
ABSTRACT
Background
Inherited retinal degeneration (IRD) associated with mutations in the Crumbs homolog 1 (CRB1) gene is associated with a severe, early-onset retinal degeneration for which no therapy currently exists. Base editing, with its capability to precisely catalyse permanent nucleobase conversion in a programmable manner, represents a novel therapeutic approach to targeting this autosomal recessive IRD, for which a gene supplementation is challenging due to the need to target three different retinal CRB1 isoforms.
Purpose
To report and classify a novel CRB1 variant and envision a possible therapeutic approach in form of base editing.
Methods
Case report.
Results
A 16-year-old male patient with a clinical diagnosis of early-onset retinitis pigmentosa (RP) and characteristic clinical findings of retinal thickening and coarse lamination was seen at the Oxford Eye Hospital. He was found to be compound heterozygous for two CRB1 variants: a novel pathogenic nonsense variant in exon 9, c.2885T>A (p.Leu962Ter), and a likely pathogenic missense change in exon 6, c.2056C>T (p.Arg686Cys). While a base editing strategy for c.2885T>A would encompass a CRISPR-pass mediated “read-through” of the premature stop codon, the resulting missense changes were predicted to be “possibly damaging” in in-silico analysis. On the other hand, the transversion missense change, c.2056C>T, is amenable to transition editing with an adenine base editor (ABE) fused to a SaCas9-KKH with a negligible chance of bystander edits due to an absence of additional Adenines (As) in the editing window.
Conclusions
This case report records a novel pathogenic nonsense variant in CRB1 and gives an example of thinking about a base editing strategy for a patient compound heterozygous for CRB1 variants.
Introduction
CRB1 gene
The Crumbs homolog 1 (CRB1) gene is located in chromosomal region 1q31.3 and is expressed as a highly conserved transmembrane protein in retina and brain of humans and other mammals (Citation1,Citation2). The retinal function of CRB1 has yet to be comprehensively understood, but it is considered to play an important role in both structural integrity and development of the retina: studies in Drosophila and humans point toward CRB1 being integral for cell-to-cell adhesion and the establishment of cellular polarity as well as photoreceptor morphogenesis and retinal maturation (Citation3–8). The phenotypic hallmark of CRB1-associated inherited retinal degeneration (IRD) supports the hypothesis of its key role in retinal development: much like an immature retina, the retina of a patient presenting with a biallelic CRB1 mutations is grossly thickened and shows loss of distinct inner retinal lamination (Citation9,Citation10). These lamination defects in humans with pathogenic CRB1 variants are also present in CRB1 knockout mouse models (Citation11).
Phenotype of CRB1-associated IRDs
Biallelic CRB1 mutations have been linked to 3–9% of autosomal recessive retinitis pigmentosa (RP) cases and 7–17% of autosomal recessive Leber congenital amaurosis (LCA) cases (Citation12–14). LCA presents at birth with severe visual impairment, absent responses on electroretinography and nystagmus and is considered to be one of the most severe forms of retinal dystrophy (Citation15). RP can become symptomatic at different ages and is phenotypically heterogeneous, although most forms are characterised by a rod-cone pattern of photoreceptor degeneration, which causes night blindness and a concentric, progressive loss of visual fields (Citation16). Both RP and LCA can be caused by a broad spectrum of pathogenic gene variants (Citation17). The characteristic morphologic hallmark of CRB1-associated IRD is the above-mentioned thickening of the retina, which stands in contrast to other molecular forms of RP or LCA, in which the outer retina progressively thins due to photoreceptor loss (Citation18). Other reported phenotypic hallmarks of CRB1-associated IRDs include cystoid macula edema, preservation of the para-arteriolar retinal pigment epithelium (RPE), nummular pigmentation, and Coats-like exudates (Citation10,Citation19).
CRB1 isoforms
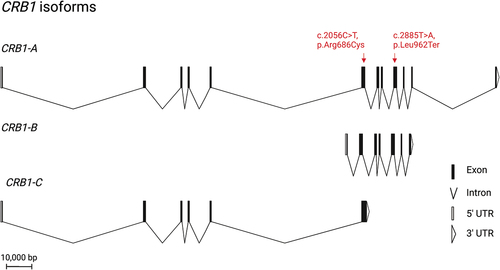
In the human retina, three isoforms of CRB1, CRB1-A, CRB1-B, and CRB1-C, are expressed at meaningful levels and differ enough in their sequence to encode a functional difference on a protein level (Citation20) (). In humans, CRB1-A has been shown to localise to photoreceptors and Müller Glia Cells (MGCs) (Citation5,Citation21). In mice, CRB1-B has been shown to localise exclusively to photoreceptors, while CRB1-A is found in MGCs (Citation20). The protein’s constitutive isoform, CRB1-A, has a large extracellular domain made up of 19 epidermal growth factor (EGF)-like and 3 laminin A-like domains, as well a highly conserved transmembrane domain followed by a short intracellular domain that contains FERM/PDZ binding motifs (Citation12). The CRB1-B isoform is the most abundant retinal isoform and has a unique C- and N-terminus, but shares the transmembrane domain as well as a large part of the extracellular domain with CRB1-A (Citation20).
Figure 1. Overview of the three retinal isoforms of CRB1 and its location. All isoforms share exon 6. CRB1-B is the most abundant retinal isoform. It has a unique N-and C-terminus while sharing the transmembrane domain as well as a large part of the extracellular domain with CRB1-A. the patient’s novel nonsense mutation affects CRB1-A and CRB1-B, while the missense mutation affects all retinal CRB1 isoforms. CRB1 is thought to play an important role in maintaining the integrity of the OLM.
Therapeutic options for CRB1-associated IRDs
Currently, there are no therapies available for CRB1-associated IRDs. Gene therapy utilizing Adeno Associated Viruses (AAVs) as vectors for retinal transgene delivery has established itself as a safe and efficacious treatment for autosomal and X-linked pathogenic mutations in humans (Citation22–24) and proof of principle studies for the treatment of CRB1-induced retinal degeneration have focused on delivering CRB2 to rescue a CRB1-induced phenotype in murine models (Citation5,Citation25). A gene supplementation approach is inherently limited by its capability to only supplement one of the three CRB1 isoforms. By targeting the endogenous genomic sequence for correction rather than providing an exogenous cDNA template, CRISPR-Cas mediated base editing, with its ability to irreversibly correct point mutations by chemical modification of nucleobases, shows great promise in addressing mutations not amenable to gene supplementation, either due to AAV packaging constraints or the presence of multiple isoforms. In previous reports, CRB1 gene supplementation and subsequent overexpression resulted in reduced retinal function an ERG, no improvement of visual function and negatively affected retinal morphology (Citation5,Citation25), making gene editing a particularly attractive alternative for this gene.
Base editing with CRISPR-Cas
The field of genome engineering was revolutionised when it was shown that the class 2 type II CRISPR-Cas9 system, an RNA-guided DNA endonuclease utilised by select bacteria as an innate immune defence (Citation26–28), could be harnessed to introduced programmed, targeted DNA cleavage in living eukaryotic cells with unprecedented ease and adaptability (Citation29).
Base editing avoids backbone cleavage of DNA and utilises naturally occurring deaminases fused to a partially or fully deactivated Cas9 to introduce single nucleotide variants (SNVs) into the human genome by directly and irreversibly chemically modifying target nucleobases (Citation30,Citation31). Cytosine base editors (CBEs) take advantage of the cytidine deaminase enzyme rAPOBEC1 to deaminate Cytosine (C) to Uracil (U), which is read as a thymine (T) by the endogenous DNA mismatch repair machinery, therefore catalysing a C:G to T:A conversion (bold letters indicate nucleobases on the edited strand/directly edited nucleobases in bold) (Citation30). Adenine base editors (ABEs) exploit the naturally occurring TadA deaminase of Escherischia coli (ecTadA) to catalyse A:T to G:C edits (Citation31). Together, CBEs and ABEs are capable of targeting all four transition mutations.
Recent optimisations of base editors have focused on decreasing genome-wide DNA and RNA off-target editing events and bystander edits (Citation32–35). Also, the targeting scope has been improved by using both SpCas9 variants with alternate PAM requirements (Citation36–39) and SpCas9 homologues, such as SaCas9 and CjCas9 with relaxed PAM requirements into the BE architecture (Citation40–42). More recently, glycosylase base editors (GBE) capable of targeting C:G to G:C transversion mutations have been reported (Citation43–45). Base editing presents a promising therapeutic avenue for pathogenic CRB1 variants, since 62% of reported pathogenic SNVs are targetable with a base editor and of these, 87% of these have a suitable PAM site (Citation46).
In this case report, we focus on envisioning a strategy of targeted single nucleotide alteration in genomic DNA via base editing as a treatment option for a patient compound heterozygous for pathogenic CRB1 variants.
Materials and methods
Infrared (IF) and autofluorescence (AF, Spectralis, Heidelberg Engineering Inc., Heidelberg, Germany) imaging as well as an optical coherence tomography (OCT, Heidelberg Engineering Inc., Heidelberg, Germany) were performed on a 16-year-old male patient. Based on the clinical diagnosis, informed consent for DNA blood sampling was taken from the patient and molecular genetic analysis for retinitis pigmentosa and RP-like phenotypes was undertaken by exome sequencing of 111 genes associated with RP and RP-like phenotypes (RP 111 gene panel) with next generation sequencing (NGS). Pathogenic variants were confirmed with Sanger sequencing. Geneious software (Version 11.0) was used to further investigate the identified mutations and their amino acid consequences as well as PAM site locations and potential guide RNA (gRNA) designs. The Leiden Open Variation Database (LOVD) and ClinVar database were used to search for previously reported CRB1 variants. Computational evidence for functional in-silico prediction of variant effects was evaluated with the following tools: Polymorphism Phenotyping version 2 (PolyPhen2), Sorting Intolerant from Tolerant (SIFT) and Mutation Taster (Citation47–49). Amino acid preservation between species was evaluated using Geneious (Version 11.0) and Mutation Taster software.
Results
Clinical results
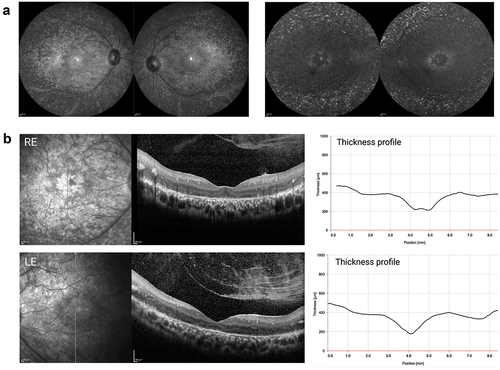
The 16-year-old male presented with a visual acuity (VA) of 6/36 (20/125 or 0.8 LogMAR) in both eyes, which improved to 6/24 (20/80 or 0.6 LogMAR) in both eyes with pinhole. He reportedly experienced night vision problems since birth and was formally diagnosed with early-onset RP at the age of 7. No systemic changes indicative of a syndromic form of RP were noted. IF and AF imaging at 55 degrees as well as OCT showed symmetric changes in both eyes. Concentric atrophy in the perifoveal region seen on IF and OCT imaging corresponded to a perifoveal ring of hypofluorescence surrounded by a hyperfluorescent halo. The fluorescence outside the vascular arcades was dominated by hyperfluorescent spots (). On OCT, a central island of remaining photoreceptors could be identified in the fovea of both eyes with some residual ellipsoid zone (EZ) signal in the right eye (). In a characteristic fashion for a CRB1-associated RP, there was gross thickening of the retina (400 µm outside the foveal depression) with loss of a distinct retinal lamination pattern disrupted by intraretinal exudates. This prevented correct identification of the retinal layers by the automated segmentation algorithm and made the manual correction challenging. The choroid appeared normal beyond the Bruch’s membrane (BM), while a prominent inner limiting membrane (ILM) faced a vitreous body with incomplete posterior vitreous detachment and vitreoschisis.
Figure 2. Clinical phenotype of patient with compound heterozygous for single nucleotide variants in CRB1. (a) Infrared (left) and autofluorescence (right) imaging shows atrophic changes in the central retina with hyperfluorescent spots outside the temporal arcades. (b) Characteristic for a CRB1-associated RP, both left and right eye show increased retinal thickness. the retinal lamination pattern is coarse, reminiscent of an immature retina and disrupted by intraretinal exudates.
Genetic testing results and ACMG classification
The exome sequencing panel revealed a novel nonsense variant, c.2885T>A (p.Leu962Ter), in exon 9 of CRB1-A. The location of the pathogenic variant corresponds to the Laminin A-like domain 3 in extracellular domain of CRB1 and affects the two most abundant retinal CRB1 isoforms, CRB-1A, and CRB1-B. A second missense change, c.2056C>T (p.Arg686Cys) was found in exon 6 of CRB1, which corresponds to a location in the extracellular EGF-like 12 domain of the CRB1 protein. The very strong criteria of pathogenicity PVS1 can be applied to the novel nonsense variant c.2885T>A (p.Leu962Ter), since it is predicted to lead to nonsense mediated decay (NMD) and therefore loss of function (LOF) in CRB1. The nonsense variant is absent from the Genome Aggregation Database (gnomAD), the Exome Aggregation Consortium (ExAC) database as well as the 1000 Genes (1000 G) databases, which makes the PM2 criteria applicable. Further, supporting criteria for pathogenicity, PP4, can be applied to this variant: and the patient’s phenotype of early onset RP with thickened retina is highly specific for a monogenetic, CRB1-associated retinal disease.
PP4 can also be applied to the missense variant c.2056C>T (p.Arg686Cys). This missense variant occurs with the novel pathogenic variant c.2885T>A (p.Leu962Ter). Paternal segregation analysis revealed the patient’s father as carrier of this nonsense variant as well as an absence of the c.2056C>T in the paternal genome, but since segregation analysis could not be performed on the mother, phase is unknown. Wang et al. (Citation50) and Liu et al. (Citation51) describe a patient compound heterozygous for with the same missense variant c.2056C>T (p.Arg686Cys) and a pathogenic frameshift variant c.2540_2541delTC (p.Phe848Glnfs *60). Phase is unknown in this patient as well. Taking this evidence together, the PM3_Moderate criteria can be applied to this mutation. When entered into three computational predictive tools, PolyPhen2, SIFT, and Mutation Taster, the c.2056C>T variant was predicted to be “probably damaging” (0.92, Sensitivity = 0.68, Specificity = 0.90), “damaging” and “deleterious” for all protein-coding CRB1 isoforms by the three respective algorithms. The high evolutionary conservation across a broad range of species further supports the PP3 evidence criteria. Importantly, a Cysteine has not been observed at the equivalent amino acid position in any species, indicating a low tolerance toward this amino acid. The missense variant has been reported a total of 9 times in the LOVD and ClinVar database, with conflicting interpretation. As mentioned above, both Wang et al. (Citation50) and Liu et al. (Citation51) describe the same patient compound heterozygous for a pathogenic frameshift variant and the c.2056C>T missense variant, which they label as “likely pathogenic”, without detailing the criteria that led to this ACMG labeling (Citation50,Citation51). Conversely, all seven independent laboratory reports in the ClinVar database and 2 out of 4 reports in the LOVD label the missense change as a VUS. Due to this conflicting variant classification, as well as the recommendation that PP5 evidence criteria be removed from the ACMG classification guidelines (Citation52), this evidence criteria could not be used to support pathogenicity. Two missense variants, c.2057 G>A (p.Arg686His) and c.2057 G>C (p.Arg686Pro), affecting the same Arginine amino acid at position 686 have been reported in ClinVar and in Gao et al. (2019), but neither of these have been classified as pathogenic, which precludes an application of the moderate pathogenicity criteria PM5 (Citation53). Lastly, the missense variant c.2056C>T has not been reported in ExAC or 100 G databases and is reported as a single heterozygous occurrence in >250,000 alleles in gnomAD. This extremely low frequency is consistent with this variant being a rare recessive variant. PM2 can be applied in this instance. In summary, as per the ACMG guidelines, the missense variant c.2056C>T is labeled as a likely pathogenic variant. For a summary of the ACMG classification of the patient’s CRB1 variants, refer to .
Table 1. Classification of the novel CRB1 c.2885T>A (p.Leu962Ter) and CRB1 c.2056C>T (p.Arg686Cys) variant using ACMG standards and guidelines.
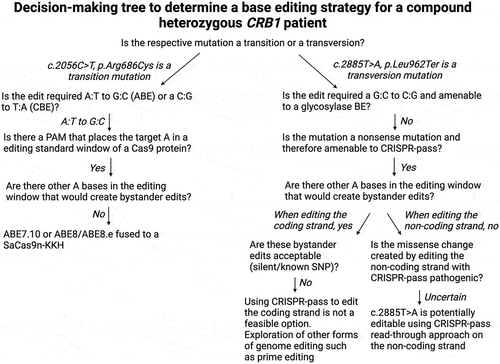
In silico approach to a CRISPR-Cas mediated base editing strategy
Editing of a premature termination codon to a missense variant
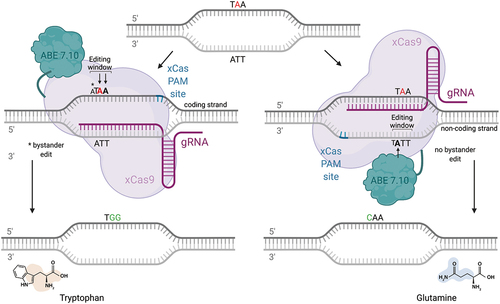
Although the patient’s novel nonsense mutation, c.2885T>A (p.Leu962Ter) is not amenable to correction with a currently available base editor, the premature termination stop codon (PTC) could be converted to a translatable codon by a base editor, thus allowing the production of a full-length protein. To this end, Lee et al. developed “CRIPSR-pass”, an ABE7.10 fused to the evolved SpCas9 variant xCas3.7 (Citation54). By targeting the coding strand, the patient’s PTC TAA would be converted to a TGG codon, which would be read as Tryptophan (Trp, W) by the cell’s translational machinery. Alternatively, by targeting the first base of the codon on the non-coding strand (ATT>GTT), the novel nonsense mutation would be converted to Glutamine (Gln,Q) (). The evaluation of the functional effect of these induced missense changes is critical to assess therapeutic success. Both missense changes are predicted to be “possibly damaging” by Polyphen2, with a pathogenicity score of 0.99 (sensitivity = 0.68, specificity = 0.97) for Trp and a pathogenicity score of 0.98 (sensitivity 0.78, specificity 0.96) for Gln. While Leu is not strongly preserved across species, the closely related, aliphatic, nonpolar and hydrophobic isoleucine and valine—not Trp or Gln—are used in place of Leu at the equivalent position. Neither Gln nor Trp bear a strong structural resemblance to Leu. While the molecular weight (MW) of the amide Gln is 146 g/mol and similar to that of Leu’s (130 g/mol), Gln is polar and hydrophilic while Leu is nonpolar and hydrophobic. Trp shares its hydrophobicity with Leu but has a higher MW (204 g/mol) and differs from Leu’s structure due to an aromatic amino acid side chain. Trp and Gln in place of Leu at position 962 are absent from the variant database gnomAD. When assessing possible bystander editing, targeting the non-coding strand would be preferable, since additional As are absent in the editing window, whereas targeting the coding sequence could result in bystander editing due to the presence of an additional A in the base editing window: AATATAATTCAGAAGCAATG (editing window underlined, bystander editing in blue, mutation in red) () (Citation55). In summary, while editing the non-coding strand is preferable due to a lower risk of accruing bystander edits, both missense changes would result in possibly damaging variants. Prime editing (PE), a more recently described form of genome editing, is unique in its capability of editing all 12 transversion and transition variants as well as small indels and could therefore also edit this patient’s novel pathogenic transversion variant. PE uses a catalytically impaired Cas9 fused to a reverse transcriptase (RT) and a prime editing gRNA (pegRNA) that both specifies the target site and encodes the desired edit (Citation56). Although PE has yet to be tested in a human clinical trial, it bears huge potential for editing pathogenic variants not targetable with traditional base editors.
Figure 3. Therapeutic “read-through” approach mediated by CRISPR-pass. While base editing with CRISPR-pass does not allow a true ribosomal read-through, nonsense mutations not amenable to conventional base editing strategies can be turned into a missense change by using an ABE-xCas base editing construct (CRISPR-pass), targeting the coding or non-coding strand. in this case, a bystander mutation would occur when targeting the coding strand due to the presence of an additional a in the editing window. the tryptophan and glutamine missense changes resulting from CRISPR-pass mediated editing are predicted to be “possibly damaging” by the in-silico tool Polyphen 2, which evaluates the functional impact of missense changes.
An ABE-mediated correction of the missense change c.2056C>T
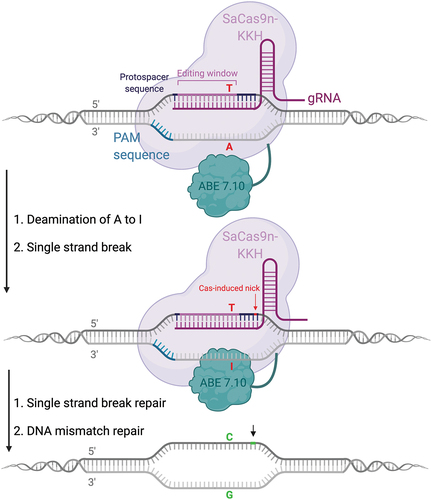
The second likely pathogenic variant present in this patient is a transition amenable to correction by an ABE targeting the non-coding strand and generating an A:T > G:C base correction (). Apart from the target A on the non-coding strand, there are no additional As present in the editing window of the protospacer motif, virtually eliminating the chance of bystander editing. This would allow us the use of an eighth-generation ABE, ABE8, which demonstrate a 6-fold increase in on-target editing efficiency but also a higher processivity (Citation57,Citation58). While ABEs are less inclined to generate DNA off-target edits than CBEs, they do generate guide-dependent and guide-independent off-target RNA editing (Citation59,Citation60). Therefore, the variant ABE7.10TadA*(F148A) would be a promising choice, since it shows a reduction in off-target RNA SNVs but retains efficient on-target editing efficiency (Citation61). A PAM site for the SpCas9 5’-NGG-3’) and its variants as well as for the Cas9 homologue SaCas9-KKH 5’-NNNRRT-3’) can be identified adjacent to the 3' end of the protospacer sequence. The SaCas9-KKH homologue would be the preferred Cas9 in this case, as it is small enough to fit into an AAV (3246 bp) and has already been shown to be compatible both with the most recent generation of ABE, ABE8, as well as shown editing efficiency in rate and mouse embyros when paired with an ABE7.10 (Citation62). In the present mutation, this PAM site 5’-CAAGGT-3’) would place the mutation at position 14 within the editing window of SaKKH, which spans from position 2–15 of the gRNA, with the first nucleotide after the PAM site being counted as 1 (Citation63).
Figure 4. Overview of base editing strategy for a C>T transition mutation. the patient could be transferred to a carrier status by using an ABE7.10 or an ABE8 fused to a SaCas9-KKH, which displays a relaxed PAM site 5’-NNNRRT-3’ and an editing window within 2–15 nucleotides upstream of the PAM site. No additional A’s are present in the editing window, greatly reducing the possibility of bystander mutations.
To summarize, a base editing strategy for the missense change c.2056C>T would entail using an ABE, either ABE7.10, ABE7.10 TadA* (F148A) or an ABE8, fused with a SaCas9-KKH to target the non-coding strand. A summary of the process of designing a base editing strategy for this patient can be found in . By correcting the c.2056C>T mutation, the patient would be transferred to a carrier status. In an autosomal recessive disease, such as CRB1-associated retinopathy, transferring the patient to a carrier status is presumed to be of therapeutic benefit.
Figure 5. Decision making tree for developing a base editing strategy for a patient with compound heterozygous mutation in CRB1.
Discussion
A 16-year-old patient with a clinical diagnosis of early-onset RP and thickened retina on OCT was found to be compound heterozygous for a novel nonsense variant, c.2885T>A (p.Leu962Ter), and a missense variant, c.2056C>T (p.Cys686Arg), in CRB1. The novel c.2885T>A nonsense variant is classified as pathogenic according to the ACMG criteria. After genetic testing as well as a database and literature review, the missense change fulfilled the ACMG evidence criteria to be labelled as likely pathogenic. Variants in this patient were determined by exome sequencing, which precludes the detection of possible structural variants. It should also be noted that only the canonical CRB1-A isoform was sequenced in this patient.
Correction of premature termination codons (PTCs) have long presented a challenge in genetic disease. In the treatment of IRDs, antisense oligonucleotide (ASOs)-mediated splicing modulation has shown promise. Translational read-through of PTCs has been achieved by using ASO to induce skipping of the USH2A exon 13 and has been reported to restore retinal function in animal models (Citation64). Sepofarsen, an ASO that corrects splicing in CEP290-mediated LCA, has shown significant improvement in VA as well as secondary endpoints in clinical trials (Citation65,Citation66). By comparison, base editing with CRISPR-pass does not allow a true ribosomal read-through by misreading of a PTC in the mRNA, but catalyses editing of a nonsense to a missense change, thus allowing translation of a full-length protein. In-silico prediction and functional evaluation of the resulting missense changes must proceed a therapeutic effort and PE should be considered as a possible gene editing option in the future.
While this paper focuses on designing a base editor construct, the delivery strategy of the optimal base editor to the target cell should also be considered. Traditionally, AAV-based vector delivery has been employed due to its proven safety profile, low immunogenicity, and the availability of many serotypes that provide cell-specific tropism (Citation67,Citation68). Due to packaging constraints of AAV as well as the residual immunogenicity that it does elicit, other non-viral nucleic acid delivery vehicles are being tested in preclinical trials (Citation69,Citation70). The benefits of these are the elimination of packaging constraints and the minimal risk of genomic integration. Traditionally, base editors have been delivered as plasmid DNA, but recently, mRNA delivery has been investigated to address a more immediate onset of transcription as well as a less prolonged expression of the base editing machinery (Citation71).
Since CRB1 is expressed in both the photoreceptors and the MCG, it raises the question which promotor could be used to target both cell types. Three common ubiquitous promotors, cytomegalovirus immediate-early promoter (CMV), human ubiquitin C promoter (UbiC), and chicken beta actin promoter (CAG), have showed a dose-dependent toxicity. This contrasts with cell-specific promotors such as rhodopsin kinase (RK), which did not show toxicity, even at high doses (Citation72). The lack of CRB1 promotor flexibility and the resulting ectopic expression might also contribute to the deleterious effect of seen in pre-clinical CRB1 gene supplementation (Citation5,Citation25). Finding a suitable, representative model for CRB1-based disease is equally important for modeling mutations and their treatment options. While several naturally occurring and engineered mouse models exist (Citation20,Citation73–76), none of these models are suitable for testing a base editing strategy. Due to the species-specific differences between humans and mice, as well as the mild phenotype of Crb1 mutant mice, human induced pluripotent stem cells (hiPSC)-derived retinal organoids from CRB1 patients may be good models for testing base and gene editing strategies, as they have been shown to have a phenotype (Citation21) and would more accurately mimic CRB1 localization and disease phenotype in humans.
In summary, since targeting the nonsense mutation c.2885T>A with the base editor construct CRISPR-pass would result in a potentially damaging missense variant, we propose transferring this patient to a carrier status by editing of the missense variant c.2056C>T (p.Arg686Cys) with an ABE fused to a SaCas9-KKH. This variant results from hydrolytic C > T deamination and is representative of over half the recorded pathogenic SNPs (Citation31,Citation77). Since there are no known cases of autosomal dominant CRB1 mutations, one healthy allele would confer haplosufficiency and therefore prevent or decelerate photoreceptor degeneration or even result in photoreceptor rescue, depending on the stage of degeneration.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- den Hollander AI, ten Brink JB, de Kok YJ, van Soest S, van den Born LI, van Driel MA, van de Pol DJR, Payne AM, Bhattacharya SS, Kellner U, et al. Mutations in a human homologue of Drosophila crumbs cause retinitis pigmentosa (RP12). Nat Genet. 1999;23(2):217–21. doi:10.1038/13848.
- den Hollander AI, Heckenlively JR, van den Born LI, de Kok YJ, van der Velde-Visser SD, Kellner U, Jurklies B, van Schooneveld MJ, Blankenagel A, Rohrschneider K, et al. Leber congenital amaurosis and retinitis pigmentosa with Coats-like exudative vasculopathy are associated with mutations in the crumbs homologue 1 (CRB1) gene. Am J Hum Genet. 2001;69(1):198–203. doi:10.1086/321263.
- Mehalow AK, Kameya S, Smith RS, Hawes NL, Denegre JM, Young JA, et al. CRB1 is essential for external limiting membrane integrity and photoreceptor morphogenesis in the mammalian retina. Hum Mol Genet. 2003;12(17):2179–89. doi:10.1093/hmg/ddg232.
- Pellikka M, Tanentzapf G, Pinto M, Smith C, McGlade CJ, Ready DF, Tepass U, et al. Crumbs, the Drosophila homologue of human CRB1/RP12, is essential for photoreceptor morphogenesis. Nature. 2002;416(6877):143–49. doi:10.1038/nature721.
- Pellissier LP, Quinn PM, Alves CH, Vos RM, Klooster J, Flannery JG, Heimel JA, Wijnholds J, et al. Gene therapy into photoreceptors and Müller glial cells restores retinal structure and function in CRB1 retinitis pigmentosa mouse models. Hum Mol Genet. 2015;24(11):3104–18. doi:10.1093/hmg/ddv062.
- Izaddoost S, Nam SC, Bhat MA, Bellen HJ, Choi KW.Drosophila crumbs is a positional cue in photoreceptor adherens junctions and rhabdomeres. Nature. 2002;416(6877):178–83. doi:10.1038/nature720.
- Stingl KT, Kuehlewein L, Weisschuh N, Biskup S, Cremers FPM, Khan MI, Kelbsch C, Peters T, Ueffing M, Wilhelm B, et al. Chromatic full-field stimulus threshold and pupillography as functional markers for late-stage, early-onset retinitis pigmentosa caused by CRB1 mutations. Transl Vis Sci Technol. 2019;8(6):45. doi:10.1167/tvst.8.6.45.
- Alves CH, Pellissier LP, Wijnholds J. The CRB1 and adherens junction complex proteins in retinal development and maintenance. Prog Retin Eye Res. 2014;40:35–52. doi:10.1016/j.preteyeres.2014.01.001.
- Jacobson SG, Cideciyan AV, Aleman TS, Pianta MJ, Sumaroka A, Schwartz SB, et al. Crumbs homolog 1 (CRB1) mutations result in a thick human retina with abnormal lamination. Hum Mol Genet. 2003;12(9):1073–78. doi:10.1093/hmg/ddg117.
- Talib M, van Schooneveld MJ, van Genderen MM, Wijnholds J, Florijn RJ, Ten Brink JB, Schalij-Delfos NE, Dagnelie G, Cremers FPM, Wolterbeek R, et al. Genotypic and phenotypic characteristics of CRB1-associated retinal Dystrophies: a long-term follow-up study. Ophthalmology. 2017;124(6):884–95. doi:10.1016/j.ophtha.2017.01.047.
- Pellissier LP, Alves CH, Quinn PM, Vos RM, Tanimoto N, Lundvig DM, Dudok JJ, Hooibrink B, Richard F, Beck SC, et al. Targeted ablation of CRB1 and CRB2 in retinal progenitor cells mimics Leber congenital amaurosis. PLoS Genet. 2013;9(12):e1003976. doi:10.1371/journal.pgen.1003976.
- Bujakowska K, Audo I, Mohand-Said S, Lancelot ME, Antonio A, Germain A, Léveillard T, Letexier M, Saraiva J-P, Lonjou C, et al. CRB1 mutations in inherited retinal dystrophies. Hum Mutat. 2012;33(2):306–15. doi:10.1002/humu.21653.
- Corton M, Tatu SD, Avila-Fernandez A, Vallespin E, Tapias I, Cantalapiedra D, Blanco-Kelly F, Riveiro-Alvarez R, Bernal S, García-Sandoval B, et al. High frequency of CRB1 mutations as cause of early-onset retinal dystrophies in the Spanish population. Orphanet J Rare Dis. 2013;8:20.
- Vallespin E, Cantalapiedra D, Riveiro-Alvarez R, Wilke R, Aguirre-Lamban J, Avila-Fernandez A, Lopez-Martinez MA, Gimenez A, Jose Trujillo-Tiebas M, Ramos C, et al. Mutation screening of 299 Spanish families with retinal dystrophies by Leber congenital amaurosis genotyping microarray. Investigative Ophthalmol Visual Sci. 2007;48(12):5653–61. doi:10.1167/iovs.07-0007.
- Daich Varela M, Cabral de Guimaraes TA, Georgiou M, Michaelides M. Leber congenital amaurosis/early-onset severe retinal dystrophy: current management and clinical trials. Br J Ophthalmol. 2022;106:445–51.
- Ferrari S, Di Iorio E, Barbaro V, Ponzin D, Sorrentino FS, Parmeggiani F.Retinitis pigmentosa: genes and disease mechanisms. Curr Genomics. 2011;12(4):238–49. doi:10.2174/138920211795860107.
- Hartong DT, Berson EL, Dryja TP.Retinitis pigmentosa. Lancet. 2006;368(9549):1795–809. doi:10.1016/S0140-6736(06)69740-7.
- Tee JJL, Yang Y, Kalitzeos A, Webster A, Bainbridge J, Michaelides M. Natural history study of retinal structure, progression, and symmetry using Ellipzoid zone metrics in RPGR-associated retinopathy. Am J Ophthalmol. 2019;198:111–23. doi:10.1016/j.ajo.2018.10.003.
- Nguyen XT, Talib M, van Schooneveld MJ, Wijnholds J, van Genderen MM, Schalij-Delfos NE, et al. CRB1-Associated retinal dystrophies: a prospective natural history study in anticipation of future clinical trials. Am J Ophthalmol. 2021;234:37–48. doi:10.1016/j.ajo.2021.07.021.
- Ray TA, Cochran K, Kozlowski C, Wang J, Alexander G, Cady MA, Spencer WJ, Ruzycki PA, Clark BS, Laeremans A, et al. Comprehensive identification of mRNA isoforms reveals the diversity of neural cell-surface molecules with roles in retinal development and disease. Nat Commun. 2020;11(1):3328. doi:10.1038/s41467-020-17009-7.
- Quinn PM, Buck TM, Mulder AA, Ohonin C, Alves CH, Vos RM, Bialecka M, van Herwaarden T, van Dijk EHC, Talib M, et al. Human iPSC-derived retinas recapitulate the fetal CRB1 CRB2 complex formation and demonstrate that photoreceptors and Müller Glia are targets of AAV5. Stem Cell Rep. 2019;12(5):906–19. doi:10.1016/j.stemcr.2019.03.002.
- Bennett J, Wellman J, Marshall KA, McCague S, Ashtari M, DiStefano-Pappas J, Elci OU, Chung DC, Sun J, Wright JF, et al. Safety and durability of effect of contralateral-eye administration of AAV2 gene therapy in patients with childhood-onset blindness caused by RPE65 mutations: a follow-on phase 1 trial. Lancet. 2016;388(10045):661–72. doi:10.1016/S0140-6736(16)30371-3.
- Cehajic-Kapetanovic J, Xue K, Martinez-Fernandez de la Camara C, Nanda A, Davies A, Wood LJ, Salvetti AP, Fischer MD, Aylward JW, Barnard AR, et al. Initial results from a first-in-human gene therapy trial on X-linked retinitis pigmentosa caused by mutations in RPGR. Nat Med. 2020;26(3):354–59. doi:10.1038/s41591-020-0763-1.
- Cideciyan AV, Hauswirth WW, Aleman TS, Kaushal S, Schwartz SB, Boye SL, Windsor EAM, Conlon TJ, Sumaroka A, Pang J-J, et al. Human RPE65 gene therapy for Leber congenital amaurosis: persistence of early visual improvements and safety at 1 year. Hum Gene Ther. 2009;20(9):999–1004. doi:10.1089/hum.2009.086.
- Buck TM, Vos RM, Alves CH, Wijnholds J. AAV-CRB2 protects against vision loss in an inducible CRB1 retinitis pigmentosa mouse model. Mol Ther Methods Clin Dev. 2021;20:423–41. doi:10.1016/j.omtm.2020.12.012.
- Wiedenheft B, Sternberg SH, Doudna JA.RNA-Guided genetic silencing systems in bacteria and archaea. Nature. 2012;482(7385):331–38. doi:10.1038/nature10886.
- Bhaya D, Davison M, Barrangou R. CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu Rev Genet. 2011;45:273–97. doi:10.1146/annurev-genet-110410-132430.
- Terns MP, Terns RM.CRISPR-Based adaptive immune systems. Curr Opin Microbiol. 2011;14(3):321–27. doi:10.1016/j.mib.2011.03.005.
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E.A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816–21. doi:10.1126/science.1225829.
- Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR.Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533(7603):420–24. doi:10.1038/nature17946.
- Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, et al. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature. 2017;551(7681):464–71. doi:10.1038/nature24644.
- Doman JL, Raguram A, Newby GA, Liu DR.Evaluation and minimization of Cas9-independent off-target DNA editing by cytosine base editors. Nat Biotechnol. 2020;38(5):620–28. doi:10.1038/s41587-020-0414-6.
- Yu Y, Leete TC, Born DA, Young L, Barrera LA, Lee SJ, Rees HA, Ciaramella G, Gaudelli NM, et al. Cytosine base editors with minimized unguided DNA and RNA off-target events and high on-target activity. Nat Commun. 2020;11(1):2052. doi:10.1038/s41467-020-15887-5.
- Gehrke JM, Cervantes O, Clement MK, Wu Y, Zeng J, Bauer DE, Pinello L, Joung JK, et al. An APOBEC3A-Cas9 base editor with minimized bystander and off-target activities. Nat Biotechnol. 2018;36(10):977–82. doi:10.1038/nbt.4199.
- Kim YB, Komor AC, Levy JM, Packer MS, Zhao KT, Liu DR.Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat Biotechnol. 2017;35(4):371–76. doi:10.1038/nbt.3803.
- Kleinstiver BP, Prew MS, Tsai SQ, Nguyen NT, Topkar VV, Zheng Z, Joung JK, et al. Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat Biotechnol. 2015;33(12):1293–98. doi:10.1038/nbt.3404.
- Chatterjee P, Jakimo N, Lee J, Amrani N, Rodriguez T, Koseki SRT, Tysinger E, Qing R, Hao S, Sontheimer EJ, et al. An engineered ScCas9 with broad PAM range and high specificity and activity. Nat Biotechnol. 2020;38(10):1154–58. doi:10.1038/s41587-020-0517-0.
- Kim HK, Lee S, Kim Y, Park J, Min S, Choi JW, Huang TP, Yoon S, Liu DR, Kim HH, et al. High-Throughput analysis of the activities of xCas9, SpCas9-NG and SpCas9 at matched and mismatched target sequences in human cells. Nat Biomed Eng. 2020;4(1):111–24. doi:10.1038/s41551-019-0505-1.
- Miller SM, Wang T, Randolph PB, Arbab M, Shen MW, Huang TP, Matuszek Z, Newby GA, Rees HA, Liu DR, et al. Continuous evolution of SpCas9 variants compatible with non-G PAMs. Nat Biotechnol. 2020;38(4):471–81. doi:10.1038/s41587-020-0412-8.
- Friedland AE, Baral R, Singhal P, Loveluck K, Shen S, Sanchez M, Marco E, Gotta GM, Maeder ML, Kennedy EM, et al. Characterization of staphylococcus aureus Cas9: a smaller Cas9 for all-in-one adeno-associated virus delivery and paired nickase applications. Genome Biol. 2015;16:257.
- Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, et al. In vivo genome editing using staphylococcus aureus Cas9. Nature. 2015;520(7546):186–91. doi:10.1038/nature14299.
- Kim E, Koo T, Park SW, Kim D, Kim K, Cho HY, Song DW, Lee KJ, Jung MH, Kim S, et al. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat Commun. 2017;8:14500.
- Kurt IC, Zhou R, Iyer S, Garcia SP, Miller BR, Langner LM, Grünewald J, Joung JK, et al. CRISPR C-to-G base editors for inducing targeted DNA transversions in human cells. Nat Biotechnol. 2021;39(1):41–46. doi:10.1038/s41587-020-0609-x.
- Zhao D, Li J, Li S, Xin X, Hu M, Price MA, Rosser SJ, Bi C, Zhang X, et al. Glycosylase base editors enable C-to-A and C-to-G base changes. Nat Biotechnol. 2021;39(1):35–40. doi:10.1038/s41587-020-0592-2.
- Chen L, Park JE, Paa P, Rajakumar PD, Prekop HT, Chew YT, Manivannan SN, Chew WL, et al. Programmable C:G to G:C genome editing with CRISPR-Cas9-directed base excision repair proteins. Nat Commun. 2021;12(1):1384. doi:10.1038/s41467-021-21559-9.
- Bellingrath JS, McClements ME, Kaukonen M, Fischer MD, MacLaren RE.In silico analysis of pathogenic CRB1 single nucleotide variants and their amenability to base editing as a potential lead for therapeutic intervention. Genes (Basel). 2021;12(12):1908. doi:10.3390/genes12121908.
- Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013. 76. Chapter 7:Unit7 20. doi:10.1002/0471142905.hg0720s76.
- Ng PC, Henikoff S.SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–14. doi:10.1093/nar/gkg509.
- Steinhaus R, Proft S, Schuelke M, Cooper DN, Schwarz JM, Seelow D.MutationTaster2021. Nucleic Acids Res. 2021;49(W1):W446–W51. doi:10.1093/nar/gkab266.
- Wang L, Zhang J, Chen N, Wang L, Zhang F, Ma Z, Li G, Yang L, et al. Application of whole exome and targeted panel sequencing in the clinical molecular diagnosis of 319 Chinese families with inherited retinal dystrophy and comparison study. Genes (Basel). 2018;9(7):360. doi:10.3390/genes9070360.
- Liu X, Tao T, Zhao L, Li G, Yang L.Molecular diagnosis based on comprehensive genetic testing in 800 Chinese families with non-syndromic inherited retinal dystrophies. Clin Exp Ophthalmol. 2021;49(1):46–59. doi:10.1111/ceo.13875.
- Biesecker LG, Harrison SM.ClinGen sequence variant interpretation working G. the ACMG/AMP reputable source criteria for the interpretation of sequence variants. Genet Med. 2018;20(12):1687–88. doi:10.1038/gim.2018.42.
- Gao FJ, Li JK, Chen H, Hu FY, Zhang SH, Qi YH, Xu P, Wang D-D, Wang L-S, Chang Q, et al. Genetic and clinical findings in a large cohort of Chinese patients with suspected retinitis pigmentosa. Ophthalmology. 2019;126(11):1549–56. doi:10.1016/j.ophtha.2019.04.038.
- Lee C, Hyun Jo D, Hwang GH, Yu J, Kim JH, Park SE, Kim J-S, Kim JH, Bae S, et al. CRISPR-Pass: gene rescue of nonsense mutations using adenine base editors. Mol Ther. 2019;27(8):1364–71. doi:10.1016/j.ymthe.2019.05.013.
- Hu JH, Miller SM, Geurts MH, Tang W, Chen L, Sun N, Zeina CM, Gao X, Rees HA, Lin Z, et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature. 2018;556(7699):57–63. doi:10.1038/nature26155.
- Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, Chen PJ, Wilson C, Newby GA, Raguram A, et al. Search-And-Replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576(7785):149–57. doi:10.1038/s41586-019-1711-4.
- Gaudelli NM, Lam DK, Rees HA, Sola-Esteves NM, Barrera LA, Born DA, Edwards A, Gehrke JM, Lee S-J, Liquori AJ, et al. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat Biotechnol. 2020;38(7):892–900. doi:10.1038/s41587-020-0491-6.
- Richter MF, Zhao KT, Eton E, Lapinaite A, Newby GA, Thuronyi BW, Wilson C, Koblan LW, Zeng J, Bauer DE, et al. Phage-Assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat Biotechnol. 2020;38(7):883–91. doi:10.1038/s41587-020-0453-z.
- Grunewald J, Zhou R, Garcia SP, Iyer S, Lareau CA, Aryee MJ, Joung JK, et al. Transcriptome-Wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature. 2019;569(7756):433–37. doi:10.1038/s41586-019-1161-z.
- Rees HA, Wilson C, Doman JL, Liu DR.Analysis and minimization of cellular RNA editing by DNA adenine base editors. Sci Adv. 2019;5(5):eaax5717. doi:10.1126/sciadv.aax5717.
- Zhou C, Sun Y, Yan R, Liu Y, Zuo E, Gu C, Han L, Wei Y, Hu X, Zeng R, et al. Off-Target RNA mutation induced by DNA base editing and its elimination by mutagenesis. Nature. 2019;571(7764):275–78. doi:10.1038/s41586-019-1314-0.
- Yang L, Zhang X, Wang L, Yin S, Zhu B, Xie L, Duan Q, Hu H, Zheng R, Wei Y, et al. Increasing targeting scope of adenosine base editors in mouse and rat embryos through fusion of TadA deaminase with Cas9 variants. Protein Cell. 2018;9(9):814–19. doi:10.1007/s13238-018-0568-x.
- Evanoff M, Komor AC, Bayley H.Base editors: modular tools for the introduction of point mutations in living cells. Emerging Topics Life Sci. 2019;3(5):483–91. doi:10.1042/ETLS20190088.
- Dulla K, Slijkerman R, van Diepen HC, Albert S, Dona M, Beumer W, Turunen JJ, Chan HL, Schulkens IA, Vorthoren L, et al. Antisense oligonucleotide-based treatment of retinitis pigmentosa caused by USH2A exon 13 mutations. Mol Ther. 2021;29(8):2441–55. doi:10.1016/j.ymthe.2021.04.024.
- Cideciyan AV, Jacobson SG, Ho AC, Garafalo AV, Roman AJ, Sumaroka A, Krishnan AK, Swider M, Schwartz MR, Girach A, et al. Durable vision improvement after a single treatment with antisense oligonucleotide sepofarsen: a case report. Nat Med. 2021;27(5):785–89. doi:10.1038/s41591-021-01297-7.
- Cideciyan AV, Jacobson SG, Drack AV, Ho AC, Charng J, Garafalo AV, Roman AJ, Sumaroka A, Han IC, Hochstedler MD, et al. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nat Med. 2019;25(2):225–28. doi:10.1038/s41591-018-0295-0.
- Zincarelli C, Soltys S, Rengo G, Rabinowitz JE.Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol Ther. 2008;16(6):1073–80. doi:10.1038/mt.2008.76.
- Charbel Issa P, De Silva SR, Lipinski DM, Singh MS, Mouravlev A, You Q, Barnard AR, Hankins MW, During MJ, MacLaren RE, et al. Assessment of tropism and effectiveness of new primate-derived hybrid recombinant AAV serotypes in the mouse and primate retina. PLoS One. 2013;8(4):e60361. doi:10.1371/journal.pone.0060361.
- Trigueros S, Domenech EB, Toulis V, Marfany G.In vitro gene delivery in retinal pigment epithelium cells by plasmid DNA-wrapped gold nanoparticles. Genes (Basel). 2019;10(4):289. doi:10.3390/genes10040289.
- Wang Y. Nanoparticles as delivery vehicles for the treatment of retinal degenerative diseases. In: Retinal degenerative diseases. Advances in experimental medicine and biology, Editor: Ash, JD. Springer (New York City), 2018:pp. 117–23. doi:10.1007/978-3-319-75402-4_15.
- Li L, Hu S, Chen X. Non-Viral delivery systems for CRISPR/Cas9-based genome editing: challenges and opportunities. Biomaterials. 2018;171:207–18. doi:10.1016/j.biomaterials.2018.04.031.
- Xiong W, Wu DM, Xue Y, Wang SK, Chung MJ, Ji X, Rana P, Zhao SR, Mai S, Cepko CL, et al. AAV cis -regulatory sequences are correlated with ocular toxicity. Proc Natl Acad Sci USA. 2019;116(12):5785–94. doi:10.1073/pnas.1821000116.
- van de Pavert SA, Kantardzhieva A, Malysheva A, Meuleman J, Versteeg I, Levelt C, Klooster J, Geiger S, Seeliger MW, Rashbass P, et al. Crumbs homologue 1 is required for maintenance of photoreceptor cell polarization and adhesion during light exposure. J Cell Sci. 2004;117(Pt 18):4169–77. doi:10.1242/jcs.01301.
- van de Pavert SA, Meuleman J, Malysheva A, Aartsen WM, Versteeg I, Tonagel F, Kamphuis W, McCabe CJ, Seeliger MW, Wijnholds J, et al. A single amino acid substitution (Cys249trp) in Crb1 causes retinal degeneration and deregulates expression of pituitary tumor transforming gene Pttg1. J Neurosci. 2007;27(3):564–73. doi:10.1523/JNEUROSCI.3496-06.2007.
- Alves CH, Boon N, Mulder AA, Koster AJ, Jost CR, Wijnholds J. CRB2 loss in rod photoreceptors is associated with progressive loss of retinal contrast sensitivity. Int J Mol Sci. 2019;20(17). doi:10.3390/ijms20174069.
- Alves CH, Sanz AS, Park B, Pellissier LP, Tanimoto N, Beck SC, Huber G, Murtaza M, Richard F, Sridevi Gurubaran I, et al. Loss of CRB2 in the mouse retina mimics human retinitis pigmentosa due to mutations in the CRB1 gene. Hum Mol Genet. 2013;22(1):35–50. doi:10.1093/hmg/dds398.
- Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44(D1):D862–8. doi:10.1093/nar/gkv1222.