the RPE followed by progressive RPE and outer retinal loss. The following investigations are recommended.
Clinical examination
The best-corrected visual acuity should be recorded as it provides an indication of foveal sparing. Colour vision assessment is useful for differentiating STGD1 from phenotypically similar cone dysfunction syndromes. Fundoscopy typically reveals retinal flecks around the macular region with or without a mid-peripheral distribution. However, by the time, a beaten bronze sign appears, the vision loss is already profound. Large peripheral pigmented lesions and even extensive bone spicules can be seen in those with childhood-onset disease (Citation17).
Multimodal retinal imaging
Multimodal imaging should include spectral domain optical coherence tomography (SD-OCT), FAF and ultra-widefield (UWF) imaging. OCT thickness and angiographic parameters were shown to be statistically different between 70 STGD1 patients as compared to 70 healthy eyes (Citation18). Therefore, SD-OCT should be performed to assess the degree and extent of outer retinal loss and RPE atrophy. OCT macular volume decline can serve as a biomarker for disease progression (Citation19). However, automated segmentation of the diseased RPE-Bruch’s membrane is unreliable and requires manual adjustment using an “adaptive” approach or automated deep learning segmentation (Citation20,Citation21). Flecks are best visualized on FAF and their growth and life cycle may serve as a biomarker for disease progression where one study has developed deep learning segmentation of flecks in STGD1 (Citation22). FAF imaging can also illustrate early macular RPE atrophy. Recent studies have demonstrated differences in atrophy between FAF images generated by near-infrared versus short-wavelength excitation light (Citation23–25). Near-infrared FAF has been recommended as an adjunct to short-wavelength FAF given the earlier detection of hypoautofluorescent lesions with excellent intra- and interobserver reliability of areas of decreased FAF. UWF retinal imaging, in both colour and AF modes, should be used in all cases as it allows for the visualization of the peripheral retina and is useful particularly in the assessment and monitoring of childhood-onset disease. Severity of UWF-FAF has been classified into three types depending on the extent of peripheral retinal involvement and atrophy (Citation12). Each of these three phenotypes was associated with increasing OCT severity.
Functional assessment
In addition to structural assessment, objective functional evaluation with electrophysiology is useful for disease monitoring, determining prognosis and patient counselling (Citation26). Of those with a normal full-field ERG at baseline only 20% showed significant progression after 10 years of follow up. Full-field ERG findings have been used to classify STGD1 into mild and severe phenotypes (Citation27) whilst ERG features have also been used to classify ABCA4 mutation severity (Citation28). Microperimetry has been used do document central visual field impairment in STGD1 (Citation29). It is being investigated as a clinical trial endpoint but its role in routine monitoring of STGD1 progression remains uncertain (Citation30). The measurement of total volume beneath the sensitivity surface of a 3-D model of the hill of vision derived from microperimetry outputs has been proposed as an alternative to average sensitivity decline (Citation31). Unfortunately, microperimetry remains challenging or impossible to perform in those with advanced disease and a preferred retinal locus outside the posterior pole. Widefield perimetry, using the Octopus or the Goldmann device, maybe the only method to measure residual peripheral vision in STDG1 with severe posterior pole disease (Citation32). However, caution is required in interpreting results in children as there is large test–retest variability and the area of intact peripheral field has been shown to increase substantially as these children age (Citation33).
Clinical presentation of STGD1
Following the identification of the causative gene (Citation1,Citation7,Citation34–39), several large case series of genetically confirmed STGD1 have reported a wide spectrum in the age of presentation and clinical manifestations. These can be divided into childhood-onset, early adult-onset and late adult-onset. Each of these groups may have their own variations in phenotype that correlate with specific ABCA4 variants. The variability in age of onset can also be found within the same family (Citation16,Citation40).
Childhood-onset STGD1
Children with biallelic severe or null-like ABCA4 variants present with central visual loss attributed to macular dysfunction alone or in combination with cone-rod dystrophy (CORD3, OMIM 604116) between the ages of five and eleven years (). The diagnosis of childhood-onset STGD1 may be challenging in the early stages of the disease when the fundus may appear normal and the full-field ERG may be unremarkable resulting in delayed diagnosis (Citation41). Up to 24% of children less than 10 years of age have shown a normal fundus appearance (Citation41). Thus, the absence of flecks or macular atrophy early in the disease is a relatively common finding and examination may not identify lesions until 3 years after the onset of symptoms, with the earliest signs being retinal pigment epithelial (RPE) alteration, bull’s eye appearance and parafoveal flecks. Therefore, children may initially be labelled with functional vision loss or treated for amblyopia (Citation42). This misdiagnosis has led to a median delay of 3 years in 50–90% of cases (Citation41,Citation42). However, given the widespread accessibility and use of OCT to detect early features of STGD1, the frequency of misdiagnosis is expected to decline.
Figure 1. Wide-field colour and autofluorescence imaging in an eleven-year-old with childhood-onset STGD1. The two alleles are effectively null. There is increased autofluorescence signal in the posterior pole with hyperautofluorescent flecks and speckled hypoautofluorescence. Optical coherence tomography demonstrates subretinal debris and outer retinal layer loss in the foveal region.
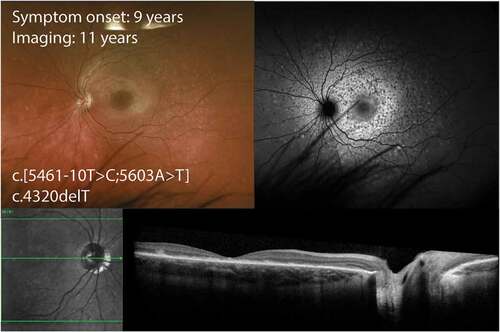
In children with no abnormalities on clinical examination, FAF has demonstrated discrete hyperautofluorescent dots confined to the foveola and even mild hyperautofluorescence in the perifoveal region (Citation43). Furthermore, a broader and more diffuse band of hyperautofluorescence with increasing retinal eccentricity has also been demonstrated (Citation43). FAF abnormalities have been reported in most patients with normal fundoscopy (Citation43). A thickened external limiting membrane (ELM) as seen on SD-OCT may provide an early biomarker for childhood-onset disease (Citation43,Citation44). This thickening demonstrated a maximal prominence at the foveola and decreased symmetrically with increasing eccentricity. These changes may represent disruption of the outer nuclear layer (ONL) within the cones and this is consistent with cone photoreceptor nuclei residing close to the ELM in the perifoveal region (Citation45). Sequential OCT images have illustrated focal collapse of the inner retinal layers, secondary to loss of the outer retinal structures (Citation43). These changes appear to preferentially affect perifoveal areas as the foveola photoreceptors are more resistant to degenerative processes (Citation43). Intraretinal pigmentations have been correlated to hyperreflective deposits in the inner layers of the fovea.
In childhood-onset STGD1, there may be a normal or near-normal pan-retinal cone and rod function on ERG however many of these patients later develop abnormal full-field ERG amplitudes (Citation3,Citation14,Citation41,Citation15,Citation46). Normal full-field ERG findings have been reported in 74% of children, with no obvious fundus abnormalities, at an average of two years (range, 0.1–27) after the onset of vision loss (Citation42). In this study, group 3 ERG recordings () were obtained in 11% of patients at 18 years (range, 6–27) after the onset of disease (Citation42). When assessing the five, an abnormal pattern ERG P50 component has been reported in children with no evidence of macular atrophy illustrating macular dysfunction (Citation43). Despite having a normal peripheral retina on fundoscopy four children have demonstrated generalized retinal dysfunction on full-field ERG (Citation43).
A form of rapid-onset chorioretinopathy (ROC), was described in a third of cases with biallelic null or severe ABCA4 mutations where the mean age of onset was seven years, and the key features were early and severe vision loss (20/200 to counting fingers), group 3 ERG findings, extensive bone spicules with attenuated retinal vasculature and markedly increased AF signal (Citation47).
A controversial manifestation is retinitis pigmentosa (RP19, OMIM 601718) (Citation48–53). It is now accepted that the RP phenotype refers to heavy pigmentation seen in the later stages of a rapidly progressive CORD3 phenotype. Close examination of “typical” RP reveals extensive macular and choroidal atrophy, with peripheral pigmentation (Citation48,Citation51,Citation53). These cases had early central visual loss that rapidly progressed to peripheral field loss and night vision impairment. This is probably the same as ROC (Citation47), except the phenotype was documented at a later stage when fundus features were dominated by peripheral atrophy, pigment plaques and bone spicules (Citation50). Such severe cases of “inverse” RP or ROC are generally associated with two null or deleterious variants with a low basal ATPase activity not stimulated by N-retinylidene-phosphatidylethanolamine (Citation54).
Early adult-onset STGD1
These patients present with central visual loss due to foveal atrophy in their second or third decade of life. At presentation, fundoscopy typically shows prominent macular atrophy with numerous paramacular and peripheral flecks. A key clinical finding is also peripapillary sparing (Citation55), although this is not specific. These patients often carry ABCA4 variants with an intermediate impact on ABCA4 function (e.g. c.6079C>T and c.[2588G>C;5603A>T]) either on both alleles or as compound heterozygous combination with a null/severe ABCA4 variant. In contrast to childhood-onset disease, their retinal flecks tend to spread centrifugally to cover most of the retina whilst the atrophy expands peripherally to only the perimacular region by the 5th decade of life (Citation56,Citation57). An exception is a subset of patients carrying the c.5882G>A, p.(Gly1961 Glu) variant who also develop symptoms in early adulthood ().
Figure 2. Wide-field color and autofluorescence imaging in a 34-year-old with early adult-onset STGD1 due to an allele with intermediate severity p.(Arg1108Leu) and the common variant p.(Gly1961Glu) with mild severity. There is a ring of hyperautofluorescence in the fovea with no lesions elsewhere in the retina. Optical coherence tomography demonstrates loss of photoreceptor in the fovea without any signs of fleck deposit.
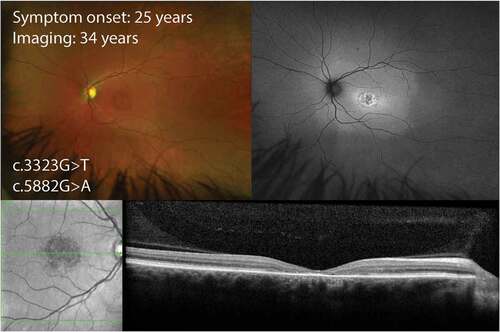
The phenotype linked to the c.5882G>A, p.(Gly1961Glu) variant (Citation5,Citation58–63) is bull’s eye maculopathy with paucity of flecks and normal full-field ERG (). The p.(Gly1961Glu) phenotype typically demonstrates a Fishman clinical grading of I or occasionally II, FAF imaging has shown a small region of decreased AF in the fovea, with or without a continuous ring of increased AF and all cases have normal dark and light-adapted full-field ERG. Those with homozygous p.(Gly1961Glu) complexed with another missense variant (His1838Asp or Asn96Lys) had childhood onset disease (4–12 years at onset) with Fishman clinical grading of III or IV and ERG group III (Citation62). Those with a deleterious mutation in trans with p.(Gly1961Glu) tended to have an earlier onset however the range was wide (Citation13,Citation28). A multicenter study of 79 cases carrying this mutation showed a greater penetrance of this gene in females, but no age difference in onset (Citation64). At presentation around a third may have a history of nyctalopia and another third have photoaversion (Citation60). Visual impairment was mild at around 0.8–0.9 in logarithm of minimum angle of resolution (logMAR) scale. Those presenting at a later age may have a larger area of RPE atrophy resembling geographic atrophy (Citation62). Quantitative FAF measurement has shown signal intensity in the 7°-9° zone falling within the 95% confidence interval for healthy eyes (Citation63). OCT typically shows localized outer segment loss resulting in an optical gap (Citation65,Citation66), but later onset disease may be accompanied by flecks. Microperimetry confirmed a localized foveal or perifoveal scotoma (Citation61,Citation62).
Late adult-onset STGD1
The definition of a “late” onset disease has varied from those ≥ 35 to those ≥ 50 years of age (Citation2,Citation67–70). Thus, patients can present from the fourth decade onwards. In contrast to childhood-onset, those with late adult-onset disease are often identified incidentally on retinal screening of family members of an affected individual, or when they become symptomatic secondary to subfoveal choroidal neovascularization. This lack of symptoms is explained by atrophy sparing the fovea. The mean age for onset of symptoms was 55 years (range 45–72 years) in one case series (Citation68). Although clinical examination may only show paracentral atrophy, FAF imaging often shows subtle abnormalities that may spread peripherally (Citation71). Hyperautofluorescent flecks are generally found surrounding the atrophy but they may extend nasally to the disc and peripherally. OCT features are marked by prominent RPE loss with co-localized outer retinal layer loss. Some patients may be misdiagnosed as having age-related macular degeneration if OCT and FAF features of flecks were not recognized at presentation.
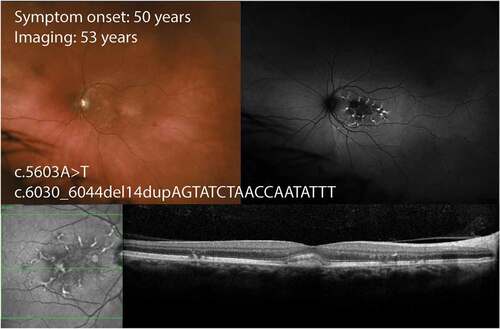
The recognition of c.5603A>T p.(Asn1868Ile) as a hypomorphic allele has facilitated the resolution of diagnosis in 80% of cases that were previously labelled as mono-allelic () (Citation72). Patients with Asn1868Ile in trans with a severe or deleterious allele had a mean age of symptom onset of 40 years, whilst those with a mild-to-moderate allele may not manifest the disease. There is also increased penetrance in females (Citation64). Those with an age of symptom onset < 30 years old tend to have non-specific visual complaints such as photoaversion and mild nyctalopia (Citation73). The mean visual acuity in the better-seeing eye at presentation was around 0.10 in logMAR (Citation73). FAF typically shows a paracentral atrophy sparing the fovea surrounded by a hyperautofluorescent zone or discrete fleck lesions without extension beyond the vascular arcades. In pre-atrophic cases the OCT may show increased reflectivity of the ELM band in the perifoveal region sparing the fovea.
Figure 3. Wide-field color and autofluorescence imaging in a 53-year-old with late adult-onset STGD1 due to a truncating allele and the common hypomorphic variant p.(Asn1868Ile). There are linear branching hyperautofluorescent flecks in the perifoveal region with no lesions outside the macula. Optical coherence tomography demonstrates subretinal deposit resembling a vitelliform lesion and intraretinal migration of the perifoveal flecks.
Variations in disease progression
The ability to predict disease progression based on genotype and the presenting phenotype is critical for both counselling and the design of clinical trials. Measurement of disease progression rates can be made by calculating the expansion rate of the area of RPE atrophy in near-infrared and short-wavelength FAF images (Citation23,Citation24,Citation74,Citation75), intensity of autofluorescence signal in regions unaffected by flecks or RPE loss (Citation76) and counting of hyperautofluorescent flecks (Citation22,Citation77). The area of definitely decreased autofluorescence was shown to decline by 0.51 (95% confidence interval, 0.42–0.61) mm2 per year (Citation75). However, area growth is highly dependent on baseline lesion size. A recent study reported a mean expansion rate of 0.69, 0.78 and 0.40 mm2 per year for children, adults with childhood-onset and adults with late-onset disease (Citation78). Growth rate based on square root transformed area of atrophy has been shown to be independent of baseline area (Citation79). This approach has allowed demonstration of the importance of ERG severity and genotype group on atrophy expansion rates in STGD1 (Citation56,Citation80). Future clinical trials should consider the use of square root transformed parameters in measuring area growth in STGD1 (Citation81). The feasibility of measuring retinal atrophy progression rate by calculating total macular volume decline (Citation19) on volumetric OCT has been described. However, the importance of genotype on the rate of macular volume loss has yet to be investigated. Mapping functional decline based on microperimetry has also been described over a one-year duration in the ProgStar cohort, although the decline was only 0.68 (95% confidence interval, 0.47 to 0.89) dB per year (Citation30). There was no association between microperimetry progression rate and genotype (Citation31).
Molecular biology of the human ABCA4 gene and protein
The ABCA4 gene
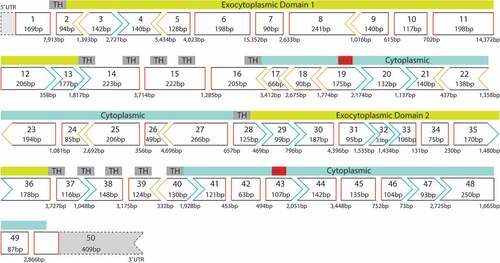
Several groups refined the ABCA4 gene location to a 2–3 cM interval (Citation36,Citation37). Allikmets et al. localized two markers that flank the ABCA4 gene, D1S3361 and D1S236, to 1p22.3–1p22.2, therefore repositioning the gene to 1p22 (Citation82). By fluorescence in situ hybridization, the chromosomal position of the human ABCA4 gene was mapped to 1p21-p22.1 (Citation83), the current location published on the National Centre for Biotechnology Information website (NCBI, https://www.ncbi.nlm.nih.gov/nuccore/NM_000350.3). The coding region of the ABCA4 gene consists of 50 exons, spanning over 100 kb (Citation39,Citation82). According to Ensembl (http://www.ensembl.org), the ABCA4 exons range in size from 33 bp to 266 bp (). There are 8 splice variants for ABCA4, three of which are protein-coding transcripts whilst the remaining are non-protein-coding. The full length, annotated ABCA4 transcript of 7328 bp, consisting of an open reading frame of 6822 bp encoding a 2273-amino-acid protein is published by NCBI (NP_000341.2).
Figure 4. The ABCA4 exon map, showing the reading frame and the functional protein domains encoded by the corresponding exons. Exon 2–13 and exon 28–36: exocytoplasmic domains, exon 16–28 and exon 40–50: cytoplasmic domains, exon 19 and exon 43: encode nucleotide-binding domains (NBDs). In-frame exons are indicated by rectangle with red sides, whereas codons disrupted by exon junctions are indicated by chevron sides in blue or orange. TH: transmembrane helix; UTR: untranslated region.
The ABCA4 protein
The ABCA4 protein is localized to the membrane and expressed in the outer segments of photoreceptors. ABCA4 is subclassed under the ABC transporter superfamily, the largest and most diverse family of transmembrane transport proteins that typically utilize the energy of ATP hydrolysis to pump various substrates across cell membranes against concentration gradients. Consistent with other ABC transporters, ABCA4 protein is composed of the typical structure of four domains for functionality: two highly conserved nucleotide-binding domains (NBDs) that provide energy for translocating substrates by binding and hydrolyzing ATP to ADP, and two highly hydrophobic transmembrane domains (TMDs) that act synergistically with NBDs ().
Figure 5. Topology of ABCA4 protein, adapted from 145. ECD: exocytoplasmic domain; TMD: transmembrane domain; S-S: disulfide bond; NBD: nucleotide-binding domain; ATP: adenosine triphosphate; ADP: adenosine diphosphate; ROS: rod outer segment.
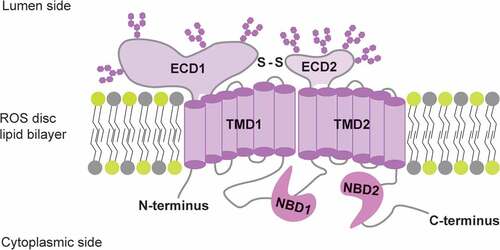
Opinions regarding the function of both NBDs differ. The complementary hydrogen/deuterium exchange studies suggested that only NBD2 is catalytically active (Citation84). This hypothesis is in line with other biochemical studies indicating that NBD2 binds and hydrolyses ATP in the presence or absence of substrates, whereas NBD1, containing a bound ADP, associates with NBD2 to play a non-catalytic role in substrate translocation (Citation85). However, this is not consistent with the results of a mutagenesis study, showing that both NDBs are catalytically active, albeit with different functions; mutations in NBD1, either alone or in combination with mutations in NBD2, abolished both basal and retinaldehyde-stimulated ATPase activity, whereas mutations in NBD2 result in inhibition of ATP hydrolysis stimulated by retinaldehyde (Citation86).
The signature motif of the prokaryotic ABC importers “EAA” was found in the N-terminal transmembrane domain (TMD) of ABCA4, revealing its identity as the only known mammalian importer (Citation87), flipping substrates from the luminal (topographically equivalent to the extracellular) to the cytoplasmic side. However, the “EAA” sequence, which interfaces the NBD and the TMD, is absent in the C-terminal half (Citation88). Two large exocytoplasmic domains (ECD1 with 600 residues and ECD2 with 300 residues) locate inside the disc lumen (Citation84,Citation89). The number of N-glycosylation sites on ECDs are controversial and the biological role of the two large ECDs remains to be determined (Citation90,Citation91). Discussion on the detailed functional and structural characterization of ABCA4 domains are beyond the scope of this review. We refer readers to comprehensive reports for more information regarding this aspect (Citation84,Citation88,Citation92–98).
ABCA4 function in the retinoid cycle
Upon photon absorption in photoreceptors, the excessively released 11-cis-retinal and its derivatives all-trans-retinal rapidly and reversibly react with phosphatidylethanolamine (PE) to form N-retinylidene-phosphatidylethanolamine (N-ret-PE). Depending on the orientation of the retinylidene-bearing head, two forms of N-ret-PE have been proposed: cytoplasmic oriented N-ret-PE and luminal N-ret-PE (Citation99). The cytoplasmic oriented N-ret-PE can be catalyzed by the all-trans-retinol dehydrogenases (all-trans-RDHs) that exist in the cytoplasm (Citation100,Citation101), producing all-trans-retinol products that re-enters the retinoid cycle. However, the luminal N-ret-PE is unable to cross the disc membrane independently (Citation102) and thus is inaccessible by RDHs. By utilizing ATP hydrolysis as an energy source, ABCA4 assists in the elimination of cytotoxic retinoid chemicals through actively flipping the luminal N-ret-PE to the cytoplasmic surface of disk membranes (Citation87,Citation103). A portion of luminal N-ret-PE that has not been flipped by ABCA4 can be further condensed with another all-trans-retinal to produce bisretinoids in the discs. Upon phagocytosis by RPE, the condensed compounds in the outer segments of photoreceptors are thought to be eventually converted to bis-retinoid N-retinylidene-N-retinylethanolamine (A2E), the major form of lipofuscin deposits in the low-pH environment of RPE phagolysosomes (Citation102). Recently, it was proposed that ABCA4 performs a similar function in the RPE endolysosomal membrane to that in photoreceptor outer segments, that is, it translocates N-ret-PE from the luminal to the cytoplasmic surface in an ATP-dependent manner (Citation99).
Molecular pathogenesis of ABCA4-STGD1
Mutation spectrum
With the advent of high-throughput next-generation sequencing, an ever-increasing number of ABCA4 variants have been consecutively reported since the linkage of STGD1 to ABCA4 in 1997 (Citation1). At the time of submission, a total of 1196 unique ABCA4 variants have been recorded in the Leiden Open Variation Database (LOVD, https://databases.lovd.nl/shared/genes/ABCA4). Cornelis et al. (Citation104) analyzed 913 unique ABCA4 variants published before 2016 and concluded that the majority are missense variants (51.59%), followed by protein-truncating variants (34.39%), complex alleles (7%), non-canonical splice site variants (3.61%), variants of non-truncating insertion/deletion (1.75%), deep-intronic variants (1.1%) and synonymous variants (0.55%). Among these, c.5882G>A p.(Gly1961Glu), c.[2588G>C;5603A>T] p.[(Gly863Ala,Gly863del;Asn1868Ile)] and c.[5461–10T>C;5603A>T] p.[Thr1821Aspfs*6,Thr1821Valfs*13;(Asn1868Ile)] are the three most frequent ABCA4 variants.
Given that ABCA4 mutations are extremely heterogeneous and several common variants are clustered in specific ethnic groups (Citation105,Citation106), correct interpretation and a consensus on the pathogenic classification of ABCA4 variants is essential for disease prognosis and potential therapeutic options. Recently, the American College of Medical Genetics and Genomics (ACMG) published a “gold standard” to standardize terminology and scoring system of variants identified in genes that cause Mendelian disorder (Citation107). A five-tier terminology system using the terms “pathogenic,” “likely pathogenic,” “uncertain significance,” “likely benign,” and “benign” is recommended based on the classification criteria provided in this report. By using this guideline, pathogenicity and its classification could be assigned to 1103 ABCA4 variants from LOVD, with 255 variants identified as pathogenic, 158 as likely pathogenic, 94 as uncertain significance, 31 as likely benign and 88 as benign. As currently developing functional assays are rapidly and constantly contributing to an update of variants in this regard, we reviewed pathogenic evidence obtained from these assays in below, for a better understanding of the molecular pathogenesis of certain variants that lead to disease.
Effect of causal missense variants
Over the decades, a series of research methods have been established to understand the effects of ABCA4 mutations on its function: using fluorescent microscopy and in situ hybridization to characterize cellular localization (Citation99), transiently expressing wild and mutant human ABCA4 cDNA into cell lines for soluble protein quantification (Citation54,Citation108), in vitro measurement of ATPase activities and retinoid substrate-binding activity (Citation87,Citation109–112) for assessing individual functional domain activities etc. Several studies investigated the effects of single amino acid substitution (Citation86,Citation108,Citation113), small in-frame deletions (Citation86,Citation113) and frameshift mutations (Citation86). Some of the mutations resulted in reduced protein levels (Citation86,Citation108), whereas others were found to compromise ATPase activity (Citation113,Citation114) and/or retinoid binding affinity (Citation108,Citation113).
As the major group of variants, missense variants directly compromise the function of protein, or alter the secondary structure that is crucial for correct localization, resulting in functional defects. For example, the amino acid change of Gly863Ala due to variant c.2588G>C, the most common mutation observed in STGD1 patients, attenuated the ATPase and CTPase activities of NBD1 and the rate of ATP hydrolysis significantly, as compared with the normal or WT NBD1 (Citation114). Missense mutation in cysteine residues within ECDs (Cys54Tyr, Cys75Gly, Cys1488Arg, Cys1490Tyr) that alter disulfide bonds, could cause ABCA4 protein to be misfolded and lead to STGD1 (Citation90). Mutations involving glycosylation sites also have impacts on protein structure thus causing diseases. One example is the variant p.Ser100Pro, a STDG1-related mutation that introduces a proline residue and prevents the occurrence of N-linked glycosylation on Asn-98 site (Citation91,Citation115). In this case, the mislocalization or incorrect insertion of ABCA4 protein contributes to photoreceptor degeneration. The retention of mutant ABCA4 protein in the inner segment of photoreceptors, likely due to misfolding, has also been reported in patients with retinal dystrophies (Citation116).
While the function of misfolded protein is compromised, it could overload the cellular processing systems, the capability of which becomes more tenuous under the elevated stress caused by lipofuscin accumulation in photoreceptor and RPE cells. Moreover, such misfolded protein could in turn exacerbate the primary loss-of-function consequence by toxic gain-of-function (Citation115), manifested by apoptotic signaling activation resulting from endoplasmic reticulum stress (Citation117) and unfolded protein response (Citation118). Patients with two non-truncating mutations presenting with greater disease severity than patients carrying two truncating mutations could be explained by such gain-of-function toxicity (Citation115). However, STGD1 remains a recessive disease despite the potential for toxic-gain of function in a carrier of such mutations.
Effect of causal splice variants
Alternative splicing has been considered to contribute to extensive transcriptomic and proteomic complexity by generating multiple transcripts from a defined genomic repertoires, and underlies significant phenotypic difference (Citation119,Citation120). It was estimated that in the retina, 13% of novel mRNA junctions were expressed at levels similar to, or higher than the reference transcripts (Citation121). However, this complexity comes at a cost (Citation122). It is now evident that missense, nonsense or even silent mutations can cause disease through effects on splicing, rather than directly through amino acid changes that impair protein function (Citation123), and for certain genes these types of mutation can be found in as many as 50% of the cases (Citation123). To characterize the functional consequences of ABCA4 splice variants, series of in vitro splice assays, including midigene assays (Citation124), patient-derived fibroblast-based assays (Citation16), assays employing the induced pluripotent stem cell (iPSC) (Citation125–128) and iPSC-derived retinal cells (Citation127,Citation129,Citation130) etc., have been developed successively.
Mutations affecting the invariant GT or AG sequences at the 5ʹ (donor) or 3ʹ (acceptor) of exons typically result in exon skipping (Citation121) due to inactivation of the canonical splice sites, although intron retention has also been reported in some cases. According to data from our laboratory, the mutation c.5835+1G>A affecting the first highly conserved “G” of the ABCA4 intron 41 disrupts the canonical splice site and leads to the retention of 12 nucleotides downstream of exon 41 and use of a cryptic donor splice site.
Recent analysis of a splice variant derived from our STGD1 cohort revealed that single or multiple exon skipping and intron retention was observed due to the variant location in the noncanonical splice sequence (see definition (Citation124)). One example is the most prevalent variant c.[5461–10T>C;5603A>T]. Patient-derived fibroblast harboring this variant show defective ABCA4 mRNA splicing, leading to reduced abundance of full-length transcripts and generation of alternatively spliced transcripts missing exon 39 or both exon 39 and 40. Such splicing defects were also validated in patient-derived progenitor cells (Citation127). However, other splice events such as partial exon skipping and exon elongation through intron retention were also reported for noncanonical splice variants (Citation131).
In recent years, mutations located in noncoding regions of ABCA4, especially deep intronic mutations are gaining more attention as they account for the missing inheritability in some cases where only monoallelic ABCA4 variant was detected (Citation121,Citation132–135). Indeed, STGD1 cases where the second allele was later identified as having a deep intronic mutation, aberrant splicing occurred due to the activation of cryptic splice sites or alteration to splice enhancer or silencer motifs. One example is the presence of two neighboring intronic mutations in intron 30 of ABCA4; (c.4539+2001G>A and c.4539+2028C>T) created an exonic splicing enhancer and resulted in a retina-specific transcript containing a 345-nt pseudoexon (Citation129). The absence of the aberrantly spliced transcript in patient-derived fibroblasts further implies that the pre-mRNA splicing in the retina is modulated under more complex splicing programs than in other tissues (Citation136), and certain splicing motifs can only be recognized by tissue-specific factors present exclusively in retinal cells (Citation135).
A significant proportion of missense, nonsense, insertion and deletion mutations may in fact exert their effects through altered splicing due to the disruption or introduction of exonic splicing enhancers and silencers or creation of novel splice sites (Citation16,Citation137), highlighting the necessity of validating the presumed effects of pathogenic variants on pre-mRNA splicing.
Impact of altered retinoid metabolism on cellular function and death
As stated above, in the well-studied retinoid cycle () (Citation138), the chromophore of visual pigments consists of 11-cis-retinal, the regeneration and the replenishment of which is mediated by the adjacent RPE cells. However, these elaborate pathways for pigment regeneration also carry potential risks, particularly when various enzymes, retinoid-binding protein and complex diffusional processes are involved.
Figure 6. A schematic showing the retinoid cycle. The chromophore (rhodopsin in this figure) consists of 11-cis-retinal and undergoes photoisomerization to all-trans-retinal. The role of ABCA4 is to actively flip N-retinylidene-phosphatidylethanolamine from the luminal side to cytoplasmic surface, where it reduced by the all-trans-retinol dehydrogenases (RDH) to all-trans-retinol. Together with that derived from the blood circulation, all-trans-retinol enters the RPE and is esterified by LART (lecithin: retinol acyltransferase) to generate all-trans retinyl esters. The retinyl ester can either be stored in the RPE cells or used as the substrate for the isomerohydrolase RPE65 to produce 11-cis-retinol, which can be later oxidized to 11-cis-retinal by NADP+, catalyzed by 11-cis-RDH. The 11-cis-retinal re-enters the outer segment of photoreceptors, where it assembles with opsin and regenerates rhodopsin. NADPH: nicotinamide adenine dinucleotide phosphate; RBP: retinoid-binding protein; TTR: transthyretin; RPE: retinal pigment epithelium; CRALBP: cellular retinol-binding protein.
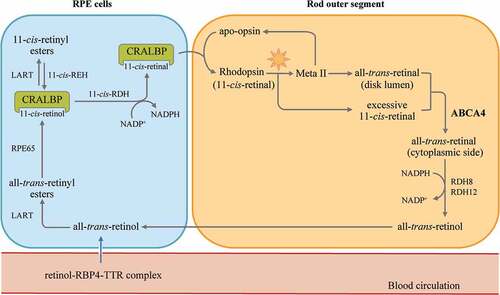
With the loss of ABCA4 transporter, the transportation and clearance of N-ret-PE is delayed and favors condensation between N-ret-PEs to generate bis-retinoids, such as N-retinylidene-phosphatidylethanolamine (APE), dihydro-N-retinylidene-N-retinylphosphatidylethanolamine (A2PE-H2) and its oxidized form (A2PE) (Citation99). By acid hydrolysis in RPE cells, non-degradable A2E is formed and progressively accumulated as lipofuscin deposits, either within the subretinal space or in the RPE cells (Citation102,Citation139).
Several studies have investigated the mechanisms of A2PE/A2E cytotoxicity. A2PE can serve as photosensitizer and itself can be photo-oxidized, contributing to photo-oxidative damage to photoreceptors (Citation140). A2E not only delays digestion of the lipid components of phagocytosed outer segments thus increasing the undigested substances in RPE (Citation141), it also increases the cytotoxicity of blue light (Citation142,Citation143). A highly reactive form of oxygen, superoxide, can be generated by irradiation of A2E, contributing to the further oxidation of A2E, resulting in photooxidative damage (Citation144). A recent study furthermore indicated that bis-retinoid compound accumulation has a role in complement system activation (Citation145). As a result, the nondegradable A2E fluorophores severely retard RPE phagocytotic and lysosomal functions, subsequently leading to RPE degeneration and loss of overlying photoreceptors.
Animal models developed to study the underlying pathogenesis of STGD1 have been available for some time (). However, differences of anatomy and disease phenotype between the mouse and human eye exist, and two important pathological features of STGD1 in humans, the accumulation of A2E in RPE cells and delayed dark adaptation, were observed in Abca4−/− animal models (Citation146–154). Other pathological features, including photoreceptor atrophy and RPE disturbance (Citation147), fundus autofluorescence (Citation147,Citation148) and relevant topical changes eg. oxidative stress response and compliment activation (Citation150–154) are also reproducible in animal models. Of note, the correlation between increased lipofuscin, mainly A2E and partly all-trans-retinal dimers (Citation155), and the increased intensity of fundus autofluorescence has been confirmed in Abca4−/− mice (Citation147,Citation156).
Table 2. Summary of current animal models for Stargardt disease
The accumulation of all-trans-retinal condensation products followed by accentuated RPE and photoreceptor degeneration at an early age were also observed in the dual Abca4 and Rdh8 knockout mouse model (Citation157). Moreover, the acute and light-induced retinopathy induced in the Abca4−/− Rdh8−/− mouse model indicates that the free all-trans-retinal rather than A2E condensation products appears to be the pathogenic factor in the retinopathy (Citation158). Consistently, Yu et al. found that under exposure of bright light, the increased all-trans-retinal detected in Abca4−/− Rdh8−/− mouse can induce rapid NADPH oxidase-mediated overproduction of intracellular reactive oxygen species (Citation159) that is involved in retinal dystrophy (Citation160).
Given that the macula is primarily affected in STGD1 patients, and mouse models are unable to fully recapitulate human STGD1 disease due to the lack of macula, recent dog models have bridged this gap. The Labrador retriever model carrying the homozygous loss-of-function mutation c.4176insC p.(Phe1393Leufs*1395) in ABCA4 presented with a clinical phenotype of photoreceptor degeneration, similar to human STGD1 (Citation161). In addition to the obvious autofluorescent lipofuscin in the RPE that was also observed in mouse models, abnormal rod and cone function as well as histopathological findings, i.e. RPE degeneration and thinning of outer nuclear layer were observed in the affected dog (Citation161). Further studies of this dog model may inform correlations among these observable pathological features, helping to explain the human STGD1 disease phenotype and support development of therapeutics as an ultimate goal.
Current therapeutic modalities for STGD1
The eye has many unique features that make it an attractive therapeutic target compared to other tissues. The optical transparency facilitates accessibility from the exterior and enables precise surgical delivery of therapeutic components to the targeted retinal cell layer. With the advances in retinal imaging techniques, non-invasive examination and functional assessment allow safe and repeated measurements of disease progression and therapeutic effects. Additionally, the relative isolation of the compartment and modified immune privilege make it possible to maintain effective therapeutic drug concentration, minimize systemic immune exposure and reduce potential toxicity to other organs that are not targeted (Citation162). This section will highlight therapeutics for ABCA4-STGD1 undergoing clinical trials.
Molecular therapies
A wide range of molecular approaches aimed at modulating secondary pathological pathways to overcome ABCA4 variant-induced metabolic dysfunction are under intense pre-clinical and clinical investigation. For example, a neuroprotection strategy is to add genes that supply naturally occurring neuroprotective factors that may prolong the lifespan of retinal cells rather than specifically addressing the mutated gene or disease pathogenesis (Citation163). Molecular pharmaceuticals under clinical evaluation for the treatment of ABCA4-STGD1 are shown in Supplemental .
Pharmacotherapy targeting vitamin A metabolism and transport
Overall, current pharmaceutical strategies lead to two approaches: generally inhibiting the retinoid cycle or intervening in the function of related transporters to decrease the formation of toxic bisretinoids. The first strategy utilizes retinoid cycle modulators to target key enzymes of the retinoid cycle and slow down chromophore regeneration, exemplified by Emixustat and isotretinoin; or disrupts the transporters of retinol, such as retinol-binding protein 4 (RBP4) antagonists including N-(4-hydroxyphenyl) retinamide (fenretinide), A1120, BPN-14136, STG-001 and LBS-008 (tinlarebant).
Emixustat hydrochloride (ACU-4429) is an orally administered small molecule that acts by inhibiting the retinoid cycle isomerase RPE65, the crucial enzyme involved in regeneration of chromophores (refer to ). By suppressing the conversion of all-trans-retinyl ester to 11-cis-retinol and concomitantly suppressing rod photoreceptor activity through decreased rhodopsin regeneration, Emixustat is thought to reduce accumulation of toxic lipid-retinoid by-products. In an early phase 1a study (Citation164), a dose-dependent suppression effect, manifested as slow recovery in rod-derived b-wave in ERG, was observed in the group treated with Emixustat. Since the suppression effect was reversible and returned to the baseline by day 7, oral administration on a daily basis was recommended. The subsequent phase 1b study (Citation165) exploring the pharmacokinetics, tolerability and safety of Emixustat that was orally administered into 40 healthy individuals revealed that systematic adverse events were minimal, while milder ocular adverse events comprising chromatopsia, followed by vision blur and reduction of visual acuity were reported in 67% of the subjects who received Emixustat doses 20 mg. However, these ocular-related side-effects were reversible and resolved within 7 to 14 days after completion of the study. In January 2017, the application of Emixustat was expanded to the treatment of STGD1, in two trials. The phase 2 clinical trial (NCT03033108) with 23 participants, aiming to study the pharmacodynamics and assess the safety of Emixustat during a one-month time frame has been completed, the outcome of which is yet to be released. Meanwhile, a phase 3 placebo-controlled trial (NCT03772665) has been activated, with the purpose to determine whether Emixustat can rescue the macular atrophy progression in subjects with STGD1.
Several other potential drugs for the treatment of STGD1 are under preclinical evaluation. Isotretinoin, a conventional drug for acne treatment, is one such drug. A side effect during acne treatment is that patients experienced delayed dark adaptation, due to its inhibitory effect on 11-cis-retinol dehydrogenase in RPE cells (Citation166). Such a side effect of slowing down rhodopsin regeneration however may reduce levels of A2E molecular precursor, thus reflecting the possible therapeutic effects in the correction of A2E accumulation (Citation167). Although isotretinoin has shown the ability to block A2E formation and lipofuscin accumulation in an Abca4−/− mouse model of STGD1 (Citation166), evidence for efficacy in human is currently lacking. Side effects, such as mild delays in dark adaptation (Citation166) and decreased night vision, observed in those under isotretinoin treatment for acne (Citation168) should be further evaluated before clinical trials in STGD1 patients.
Studies have shown that biosynthesis of bisretinoids depends on the influx of serum retinol (Citation169), and dietary supplementation with retinol increases lipofuscin accumulation in both liver and RPE cells of Abca4−/− mice (Citation152). Accordingly, methods of reducing the circulating retinol are hypothesized to reduce bisretinoid levels. After being secreted from the liver, retinol from the diet binds to specific retinol-binding protein 4 (RBP4) and transthyretin (TTR) and is transported to extrahepatic organs and tissues. The formation of a tertiary complex increases the molecular weight in circulation, therefore avoiding its loss through rapid glomerular filtration. Inhibition of the retinol-induced interaction of RBP4 and transthyretin (TTR) may compromise the uptake of serum retinol to the retina, thereby decreasing the formation of lipofuscin fluorophores (Citation170).
A synthetic retinoic acid analogue, fenretinide, or N-(4-hydroxyphenyl) retinamide (HPR), can efficiently displace retinol from RBP4 under physiological conditions, thereby accelerating the clearance of retinol in serum. In animal models, administration of fenretinide produced commensurate reduction in retinol level in both serum and retinoid cycle and lowered subsequent lipofuscin accumulation in RPE cells (Citation169). Although a modest delay in dark adaptation was observed in the study (Citation169), other physiological indicators such as light sensitivity of photoreceptors, 11-cis-retinal regeneration kinetics, and phototransduction process remained normal, suggesting that fenretinide may not interfere with retinoid cycle rates while reducing the accumulation of lipofuscin fluorophores as a therapeutic effects. However, fenretinide at higher concentrations was observed to induce apoptosis of human retinal pigment epithelial (ARPE-19) cells cultured in vitro, through RAR mediated generation of reactive oxygen species, expression of DNA damage-inducible transcription factor 153 and stress response protein (Citation171).
A1120 is another molecule that aims to lower the serum retinol levels. Like fenretinide, A1120 displaces retinol from RBP4, but does not act as an agonist to RAR, sparing patients from RAR-mediated side effects (Citation170). Nicoleta et al. observed that after oral administration of A1120 (30 mg/kg) to Abca4−/− mice for a period of 6 weeks, certain retinoid cycle retinoids were depleted and A2E accumulation decreased in A1120-treated group (Citation170). Unlike other retinoid cycle modulators, the capability of A1120 for reducing lipofuscin bisretinoid production in the retina is not associated with measurable suppression of the retinoid cycle, which shows the favorable safety profile of this non-retinoid RBP4 antagonist (Citation170). In addition to the therapeutic benefits as with A1120, BPN-14136, another non-retinoid RBP4 antagonist was able to normalize the dysregulated complement system in the retina of Abca4−/− mice after 12 weeks of oral administration at a daily basis (Citation172). No trials to date, however, involve these two RBP4 antagonists for the treatment of STGD1. In contrast, STG-001 is another RBP4 inhibitor which is currently in phase 2a trial comparing two doses for 28 days (NCT04489511). Similarly, LBS-008 (tinlarebant), another inhibitor of RBP4 has been given to healthy subjects in a phase 1 trial (ACTRN12618001823268).
Inhibitors of lipofuscin production
Reduction of toxic by-products can also be achieved by intervening in the chemical reactions underlying formation of condensed compounds. By replacing the C20 hydrogen atoms of vitamin A with deuterium, the binding strength of the C20 carbon-hydrogen bond of retinaldehyde-PE, a compound formed between retinaldehyde and PE is strengthened. Since the cleavage of the C20-H bond is the rate-limiting step in synthesis of vitamin A dimers, the deuterated form of vitamin A is therefore less able to form dimers and cytotoxic lipofuscin (Citation173). Ma et al. showed that compared with the control group fed vitamin A at its natural isotopic abundance, Abca4−/− mutant albino mice raised on diets containing C20-D3-vitamin A exhibited reduced A2E levels after three months (Citation155). In addition, decreased levels of lipofuscin granules were observed in Abca4−/− mice after treatment with C20-D3-vitamin A for 6 months, and the function of the retina improved after a year compared with untreated animals (Citation155). Similar therapeutic potential was also observed in another study, which further showed that administration of C20-D3-vitamin A normalized the dysregulated complement system without impairing retinal function (Citation174). After completing the phase 1 safety study of C20-D3-vitamin A in 40 healthy volunteers (NCT02230228), a phase 2 clinical trial (NCT02402660) involving STGD1 patients was initiated, aiming to determine its long-term safety and tolerability. At the time of submission, no results have been released from these trials.
Neuroprotective strategies
Neurotrophic agents delivered into the eye, particularly into the vitreous or subretinal space, may preserve photoreceptors and RPE cells. The factors include ciliary neurotrophic factor (CNTF), glial cell line-derived neurotrophic factor, brain derived neurotrophic factor, fibroblast and lens-epithelium-derived growth factors, pigment epithelium-derived factor, X-linked inhibitor of apoptosis, erythropoietin and its derivatives, heme oxygenase 1, superoxide dismutase and catalase, synthetic bile acids, progesterone, dopamine-based therapies and others (Citation163,Citation175–179). Among these factors, CNTF has had the fastest trajectory to clinical trials for the treatment of retinal degeneration.
After obtaining safety and efficacy data from animal studies (Citation180), a phase 1 clinical trial (Citation181) and two subsequent phase 2 trials (Citation182) using CNTF expressing cell implants for RP were conducted. While encapsulated cell implants, as a mode of administration have shown promise in terms of delivering viable cells secreting CNTF (Citation181), therapeutic benefits on visual acuity were not observed in RP patients, either short-term (Citation182) or years after the CNTF treatments (Citation183). Considering that the efficacy for treatments that delay photoreceptor degeneration may be questionable, use of neurotrophic agents also lack guidance regarding optimal dosing, timing, route of administration and combinations of neurotrophic agents. In addition, the exogenous properties inevitably raise safety concerns, and all of these issues have limited the further progression of neurotrophic agents in clinical trials.
As complement-associated inflammation contributes to the progression of STGD1 (Citation184), its modulation provides therapeutic potential. Subretinal injection of recombinant adeno-associated virus encoding complement receptor 1-like protein y (CRRY), a significant complement negative regulatory protein of mouse that prevents host cells from complement attacks by impeding the formation of the cytolytic membrane attack complex, has been shown to increase visual chromophore levels while reducing the level of bisretinoid in a STGD1 mouse model (Citation185). More importantly, photoreceptor degeneration was delayed in the treated mice. This result not only implicates inappropriate complement activation in the pathogenesis of STGD1 but also presents an alternative treatment for STGD1 and other retinal diseases associated with dysregulated complement signaling. The complement factor C5 inhibitor (Zimura) has recently entered clinical trials (NCT03364153) for safety and efficacy evaluation in STGD1 patients (Citation186). The primary outcome presented by the mean rate of change in the area of ellipsoid zone defect measured by en face SD-OCT within a time frame of 18 months was submitted, and the results are highly anticipated.
Saffron also works as a neuroprotectant. The constituents, crocetin and crocin, have been suggested to counteract retinal oxidative damage and protect retinal cells from apoptosis (Citation187). In a clinical trial (NCT01278277) of oral saffron versus placebo for six months, saffron was well tolerated among the 31 recruited patients with ABCA4-STGD1, but no measurable improvement was recorded, warranting a long-term study to evaluate therapeutic effects on the progression of retinal dystrophy in STGD1 (Citation187).
Preclinical studies aiming to find a treatment for STGD1 are emerging, including retinylamine and its derivatives (Citation188,Citation189), G protein-coupled receptor modulators (Citation190) and dietary supplements (Citation191), and have been reviewed by other authors (Citation192,Citation193).
ABCA4 gene- or mutation-specific therapies
With the broad aim of introducing therapeutic nucleic acids into targeted cells, gene therapy approaches can be classified into (Citation1) gene replacement that introduces a functional copy of the causative gene into affected cells, without addressing the mutation itself and is generally applicable for diseases caused by loss-of-function mutations (Citation194; Citation2) gene editing utilizes highly specific nucleases to repair the pathogenic mutation in the endogenous affected allele, with the aim of restoring the wild-type DNA sequence and functional protein expression (Citation195; Citation3) splice modulation that induces alternatively spliced transcripts via administration of antisense oligonucleotides or specialized small molecules. By targeting selected splicing motifs as needed, a disrupted reading frame due to mutations can be restored, or exons carrying disease-causing mutations can be removed during pre-mRNA splicing process, with the aim to generate functional protein (Citation162).
Gene replacement therapies
There are presently two major vector platforms for gene replacement therapy, viral and non-viral. A number of viral vectors have demonstrated tropism for specific ocular cells in tissue culture and animal models (Citation196), among which, adenovirus, adeno-associated virus and lentivirus are the most common vectors used to deliver therapeutic cargoes. Unlike viral systems, non-viral delivery approaches involve administration of naked DNA, or in combination with chemical substances such as cationic lipids, peptides, polymers and nanoparticles, or physiochemical methods such as electroporation, iontophoresis and microinjection (Citation197).
The minimal functional size of ABCA4 cDNA is 6.8 kb and hence exceeds the cargo capacity of recombinant adeno-associated virus (rAAV, ≤ 4.7 kilobases). For years, efforts have focused on artificially expanding AAV packaging capacity. In 2008, a landmark publication reported that expression of murine Abca4 protein was localized to rod outer segments following transduction with recombinant AAV2/5 packaging a large expression cassette containing single-stranded Abca4 (8.9 kilobases) (Citation198). Furthermore, subretinal delivery of the vector in the Abca4−/− mouse model resulted in significant reduction of lipofuscin granules (mainly fluorophore A2E) accumulated in RPE, and the ability of photoreceptors to recover from light desensitization was significantly improved (Citation198).
Although three independent research groups later noticed the discrepancy between heterogeneously partially packaged sequences (no larger than 5.2 kilobases) and full gene expression cassettes (> 5 kilobases) (Citation199–202), the ability of ABCA4 gene fragments to recombine once inside the target cells later inspired dual AAV vector gene replacement approaches. In this way, ABCA4 gene fragments are assembled through inverted terminal repeat-mediated concatemerization (the trans-splicing approach) (Citation203), homologous overlapping sequence-mediated extension (the overlapping approach) (Citation204), or a combination of the two (the dual hybrid AAV approach) (Citation205). Trapani and colleagues have compared the efficiency of dual AAV strategies and AAV oversize vectors to deliver EGFP, ABCA4 and MYO7A in vitro, as well as in the retina of mouse and pig (Citation206). In their study, dual AAV strategies outperform AAV oversize vectors in terms of transduction levels both in vitro and in vivo. Dual AAV trans-splicing vectors and AAV hybrid vectors containing alkaline phosphatase recombinogenic sequence efficiently transduced both photoreceptors and RPE cells, albeit less efficiently compared to the single normal-sized AAV vectors. However, only dual AAV overlapping vectors efficiently transduced mouse RPE cells rather than photoreceptors. Notably, subretinal delivery of dual AAV trans-splicing and hybrid vectors improves the retinal defects of the Abca4−/− mouse model, with neither electroretinography (ERG) nor retinal histological abnormalities detected during 3–8 months follow-up post-treatment. Other studies also reported efficient transduction using dual AAV vectors in photoreceptors of Abca4−/− mouse and pigs (Citation207–209), and methods to improve transduction efficiency of vectors delivering ABCA4 have been discussed elsewhere (Citation208,Citation210).
Lentiviral vectors have gained attention for the delivery of large genes such as ABCA4 due to its ~8 kb cargo capacity. Kong et al. showed that delivery of wild-type ABCA4 cDNA via subretinal injection of equine infectious anemia virus (EIAV)-derived lentiviral vectors into newborn Abca4−/− mice reduced A2E accumulation (Citation211). Although the transduction efficiency of photoreceptors in the injected area was low (from 5%-20%), ERG evaluation indicated that the treated group benefited from subretinal injection of the ABCA4 lentiviral vectors, suggesting that this level of photoreceptor transduction was sufficient to rescue phenotypes in the Abca4−/− mouse model (Citation211). Another preclinical safety study demonstrated that a single injection of EIVA-derived lentiviral vector is safe and well-tolerated in rabbits and macaques, though a slight and transient cellular inflammatory response was observed in the treated eyes (Citation212). A phase 1/2 clinical trial (NCT01367444) employing EIAV-derived lentivirus pseudo-typed using vesicular stomatitis virus glycoprotein (drug SAR422459) for subretinal delivery of ABCA4 cDNA was been terminated early this year, and no efficacy data was published. Its phase 1/2 follow-up trial (NCT01736592) aiming to evaluate the long-term tolerability of SAR422459 in patients with STGD1 is activate, but yet to recruit patients (Supplemental ). Non-viral delivery of exogenous nucleic acids, either naked or conjugated with chemical carriers, is an alternative approach that theoretically should allow the delivery of much larger nucleotide fragments without triggering severe immune responses, compared to viral administration. Han et al. delivered a DNA construct compacted with polyethylene glycol-substituted 30-mer lysine peptides (CK30-PEG) carrying the human ABCA4 cDNA (6.8 kilobases) to the subretinal region in eyes of Abca4−/− mice (Citation213,Citation214). Transgene expression was persistent for up to 8 months, and both functional (delayed dark adaptation indicated by ERG) and structural (fundus lipofuscin accumulation) phenotypes of STGD1 were significantly improved (Citation213,Citation214). This study provided the first evidence of non-viral delivery of human ABCA4 to photoreceptors.
Beneficial features of the compacted nanoparticles have been discussed in various studies, including superior tolerance even after repeated injection (Citation215), persistent gene expression (Citation216), the absence of adverse events such as ocular/systemic toxicity and insertional mutagenesis (Citation217,Citation218), and absence of significant local inflammatory responses or toxicity (Citation219).
Gene editing
In contrast to gene replacement therapy, gene editing targets the mutant gene in order to restore the wild-type sequence. Gene editing technologies exploit programmable nucleases including Meganucleases (Citation220), transcription activator-like effector nucleases (TALEN) (Citation221), zinc finger nucleases (ZFN) (Citation222) or clustered regularly interspaced short palindromic repeats (CRISPR)–associated nuclease Cas9 (Citation223). The recent developments of CRISPR-Cas9 have revolutionized various fields of biotechnology and biomedicine. Since first utilized in the early-1990s (Citation224), the highly specific endonuclease platform has been developed to enable precise genome editing by introducing DNA double stand breaks (DSBs), single-strand breaks (nicks) (Citation225) or base editing (Citation226).
Overall, the development of gene editing for retinal disease splits in two directions. In the ex vivo approach, gene editing is performed in patient-derived cells to correct mutations, followed by reprogramming cells into iPSCs and differentiating to RPE or photoreceptors for subsequent transplantation. In contrast, the in vivo approach is to target mutations in retinal cells in situ (Citation195,Citation227–231) through delivery of editing machinery and repair templates using the gene delivery vectors discussed above. Once all elements, such as endonucleases, the template of the gene of interest and various modifying agents, are delivered to the target cells via intravitreal or subretinal injection, DSBs can be induced at the targeted genomic locations by endonucleases. Subsequently, endogenous host proteins detect and repair the DSB through one of the three different pathways: the error prone non-homologous end-joining (NHEJ) pathway, or with high fidelity through homology directed repair (HDR), or microhomology mediated end-joining pathway (MMEJ) (Citation195). NHEJ, during the whole cell cycle, repairs the lesion by directly reconnecting the two DSB ends in a process that does not require templates, whereas HDR requires an exogenous DNA template with sequence homology to the lesion site to integrate into the DSB during repair in the S- and G2- phase of the cell cycle (Citation232). Although NHEJ-mediated DSB repair can be rapid and accurate, it can result in small deletions, insertions or substitutions that may cause frameshifts, leading to mRNA degradation or production of non-functional truncated proteins (Citation227). MMEJ, as an alternative pathway to the classic NHEJ, aligns the micro-homologous sequence with the DSB ends for repairing small deletions, insertions and chromosome translocations (Citation233). Komor et al. engineered fusions of CRISPER/Cas9 and a cytidine deaminase that can mediate the direct conversion of cytidine to uridine without cleavage of double-stranded DNA (Citation226). This “base editor” converts cytidines within a window of approximately five nucleotides and has shown editing efficiencies of 15–75% with typically ≤ 1% insertion/deletion formation in mammalian cell lines. Soon afterwards, a new class of adenine base editors (ABEs) was developed to convert A or T to G or C in DNA, with approximately 50% efficiency and ≤ 1% incidence of insertion/deletion in human cells (Citation234).
Gene editing is contingent upon the safe and efficient viral- or non-viral delivery of DNA, RNA or protein to the target of interest. Before processing to clinical development as a treatment for ABCA4-related retinopathy, gene editing faces several hurdles including inefficient delivery of the expression cassette, and integration of endonucleases and the repair template. The potential adverse and off-target effects must also be considered. Of note, recent studies point out human pluripotent stem cells with a functional TP53 gene (encoding P53) have severely reduced efficiency of precise genome editing (Citation235), and such efficiency can be improved by p53 inhibition (Citation235,Citation236). Given that P53 inhibition may expose targeted cells to severe adverse events, such as chromosomal rearrangement, off-target mutations and tumorigenic mutations, both risks and benefits must be cautiously evaluated when developing CRISPR-Cas9-mediated therapies (Citation235,Citation236).
Splice modulation methods
Antisense oligonucleotide (AONs) mediated splice modulation is gaining increasing attention as a strategy to overcome several specific disease-causing mutations. AONs are short (8–50 nucleotide) single-stranded nucleic acids or nucleic acid analogues, complementary to the target sequence, according to the Watson–Crick base pairing principle (Citation162). The use of AONs has been expanded to invoke various mechanisms to modify gene expression at the mRNA level, including 1) induce exon skipping to bypass nonsense mutations and mutations that cause reading frame shift; 2) block cryptic splice sites that are abnormally activated; 3) impede splice silencers located near exons in such a way as to enhance exon recognition (Citation237).
Gérard et al. have provided in vivo evidence generated from mouse that intravitreal injection of 10 nanomoles of Abca4-specific AONs targeting the exonic splice enhancer near exon 10 resulted in a shortened transcript, lacking a 104 bp sequence that partially overlaps exon 9 and 10 (Citation238). The splicing-modulation effect was maintained for 10 days after treatment (Citation238). Albert and Garanto et al. reported that in STGD1 patient-derived photoreceptor progenitor cells, AON administration can prevent inclusion of the aberrantly activated 345-nucleotide pseudo-exon caused by two neighboring deep-intronic ABCA4 mutations (c.4539+2001G>A and c.4539+2028C>T) (Citation129,Citation130). Other in vitro splice intervention studies also demonstrated the ability of AONs to eliminate pseudo-exons caused by intronic ABCA4 mutations (Citation132,Citation135). A clinical trial employing AONs to target the deep-intronic mutation of CEP290 was initiated (NCT03140969). Recently published outcomes showed vision improvement without adverse events after intravitreal injection of AONs (Citation239). As increasing numbers of splice-affecting variants in ABCA4 are being revealed, AON intervention is anticipated to yield additional promising therapeutic outcomes by addressing aberrant splicing events. However, the drawback of AON strategies is the lack of one-drug-fits-all approach and individualized AON design will be required for many mutations, therefore limiting the broad application of AONs-mediated therapy to patients with uncommon mutations (Citation131). On the other hand, AONs are offering new hope to patients with rare diseases, with the recent FDA approval of “Milasen” for the treatment of a rare form of Batten’s disease, occurring within record breaking time (a year from the laboratory to get to the patients) (Citation240), highlighting AON intervention as potential precision medicine for inherited retinal diseases.
Cell replacement, neuromodulation and bionic vision
Retinal pigment epithelium and photoreceptor transplantation
Due to the extreme lack of donor cell resources and post-transplant safety issues, the development of RPE and photoreceptor cells transplantation as therapeutics has been limited. Pluripotent stem cells developed in recent decades hold great potential for the treatment of retinal degenerative diseases. Current promising cell transplantation strategies that underwent clinical trials relied on two donor cell sources, human embryonic stem cells (hESCs) and somatic cell reprogrammed induced pluripotent stem cells (iPSCs) (Supplemental Table 3).
For effective RPE cell replacement therapy, key features of RPE in terms of gene and protein expression profiles as well as morphological, behavioral and physiological capabilities must be achieved. Both hESC- and iPSC-derived RPE cells are well suited for large-scale production of human RPE cells that display the morphological and functional similarities of primary RPE cells. These cells grow as monolayers comprised hexagonal, pigmented cells with apical microvilli (Citation241–243), present correct apical-basal polarity with secretion of growth factors (Citation244–246) and are capable of phagocytosis of outer segments (Citation242,Citation247–249). The gene expression profiles of ESC- and iPSC-derived RPE cells are similar to adult primary RPE cells (Citation248,Citation250), though ESC-derived RPE cells show variability in the expression of adhesion junction and membrane transport genes (Citation251). Variability between human ECS- and iPSC-derived RPE is also seen in the genomic DNA methylation patterns, which is dynamically regulated during RPE differentiation (Citation252), and in addition, iPSC-derived RPE cells may retain the epigenetic markers associated with the cell types they were originally derived from (Citation253).
The properties and functions of transplanted ESC- and iPSC-derived RPE cell have been investigated in animal models. Transplanted cells retain morphological and functional features similar to native RPE-like tissues in vivo, such as “cobblestone” morphology with pigmentation and the ability of phagocytosis capability (Citation254–257). Maintenance of photoreceptors and visual function has been observed in rat models, in which stem cell-derived RPE cells were transplanted under the retina (Citation257,Citation258).
Translational application of human ESCs-derived cells is now being transitioned to phase 1/2 clinical trials (Supplemental Table 3). The first priority of these clinical trials concern safety, including assessments of serial vital signs and adverse events; monitoring of transplantation tolerance, integrity and rejection; local and systemic infection; and tumorigenic transformation. In 2005, the preliminary results of a clinical study (NCT01345006) aiming to evaluate safety and tolerability of subretinal injection of human ESCs-derived RPE in patients with STGD1 were released (Citation259). During a median follow-up observation period of 22 months (up to 37 months), the data showed increased and stable best-corrected visual acuity as well as improved vision-related quality-of-life measures (Citation260). No signs of hyperproliferation, tumorigenicity, ectopic tissue formation, transplant rejection or inflammation were observed. A phase 1/2 trial (NCT01469832) involving 12 patients with advanced STGD1 attempted to study the retinal structure and function in the treated region after subretinal transplantation of human ESC-derived RPE cells. No obvious benefit was observed in best-corrected visual acuity and microperimetry, and hyperpigmentation of focal areas and localized retinal thinning and sensitivity reduction may suggest potential harm arising from the treatment (Citation261). Although the evidence to date suggested many outstanding merits, the ethical controversies surrounding the use of hESC have hampered their widespread investigation. Moreover, despite hESC and its differentiated derivatives being less susceptible to immune rejection (Citation262), patients who received the allograft transplantation, differentiated from stem cells, may also need to take lifelong immunosuppressive therapies.
The loss of photoreceptors is one of the pathological features of STGD1. Therefore, replacement of these cells by transplantation could offer a potential treatment. For effective photoreceptor transplantation, a precise host synaptic connection with the transplanted photoreceptors must be established (Citation263), in order to restore the light sensitive function of the recipient retina. Among various cell transplantation approaches that have been investigated during the last three decades, transplantation of photoreceptor precursor cells is most promising.
Precursor cells have been obtained from postnatal mouse retina or isolated from retinal organoids derived from hESC and iPSC and transplanted in retinal degeneration animal models (Citation264). Precursor cells sourced from early postnatal mouse were capable of integrating into the outer nuclear layer of adult retina, forming synaptic connections, and differentiating to acquire specialized features possessed by mature rods as well as restoring some visual function (Citation265). Integration of up to 26,000 rods taken from postnatal Nrl-GFP donor mice (Citation266) were distributed across more than 50% of the retina in the Gnat1−/− mouse model (Citation267), and light responses of transplanted photoreceptors were also recorded (Citation267). The improvement of some other visual activities after transplantation of photoreceptor precursor cells was reported in other studies (Citation268–270). These preclinical outcomes indicate the feasibility of photoreceptor as a potential treatment for retinal dystrophies.
The technology of in vitro expandable pluripotent stem cells would further facilitate scalable generation of specific cell types, including RPE cells, photoreceptors and 3D retinal organoids. Protocols for derivation of specific retinal cells and retinal organoids from pluripotent stem cells have been reviewed elsewhere (Citation264,Citation271). Efforts in recent decades have led to the accumulation of new knowledge on pluripotent stem cells, bringing hope for the application of these cells in clinical development.
Cell-based preservation therapy
Numerous types of cells have been investigated for their capacity to promote the survival of retinal cells through their paracrine trophic effect, including mesenchymal stem cells, vascular precursor cells, adipose stromal cells and neural stem/progenitor cells derived from various sources (Citation241,Citation272,Citation273). Nonetheless, only few of these strategies have been clinically tested on patients with retinopathy or ophthalmic syndromes.
CD34+ haematopoietic stem cells have been explored as therapeutics in animal models of retinal ischemia and degeneration. A landmark observation unveiled that systemic circulation makes these cells to be recruited in response to tissue injury, thus facilitating tissue regeneration and angiogenesis (Citation274). Preclinical data indicate that the preservative therapeutic effects of CD34+ cells also depend on their paracrine trophic effects, by secreting neurotrophic and proangiogenic factors (reviewed by Susanna et al (Citation275)). A phase 1 trial using autologous bone marrow-derived CD34+ cells is currently being tested as a preservation treatment involving STGD1 patients (NCT01736059). Although the number of participants of this trial being extremely limited (only 2 STGD1 patients), preliminary results indicated that the intravitreal injection of autologous bone marrow-derived CD34+ cells was well tolerated by the patients. Over 18 months, no ocular or systemic adverse effects or hyperproliferation were observed, suggesting the safety and feasibility of this approach (Citation276). However, a recent report highlighted the risk of adipose stem cells implantation (Citation277). In this report, three patients with AMD experienced severe bilateral visual loss, associated with serious complications including ocular hypertension, haemorrhagic retinopathy, vitreous haemorrhage, combined traction, rhegmatogenous retinal detachment, or lens dislocation, after they received intravitreal injection of autologous adipose tissue-derived “stem cells.” The visual acuity of the three patients ranged from 20/200 to no light perception after one year of transplantation.
It is worth mentioning that an appreciable number of functional photoreceptors are necessary for both cell-based replacement and preservation therapies. Either transplanting RPE cells too early (host RPE is still predominant) or too late (host RPE has gone) may lead to potential complications (Citation241). It has also been suggested that genotypes of patients, as well as retinal structural evidence measured by optical coherence tomography and en face autofluorescence, should be considered as the inclusion criteria in cell transplantation trials (Citation278).
Optogenetics
For patients with nearly complete loss of photoreceptors, there is very little chance of benefit from other treatments such as neuroprotective strategies and mutation-specific therapies. In such case, optogenetics can provide an alternative therapeutic strategy (Citation279). Optogenetics is generally based on the idea that, by genetically introducing light-sensitive protein (mainly opsin-based protein) into cell membrane, those inner retinal neurons that are intrinsically light insensitive and temporarily exempted from the primary damage in retinal diseases can be artificially photosensitized. The optogenetics toolkit at its present stage comprises two distinct families: animal opsin and microbial opsin. Lin et al. reported that expression of the light-sensitive protein melanopsin in retinal ganglion cells, a type of retinal neuron that is intrinsically photosensitive yet contribute little to vision, can enhance visual function regarding behavioral light avoidance response and discrimination of light stimulus in rd1 mice (Citation280). Later, Cehajic-kapetanovic et al. ectopically expressed human opsin in the rd1 mouse model and effectively restored the visual responses induced by simple light pulses, luminance increases and naturalistic movies, and behavioral responses to light pulses at the intensity levels as the natural indoor environment (Citation281). Though the reliability of these promising results obtained in animal models await further evaluation in humans, vision restoration through light stimulation including optogenetics is rapidly evolving from an idea into reality (Citation282).
Microbial opsin, compared to animal opsins, owns less complexity and requires a lesser collaborative neuron network for photoresponses. Microbial opsins are usually light-activated ion channels or pumps, such as channelrhodopsin, halorhodopsin, bacteriorhodopsin and proton pumps (comprehensively reviewed in (Citation282)). Based on the very first report showing that ectopic expression of channelrhodopsin-2 in the residual inner retinal neurons in photoreceptor-deficient rd1 mice facilitated the restoration of vision (Citation283), further advancement of optogenetics has aimed at inducing specific expression of opsins in selected target cell types. Busskamp et al. reported that expression of archaebacterial halorhodopsin can replace the native phototransduction cascade and reactivate light-insensitive cones in mouse model of RP (Citation284). The resensitized photoreceptors were able to activate retinal cone pathways and drive the sophisticated secondary retinal circuit functions (Citation284). By using human ex vivo retina, experiments further showed that defective photoreceptors can regain light sensitivity with the expression of halorhodopsin (Citation284). It is also suggested that patients with cones, light-insensitive but with cell bodies remaining in the central area, can be considered as candidates for the corresponding therapy (Citation284).
Varying degrees of visual improvement were also observed in other studies that performed optogenetic stimulations of inner retinal neurons in animal models of retinal dystrophy (Citation285,Citation286). At present, the translation of optogenetics combined with gene therapy to the clinical realm is in progress for RP (NCT02556736 and NCT03326336). Last year, Garita-Hernandez reported that after transplanting optogenetically transformed photoreceptor precursor cells, partial visual function was restored in blind mice (Citation287). Additionally, the transplantation of cones differentiated from human iPSCs, expressing the chloride pump, contributed to light-driven responses in both photoreceptors and ganglion cells in blind mice, indicating that by integration of stem cell technology and optogenetics, structure and function of retina can be restored (Citation287).
Artificial retinal prosthesis
Similar to optogenetics, retinal prosthesis is another strategy that stimulates inner retinal neurons that remain functional in cases with demise of photoreceptors and RPE cells, thereby enabling the signals to be transmitted along the intact visual pathway to the corresponding cortex for processing. The difference is that retinal prostheses work by electrical stimulation. According to the location of the implants, retinal prosthetics can be categorized into epiretinal prosthetics, subretinal prosthetics and suprachoroidal prosthetics. The advances of each prostheses in the related retinal research, with particular emphasis on the engineering and clinical specifics has been covered elsewhere (Citation288). Another review discussed the principles and challenges of each bioelectrical approach (Citation282).
At present, the most successful device is the Argus II Retinal Prosthesis System. With the approval from Conformité Européenne in 2011 and the U.S. Food and Drug Administration in 2013, worldwide commercial implantation of Argus II has commenced in patients with retinal diseases, including RP and to a lesser extent, AMD and choroideremia (Citation288). The preclinical animal and human studies that led to the development of Argus II and the outcomes of these studies in terms of safety profile and function improvement have been comprehensively reviewed (Citation289). Recently, Endo et al. reported that after implantation with a suprachoroidal-transretinal stimulation (STS) retinal prosthesis in the right eye of a patient with advanced STGD1 with residual vision, the performance of a targeted reaching movement was consistently improved during one-year follow-up period (Citation290). This study brings hope for integrating retinal prosthesis with the treatment for STGD1.
Conclusion remarks and future directions
STGD1 is currently incurable, and once diagnosed, is progressive and irreversible. The large variability in phenotype and disease progression rates even among family members carrying the same mutations remain a major obstacle for clinical trial design and patient selection for clinical trials. The therapies currently applied towards the treatment of STGD1 attempt to delay or halt vision loss in patients, but no treatment has been proven to restore vision. The search for effective and safe therapeutic options has led to the development of two branches of studies aiming to model human STGD1. One is the development of animal models, and the other one is to isolate the appropriate cell lines of interest. The currently available animal models cannot fully mimic human STGD1 (Citation291). Thus far, some of the potential therapeutic strategies such as gene therapy, cell-based therapeutic platforms and pharmaceuticals as discussed in this review have shown great promise, though various hurdles remain to be addressed in order to achieve safe and effective therapies. In this context, the uncertainties such as the impact of high mutational heterogeneity of ABCA4 for certain therapies, and difficulties including the lack of consensus in selecting the optimal treatment window, administration route, sites and dosages; the lack of a stronger evidence for the molecular mechanisms underlying ABCA4-associated STGD1, based on animal models or cell lines; the lack of sufficient clinical evidence supporting therapeutic longevity, safety and efficacy; and the lack of long term observation of side effects or potential deleterious events during treatment etc., have hindered progress and require ongoing efforts of generations of researchers.
To conclude, it is important to reflect on several key points. Firstly, the cautious pathogenicity assessment of ever-increasing numbers of ABCA4 variants and a better understanding of their molecular consequences is essential for developing correlations between genotype and phenotype, as well as exploring mutation mechanism-dependent treatment strategies, regardless of whether they are targeting the cause or the consequences of STGD1. In addition, the combination of current therapeutic options for STGD1 with the current advanced ophthalmological surgery techniques could be the key to increasing the success in maintaining, or ideally restoring visions in patients with STGD1. Last but not least, in order to achieve optimal maintenance or improvement of vision, attempts to develop neuroprotective treatments for STGD1 using mutation-dependent and/or -independent strategies should continue to be investigated. In addition to their therapeutic effects, the safety profiles of these treatments for various phenotypes of STGD1 warrant careful consideration, given the large number of mutations and the varied impact of these on ABCA4 splicing and ABCA4 protein function.
Acknowledgments
We thank the support from the Australian Inherited Retinal Disease Registry and DNA bank. We thank Wiley publisher for allowing us to use a figure modified from previous publication.
Supplementary Material
Download PDF (192.3 KB)Supplemental data
Supplemental data for this article can be accessed on the publisher’s website.
Disclosure Statement
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Additional information
Funding
References
- Allikmets R, Singh N, Sun H, Shroyer NF, Hutchinson A, Chidambaram A, Gerrard B, Baird L, Stauffer D, Peiffer A, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997;15(3):236–46. doi:10.1038/ng0397-236.
- Armstrong JD, Meyer D, Xu S, Elfervig JL. 1998. Long-term follow-up of Stargardt’s disease and fundus flavimaculatus. Ophthalmology. 105(3):448–58. doi:10.1016/S0161-6420(98)93026-3
- Fujinami K, Lois N, Mukherjee R, McBain VA, Tsunoda K, Tsubota K, Stone EM, Fitzke FW, Bunce C, Moore AT, et al. A longitudinal study of Stargardt disease: quantitative assessment of fundus autofluorescence, progression, and genotype correlations. Invest Ophthalmol Vis Sci. 2013;54(13):8181–90. doi:10.1167/iovs.13-12104.
- Heath Jeffery RC, Mukhtar SA, McAllister IL, Morgan WH, Mackey DA, Chen FK. Inherited retinal diseases are the most common cause of blindness in the working-age population in Australia. Ophthalmic Genet. 2021;1–9. doi:10.1080/13816810.2021.1913610.
- Simonelli F, Testa F, Zernant J, Nesti A, Rossi S, Allikmets R, Rinaldi E. 2005. Genotype-phenotype correlation in Italian families with Stargardt disease. Ophthalmic Res. 37(3):159–67. doi:10.1159/000086073.
- Walia S, Fishman GA. 2009. Natural history of phenotypic changes in Stargardt macular dystrophy. Ophthalmic Genet. 30(2):63–68. doi:10.1080/13816810802695550.
- Anderson KL, Baird L, Lewis RA, Chinault AC, Otterud B, Leppert M, Lupski JR.A YAC contig encompassing the recessive Stargardt disease gene (STGD) on chromosome 1p. Am J Hum Genet. 1995;57(6):1351–63.
- Fishman GA.Fundus flavimaculatus. A clinical classification. Arch Ophthalmol. 1976;94(12):2061–67.
- Cukras CA, Wong WT, Caruso R, Cunningham D, Zein W, Sieving PA. 2012. Centrifugal expansion of fundus autofluorescence patterns in Stargardt disease over time. Arch Ophthalmol. 130(2):171–79. doi:10.1001/archophthalmol.2011.332
- Sparrow JR, Marsiglia M, Allikmets R, Tsang S, Lee W, Duncker T, Zernant J. 2015. Flecks in Recessive Stargardt Disease: short-Wavelength Autofluorescence, Near-Infrared Autofluorescence, and Optical Coherence Tomography. Invest Ophthalmol Vis Sci. 56(8):5029–39. doi:10.1167/iovs.15-16763
- McBain VA, Townend J, Lois N. 2012. Progression of retinal pigment epithelial atrophy in stargardt disease. Am J Ophthalmol. 154(1):146–54. doi:10.1016/j.ajo.2012.01.019
- Klufas MA, Tsui I, Sadda SR, Hosseini H, Schwartz SD. 2018. ULTRAWIDEFIELD AUTOFLUORESENCE IN ABCA4 STARGARDT DISEASE. Retina. 38(2):403–15. doi:10.1097/iae.0000000000001567
- Fakin A, Robson AG, Fujinami K, Moore AT, Michaelides M, Pei-Wen Chiang J, Webster GEH. 2016. AR. Phenotype and Progression of Retinal Degeneration Associated With Nullizigosity of ABCA4. Invest Ophthalmol Vis Sci. 57(11):4668–78. doi:10.1167/iovs.16-19829
- Lois N, Holder GE, Bunce C, Fitzke FW, Bird AC.Phenotypic subtypes of Stargardt macular dystrophy-fundus flavimaculatus. Arch Ophthalmol. 2001;119(3):359–69.
- Fujinami K, Zernant J, Chana RK, Wright GA, Tsunoda K, Ozawa Y, Tsubota K, Robson AG, Holder GE, Allikmets R, et al. Clinical and molecular characteristics of childhood-onset Stargardt disease. Ophthalmology. 2015;122(2):326–34. doi:10.1016/j.ophtha.2014.08.012.
- Huang D, Thompson JA, Charng J, Chelva E, McLenachan S, Chen SC, Zhang D, McLaren TL, Lamey TM, Constable IJ, et al. Phenotype-genotype correlations in a pseudodominant Stargardt disease pedigree due to a novel ABCA4 deletion-insertion variant causing a splicing defect. Mol Genet Genomic Med. 2020;8(7):e1259. doi:10.1002/mgg3.1259.
- Al-Ani HH, Sheck L, Vincent AL. Peripheral pigmented lesions in ABCA4-associated retinopathy. Ophthalmic Genet. 2021;1–9. doi:10.1080/13816810.2021.1897850.
- Arrigo A, Grazioli A, Romano F, Aragona E, Marchese A, Bordato A, Di Nunzio C, Sperti A, Bandello F, Parodi MB. 2020. Multimodal evaluation of central and peripheral alterations in Stargardt disease. A Pilot Study Br J Ophthalmol. 104(9):1234–38. doi:10.1136/bjophthalmol-2019-315148
- Strauss RW, Muñoz B, Wolfson Y, Sophie R, Fletcher E, Bittencourt MG, Scholl HPN. 2016. Assessment of estimated retinal atrophy progression in Stargardt macular dystrophy using spectral-domain optical coherence tomography. Br J Ophthalmol. 100(7):956–62. doi:10.1136/bjophthalmol-2015-307035
- Velaga SB, Nittala MG, Jenkins D, Melendez J, Ho A, Strauss RW, Scholl HP, Sadda SR. 2019. Impact of segmentation density on spectral domain optical coherence tomography assessment in Stargardt disease. Graefes Arch Clin Exp Ophthalmol. 257(3):549–56. doi:10.1007/s00417-018-04229-3
- Kugelman J, Alonso-Caneiro D, Chen Y, Arunachalam S, Huang D, Vallis N, Collins MJ, Chen FK. 2020. Retinal Boundary Segmentation in Stargardt Disease Optical Coherence Tomography Images Using Automated Deep Learning. Transl Vis Sci Technol. 9(11):12. doi:10.1167/tvst.9.11.12
- Charng J, Xiao D, Mehdizadeh M, Attia MS, Arunachalam S, Lamey TM, Thompson JA, McLaren TL, De Roach JN, Mackey DA, et al. Deep learning segmentation of hyperautofluorescent fleck lesions in Stargardt disease. Sci Rep. 2020;10(1):16491. doi:10.1038/s41598-020-73339-y.
- Cicinelli MV, Rabiolo A, Brambati M, Viganò C, Bandello F, Battaglia Parodi M. 2020. Factors Influencing Retinal Pigment Epithelium-Atrophy Progression Rate in Stargardt Disease. Transl Vis Sci Technol. 9(7):33. doi:10.1167/tvst.9.7.33.
- Jauregui R, Nuzbrokh Y, Su P-Y, Zernant J, Allikmets R, Tsang SH, Sparrow JR.Retinal Pigment Epithelium Atrophy in Recessive Stargardt Disease as Measured by Short-Wavelength and Near-Infrared Autofluorescence. Transl Vis Sci Technol. 2021;10(1):3.
- Müller PL, Birtel J, Herrmann P, Holz FG, Charbel Issa P, Functional Relevance GM. 2019. Structural Correlates of Near Infrared and Short Wavelength Fundus Autofluorescence Imaging in ABCA4-Related Retinopathy. Transl Vis Sci Technol. 8(6):46. doi:10.1167/tvst.8.6.46.
- Fujinami K, Lois N, Davidson AE, Mackay DS, Hogg CR, Stone EM, Tsunoda K, Tsubota K, Bunce C, Robson AG, et al. A longitudinal study of stargardt disease: clinical and electrophysiologic assessment, progression, and genotype correlations. Am J Ophthalmol. 2013;155(6):1075–88. doi:10.1016/j.ajo.2013.01.018.
- Simonelli F, Testa F, de Crecchio G, Rinaldi E, Hutchinson A, Atkinson A, Dean M, D’Urso M, New AR.ABCR mutations and clinical phenotype in Italian patients with Stargardt disease. Invest Ophthalmol Vis Sci. 2000;41(3):892–97.
- Fakin A, Robson AG, Chiang JP, Fujinami K, Moore AT, Michaelides M, Holder GE, Webster AR. 2016. The Effect on Retinal Structure and Function of 15 Specific ABCA4 Mutations: a Detailed Examination of 82 Hemizygous Patients. Invest Ophthalmol Vis Sci. 57(14):5963–73. doi:10.1167/iovs.16-20446.
- Pfau M, Holz FG, Müller PL. 2021. Retinal light sensitivity as outcome measure in recessive Stargardt disease. Br J Ophthalmol. 105(2):258–64. doi:10.1136/bjophthalmol-2020-316201.
- Schönbach EM, Strauss RW, Muñoz B, Wolfson Y, Ibrahim MA, Birch DG, Zrenner E, Sunness JS, Ip MS, Sadda SR, et al. Longitudinal Microperimetric Changes of Macular Sensitivity in Stargardt Disease After 12 Months: progStar Report No. 13. JAMA Ophthalmol. 2020;138(7):1–8. doi:10.1001/jamaophthalmol.2020.1735.
- Schönbach EM, Janeschitz-Kriegl L, Strauss RW, Cattaneo M, Fujinami K, Birch DG, Cideciyan AV, Sunness JS, Weleber RG, Ip MS, et al. The Progression of Stargardt Disease using Volumetric Hill of Vision Analyses Over 24 Months: progStar Report No.15. Am J Ophthalmol. 2021; doi:10.1016/j.ajo.2021.04.015.
- Zahid S, Peeler C, Khan N, Davis J, Mahmood M, Heckenlively JR, Jayasundera T. Digital quantification of Goldmann visual fields (GVFs) as a means for genotype-phenotype comparisons and detection of progression in retinal degenerations. Adv Exp Med Biol. 2014;801:131–37. doi:10.1007/978-1-4614-3209-8_17.
- Dedania VS, Liu JY, Schlegel D, Andrews CA, Branham K, Khan NW, Musch DC, Heckenlively JR, Jayasundera KT. 2018. Reliability of kinetic visual field testing in children with mutation-proven retinal dystrophies: implications for therapeutic clinical trials. Ophthalmic Genet. 39(1):22–28. doi:10.1080/13816810.2017.1329447.
- Kaplan J, Gerber S, Larget-Piet D, Rozet JM, Dollfus H, Dufier JL, Odent S, Postel-Vinay A, Janin N, Briard ML, et al. A gene for Stargardt’s disease (fundus flavimaculatus) maps to the short arm of chromosome 1. Nat Genet. 1993;5(3):308–11. doi:10.1038/ng1193-308.
- Gerber S, Rozet JM, Bonneau D, Souied E, Camuzat A, Dufier JL, Amalric P, Weissenbach J, Munnich A, Kaplan J.A gene for late-onset fundus flavimaculatus with macular dystrophy maps to chromosome 1p13. Am J Hum Genet. 1995;56(2):396–99.
- Hoyng CB, Poppelaars F, Van De Pol TJ, Kremer H, Pinckers AJ, Deutman AF, Cremers FP. 1996. Genetic fine mapping of the gene for recessive Stargardt disease. Hum Genet. 98(4):500–04. doi:10.1007/s004390050247
- Weber BH, Sander S, Kopp C, Walker D, Eckstein A, Wissinger B, Zrenner E, Grimm T.Analysis of 21 Stargardt’s disease families confirms a major locus on chromosome 1p with evidence for non-allelic heterogeneity in a minority of cases. Br J Ophthalmol. 1996;80(8):745–49.
- Azarian SM, Megarity CF, Weng J, Horvath DH, Travis GH. 1998. The human photoreceptor rim protein gene (ABCR): genomic structure and primer set information for mutation analysis. Hum Genet. 102(6):699–705. doi:10.1007/s004390050765
- Gerber S, Rozet JM, Van De Pol TJ, Hoyng CB, Munnich A, Blankenagel A, Kaplan J, Cremers FP. 1998. Complete exon-intron structure of the retina-specific ATP binding transporter gene (ABCR) allows the identification of novel mutations underlying Stargardt disease. Genomics. 48(1):139–42. doi:10.1006/geno.1997.5164
- Huckfeldt RM, East JS, Stone EM, Sohn EH. Phenotypic Variation in a Family With Pseudodominant Stargardt Disease. JAMA Ophthalmol. 2016. doi:10.1001/jamaophthalmol.2015.5471.
- Lambertus S, RAC VH, Bax NM, Hoefsloot LH, Cremers FPM, Boon CJF, Klevering BJ, Hoyng CB. 2015. Early-onset stargardt disease: phenotypic and genotypic characteristics. Ophthalmology. 122(2):335–44. doi:10.1016/j.ophtha.2014.08.032
- Bax NM, Lambertus S, Cremers FPM, Klevering BJ, Hoyng CB. The absence of fundus abnormalities in Stargardt disease. Graefe’s Archive for Clinical and Experimental Ophthalmology. 2019;257(6):1147–57. doi:10.1007/s00417-019-04280-8.
- Khan KN, Kasilian M, Mahroo OAR, Tanna P, Kalitzeos A, Robson AG, Tsunoda K, Iwata T, Moore AT, Fujinami K, et al. Early Patterns of Macular Degeneration in ABCA4-Associated Retinopathy. Ophthalmology. 2018;125(5):735–46. doi:10.1016/j.ophtha.2017.11.020.
- Lee W, Nõupuu K, Oll M, Duncker T, Burke T, Zernant J, Bearelly S, Tsang SH, Sparrow JR, Allikmets R. 2014. The external limiting membrane in early-onset Stargardt disease. Invest Ophthalmol Vis Sci. 55(10):6139–49. doi:10.1167/iovs.14-15126
- Carter-Dawson LD, LaVail MM. 1979. Rods and cones in the mouse retina. I. Structural analysis using light and electron microscopy. J Comp Neurol. 188(2):245–62. doi:10.1002/cne.901880204
- Zahid S, Jayasundera T, Rhoades W, Branham K, Khan N, Niziol LM, Musch DC, Heckenlively JR. 2013. Clinical phenotypes and prognostic full-field electroretinographic findings in Stargardt disease. Am J Ophthalmol. 155(3):465–73.e3. doi:10.1016/j.ajo.2012.09.011
- Tanaka K, Lee W, Zernant J, Schuerch K, Ciccone L, Tsang SH, Sparrow JR, Allikmets R. 2018. The Rapid-Onset Chorioretinopathy Phenotype of ABCA4 Disease. Ophthalmology. 125(1):89–99. doi:10.1016/j.ophtha.2017.07.019
- Cremers FP, Van De Pol DJ, van Driel M, Den Hollander AI, van Haren FJ, Knoers NV, Tijmes N, Bergen AA, Rohrschneider K, Blankenagel A, et al. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt’s disease gene ABCR. Hum Mol Genet 1998;7:355–62.
- Klevering BJ, Yzer S, Rohrschneider K, Zonneveld M, Allikmets R, Van Den Born LI, Maugeri A, Hoyng CB, Cremers FP. 2004. Microarray-based mutation analysis of the ABCA4 (ABCR) gene in autosomal recessive cone-rod dystrophy and retinitis pigmentosa. Eur J Hum Genet. 12(12):1024–32. doi:10.1038/sj.ejhg.5201258
- Lorenz B, Preising MN. 2005. Age matters–thoughts on a grading system for ABCA4 mutations. Graefes Arch Clin Exp Ophthalmol. 243(2):87–89. doi:10.1007/s00417-004-1078-5
- Martinez-Mir A, Bayes M, Vilageliu L, Grinberg D, Ayuso C, Del Rio T, Garcia-Sandoval B, Bussaglia E, Baiget M, Gonzalez-Duarte R, et al. A new locus for autosomal recessive retinitis pigmentosa (RP19) maps to 1p13-1p21. Genomics 1997;40:142–46.
- Martínez-Mir A, Paloma E, Allikmets R, Ayuso C, Río T, Dean M, Vilageliu L, Gonzàlez-Duarte R, Balcells S. 1998. Retinitis pigmentosa caused by a homozygous mutation in the Stargardt disease gene ABCR. Nat Genet. 18(1):11–12. doi:10.1038/ng0198-11
- Rozet JM, Gerber S, Ghazi I, Perrault I, Ducroq D, Souied E, Cabot A, Dufier JL, Munnich A, Kaplan J.Mutations of the retinal specific ATP binding transporter gene (ABCR) in a single family segregating both autosomal recessive retinitis pigmentosa RP19 and Stargardt disease: evidence of clinical heterogeneity at this locus. J Med Genet. 1999;36(6):447–51.
- Sb C, Ll M, Fa G, Rs M. Functional Analysis and Classification of Homozygous and Hypomorphic ABCA4 Variants Associated with Stargardt Macular Degeneration. Hum Mutat. 2020. doi:10.1002/humu.24100.
- Cideciyan AV, Swider M, Aleman TS, Sumaroka A, Schwartz SB, Roman MI, Milam AH, Bennett J, Stone EM, Jacobson SG. 2005. ABCA4-associated retinal degenerations spare structure and function of the human parapapillary retina. Invest Ophthalmol Vis Sci. 46(12):4739–46. doi:10.1167/iovs.05-0805
- Heath Jeffery RC, Thompson JA, Lo J, Lamey TM, McLaren TL, McAllister IL, Mackey DA, Constable IJ, De Roach JN, Chen FK. 2021. Atrophy Expansion Rates in Stargardt Disease Using Ultra-Widefield Fundus Autofluorescence. Ophthalmology Science. 1(1):100005. doi:10.1016/j.xops.2021.100005
- Heath Jeffery RC, Thompson JA, Lamey TM, McLaren TL, McAllister IL, Constable IJ, Mackey DA, De Roach JN, Chen FK. Classifying ABCA4 mutation severity using age-dependent ultra-widefield fundus autofluorescence-derived total lesion size. Retina. 2021. doi:10.1097/iae.0000000000003227.
- Fishman GA, Stone EM, Grover S, Derlacki DJ, Haines HL, Hockey RR. 1999. Variation of clinical expression in patients with Stargardt dystrophy and sequence variations in the ABCR gene. Archives of Ophthalmology (Chicago, Ill: 1960). 117(4):504–10. doi:10.1001/archopht.117.4.504
- Allikmets R. 2000. Simple and complex ABCR: genetic predisposition to retinal disease. Am J Hum Genet. 67(4):793–99. doi:10.1086/303100
- Genead MA, Fishman GA, Stone EM, Allikmets R. 2009. The natural history of stargardt disease with specific sequence mutation in the ABCA4 gene. Invest Ophthalmol Vis Sci. 50(12):5867–71. doi:10.1167/iovs.09-3611
- Cella W, Greenstein VC, Zernant-Rajang J, Smith TR, Barile G, Allikmets R, Tsang SH. 2009. G1961E mutant allele in the Stargardt disease gene ABCA4 causes bull’s eye maculopathy. Exp Eye Res. 89(1):16–24. doi:10.1016/j.exer.2009.02.001
- Burke TR, Fishman GA, Zernant J, Schubert C, Tsang SH, Smith RT, Ayyagari R, Koenekoop RK, Umfress A, Ciccarelli ML, et al. Retinal phenotypes in patients homozygous for the G1961E mutation in the ABCA4 gene. Invest Ophthalmol Vis Sci. 2012;53(8):4458–67. doi:10.1167/iovs.11-9166.
- Lee W, Schuerch K, Zernant J, Collison FT, Bearelly S, Fishman GA, Tsang SH, Sparrow JR, Allikmets R. 2017. Genotypic spectrum and phenotype correlations of ABCA4-associated disease in patients of south Asian descent. Eur J Hum Genet. 25(6):735–43. doi:10.1038/ejhg.2017.13
- Eh R, Khan M, Ss C, Roosing S, Del Pozo-Valero M, Tm L, Liskova P, Roberts L, Stöhr H, CCW K, et al. Association of Sex With Frequent and Mild ABCA4 Alleles in Stargardt Disease. JAMA Ophthalmol. 2020;doi:10.1001/jamaophthalmol.2020.2990.
- Leng T, Marmor MF, Kellner U, Thompson DA, Renner AB, Moore W, Sowden JC. 2012. Foveal cavitation as an optical coherence tomography finding in central cone dysfunction. Retina. 32(7):1411–19. doi:10.1097/IAE.0b013e318236e4ea
- Nõupuu K, Lee W, Zernant J, Tsang SH, Allikmets R. 2014. Structural and genetic assessment of the ABCA4-associated optical gap phenotype. Invest Ophthalmol Vis Sci. 55(11):7217–26. doi:10.1167/iovs.14-14674
- Noble KG, Carr RE.Stargardt’s disease and fundus flavimaculatus. Arch Ophthalmol. 1979;97(7):1281–85.
- Westeneng-van Haaften SC, Boon CJF, Cremers FPM, Hoefsloot LH, Den Hollander AI, Hoyng CB. 2012. Clinical and genetic characteristics of late-onset Stargardt’s disease. Ophthalmology. 119(6):1199–210. doi:10.1016/j.ophtha.2012.01.005
- Yatsenko AN, Shroyer NF, Lewis RA, Lupski JR.Late-onset Stargardt disease is associated with missense mutations that map outside known functional regions of ABCR (ABCA4). Hum Genet. 2001;108(4):346–55.
- Lambertus S, Lindner M, Bax NM, Mauschitz MM, Nadal J, Schmid M, Schmitz-Valckenberg S, Den Hollander AI, Weber BHF, Holz FG, et al. rogression of Late-Onset Stargardt Disease. Invest Ophthalmol Vis Sci. 2016;57(13):5186–91. doi:10.1167/iovs.16-19833.
- Fujinami K, Sergouniotis PI, Davidson AE, Wright G, Chana RK, Tsunoda K, Tsubota K, Egan CA, Robson AG, Moore AT, et al. Clinical and molecular analysis of Stargardt disease with preserved foveal structure and function. Am J Ophthalmol. 2013;156(3):487–501.e1. doi:10.1016/j.ajo.2013.05.003.
- Zernant J, Lee W, Collison FT, Fishman GA, Sergeev YV, Schuerch K, Sparrow JR, Tsang SH, Allikmets R. 2017. Frequent hypomorphic alleles account for a significant fraction of ABCA4 disease and distinguish it from age-related macular degeneration. J Med Genet. 54(6):404–12. doi:10.1136/jmedgenet-2017-104540
- Collison FT, Lee W, Fishman GA, Park JC, Zernant J, McAnany JJ, Allikmets R. 2019. CLINICAL CHARACTERIZATION OF STARGARDT DISEASE PATIENTS WITH THE p.N1868I ABCA4 MUTATION. Retina. 39(12):2311–25. doi:10.1097/iae.0000000000002316
- Chen B, Tosha C, Gorin MB, Nusinowitz S. 2010. Analysis of autofluorescent retinal images and measurement of atrophic lesion growth in Stargardt disease. Exp Eye Res. 91(2):143–52. doi:10.1016/j.exer.2010.03.021
- Strauss RW, Muñoz B, Ho A, Jha A, Michaelides M, Cideciyan AV, Audo I, Birch DG, Hariri AH, Nittala MG, et al. Progression of Stargardt Disease as Determined by Fundus Autofluorescence in the Retrospective Progression of Stargardt Disease Study (ProgStar Report No. 9). JAMA Ophthalmol. 2017;135(11):1232–41. doi:10.1001/jamaophthalmol.2017.4152.
- Burke TR, Duncker T, Woods RL, Greenberg JP, Zernant J, Tsang SH, Smith RT, Allikmets R, Sparrow JR, Delori FC. 2014. Quantitative fundus autofluorescence in recessive Stargardt disease. Invest Ophthalmol Vis Sci. 55(5):2841–52. doi:10.1167/iovs.13-13624
- Quellec G, Russell SR, Scheetz TE, Stone EM, Abràmoff MD. 2011. Computational quantification of complex fundus phenotypes in age-related macular degeneration and Stargardt disease. Invest Ophthalmol Vis Sci. 52(6):2976–81. doi:10.1167/iovs.10-6232
- Georgiou M, Kane T, Tanna P, Bouzia Z, Singh N, Kalitzeos A, Strauss RW, Fujinami K, Michaelides M. Prospective Cohort Study of Childhood-Onset Stargardt Disease: fundus Autofluorescence Imaging, Progression, Comparison with Adult-Onset Disease, and Disease Symmetry. Am J Ophthalmol. 2020;211:159–75. doi:10.1016/j.ajo.2019.11.008.
- Heath Jeffery RC, Chen FK. Stargardt disease: multimodal imaging-A review. Clin Exp Ophthalmol. 2021. doi:10.1111/ceo.13947.
- Müller PL, Pfau M, Treis T, Pascual-Camps I, Birtel J, Lindner M, Herrmann P, Holz FG. 2020. PROGRESSION OF ABCA4-RELATED RETINOPATHY: prognostic value of demographic, functional, genetic, and imaging parameters. Retina. 40(12):2343–56. doi:10.1097/iae.0000000000002747
- Shen LL, Sun M, Grossetta Nardini HK, Del Priore LV. 2019. Natural History of Autosomal Recessive Stargardt Disease in Untreated Eyes: a Systematic Review and Meta-analysis of Study- and Individual-Level Data. Ophthalmology. 126(9):1288–96. doi:10.1016/j.ophtha.2019.05.015
- Allikmets R, Wasserman WW, Hutchinson A, Smallwood P, Nathans J, Rogan PK, Schneider TD, Dean M.Organization of the ABCR gene: analysis of promoter and splice junction sequences. Gene. 1998;215(1):111–22.
- Nasonkin I, Illing M, Koehler MR, Schmid M, Molday RS, Weber BH.Mapping of the rod photoreceptor ABC transporter (ABCR) to 1p21-p22.1 and identification of novel mutations in Stargardt’s disease. Hum Genet. 1998;102(1):21–26.
- Tsybovsky Y, Orban T, Molday RS, Taylor D, Palczewski K. 2013. Molecular organization and ATP-induced conformational changes of ABCA4, the photoreceptor-specific ABC transporter. Structure (London, England: 1993). 21(5):854–60. doi:10.1016/j.str.2013.03.001
- Ahn J, Beharry S, Molday LL, Molday RS. 2003. Functional interaction between the two halves of the photoreceptor-specific ATP binding cassette protein ABCR (ABCA4). Evidence for a non-exchangeable ADP in the first nucleotide binding domain. J Biol Chem. 278(41):39600–08. doi:10.1074/jbc.M304236200
- Sun H, Smallwood PM, Nathans J.Biochemical defects in ABCR protein variants associated with human retinopathies. Nat Genet. 2000;26(2):242.
- Quazi F, Lenevich S, Molday RS. ABCA4 is an N-retinylidene-phosphatidylethanolamine and phosphatidylethanolamine importer. Nat Commun. 2012;3:925. doi:10.1038/ncomms1927.
- Tsybovsky Y, Molday RS, Palczewski K. The ATP-Binding Cassette Transporter ABCA4: structural and Functional Properties and Role in Retinal Disease. Adv Exp Med Biol. 2010;703:105–25. doi:10.1007/978-1-4419-5635-4_8.
- Molday RS, Molday LL.Identification and characterization of multiple forms of rhodopsin and minor proteins in frog and bovine rod outer segment disc membranes. Electrophoresis, lectin labeling, and proteolysis studies. J Biol Chem. 1979;254(11):4653–60.
- Bungert S, Molday LL, Molday RS. 2001. Membrane topology of the ATP binding cassette transporter ABCR and its relationship to ABC1 and related ABCA transporters: identification of N-linked glycosylation sites. J Biol Chem. 276(26):23539–46. doi:10.1074/jbc.M101902200
- Tsybovsky Y, Wang B, Quazi F, Molday RS, Palczewski K. 2011. Posttranslational modifications of the photoreceptor-specific ABC transporter ABCA4. Biochemistry. 50(32):6855–66. doi:10.1021/bi200774w
- Davidson AL, Chen J. ATP-binding cassette transporters in bacteria. Annu Rev Biochem. 2004;73:241–68. doi:10.1146/annurev.biochem.73.011303.073626.
- Fetsch EE, Davidson AL Vanadate-catalyzed photocleavage of the signature motif of an ATP-binding cassette (ABC) transporter. Proc Natl Acad Sci U S A. 2002;99:9685–90. doi:10.1073/pnas.152204499.
- Hunke S, Mourez M, Jehanno M, Dassa E, Schneider E.ATP modulates subunit-subunit interactions in an ATP-binding cassette transporter (MalFGK2) determined by site-directed chemical cross-linking. J Biol Chem. 2000;275(20):15526–34.
- Rees DC, Johnson E, Lewinson O. 2009. ABC transporters: the power to change. Nat Rev Mol Cell Biol. 10(3):218–27. doi:10.1038/nrm2646
- Kos V, Ford RC. 2009. The ATP-binding cassette family: a structural perspective. Cellular and molecular life sciences. CMLS. 66(19):3111–26. doi:10.1007/s00018-009-0064-9
- Linton KJ, Higgins CF. 2007. Structure and function of ABC transporters: the ATP switch provides flexible control. Pflugers Archiv. European Journal of Physiology. 453(5):555–67. doi:10.1007/s00424-006-0126-x
- Tsybovsky Y, Palczewski K. Expression, purification and structural properties of ABC transporter ABCA4 and its individual domains. Protein Expr Purif. 2014;97:50–60. doi:10.1016/j.pep.2014.02.010.
- Lenis TL, Hu J, Ng SY, Jiang Z, Sarfare S, Lloyd MB, Esposito NJ, Samuel W, Jaworski C, Bok D, et al. Expression of ABCA4 in the retinal pigment epithelium and its implications for Stargardt macular degeneration. Proc Natl Acad Sci U S A. 2018;115(47):E11120–e7. doi:10.1073/pnas.1802519115.
- Rattner A, Smallwood PM, Nathans J.Identification and characterization of all-trans-retinol dehydrogenase from photoreceptor outer segments, the visual cycle enzyme that reduces all-trans-retinal to all-trans-retinol. J Biol Chem. 2000;275(15):11034–43.
- Chen C, Thompson DA, Koutalos Y. 2012. Reduction of all-trans-retinal in vertebrate rod photoreceptors requires the combined action of RDH8 and RDH12. J Biol Chem. 287(29):24662–70. doi:10.1074/jbc.M112.354514
- Mata NL, Weng J, Travis GH.Biosynthesis of a major lipofuscin fluorophore in mice and humans with ABCR-mediated retinal and macular degeneration. Proceedings of the National Academy of Sciences. 2000;97(13):7154–59.
- Quazi F, Molday RS. 2014. ATP-binding cassette transporter ABCA4 and chemical isomerization protect photoreceptor cells from the toxic accumulation of excess 11-cis-retinal. Proc Natl Acad Sci U S A. 111(13):5024–29. doi:10.1073/pnas.1400780111
- Cornelis SS, Bax NM, Zernant J, Allikmets R, Fritsche LG, den Dunnen JT, Ajmal M, Hoyng CB, Cremers FP. 2017. In Silico Functional Meta-Analysis of 5,962 ABCA4 Variants in 3,928 Retinal Dystrophy Cases. Hum Mutat. 38(4):400–08. doi:10.1002/humu.23165
- Zernant J, Schubert C, Im KM, Burke T, Brown CM, Fishman GA, Tsang SH, Gouras P, Dean M, Allikmets R. 2011. Analysis of the ABCA4 gene by next-generation sequencing. Invest Ophthalmol Vis Sci. 52(11):8479–87. doi:10.1167/iovs.11-8182
- Jiang F, Pan Z, Xu K, Tian L, Xie Y, Zhang X, Chen J, Dong B, Li Y. 2016. Screening of ABCA4 Gene in a Chinese Cohort With Stargardt Disease or Cone-Rod Dystrophy With a Report on 85 Novel Mutations. Invest Ophthalmol Vis Sci. 57(1):145–52. doi:10.1167/iovs.15-18190
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24. doi:10.1038/gim.2015.30.
- Garces F, Jiang K, Molday LL, Stöhr H, Weber BH, Lyons CJ, Maberley D, Molday RS. 2018. Correlating the Expression and Functional Activity of ABCA4 Disease Variants With the Phenotype of Patients With Stargardt Disease. Invest Ophthalmol Vis Sci. 59(6):2305–15. doi:10.1167/iovs.17-23364
- Garwin GG, Saari JC. High-performance liquid chromatography analysis of visual cycle retinoids. Methods Enzymol. 2000;316:313–24. doi:10.1016/s0076-6879(00)16731-x.
- Zhong M, Molday RS. Binding of retinoids to ABCA4, the photoreceptor ABC transporter associated with Stargardt macular degeneration. Methods Mol Biol. 2010;652:163–76. doi:10.1007/978-1-60327-325-1_9.
- Sun H, Molday RS, Nathans J. 1999. Retinal stimulates ATP hydrolysis by purified and reconstituted ABCR, the photoreceptor-specific ATP-binding cassette transporter responsible for Stargardt disease. J Biol Chem. 274(12):8269–81. doi:10.1074/jbc.274.12.8269
- Beharry S, Zhong M, Molday RS. 2004. N-retinylidene-phosphatidylethanolamine is the preferred retinoid substrate for the photoreceptor-specific ABC transporter ABCA4 (ABCR). J Biol Chem. 279(52):53972–79. doi:10.1074/jbc.M405216200
- Zhong M, Molday LL, Molday RS. 2009. Role of the C terminus of the photoreceptor ABCA4 transporter in protein folding, function, and retinal degenerative diseases. J Biol Chem. 284(6):3640–49. doi:10.1074/jbc.M806580200
- Suárez T, Biswas SB, Biswas EE. 2002. Biochemical defects in retina-specific human ATP binding cassette transporter nucleotide binding domain 1 mutants associated with macular degeneration. J Biol Chem. 277(24):21759–67. doi:10.1074/jbc.M202053200
- Cideciyan AV, Swider M, Aleman TS, Tsybovsky Y, Schwartz SB, Windsor EA, Roman AJ, Sumaroka A, Steinberg JD, Jacobson SG, et al. ABCA4 disease progression and a proposed strategy for gene therapy. Hum Mol Genet. 2009;18(5):931–41. doi:10.1093/hmg/ddn421.
- Wiszniewski W, Zaremba CM, Yatsenko AN, Jamrich M, Wensel TG, Lewis RA, Lupski JR. 2005. ABCA4 mutations causing mislocalization are found frequently in patients with severe retinal dystrophies. Hum Mol Genet. 14(19):2769–78. doi:10.1093/hmg/ddi310
- Gregersen N, Bross P, Vang S, Christensen JH. Protein misfolding and human disease. Annu Rev Genomics Hum Genet. 2006;7:103–24. doi:10.1146/annurev.genom.7.080505.115737.
- Schröder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–89. doi:10.1146/annurev.biochem.73.011303.074134.
- Barbosa-Morais NL, Irimia M, Pan Q, Xiong HY, Gueroussov S, Lee LJ, Slobodeniuc V, Kutter C, Watt S, Colak R, et al. The evolutionary landscape of alternative splicing in vertebrate species. Science. 2012;338(6114):1587–93. doi:10.1126/science.1230612.
- Gueroussov S, Gonatopoulos-Pournatzis T, Irimia M, Raj B, Lin ZY, Gingras AC, Blencowe BJ. 2015. An alternative splicing event amplifies evolutionary differences between vertebrates. Science. 349(6250):868–73. doi:10.1126/science.aaa8381
- Schulz HL, Grassmann F, Kellner U, Spital G, Rüther K, Jägle H, Hufendiek K, Rating P, Huchzermeyer C, Baier MJ, et al. Mutation Spectrum of the ABCA4 Gene in 335 Stargardt Disease Patients From a Multicenter German Cohort-Impact of Selected Deep Intronic Variants and Common SNPs. Invest Ophthalmol Vis Sci. 2017;58(1):394–403. doi:10.1167/iovs.16-19936.
- Li D, Mastaglia FL, Fletcher S, Wilton SD. 2018. Precision Medicine through Antisense Oligonucleotide-Mediated Exon Skipping. Trends Pharmacol Sci. 39(11):982–94. doi:10.1016/j.tips.2018.09.001
- López-Bigas N, Audit B, Ouzounis C, Parra G, Guigó R. 2005. Are splicing mutations the most frequent cause of hereditary disease? FEBS Lett. 579(9):1900–03. doi:10.1016/j.febslet.2005.02.047
- Sangermano R, Khan M, Cornelis SS, Richelle V, Albert S, Garanto A, Elmelik D, Qamar R, Lugtenberg D, Van Den Born LI, et al. ABCA4 midigenes reveal the full splice spectrum of all reported noncanonical splice site variants in Stargardt disease. Genome Res. 2018;28(1):100–10. doi:10.1101/gr.226621.117.
- Riera M, Patel A, Burés-Jelstrup A, Corcostegui B, Chang S, Pomares E, Corneo B, Sparrow JR. Generation of two iPS cell lines (FRIMOi003-A and FRIMOi004-A) derived from Stargardt patients carrying ABCA4 compound heterozygous mutations. Stem Cell Res. 2019;36:101389. doi:10.1016/j.scr.2019.101389.
- Claassen JN, Zhang D, Chen S-C, Moon SY, Lamey T, Thompson JA, McLaren T, De Roach JN, McLenachan S, Chen FK. Generation of the induced pluripotent stem cell line from a patient with autosomal recessive ABCA4-mediated Stargardt Macular Dystrophy. Stem Cell Res. 2019;34:101352. doi:10.1016/j.scr.2018.11.013.
- Sangermano R, Bax NM, Bauwens M, Van Den Born LI, De Baere E, Garanto A, Collin RW, Goercharn-Ramlal AS, den Engelsman-van Dijk AH, Rohrschneider K, et al. Photoreceptor Progenitor mRNA Analysis Reveals Exon Skipping Resulting from the ABCA4 c.5461-10T–>C Mutation in Stargardt Disease. Ophthalmology. 2016;123(6):1375–85. doi:10.1016/j.ophtha.2016.01.053.
- Jennings L, Zhang D, Chen SC, Moon SY, Lamey T, Thompson JA, McLaren T, De Roach JN, Chen FK, McLenachan S. Generation of two induced pluripotent stem cell lines from a patient with Stargardt Macular Dystrophy caused by the c.768G>T and c.6079C>T mutations in ABCA4. Stem Cell Res. 2020;48:101947. doi:10.1016/j.scr.2020.101947.
- Albert S, Garanto A, Sangermano R, Khan M, Bax NM, Hoyng CB, Zernant J, Lee W, Allikmets R, Collin RWJ, et al. Identification and Rescue of Splice Defects Caused by Two Neighboring Deep-Intronic ABCA4 Mutations Underlying Stargardt Disease. Am J Hum Genet. 2018;102(4):517–27. doi:10.1016/j.ajhg.2018.02.008.
- Garanto A, Duijkers L, Tomkiewicz TZ, Collin RWJ.Antisense Oligonucleotide Screening to Optimize the Rescue of the Splicing Defect Caused by the Recurrent Deep-Intronic ABCA4 Variant c.4539+2001G>A in Stargardt Disease. Genes (Basel). 2019;10(6). doi:10.3390/genes10060452
- Cremers FPM, Lee W, Collin RWJ, Allikmets R. Clinical spectrum, genetic complexity and therapeutic approaches for retinal disease caused by ABCA4 mutations. Prog Retin Eye Res. 2020;79:100861. doi:10.1016/j.preteyeres.2020.100861.
- Bauwens M, Garanto A, Sangermano R, Naessens S, Weisschuh N, De Zaeytijd J, Khan M, Sadler F, Balikova I, Van Cauwenbergh C, et al. ABCA4-associated disease as a model for missing heritability in autosomal recessive disorders: novel noncoding splice, cis-regulatory, structural, and recurrent hypomorphic variants. Genetics in Medicine: Official Journal of the American College of Medical Genetics. 2019;21(8):1761–71. doi:10.1038/s41436-018-0420-y.
- Braun TA, Mullins RF, Wagner AH, Andorf JL, Johnston RM, Bakall BB, Deluca AP, Fishman GA, Lam BL, Weleber RG, et al. Non-exomic and synonymous variants in ABCA4 are an important cause of Stargardt disease. Hum Mol Genet. 2013;22(25):5136–45. doi:10.1093/hmg/ddt367.
- Zernant J, Xie Y, Ayuso C, Riveiro-Alvarez R, M-a L-M, Simonelli F, Testa F, Gorin MB, Strom SP, Bertelsen M, et al. Analysis of the ABCA4 genomic locus in Stargardt disease. Hum Mol Genet. 2014;23(25):6797–806. doi:10.1093/hmg/ddu396.
- Sangermano R, Garanto A, Khan M, Runhart EH, Bauwens M, Bax NM, Van Den Born LI, Khan MI, Cornelis SS, Verheij JBGM, et al. Deep-intronic ABCA4 variants explain missing heritability in Stargardt disease and allow correction of splice defects by antisense oligonucleotides. Genetics in Medicine: Official Journal of the American College of Medical Genetics. 2019;21(8):1751–60. doi:10.1038/s41436-018-0414-9.
- Murphy D, Cieply B, Carstens R, Ramamurthy V, The Musashi SP. 2016. 1 Controls the Splicing of Photoreceptor-Specific Exons in the Vertebrate Retina. PLoS Genet. 12(8):e1006256–e. doi:10.1371/journal.pgen.1006256
- Sterne-Weiler T, Sanford JR. 2014. Exon identity crisis: disease-causing mutations that disrupt the splicing code. Genome Biol. 15(1):201. doi:10.1186/gb4150
- Saari JC. Vitamin A metabolism in rod and cone visual cycles. Annu Rev Nutr. 2012;32:125–45. doi:10.1146/annurev-nutr-071811-150748.
- Molday RS. 2007. ATP-binding cassette transporter ABCA4: molecular properties and role in vision and macular degeneration. J Bioenerg Biomembr. 39(5–6):507–17. doi:10.1007/s10863-007-9118-6
- Kim SR, Nakanishi K, Itagaki Y, Sparrow JR. 2006. Photooxidation of A2-PE, a photoreceptor outer segment fluorophore, and protection by lutein and zeaxanthin. Exp Eye Res. 82(5):828–39. doi:10.1016/j.exer.2005.10.004
- Finnemann SC, Leung LW, Rodriguez-Boulan E. The lipofuscin component A2E selectively inhibits phagolysosomal degradation of photoreceptor phospholipid by the retinal pigment epithelium. Proc Natl Acad Sci U S A. 2002;99(6):3842–47. doi:10.1073/pnas.052025899.
- Schutt F, Davies S, Kopitz J, Holz FG, Boulton ME.Photodamage to human RPE cells by A2-E, a retinoid component of lipofuscin. Invest Ophthalmol Vis Sci. 2000;41(8):2303–08.
- Sparrow JR, Nakanishi K, Parish CA.The lipofuscin fluorophore A2E mediates blue light-induced damage to retinal pigmented epithelial cells. Invest Ophthalmol Vis Sci. 2000;41(7):1981–89.
- Kim SR, Jockusch S, Itagaki Y, Turro NJ, Sparrow JR.Mechanisms involved in A2E oxidation. Exp Eye Res. 2008;86(6):975–82.
- Zhou J, Jang YP, Kim SR, Sparrow JR. Complement activation by photooxidation products of A2E, a lipofuscin constituent of the retinal pigment epithelium. Proc Natl Acad Sci U S A. 2006;103(44):16182–87. doi:10.1073/pnas.0604255103.
- Weng J, Mata NL, Azarian SM, Tzekov RT, Birch DG, Travis GH. 1999. Insights into the Function of Rim Protein in Photoreceptors and Etiology of Stargardt’s Disease from the Phenotype in abcr Knockout Mice. Cell. 98(1):13–23. doi:10.1016/s0092-8674(00)80602-9
- Charbel Issa P, Barnard AR, Singh MS, Carter E, Jiang Z, Radu RA, Schraermeyer U, MacLaren RE. 2013. Fundus autofluorescence in the Abca4(-/-) mouse model of Stargardt disease–correlation with accumulation of A2E, retinal function, and histology. Invest Ophthalmol Vis Sci. 54(8):5602–12. doi:10.1167/iovs.13-11688
- Sparrow JR, Blonska A, Flynn E, Duncker T, Greenberg JP, Secondi R, Ueda K, Delori FC. 2013. Quantitative fundus autofluorescence in mice: correlation with HPLC quantitation of RPE lipofuscin and measurement of retina outer nuclear layer thickness. Invest Ophthalmol Vis Sci. 54(4):2812–20. doi:10.1167/iovs.12-11490
- Mata NL, Tzekov RT, Liu X, Weng J, Birch DG, Travis GH.Delayed dark-adaptation and lipofuscin accumulation in abcr± mice: implications for involvement of ABCR in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2001;42(8):1685–90.
- Radu RA, Mata NL, Bagla A, Travis GH. 2004. Light exposure stimulates formation of A2E oxiranes in a mouse model of Stargardt’s macular degeneration. Proc Natl Acad Sci U S A. 101(16):5928–33. doi:10.1073/pnas.0308302101
- Radu RA, Hu J, Yuan Q, Welch DL, Makshanoff J, Lloyd M, McMullen S, Travis GH, Bok D. 2011. Complement system dysregulation and inflammation in the retinal pigment epithelium of a mouse model for Stargardt macular degeneration. J Biol Chem. 286(21):18593–601. doi:10.1074/jbc.M110.191866
- Radu RA, Yuan Q, Hu J, Peng JH, Lloyd M, Nusinowitz S, Bok D, Travis GH. 2008. Accelerated accumulation of lipofuscin pigments in the RPE of a mouse model for ABCA4-mediated retinal dystrophies following Vitamin A supplementation. Invest Ophthalmol Vis Sci. 49(9):3821–29. doi:10.1167/iovs.07-1470
- Wu L, Nagasaki T, Sparrow JR. Photoreceptor cell degeneration in Abcr (-/-) mice. Adv Exp Med Biol. 2010;664:533–39. doi:10.1007/978-1-4419-1399-9_61.
- Taubitz T, Tschulakow AV, Tikhonovich M, Illing B, Fang Y, Biesemeier A, Julien-Schraermeyer S, Schraermeyer U. Ultrastructural alterations in the retinal pigment epithelium and photoreceptors of a Stargardt patient and three Stargardt mouse models: indication for the central role of RPE melanin in oxidative stress. PeerJ. 2018;6:e5215. doi:10.7717/peerj.5215.
- Ma L, Kaufman Y, Zhang J, Washington I. 2011. C20-D3-vitamin A slows lipofuscin accumulation and electrophysiological retinal degeneration in a mouse model of Stargardt disease. J Biol Chem. 286(10):7966–74. doi:10.1074/jbc.M110.178657
- Molday LL, Wahl D, Sarunic MV, Molday RS. 2018. Localization and functional characterization of the p.Asn965Ser (N965S) ABCA4 variant in mice reveal pathogenic mechanisms underlying Stargardt macular degeneration. Hum Mol Genet. 27(2):295–306. doi:10.1093/hmg/ddx400
- Maeda A, Maeda T, Golczak M, Palczewski K. 2008. Retinopathy in mice induced by disrupted all-trans-retinal clearance. J Biol Chem. 283(39):26684–93. doi:10.1074/jbc.M804505200
- Maeda A, Maeda T, Golczak M, Chou S, Desai A, Hoppel CL, Matsuyama S, Palczewski K. 2009. Involvement of all-trans-retinal in acute light-induced retinopathy of mice. J Biol Chem. 284(22):15173–83. doi:10.1074/jbc.M900322200
- Chen Y, Okano K, Maeda T, Chauhan V, Golczak M, Maeda A, Palczewski K. 2012. Mechanism of all-trans-retinal toxicity with implications for stargardt disease and age-related macular degeneration. J Biol Chem. 287(7):5059–69. doi:10.1074/jbc.M111.315432
- Usui S, Oveson BC, Lee SY, Jo YJ, Yoshida T, Miki A, Miki K, Iwase T, Lu L, Campochiaro PA. 2009. NADPH oxidase plays a central role in cone cell death in retinitis pigmentosa. J Neurochem. 110(3):1028–37. doi:10.1111/j.1471-4159.2009.06195.x
- Mäkeläinen S, Gòdia M, Hellsand M, Viluma A, Hahn D, Makdoumi K, Zeiss CJ, Mellersh C, Ricketts SL, Narfström K, et al. An ABCA4 loss-of-function mutation causes a canine form of Stargardt disease. PLoS Genet. 2019;15(3):e1007873. doi:10.1371/journal.pgen.1007873.
- Huang D, Fletcher S, Wilton S, Palmer N, McLenachan S, Mackey D, Chen F. 2017. Inherited Retinal Disease Therapies Targeting Precursor Messenger Ribonucleic Acid. Vision. 1(3):22. doi:10.3390/vision1030022
- Colella P, Auricchio A. 2010. AAV-Mediated Gene Supply for Treatment of Degenerative and Neovascular Retinal Diseases. Curr Gene Ther. 10(5):371–80. doi:10.2174/156652310793180670
- Kubota R, Boman NL, David R, Mallikaarjun S, Patil S, Birch D.Safety and effect on rod function of ACU-4429, a novel small-molecule visual cycle modulator. Retina (Philadelphia, Pa). 2012;32(1):183–88.
- Kubota R, Al-Fayoumi S, Mallikaarjun S, Patil S, Bavik C, Chandler JW.Phase 1, dose-ranging study of emixustat hydrochloride (ACU-4429), a novel visual cycle modulator, in healthy volunteers. Retina (Philadelphia, Pa). 2014;34(3):603–09.
- Radu RA, Mata NL, Nusinowitz S, Liu X, Sieving PA, Travis GH. Treatment with isotretinoin inhibits lipofuscin accumulation in a mouse model of recessive Stargardt’s macular degeneration. Proc Natl Acad Sci U S A. 2003;100(8):4742–47. doi:10.1073/pnas.0737855100.
- Sparrow JR. 2003. Therapy for macular degeneration: insights from acne. Proc Natl Acad Sci U S A. 100(8):4353–54. doi:10.1073/pnas.1031478100
- Weleber RG, Denman ST, Hanifin JM, Cunningham WJ. 1986. Abnormal retinal function associated with isotretinoin therapy for acne. Arch Ophthalmol. 104(6):831–37. doi:10.1001/archopht.1986.01050180065031
- Radu RA, Han Y, Bui TV, Nusinowitz S, Bok D, Lichter J, Widder K, Travis GH, Mata NL. 2005. Reductions in serum vitamin A arrest accumulation of toxic retinal fluorophores: a potential therapy for treatment of lipofuscin-based retinal diseases. Invest Ophthalmol Vis Sci. 46(12):4393–401. doi:10.1167/iovs.05-0820
- Dobri N, Qin Q, Kong J, Yamamoto K, Liu Z, Moiseyev G, Ma JX, Allikmets R, Sparrow JR, Petrukhin K. 2013. A1120, a nonretinoid RBP4 antagonist, inhibits formation of cytotoxic bisretinoids in the animal model of enhanced retinal lipofuscinogenesis. Invest Ophthalmol Vis Sci. 54(1):85–95. doi:10.1167/iovs.12-10050
- Samuel W, Kutty RK, Nagineni S, Vijayasarathy C, Chandraratna RA, Wiggert B. 2006. N-(4-hydroxyphenyl)retinamide induces apoptosis in human retinal pigment epithelial cells: retinoic acid receptors regulate apoptosis, reactive oxygen species generation, and the expression of heme oxygenase-1 and Gadd153. J Cell Physiol. 209(3):854–65. doi:10.1002/jcp.20774
- Racz B, Varadi A, Kong J, Allikmets R, Pearson PG, Johnson G, Cioffi CL, Petrukhin K. A non-retinoid antagonist of Retinol-Binding Protein 4 rescues phenotype in a model of Stargardt disease without inhibiting the visual cycle. J Biol Chem. 2018. doi:10.1074/jbc.RA118.002062.
- Kaufman Y, Ma L, Washington I. 2011. Deuterium enrichment of vitamin A at the C20 position slows the formation of detrimental vitamin A dimers in wild-type rodents. J Biol Chem. 286(10):7958–65. doi:10.1074/jbc.M110.178640
- Charbel Issa P, Barnard AR, Herrmann P, Washington I, MacLaren RE. Rescue of the Stargardt phenotype in Abca4 knockout mice through inhibition of vitamin A dimerization. Proc Natl Acad Sci U S A. 2015;112(27):8415–20. doi:10.1073/pnas.1506960112.
- Boye SE, Boye SL, Lewin AS, Hauswirth WW. 2013. A comprehensive review of retinal gene therapy. Mol Ther. 21(3):509–19. doi:10.1038/mt.2012.280
- Pardue MT, Allen RS. Neuroprotective strategies for retinal disease. Prog Retin Eye Res. 2018;65:50–76. doi:10.1016/j.preteyeres.2018.02.002.
- Zhang N, Tsybovsky Y, Kolesnikov AV, Rozanowska M, Swider M, Schwartz SB, et al. Protein misfolding and the pathogenesis of ABCA4-associated retinal degenerations. Hum Mol Genet. 2015;24(11):3220–37. doi:10.1093/hmg/ddv073
- Trapani I, Puppo A, Auricchio A. Vector platforms for gene therapy of inherited retinopathies. Prog Retin Eye Res. 2014;43:108–28. doi:10.1016/j.preteyeres.2014.08.001.
- Wen R, Tao W, Li Y, Sieving PA. CNTF. and retina. Prog Retin Eye Res. 2012;31(2):136–51. doi:10.1016/j.preteyeres.2011.11.005.
- Tao W, Wen R, Goddard MB, Sherman SD, O’Rourke PJ, Stabila PF, Bell WJ, Dean BJ, Kauper KA, Budz VA, et al. Encapsulated cell-based delivery of CNTF reduces photoreceptor degeneration in animal models of retinitis pigmentosa. Invest Ophthalmol Vis Sci 2002;43:3292–98.
- Sieving PA, Caruso RC, Tao W, Coleman HR, Thompson DJ, Fullmer KR, Bush RA. Ciliary neurotrophic factor (CNTF) for human retinal degeneration: phase I trial of CNTF delivered by encapsulated cell intraocular implants. Proc Natl Acad Sci U S A. 2006;103(10):3896–901. doi:10.1073/pnas.
- Birch DG, Weleber RG, Duncan JL, Jaffe GJ, Tao W. 2013. Randomized trial of ciliary neurotrophic factor delivered by encapsulated cell intraocular implants for retinitis pigmentosa. Am J Ophthalmol. 156(2):283–92.e1. doi:10.1016/j.ajo.2013.03.021
- Birch DG, Bennett LD, Duncan JL, Weleber RG, Pennesi ME. Long-term Follow-up of Patients With Retinitis Pigmentosa Receiving Intraocular Ciliary Neurotrophic Factor Implants. Am J Ophthalmol. 2016;170:10–14. doi:10.1016/j.ajo.2016.07.013.
- Kassa E, Ciulla TA, Hussain RM, Dugel PU. 2019. Complement inhibition as a therapeutic strategy in retinal disorders. Expert Opin Biol Ther. 19(4):335–42. doi:10.1080/14712598.2019.1575358
- Lenis TL, Sarfare S, Jiang Z, Lloyd MB, Bok D, Radu RA. Complement modulation in the retinal pigment epithelium rescues photoreceptor degeneration in a mouse model of Stargardt disease. Proc Natl Acad Sci U S A. 2017;114(15):3987–92. doi:10.1073/pnas.1620299114.
- Drolet DW, Green LS, Gold L, Janjic N. 2016. Fit for the Eye: aptamers in Ocular Disorders. Nucleic Acid Ther. 26(3):127–46. doi:10.1089/nat.2015.0573
- Piccardi M, Fadda A, Martelli F, Marangoni D, Magli A, Minnella AM, Bertelli M, Di Marco S, Bisti S, Antioxidant Saffron FB. Central Retinal Function in ABCA4-Related Stargardt Macular Dystrophy. Nutrients. 2019;11:10. doi:10.3390/nu11102461.
- Puntel A, Maeda A, Golczak M, Gao SQ, Yu G, Palczewski K, Lu ZR. Prolonged prevention of retinal degeneration with retinylamine loaded nanoparticles. Biomaterials. 2015;44:5. doi:10.1016/j.biomaterials.2014.12.019.
- Yu G, Wu X, Ayat N, Maeda A, Gao SQ, Golczak M, Palczewski K, Lu ZR. 2014. Multifunctional PEG retinylamine conjugate provides prolonged protection against retinal degeneration in mice. Biomacromolecules. 15(12):4570–78. doi:10.1021/bm501352s
- Orban T, Leinonen H, Getter T, Dong Z, Sun W, Gao S, Veenstra A, Heidari-Torkabadi H, Kern TS, Kiser PD, et al. A Combination of G Protein-Coupled Receptor Modulators Protects Photoreceptors from Degeneration. J Pharmacol Exp Ther. 2018;364(2):207–20. doi:10.1124/jpet.117.245167.
- Prokopiou E, Kolovos P, Kalogerou M, Neokleous A, Nicolaou O, Sokratous K, Kyriacou K, Georgiou T. 2018. Omega-3 Fatty Acids Supplementation: therapeutic Potential in a Mouse Model of Stargardt Disease. Invest Ophthalmol Vis Sci. 59(7):2757–67. doi:10.1167/iovs.17-23523
- Saad L, Can Vitamin WI. A be Improved to Prevent Blindness due to Age-Related Macular Degeneration, Stargardt Disease and Other Retinal Dystrophies? Adv Exp Med Biol. 2016;854:355–61. doi:10.1007/978-3-319-17121-0_47.
- Hussain RM, Gregori NZ, Ciulla TA, Lam BL. 2018. Pharmacotherapy of retinal disease with visual cycle modulators. Expert Opin Pharmacother. 19(5):471–81. doi:10.1080/14656566.2018.1448060
- Lee JH, Wang JH, Chen J, Li F, Edwards TL, Hewitt AW, Liu GS. Gene therapy for visual loss: opportunities and concerns. Prog Retin Eye Res. 2018. doi:10.1016/j.preteyeres.2018.08.003.
- Yanik M, Muller B, Song F, Gall J, Wagner F, Wende W, Lorenz B, Stieger K. In vivo genome editing as a potential treatment strategy for inherited retinal dystrophies. Prog Retin Eye Res. 2017;56:1–18. doi:10.1016/j.preteyeres.2016.09.001.
- Lipinski DM, Thake M, MacLaren RE. Clinical applications of retinal gene therapy. Prog Retin Eye Res. 2013;32:22–47. doi:10.1016/j.preteyeres.2012.09.001.
- Tamboli V, Mishra GP, Mitrat AK. 2011. Polymeric vectors for ocular gene delivery. Ther Deliv. 2(4):523–36. doi:10.4155/tde.11.20
- Allocca M, Doria M, Petrillo M, Colella P, Garcia-Hoyos M, Gibbs D, Kim SR, Maguire A, Rex TS, Di Vicino U, et al. Serotype-dependent packaging of large genes in adeno-associated viral vectors results in effective gene delivery in mice. J Clin Invest. 2008;118(5):1955–64. doi:10.1172/JCI34316.
- Dong B, Nakai H, Xiao W. 2010. Characterization of genome integrity for oversized recombinant AAV vector. Molecular Therapy: The Journal of the American Society of Gene Therapy. 18(1):87–92. doi:10.1038/mt.2009.258
- Lai Y, Yue Y, Duan D. 2010. Evidence for the failure of adeno-associated virus serotype 5 to package a viral genome > or = 8.2 kb. Molecular therapy. The Journal of the American Society of Gene Therapy. 18(1):75–79. doi:10.1038/mt.2009.256
- Wu Z, Yang H, Colosi P. 2010. Effect of genome size on AAV vector packaging. Molecular Therapy: The Journal of the American Society of Gene Therapy. 18(1):80–86. doi:10.1038/mt.2009.255
- Hirsch ML, Agbandje-McKenna M, Samulski RJ. 2010. Little vector, big gene transduction: fragmented genome reassembly of adeno-associated virus. Molecular Therapy: The Journal of the American Society of Gene Therapy. 18(1):6–8. doi:10.1038/mt.2009.280
- Yan Z, Zhang Y, Duan D, Engelhardt JF. Trans-splicing vectors expand the utility of adeno-associated virus for gene therapy. Proc Natl Acad Sci U S A. 2000;97:6716–21. doi:10.1073/pnas.97.12.6716
- Duan D, Yue Y, Engelhardt JF. 2001. Expanding AAV packaging capacity with trans-splicing or overlapping vectors: a quantitative comparison. Molecular Therapy: The Journal of the American Society of Gene Therapy. 4(4):383–91. doi:10.1006/mthe.2001.0456
- Ghosh A, Yue Y, Lai Y, Duan D. 2008. A hybrid vector system expands adeno-associated viral vector packaging capacity in a transgene-independent manner. Molecular Therapy: The Journal of the American Society of Gene Therapy. 16(1):124–30. doi:10.1038/sj.mt.6300322
- Trapani I, Colella P, Sommella A, Iodice C, Cesi G, De Simone S, Marrocco E, Rossi S, Giunti M, Palfi A, et al. Effective delivery of large genes to the retina by dual AAV vectors. EMBO Mol Med. 2014;6(2):194–211. doi:10.1002/emmm.201302948.
- Dual TI. AAV Vectors for Stargardt Disease. Methods Mol Biol. 2018;1715:153–75. doi:10.1007/978-1-4939-7522-8_11.
- Trapani I, Toriello E, De Simone S, Colella P, Iodice C, Polishchuk EV, Sommella A, Colecchi L, Rossi S, Simonelli F, et al. Improved dual AAV vectors with reduced expression of truncated proteins are safe and effective in the retina of a mouse model of Stargardt disease. Hum Mol Genet. 2015;24(23):6811–25. doi:10.1093/hmg/ddv386.
- Colella P, Trapani I, Cesi G, Sommella A, Manfredi A, Puppo A, Iodice C, Rossi S, Simonelli F, Giunti M, et al. Efficient gene delivery to the cone-enriched pig retina by dual AAV vectors. Gene Ther. 2014;21(4):450–56. doi:10.1038/gt.2014.8.
- De Silva SR, Charbel Issa P, Singh MS, Lipinski DM, Barnea-Cramer AO, Walker NJ, Barnard AR, Hankins MW, MacLaren RE. 2016. Single residue AAV capsid mutation improves transduction of photoreceptors in the Abca4(-/-) mouse and bipolar cells in the rd1 mouse and human retina ex vivo. Gene Ther. 23(11):767–74. doi:10.1038/gt.2016.54
- Kong J, Kim SR, Binley K, Pata I, Doi K, Mannik J, Zernant-Rajang J, Kan O, Iqball S, Naylor S, et al. Correction of the disease phenotype in the mouse model of Stargardt disease by lentiviral gene therapy. Gene Ther. 2008;15(19):1311–20. doi:10.1038/gt.2008.78.
- Binley K, Widdowson P, Loader J, Kelleher M, Iqball S, Ferrige G, de Belin J, Carlucci M, Angell-Manning D, Hurst F, et al. Transduction of photoreceptors with equine infectious anemia virus lentiviral vectors: safety and biodistribution of StarGen for Stargardt disease. Invest Ophthalmol Vis Sci. 2013;54(6):4061–71. doi:10.1167/iovs.13-11871.
- Han Z, Conley SM, Makkia RS, Cooper MJ, Naash MI. 2012. DNA nanoparticle-mediated ABCA4 delivery rescues Stargardt dystrophy in mice. J Clin Invest. 122(9):3221–26. doi:10.1172/JCI64833
- Han Z, Conley SM, Naash MI. Gene therapy for Stargardt disease associated with ABCA4 gene. Adv Exp Med Biol. 2014;801:719–24. doi:10.1007/978-1-4614-3209-8_90.
- Cai X, Conley SM, Nash Z, Fliesler SJ, Cooper MJ, Naash MI. 2010. Gene delivery to mitotic and postmitotic photoreceptors via compacted DNA nanoparticles results in improved phenotype in a mouse model of retinitis pigmentosa. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology. 24(4):1178–91. doi:10.1096/fj.09-139147
- Han Z, Conley SM, Makkia R, Guo J, Cooper MJ, Naash MI. 2012. Comparative analysis of DNA nanoparticles and AAVs for ocular gene delivery. PloS One. 7(12):e52189. doi:10.1371/journal.pone.0052189
- Farjo R, Skaggs J, Quiambao AB, Cooper MJ, Naash MI. Efficient non-viral ocular gene transfer with compacted DNA nanoparticles. PloS One. 2006;1:e38. doi:10.1371/journal.pone.0000038.
- Conley SM, Naash MI. 2010. Nanoparticles for retinal gene therapy. Prog Retin Eye Res. 29(5):376–97. doi:10.1016/j.preteyeres.2010.04.004
- Ding XQ, Quiambao AB, Fitzgerald JB, Cooper MJ, Conley SM, Naash MI. Ocular delivery of compacted DNA-nanoparticles does not elicit toxicity in the mouse retina. PloS One. 2009;4(10):e7410. doi:10.1371/journal.pone.0007410.
- Paques F, Meganucleases DP.DNA double-strand break-induced recombination: perspectives for gene therapy. Curr Gene Ther. 2007;7(1):49–66.
- Bogdanove AJ, Voytas DF. 2011. TAL effectors: customizable proteins for DNA targeting. Science (New York, NY). 333(6051):1843–46. doi:10.1126/science.1204094
- Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. 2010. Genome editing with engineered zinc finger nucleases. Nat Rev Genet. 11(9):636–46. doi:10.1038/nrg2842
- Hsu PD, Lander ES, Zhang F. 2014. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 157(6):1262–78. doi:10.1016/j.cell.2014.05.010
- Rouet P, Smih F, Jasin M.Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol Cell Biol. 1994;14(12):8096–106.
- Davis L, Maizels N. Homology-directed repair of DNA nicks via pathways distinct from canonical double-strand break repair. Proc Natl Acad Sci U S A. 2014;111:E924–32. doi:10.1073/pnas.1400236111.
- Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. 2016. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 533(7603):420–24. doi:10.1038/nature17946
- Cox DB, Platt RJ, Zhang F. 2015. Therapeutic genome editing: prospects and challenges. Nat Med. 21(2):121–31. doi:10.1038/nm.3793
- Chuang K, Fields MA, Del Priore LV.Potential of Gene Editing and Induced Pluripotent Stem Cells (iPSCs) in Treatment of Retinal Diseases. Yale J Biol Med. 2017;90(4):635–42.
- Hung SSC, McCaughey T, Swann O, Pebay A, Hewitt AW. Genome engineering in ophthalmology: application of CRISPR/Cas to the treatment of eye disease. Prog Retin Eye Res. 2016;53:1–20. doi:10.1016/j.preteyeres.2016.05.001.
- Er B, JC G, Ja C, Jr T, Lr B, Kr C, Av D, Jh F, Ks W, La W, et al. CRISPR-Cas9 genome engineering: treating inherited retinal degeneration. Prog Retin Eye Res. 2018; doi:10.1016/j.preteyeres.2018.03.003.
- Moore CBT, Christie KA, Marshall J, Nesbit MA. Personalised genome editing - The future for corneal dystrophies. Prog Retin Eye Res. 2018. doi:10.1016/j.preteyeres.2018.01.004.
- Lieber MR. 2008. The mechanism of human nonhomologous DNA end joining. J Biol Chem. 283(1):1–5. doi:10.1074/jbc.R700039200
- Sfeir A, Symington LS. 2015. Microhomology-Mediated End Joining: a Back-up Survival Mechanism or Dedicated Pathway? Trends Biochem Sci. 40(11):701–14. doi:10.1016/j.tibs.2015.08.006
- Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, Liu DR. 2017. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature. 551(7681):464–71. doi:10.1038/nature24644
- Ihry RJ, Worringer KA, Salick MR, Frias E, Ho D, Theriault K, Kommineni S, Chen J, Sondey M, Ye C, et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat Med. 2018; doi:10.1038/s41591-018-0050-6.
- Haapaniemi E, Botla S, Persson J, Schmierer B, Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat Med. 2018. doi:10.1038/s41591-018-0049-z.
- Wilton SD, Fletcher S.RNA splicing manipulation: strategies to modify gene expression for a variety of therapeutic outcomes. Curr Gene Ther. 2011;11(4):259–75.
- Gerard X, Perrault I, Munnich A, Kaplan J, Rozet JM. Intravitreal Injection of Splice-switching Oligonucleotides to Manipulate Splicing in Retinal Cells. Mol Ther Nucleic Acids. 2015;4:e250. doi:10.1038/mtna.2015.24.
- Cideciyan AV, Jacobson SG, Drack AV, Ho AC, Charng J, Garafalo AV, Roman AJ, Sumaroka A, Han IC, Hochstedler MD, et al. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nat Med. 2019;25(2):225–28. doi:10.1038/s41591-018-0295-0.
- Kim J, Hu C, Moufawad El Achkar C, Le B, Douville J, Larson A, Mk P, Sf G, Ea L, Kuniholm A, et al. Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease. N Engl J Med. 2019;381(17):1644–52. doi:10.1056/NEJMoa1813279.
- Jones MK, Lu B, Girman S, Wang S. Cell-based therapeutic strategies for replacement and preservation in retinal degenerative diseases. Prog Retin Eye Res. 2017;58:1–27. doi:10.1016/j.preteyeres.2017.01.004.
- Ramsden CM, Powner MB, Carr AJ, Smart MJ, Da Cruz L, Coffey PJ. 2013. Stem cells in retinal regeneration: past, present and future. Development (Cambridge, England). 140(12):2576–85. doi:10.1242/dev.092270
- Brandl C, Zimmermann SJ, Milenkovic VM, Rosendahl SM, Grassmann F, Milenkovic A, Hehr U, Federlin M, Wetzel CH, Helbig H, et al. In-depth characterisation of Retinal Pigment Epithelium (RPE) cells derived from human induced pluripotent stem cells (hiPSC). Neuromolecular Med. 2014;16(3):551–64. doi:10.1007/s12017-014-8308-8.
- Vugler A, Carr AJ, Lawrence J, Chen LL, Burrell K, Wright A, Lundh P, Semo M, Ahmado A, Gias C, et al. Elucidating the phenomenon of HESC-derived RPE: anatomy of cell genesis, expansion and retinal transplantation. Exp Neurol. 2008;214(2):347–61. doi:10.1016/j.expneurol.2008.09.007.
- Kokkinaki M, Sahibzada N, Golestaneh N. 2011. Human induced pluripotent stem-derived retinal pigment epithelium (RPE) cells exhibit ion transport, membrane potential, polarized vascular endothelial growth factor secretion, and gene expression pattern similar to native RPE. Stem Cells (Dayton, Ohio). 29(5):825–35. doi:10.1002/stem.635
- Zhu D, Deng X, Spee C, Sonoda S, Hsieh CL, Barron E, Pera M, Hinton DR. 2011. Polarized secretion of PEDF from human embryonic stem cell-derived RPE promotes retinal progenitor cell survival. Invest Ophthalmol Vis Sci. 52(3):1573–85. doi:10.1167/iovs.10-6413
- Carr AJ, Vugler A, Lawrence J, Chen LL, Ahmado A, Chen FK, Semo M, Gias C, Da Cruz L, Moore HD, et al. Molecular characterization and functional analysis of phagocytosis by human embryonic stem cell-derived RPE cells using a novel human retinal assay. Mol Vis. 2009;15:283–95.
- Idelson M, Alper R, Obolensky A, Ben-Shushan E, Hemo I, Yachimovich-Cohen N, Khaner H, Smith Y, Wiser O, Gropp M, et al. Directed differentiation of human embryonic stem cells into functional retinal pigment epithelium cells. Cell Stem Cell. 2009;5(4):396–408. doi:10.1016/j.stem.2009.07.002.
- Fields M, Cai H, Gong J, Del Priore L. Potential of Induced Pluripotent Stem Cells (iPSCs) for Treating Age-Related Macular Degeneration (AMD). Cells. 2016;5:4. doi:10.3390/cells5040044.
- Klimanskaya I, Hipp J, Rezai KA, West M, Atala A, Lanza R. 2004. Derivation and comparative assessment of retinal pigment epithelium from human embryonic stem cells using transcriptomics. Cloning Stem Cells. 6(3):217–45. doi:10.1089/clo.2004.6.217
- Peng S, Gan G, Qiu C, Zhong M, An H, Adelman RA, Rizzolo LJ. 2013. Engineering a blood-retinal barrier with human embryonic stem cell-derived retinal pigment epithelium: transcriptome and functional analysis. Stem Cells Transl Med. 2(7):534–44. doi:10.5966/sctm.2012-0134
- Liu Z, Jiang R, Yuan S, Wang N, Feng Y, Hu G, Zhu X, Huang K, Ma J, Xu G, et al. Integrated analysis of DNA methylation and RNA transcriptome during in vitro differentiation of human pluripotent stem cells into retinal pigment epithelial cells. PLoS One. 2014;9(3):e91416. doi:10.1371/journal.pone.0091416.
- Kim K, Doi A, Wen B, Ng K, Zhao R, Cahan P, Kim J, MJ A, Ji H, Ehrlich LI, et al. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467(7313):285–90. doi:10.1038/nature09342.
- Haruta M, Sasai Y, Kawasaki H, Amemiya K, Ooto S, Kitada M, Suemori H, Nakatsuji N, Ide C, Honda Y, et al. In vitro and in vivo characterization of pigment epithelial cells differentiated from primate embryonic stem cells. Invest Ophthalmol Vis Sci 2004;45:1020–25.
- Westenskow PD, Bucher F, Bravo S, Kurihara T, Feitelberg D, Paris LP, Aguilar E, Lin JH, Friedlander M. iPSC-Derived Retinal Pigment Epithelium Allografts Do Not Elicit Detrimental Effects in Rats: a Follow-Up Study. Stem Cells Int. 2016;2016:8470263. doi:10.1155/2016/8470263.
- Li Y, Tsai YT, Hsu CW, Erol D, Yang J, Wu WH, Davis RJ, Egli D, Tsang SH. Long-term safety and efficacy of human-induced pluripotent stem cell (iPS) grafts in a preclinical model of retinitis pigmentosa. Molecular Medicine (Cambridge, Mass). 2012;18:1312–19. doi:10.2119/molmed.2012.00242.
- Carr AJ, Vugler AA, Hikita ST, Lawrence JM, Gias C, Chen LL, Buchholz DE, Ahmado A, Semo M, Smart MJ, et al. Protective effects of human iPS-derived retinal pigment epithelium cell transplantation in the retinal dystrophic rat. PLoS One. 2009;4(12):e8152. doi:10.1371/journal.pone.0008152.
- Lund RD, Wang S, Klimanskaya I, Holmes T, Ramos-Kelsey R, Lu B, Girman S, Bischoff N, Sauve Y, Lanza R. 2006. Human embryonic stem cell-derived cells rescue visual function in dystrophic RCS rats. Cloning Stem Cells. 8(3):189–99. doi:10.1089/clo.2006.8.189
- Schwartz SD, Hubschman J-P, Heilwell G, Franco-Cardenas V, Pan CK, Ostrick RM, Mickunas E, Gay R, Klimanskaya I, Lanza R. 2012. Embryonic stem cell trials for macular degeneration: a preliminary report. The Lancet. 379(9817):713–20. doi:10.1016/s0140-6736(12)60028-2
- Schwartz SD, Regillo CD, Lam BL, Eliott D, Rosenfeld PJ, Gregori NZ, Hubschman J-P, Davis JL, Heilwell G, Spirn M, et al. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt’s macular dystrophy: follow-up of two open-label phase 1/2 studies. The Lancet. 2015;385(9967):509–16. doi:10.1016/s0140-6736(14)61376-3.
- Mehat MS, Sundaram V, Ripamonti C, Robson AG, Smith AJ, Borooah S, Robinson M, Rosenthal AN, Innes W, Weleber RG, et al. Transplantation of Human Embryonic Stem Cell-Derived Retinal Pigment Epithelial Cells in Macular Degeneration. Ophthalmology. 2018;125(11):1765–75. doi:10.1016/j.ophtha.2018.04.037.
- Drukker M, Katchman H, Katz G, Even-Tov Friedman S, Shezen E, Hornstein E, Mandelboim O, Reisner Y, Benvenisty N. 2006. Human embryonic stem cells and their differentiated derivatives are less susceptible to immune rejection than adult cells. Stem Cells. 24(2):221–29. doi:10.1634/stemcells.2005-0188
- Cuevas E, Parmar P, Sowden JC. Restoring Vision Using Stem Cells and Transplantation. Adv Exp Med Biol. 2019;1185:563–67. doi:10.1007/978-3-030-27378-1_92.
- Jin ZB, Gao ML, Deng WL, Wu KC, Sugita S, Mandai M, Takahashi M. Stemming retinal regeneration with pluripotent stem cells. Prog Retin Eye Res. 2019;69:38–56. doi:10.1016/j.preteyeres.2018.11.003.
- MacLaren RE, Pearson RA, MacNeil A, Douglas RH, Salt TE, Akimoto M, Swaroop A, Sowden JC, Ali RR. Retinal repair by transplantation of photoreceptor precursors. Nature. 2006;444(7116):203–07. doi:10.1038/nature05161.
- Akimoto M, Cheng H, Zhu D, Brzezinski JA, Khanna R, Filippova E, Oh EC, Jing Y, Linares JL, Brooks M, et al. Targeting of GFP to newborn rods by Nrl promoter and temporal expression profiling of flow-sorted photoreceptors. Proc Natl Acad Sci U S A. 2006;103(10):3890–95. doi:10.1073/pnas.0508214103.
- Pearson RA, Barber AC, Rizzi M, Hippert C, Xue T, West EL, Duran Y, Smith AJ, Chuang JZ, Azam SA, et al. Restoration of vision after transplantation of photoreceptors. Nature. 2012;485(7396):99–103. doi:10.1038/nature10997.
- Barber AC, Hippert C, Duran Y, West EL, Bainbridge JW, Warre-Cornish K, Luhmann UF, Lakowski J, Sowden JC, Ali RR, et al. Repair of the degenerate retina by photoreceptor transplantation. Proc Natl Acad Sci U S A. 2013;110(1):354–59. doi:10.1073/pnas.1212677110.
- Santos-Ferreira T, Postel K, Stutzki H, Kurth T, Zeck G, Ader M. 2015. Daylight vision repair by cell transplantation. Stem Cells. 33(1):79–90. doi:10.1002/stem.1824
- Singh MS, Balmer J, Barnard AR, Aslam SA, Moralli D, Green CM, Barnea-Cramer A, Duncan I, MacLaren RE. Transplanted photoreceptor precursors transfer proteins to host photoreceptors by a mechanism of cytoplasmic fusion. Nat Commun. 2016;7:13537. doi:10.1038/ncomms13537.
- Llonch S, Carido M, Ader M. 2018. Organoid technology for retinal repair. Dev Biol. 433(2):132–43. doi:10.1016/j.ydbio.2017.09.028
- McLenachan S, Zhang D, Hao E, Zhang L, Chen SC, Chen FK. 2017. Human limbal neurospheres prevent photoreceptor cell death in a rat model of retinal degeneration. Clin Exp Ophthalmol. 45(6):613–24. doi:10.1111/ceo.12940
- Park SS. 2016. Cell Therapy Applications for Retinal Vascular Diseases: diabetic Retinopathy and Retinal Vein Occlusion. Invest Ophthalmol Vis Sci. 57(5):ORSFj1–ORSFj10. doi:10.1167/iovs.15-17594
- Asahara T, Murohara T, Sullivan A, Silver M, Van Der Zee R, Li T, Witzenbichler B, Schatteman G, Isner JM.Isolation of putative progenitor endothelial cells for angiogenesis. Science (New York, NY). 1997;275(5302):964–67.
- Park SS, Moisseiev E, Bauer G, Anderson JD, Grant MB, Zam A, Zawadzki RJ, Werner JS, Nolta JA. Advances in bone marrow stem cell therapy for retinal dysfunction. Prog Retin Eye Res. 2017;56:148–65. doi:10.1016/j.preteyeres.2016.10.002.
- Park SS, Bauer G, Abedi M, Pontow S, Panorgias A, Jonnal R, Zawadzki RJ, Werner JS, Nolta J.Intravitreal autologous bone marrow CD34+ cell therapy for ischemic and degenerative retinal disorders: preliminary phase 1 clinical trial findings. Invest Ophthalmol Vis Sci. 2015;56(1):81–89.
- Kuriyan AE, Albini TA, Townsend JH, Rodriguez M, Pandya HK, Leonard RE 2nd, Parrott MB, Rosenfeld PJ, Flynn HW Jr., Goldberg JL. 2017. Vision Loss after Intravitreal Injection of Autologous “Stem Cells” for AMD. N Engl J Med. 376(11):1047–53. doi:10.1056/NEJMoa1609583
- Jacobson SG, Matsui R, Sumaroka A, Cideciyan AV. 2016. Retinal Structure Measurements as Inclusion Criteria for Stem Cell-Based Therapies of Retinal Degenerations. Invest Ophthalmol Vis Sci. 57(5):ORSFn1–9. doi:10.1167/iovs.15-17654
- Dalkara D, Goureau O, Marazova K, Sahel JA. 2016. Let There Be Light: gene and Cell Therapy for Blindness. Hum Gene Ther. 27(2):134–47. doi:10.1089/hum.2015.147
- Lin B, Koizumi A, Tanaka N, Panda S, Masland RH. 2008. Restoration of visual function in retinal degeneration mice by ectopic expression of melanopsin. Proc Natl Acad Sci U S A. 105(41):16009–14. doi:10.1073/pnas.0806114105
- Cehajic-Kapetanovic J, Eleftheriou C, Allen AE, Milosavljevic N, Pienaar A, Bedford R, Davis KE, Bishop PN, Lucas RJ. 2015. Restoration of Vision with Ectopic Expression of Human Rod Opsin. Curr Biol. 25(16):2111–22. doi:10.1016/j.cub.2015.07.029
- Yue L, Weiland JD, Roska B, Humayun MS. Retinal stimulation strategies to restore vision: fundamentals and systems. Prog Retin Eye Res. 2016;53:21–47. doi:10.1016/j.preteyeres.2016.05.002.
- Bi A, Cui J, Ma YP, Olshevskaya E, Pu M, Dizhoor AM, Pan ZH. 2006. Ectopic expression of a microbial-type rhodopsin restores visual responses in mice with photoreceptor degeneration. Neuron. 50(1):23–33. doi:10.1016/j.neuron.2006.02.026
- Busskamp V, Duebel J, Balya D, Fradot M, Viney TJ, Siegert S, Groner AC, Cabuy E, Forster V, Seeliger M, et al. Genetic reactivation of cone photoreceptors restores visual responses in retinitis pigmentosa. Science. 2010;329(5990):413–17. doi:10.1126/science.1190897.
- Doroudchi MM, Greenberg KP, Liu J, Silka KA, Boyden ES, Lockridge JA, Arman AC, Janani R, Boye SE, Boye SL, et al. Virally delivered channelrhodopsin-2 safely and effectively restores visual function in multiple mouse models of blindness. Mol Ther. 2011;19(7):1220–29. doi:10.1038/mt.2011.69.
- Macé E, Caplette R, Marre O, Sengupta A, Chaffiol A, Barbe P, Desrosiers M, Bamberg E, Sahel JA, Picaud S, et al. Targeting channelrhodopsin-2 to ON-bipolar cells with vitreally administered AAV Restores ON and OFF visual responses in blind mice. Mol Ther. 2015;23(1):7–16. doi:10.1038/mt.2014.154.
- Garita-Hernandez M, Lampič M, Chaffiol A, Guibbal L, Routet F, Santos-Ferreira T, Gasparini S, Borsch O, Gagliardi G, Reichman S, et al. Restoration of visual function by transplantation of optogenetically engineered photoreceptors. Nat Commun. 2019;10(1):4524. doi:10.1038/s41467-019-12330-2.
- Cheng DL, Greenberg PB, Borton DA. 2017. Advances in Retinal Prosthetic Research: a Systematic Review of Engineering and Clinical Characteristics of Current Prosthetic Initiatives. Curr Eye Res. 42(3):334–47. doi:10.1080/02713683.2016.1270326
- YH L, Da Cruz L. The Argus((R)) II Retinal Prosthesis System. Prog Retin Eye Res. 2016;50:89–107. doi:10.1016/j.preteyeres.2015.09.003.
- Endo T, Fujikado T, Hirota M, Kanda H, Morimoto T, Nishida K. 2018. Light localization with low-contrast targets in a patient implanted with a suprachoroidal-transretinal stimulation retinal prosthesis. Graefes Arch Clin Exp Ophthalmol. 256(9):1723–29. doi:10.1007/s00417-018-3982-0
- Slijkerman RW, Song F, Astuti GD, Huynen MA, van Wijk E, Stieger K, Collin RW. The pros and cons of vertebrate animal models for functional and therapeutic research on inherited retinal dystrophies. Prog Retin Eye Res. 2015;48:137–59. doi:10.1016/j.preteyeres.2015.04.004.