
ABSTRACT
Introduction
Most bispecific antibody (BsAb) therapies focus on stimulating the adaptive immune system, in particular T cells, to promote tumor cell killing. Another method to promote tumor eradication is through the engagement of myeloid cells, including macrophages and neutrophils, which are abundantly present and possess intrinsic cytotoxic mechanisms for tumor cell killing, making them interesting effector cells to recruit for BsAb therapy.
Areas covered
In this review, we describe the evolving knowledge of the role of macrophages and neutrophils in cancer in scientific literature. Moreover, we address the BsAbs that have been developed over the years to recruit these cell types as effector cells in immunotherapy of cancer. This includes the discussion of BsAbs that target Fc receptors (i.e. FcγR and FcαRI) to induce antibody-dependent cellular phagocytosis (ADCP) by macrophages or trogoptosis via neutrophils, as well as BsAbs that interfere with checkpoint inhibition, including the SIRPα-CD47 pathway.
Expert opinion
Elucidating the complexity of macrophage and neutrophil heterogeneity in cancer may help to specifically enlist the cytotoxic ability of these cells through targeting Fc receptors and checkpoint pathways, which may further enhance anti-cancer immunity.
1. Introduction
Cancer is one of the leading causes of death globally [Citation1] and is a highly complex disease. Currently, eight hallmarks of cancer have been identified, including limitless proliferation, resistance to cell death, and the ability to evade destruction by the immune system [Citation2]. Immune cells play an important role during different phases of cancer development. In early stages, immune cells have a function in immunosurveillance, resulting in eradication of tumor cells [Citation3]. However, during development tumor cells gain characteristics that enable them to evade the immune system. For instance, upregulation of immune checkpoints or downregulation of MHC class I are often observed [Citation4,Citation5]. Furthermore, secretion of immunosuppressive cytokines and selective attraction of immunoregulatory cells, such as regulatory T cells and myeloid-derived suppressor cells, are frequently present [Citation6–8]. Thus, as cancer progresses, an immunosuppressive tumor microenvironment (TME) is induced, preventing activation and recruitment of cytotoxic immune cells to kill tumor cells. Immunotherapy was developed to overcome immunosuppression as it aims to reeducate the immune system of patients, ultimately resulting in eradication of cancer cells [Citation9].
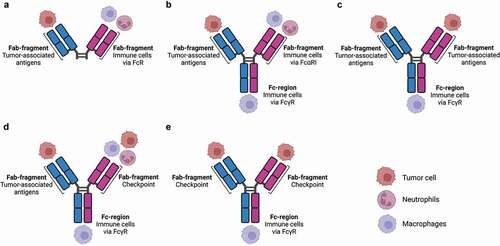
Monoclonal antibody (mAb) therapy represents a promising way of activating the immune system [Citation10,Citation11]. Antibodies can bridge targets (e.g. bacteria, tumor cells) after binding of the fragment antigen-binding (Fab) domains to effector immune cells through the Fc tail, as many immune cells express Fc receptors (FcRs) [Citation11,Citation12]. Most antitumor-associated antigen (TAA) mAbs that are used in the clinic are of the IgG isotype [Citation11]. By targeting TAA, monocytes, macrophages, and natural killer cells are recruited through binding to Fcy receptors, after which they are able to exert effector functions such as complement-dependent cytotoxicity (CDC), antibody-dependent cellular cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP) [Citation10,Citation13,Citation14]. By contrast, neither neutrophils nor T cells are effectively engaged as effector cells in IgG mAb therapy [Citation12,Citation15]. To increase the efficacy of mAb therapy, the use of bispecific antibody (BsAb) formats has been proposed, which can recognize tumor antigens with one Fab, while recruiting and activating immune cells with the other Fab [Citation16,Citation17]. Over 100 BsAb formats have been created, which can bind different targets on either tumor or immune cells (). Most of the current BsAbs focus on activating and recruiting T cells [Citation18–22]. However, other cell populations can also act as potential effector cells to induce BsAb-mediated tumor regression. In this review, we will focus on myeloid cells, including macrophages and neutrophils, as effector cells in BsAb therapy.
Figure 1. Different formats of bispecific antibodies targeting myeloid cells.
2. Myeloid cells in cancer
Myeloid cells constitute a heterogeneous population of bone marrow-derived cells, including neutrophils and macrophages [Citation23]. Myeloid cells are actively recruited to the tumor microenvironment, where they play an important role during tumor development [Citation24]. Significant myeloid cell infiltrates can be found in malignant tumors. Under influence of the TME, most myeloid cells differentiate into immunosuppressive phenotypes, including tumor-associated macrophages (TAM) or neutrophils (TAN) and myeloid-derived suppressor cells [Citation25–27].
2.1. Tumor-associated macrophages
Macrophages are plastic cells that exert different roles depending on the environmental cues. They have important functions in immunity as they are able to engulf pathogens or cell debris, and release pro-inflammatory cytokines/chemokines that can activate the adaptive immune system [Citation28]. Alternatively, they have an essential role in wound healing and immunomodulation. It has been shown that many cancers contain prominent macrophage populations, which are referred to as TAMs [Citation28]. TAMs are derived from circulating monocytes or locally derived tissue-resident macrophages. For instance, it was demonstrated in the MMTV-PyMT (PyMT) mouse mammary tumor model that bone marrow-derived Ly6C+ monocytes were recruited from the circulation into tumors [Citation29]. The TME can affect the phenotype of TAMs, i.e. they can either promote tumor growth or kill tumor cells [Citation30]. Based on this dichotomy, previously a classification into classically activated or ‘M1’ versus alternatively activated or ‘M2’ phenotypes was proposed [Citation31,Citation32]. On the one hand, M1 macrophages are induced by interferon-γ (IFN-γ), tumor necrosis factor-alpha (TNF-α) and lipopolysaccharide (LPS) [Citation28,Citation31]. M1 activation leads to increased expression of MHC class II and release of high levels of pro-inflammatory cytokines (TNF-α, interleukin (IL)-1, IL-12 or IL-23) [Citation28,Citation31]. M1 macrophages are able to kill pathogens and tumor cells, and as such they have an antitumor phenotype [Citation31]. On the other hand, M2 macrophages, which can be induced via IL-4/IL-10/IL-13 downregulate MHC class II and IL-12 production. By contrast, increased expression of the anti-inflammatory cytokine IL-10 was observed [Citation31]. M2 macrophages thus exert pro-tumor effects by, e.g. downregulating antitumor immune responses. M2 macrophages have been further subdivided into three sub-groups: i.e. M2a, M2b, and M2c [Citation33,Citation34]. Nonetheless, the M1 versus M2 classification is an oversimplification, and represents the two extremes of a scale of functional phenotypes in vivo [Citation28,Citation33]. Other ways of grouping macrophage populations have been proposed as well, for instance based on roles in host defense, wound healing or immune regulation [Citation35]. Although all classifications remain oversimplifications, TAMs likely express an M1-like phenotype in early-stage tumors, which can lead to eradication of tumor cells. Thereafter, in later tumor stages macrophages are skewed toward more M2-like phenotypes by signals derived from the TME. M2-like macrophages then promote tumor progression by stimulating angiogenesis, invasion, and tumor cell metastasis [Citation32,Citation36]. Additionally, they have an immunoregulatory phenotype, thereby suppressing antitumor immune responses [Citation37,Citation38]. Presence of macrophages has been correlated with poor prognosis in many types of malignancies [Citation30,Citation39,Citation40]. For example, increased infiltration of macrophages was linked to poor patient outcome in invasive breast carcinoma patients [Citation30,Citation32,Citation41]. In contrast, patients that were diagnosed with colorectal cancer had a better prognosis when colon tumors were infiltrated with high TAM numbers [Citation39,Citation42]. This suggests that depending on the type of cancer, the number of infiltrated macrophages is either associated with bad or good prognosis in patients. Considering the different roles during tumor development and progression, reprogramming TAMs into more classically activated or M1-like cells represents an interesting strategy as this could contribute to the induction of antitumor responses. Monocytes can infiltrate the tumor and mature and polarize into TAMs depending on the immune state of the tumor. The use of BsAbs may affect the state of macrophages by differentiating suppressor TAMs into a pro-inflammatory phenotype including cytotoxic activity toward tumor cells, but this needs to be investigated in more detail [Citation43].
2.2. Neutrophils in cancer
Neutrophils represent the ‘first-line of defense’ against invading pathogens and are the first cells that are recruited to a site of infection where they are able to phagocytose pathogens or debris [Citation23]. They are short-lived cells and are therefore continuously replenished from the bone marrow [Citation44–46]. In addition to their role in host defense, it is becoming increasingly clear that neutrophils also play an important role in cancer [Citation47,Citation48]. The nomenclature of the heterogeneity of neutrophils related to cancer is somewhat confusing. Neutrophils infiltrating the tumor can be referred to as TANs, which are usually regarded as pro-tumorigenic, as the presence of neutrophils in the tumor often correlates with poor prognosis [Citation49–51]. Furthermore, neutrophils with an immunosuppressive phenotype have been defined as granulocytic-myeloid derived suppressor cells [Citation26]. Neutrophils have also been classified based on pro- or antitumor phenotypes. Similar to the M1/M2 classification of macrophages, ‘N1’ neutrophils are regarded as pro-inflammatory and antitumor, while ‘N2’ neutrophils are considered immunosuppressive and pro-tumorigenic [Citation51,Citation52]. A high neutrophil-to-lymphocyte ratio has been associated with poor prognosis and can therefore serve as a prognostic marker [Citation53,Citation54]. Nonetheless, different studies have shown that neutrophils are able to eliminate tumor cells, in vitro and in vivo [Citation55]. Considering the sheer number of neutrophils, recruiting them as effector cells in BsAb therapy may therefore represent an attractive option for potent tumor eradication.
3. Targeting of macrophages to improve tumor killing
3.1. Engaging macrophages via FcγR targeting
Phagocytosis of tumor cells by macrophages is greatly enhanced in the presence of antibodies [Citation10,Citation32,Citation56]. This was demonstrated in vitro with human effector cells and tumor cells in the presence of mAbs directed against TAA such as HER-2, carcinoembryonic antigen (CEA), epithelial cell adhesion molecule (Ep-CAM), EGFR, human epithelial mucin (MUC)-1, CD20, CD30, and CD40 [Citation10,Citation57–66]. After engulfment, a phagosome is formed, which fuses with lysosomes leading to acidification of the phagosome and degradation of the target cell () [Citation67]. Human macrophages express several Fcγ receptors such as FcyRI (CD64), FcγRIIa (CD32a), and FcγRIIIa (CD16a) as well as the inhibitory receptor FcyRIIb (CD32b) [Citation10,Citation68,Citation69]. Mouse macrophages express activating receptors FcyRI, FcγRIII, and FcγRIV as well as the inhibitory receptor FcγRII. Mice lacking FcγRII had increased antitumor responses after mAb therapy, which led to prevention of tumor development [Citation70]. IgG can also bind to the inhibitory FcyRIIb on human macrophages. To overcome binding of IgG to FcyRIIb, and concomitant suppression of effector functions, BsAbs that target specific Fc receptors were developed [Citation10,Citation65,Citation71]. Multiple studies have shown that BsAbs targeting macrophages could lead to enhanced phagocytosis (). An overview of the BsAbs in preclinical or clinical stages is provided in . For example, the humanized BsAb CD30xFcγRI (i.e. H22xKi4) consists of two different Fabs. One Fab arm binds to CD30, which is expressed on lymphoma cells. The other Fab arm targets FcγRI [Citation71]. When macrophages were co-cultured in vitro with the L540 cell line that overexpresses CD30 in the presence of 0.1 or 1.0 µg/ml H22xKi4, efficient ADCP was observed. MDX-210 is a BsAb that is directed against HER-2/neu and FcγRI. HER-2/neu is overexpressed in a subgroup of patients with breast cancer. When human macrophages were co-cultured with HER-2/neu expressing cancer cells and 0.1 µg/ml MDX-210 in vitro, approximately 80–95% phagocytosis was observed in the presence of MDX-210 [Citation65,Citation72]. On the contrary, cells expressing low levels of HER-2/neu, e.g. A431, were not susceptible to MDX-H210-mediated killing [Citation72]. In vitro, it was shown that incubation with BsAb in combination with GM-CSF led to more efficient phagocytosis of tumor cells by human macrophages compared to addition of BsAb alone or the combination of BsAb and IFN-γ. This suggests that IFN-y is not as effective as GM-CSF in activating macrophages [Citation73].
Figure 2. Antibody-dependent cellular phagocytosis by macrophages.
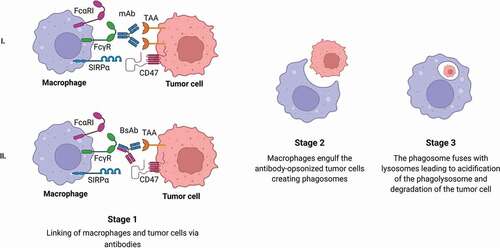
Table 1. Overview of bispecific antibodies targeting FcγR, FcαRI, or CD47/SIRPα on myeloid cells.
Amivantamab (JNJ-61186372) is a BsAb targeting both c-MET and endothelial growth factor receptor (EGFR), which are expressed in non-small cell lung cancer (NSCLC). Since it has a fully functional human IgG1 Fc tail, amivantamab can engage macrophages and NK cells via Fcγ receptors. It was shown that amivantamab (at concentrations ranging 0.2–20 nM) induced phagocytosis of the NSCLC tumor cell lines H1975 and HCC-827 (expressing c-MET and EGFR) by human macrophages in vitro [Citation74]. Additionally, 10 µg/ml amivantamab induced trogocytosis – a mechanism whereby ligands or membrane fragments of the tumor cells are transferred to either M1 or M2c macrophages [Citation75]. When macrophages were depleted in an H1975 xenograft model in immunodeficient mice, efficacy of BsAb treatment was reduced, supporting that macrophages play a role in preventing tumor growth after amivantamab therapy [Citation75].
3.2. Stimulation of macrophages via FcαRI targeting
In addition to FcγR, macrophages also express the IgA receptor FcαRI (CD89) [Citation76], and the use of IgA as alternative in antibody therapy has been previously proposed. However, clinical development of IgA antibodies is challenging as IgA antibodies have significantly reduced half-life in vivo compared to IgG. Moreover, IgA is a heavily glycosylated molecule, which is difficult to exactly and consistently mimic during production. One way to circumvent these obstacles is the use of IgG BsAb in which one Fab arm targets FcαRI and the other a TAA. Still, testing anti-FcαRI BsAb in vivo in wild type mice is precluded, as mice do not express an FcαRI homologue [Citation77]. To allow testing, FcαRI transgenic (Tg) mice were created. Several transgenic mouse models have been described in which the role of FcαRI in vivo can be assessed. In one model, the CD11b promoter was used, resulting in high human FcαRI expression levels on monocytes and macrophages [Citation78]. In a second model, a human FcαRI Tg mouse strain was generated using a murine CD14 promoter restricting FcαRI expression to monocytes and tissue macrophages [Citation79]. A third model was created with the use of a cosmid clone that contained the human FcαRI gene [Citation80,Citation81]. Comparable to humans, the latter FcαRI Tg mice express FcαRI on cells of the myeloid lineage, most notably on neutrophils, and (subpopulations) of macrophages. Various anti-FcαRI BsAbs have been created and are currently tested.
For instance, the A77xKi-4 BsAbs targets CD30 and FcαRI. When human macrophages were co-cultured with L540 Hodgkin’s cells in the presence of 0.1 µg/ml A77xKi4 BsAbs, efficient phagocytosis was observed [Citation71]. Additionally, BsAbs that target FcαRI and HER-2/neu (A77-520C9) have been developed. A77x520C9 (0.01–1.0 µg/ml) induced similar phagocytosis of HER-2/neu breast cancer cells by human macrophages compared to anti-FcγRI BsAb (H22x520C9; 0.01–1.0 µg/ml) [Citation73]. Treatment of human macrophages with IFN-γ did not lead to increased phagocytosis. However, when macrophages were treated with GM-CSF, phagocytosis of breast cancer cells was more efficient in the presence of 1.0 µg/ml A77x520C9 compared to 1.0 µg/ml H22x520C9 [Citation73].
A BsAb targeting CD20 and FcαRI has also been designed, which has a somewhat different format. This BsAb only contains one Fab arm targeting FcαRI, whereas the Fc-region contains a CD20 binding domain, hampering Fc-mediated functions [Citation79]. FcαRIxCD20 BsAbs were tested in a human FcαRI Tg model under the CD14 promoter. Macrophages from FcαRI Tg mice phagocytosed human Raji tumor cells more efficiently in the presence of 1.0 µg/ml FcαRIxCD20 BsAbs compared to IgG anti-CD20 mAbs in vitro. To investigate in vivo efficacy, murine Lewis lung cancer (LLC) cells were transduced with human CD20 (LLC-hCD20). Therapy was ineffective in wild-type mice (which lack FcαRI expression), but significantly more regression of LLC-hCD20 tumors was observed when FcαRI Tg mice were treated with FcαRIxCD20 BsAbs (10 mg/kg) compared to IgG anti-CD20 mAbs. When FcαRI Tg TAMs were isolated from LLC-hCD20 tumors efficient in vitro phagocytosis of human Raji tumor cells (Burkitt’s lymphoma) was observed in the presence of 1.0 µg/ml FcαRIxCD20 BsAbs [Citation79].
Recently, we developed a BsAb, with a functional IgG Fc tail, that targets a TAA and FcαRI [Citation82]. This anti-FcαRI BsAb consists of three binding components: (1) a tumor-antigen targeting Fab, e.g. EGFR or gp75; (2) a FcαRI targeting Fab; and (3) a functional Fc tail that can interact with FcγR. Phagocytosis of A431 tumor cells in the presence of 1 µg/ml FcαRIxEGFR BsAbs induced efficient phagocytosis, which was comparable to ADCP in the presence of the parental anti-EGFR IgG mAb. Since ADCP was less efficient in the presence of IgA anti-EGFR mAbs, it is likely that the IgG Fc tail, and not the anti-FcαRI was responsible for induction of phagocytosis by macrophages. Additionally, murine B16F10 tumor cells that expressed gp75 (B16F10gp75) were effectively taken up in the presence of FcαRIxgp75 BsAbs. When mice were treated with 50 µg FcαRIxgp75 BsAbs, significant reduced outgrowth of B16F10gp75 melanoma was observed, which was similar to treatment with IgG anti-gp75 mAbs in non-transgenic mice. However, in FcαRI Tg mice (expression on myeloid cells) BsAb treatment was most effective. When macrophages were depleted, the efficacy of BsAb therapy was abrogated, supporting that macrophages played an important role in the elimination of tumor cells [Citation82].
To combine functions of IgA and IgG, antibodies were developed with a cross-isotype Fc domain in which residues from IgG1 and IgA were merged (IgGA) [Citation83]. The fab domains of the anti-HER-2 mAb trastuzumab were fused to the IgGA Fc region (trastuzumab-IgGA). Human macrophages showed more effective phagocytosis of MDA-MB-453 breast cancer cells (expressing HER-2) in the presence of trastuzumab-IgGA (0.05–5 µg/ml) compared to trastuzumab-IgG [Citation83]. Similarly, enhanced phagocytosis of SK-BR-3 HER-2+ and MDA-MB-453 HER-2+ cells was observed in the presence of anti-HER-2-IgG1/IgA2 chimeric antibodies compared to IgG1 anti-HER-2 mAbs in vitro [Citation84].
Another cross-isotype antibody is the anti-CD20-IgGA [Citation85]. Macrophages that were isolated from FcαRI Tg mice phagocytosed Raji lymphoma cells more efficiently in the presence of 10 µg/ml anti-CD20-IgGA cross antibodies or IgA anti-CD20 mAbs compared to IgG anti-CD20 mAbs. Treatment of FcαRI Tg mice (FcαRI expression restricted to macrophages) with 5 mg/kg anti-CD20-IgGA induced significant LLC-CD20 tumor regression compared to treatment with anti-CD20-IgA or anti-CD20-IgG mAbs. Treatment with IgA anti-CD20 mAbs had no therapeutic efficacy in WT mice. Additionally, no differences were observed between IgG anti-CD20 mAb or anti-CD20-IgGA treatment, suggesting that enhanced in vivo antitumor activity of anti-CD20-IgGA therapy was dependent on FcαRI [Citation85]. Taken together, targeting FcαRI triggers phagocytosis by macrophages, supporting that macrophages will be recruited during anti-FcαRI BsAb therapy.
3.3. Interfering with the SIRPα inhibitory pathway in macrophages
Macrophages express the signal regulatory protein alpha (SIRPα), which is an inhibitory receptor [Citation86]. SIRPα can bind to CD47, which is overexpressed in many types of cancer [Citation86,Citation87]. Binding of SIRPα to CD47 leads to a ‘don’t eat me’ signal, which can result in tumor cell escape. Thereby, cancer cells are able to avoid immune surveillance [Citation56,Citation86]. CD47-SIRPα is the best studied innate immune checkpoint pathway, and currently multiple therapeutical biologics are entering clinical phase studies with promising early-stage results [Citation88]. It has been shown that anti-CD47 mAbs enhanced phagocytosis of tumor cells including acute myeloid leukemia, non-Hodgkin lymphoma and bladder cancer cells in vitro by human and mouse macrophages [Citation87,Citation89]. When non-Hodgkin lymphoma cells were incubated with human or mouse macrophages in the presence of the anti-CD20 mAb rituximab (10 µg/ml) in combination with anti-CD47 mAbs (5 µg/ml) significant increased phagocytosis was observed compared to either antibodies alone. Similarly, this combination treatment led to elimination of tumors when immunodeficient mice were engrafted with human non-Hodgkin lymphoma cells. When macrophages were depleted therapeutic efficacy was significantly decreased, indicating that macrophages contributed to the killing of tumor cells [Citation87,Citation89]. Thus, simultaneous blocking of CD47 during antibody-based immunotherapy might lead to more effective tumor elimination. However, CD47 is widely expressed on all normal cell types, including on hematopoietic stem cells, erythrocytes, and platelets [Citation86]. As such, this can lead to binding of anti-CD47 antibodies to normal cells, which could lead to autoimmunity. To decrease unwanted side effects, anti-CD47 BsAb that target TAA for local delivery have been developed [Citation90].
The CD47xEGFR-IgG1 BsAb has one Fab directed against EGFR, which is a target in head and neck cancer, metastasized colorectal cancer, NSCLC, and in pancreatic cancer [Citation91], and one Fab directed against CD47 [Citation90]. Moreover, this BsAb contains a fully functional human IgG1 Fc effector domain. Incubation of human macrophages with EGFR-transfected/CD47+ K562 tumor cells and 1 µg/ml CD47xEGFR-IgG1 BsAbs led to enhanced phagocytosis in vitro. Increased uptake was shown for multiple CD47-positive cancer cell lines overexpressing EGFR, including NCI-H292, A431, and FaDu cells [Citation90]. It was also investigated if BsAbs could promote cross-talk between macrophages and T cells. First, human macrophages were incubated with K562 tumor cells expressing EGFR and CD47 in the presence of 1 µg/ml CD47xEGFR-IgG1 BsAbs to induce phagocytosis, after which tumor cells were removed and human CD8+ T cells were added [Citation90]. Upregulation of CD137 on CD8+ T cells and enhanced secretion of IFN-γ was observed. Macrophages were necessary for this process, as blocking the interaction between macrophages and T cells decreased T cell activation, suggesting that cross-presentation was promoted in a FcR-dependent manner [Citation90]. As hematopoietic cells highly express CD47, binding of CD47xEGFR-IgG1 BsAb to erythrocytes and tumor cells was determined. Binding of BsAb to erythrocytes was significantly lower compared to anti-CD47 IgG1. In addition, binding of CD47xEGFR-IgG1 BsAb to EGFR-expressing tumor cells was only slightly affected by addition of whole blood, indicating that most BsAb will target tumor cells and not erythrocytes, which may be an advantage over the use of anti-CD47 mAbs [Citation90]. Results with a BsAb that targets CD47 and CD19 (NI-1701) are also promising. CD19 is expressed on B cells and is a target to treat B cell malignancies. Addition of 10 µg/ml NI-1701 to co-cultures of Raji cells and human macrophages resulted in activation of macrophages and phagocytosis of tumor cells in vitro [Citation92,Citation93]. However, since normal B cells express CD19 and CD47 as well, treatment with this BsAb will presumably not only result in ADCP of malignant B cells but also of normal B cells.
In addition, CD47 targeting can be combined with blockade of a second checkpoint. For example, a BsAb that targets CD47 and PD-L1 has been described. When human macrophages were co-cultured with human CD47+PD-L1+ tumor cells (HT-1080, CCRF-CEM, Raji cells, or H292 cell lines) CD47xPD-L1 BsAbs enhanced phagocytosis compared IgG anti-CD47 or PD-L1 mAbs alone [Citation94,Citation95]. To study crosstalk between the innate and adaptive immunity, mixed lymphocyte reaction assays were performed to investigate dendritic cell (DC) mediated T cell activation. The presence of anti-CD47 mAbs in co-cultures led to reduced IL-2 secretion, while addition of anti-PD-L1 mAbs alone or in combination with anti-CD47 treatment resulted in increased IL-2 release. Interestingly, incubation with 200 nM CD47xPD-L1 BsAb significantly increased IFN-γ and IL-2 release compared to single antibodies or combination treatment [Citation94], suggesting that BsAbs contributed to the interaction between DCs and T cells. To assess potential adverse events, binding of the CD47xPD-L1 BsAb to tumor cells or erythrocytes was assessed in vitro and in vivo. In vitro, erythrocytes were added to human cancer cells, to determine binding selectivity. To assess antibody distribution in vivo, blood and tumor cells were harvested and analyzed for BsAb binding. CD47xPD-L1 BsAb preferentially bound CD47+PD-L1+ tumor cells, and only bound CD47+PD-L1− erythrocytes at higher antibody concentrations (4 nM) [Citation94]. This indicates that administration of lower antibody concentrations may be sufficient to achieve antitumor effects, while erythrocytes remain unaffected at these concentrations.
Instead of targeting CD47, BsAbs that are directed against SIRPα have also been developed. Human CD70 is an antigen that is overexpressed in several malignancies, such as non-Hodgkin lymphoma, multiple myeloma, renal carcinoma, and glioblastoma [Citation96,Citation97]. Phagocytosis of various renal carcinoma cell lines, including RCC4, RCC10, TK10, and CAKi-1 by macrophages was significantly enhanced in the presence of 10 µg/ml CD70xSIRPα BsAbs in vitro, which was not observed in the presence of anti-SIRPα or anti-CD70 mAbs alone [Citation98]. Furthermore, treatment with 10 mg/kg CD70xSIRPα BsAbs reduced human Burkitt’s lymphoma cell growth in immunodeficient mice, which was comparable to co-treatment with anti-SIRPα and anti-CD70 mAbs. In summary, targeting either CD47 or SIRPα with BsAbs can stimulate macrophages, which may lead to enhanced eradication of tumor cells.
4. Potentiating neutrophil effector functions with BsAbs
4.1. Stimulating neutrophils through Fcγ receptors
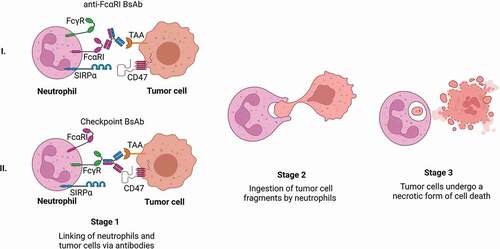
Neutrophils can kill tumor cells via different modes of action. For killing to occur, an immunological synapse needs to be established between neutrophils and tumor cells [Citation99,Citation100]. These cell–cell contacts are dependent on the expression of complement receptor 3 (CR3; i.e. Mac-1), which consists of CD11b and CD18 [Citation100]. After the formation of an immunological synapse, different cytotoxic mechanisms can be initiated, including phagocytosis, degranulation, NET release, and trogocytosis [Citation47]. It has been demonstrated that neutrophils are able to completely encapsulate small tumor cells, i.e. B cell chronic lymphocytic leukemia (B-CLL) cells [Citation59], after which the phagocytosed particles are degraded in a phagolysosome. Nonetheless, most tumor cells are too big to be taken up by neutrophils and other neutrophil effector functions play a role in the elimination of these cancer cells. Neutrophils contain many granules, which store different cytotoxic components. Based on their contents, granules can be divided into primary, secondary, and tertiary granules. Primary granules are considered the most proinflammatory and cytotoxic, as they contain potent hydrolytic enzymes (e.g. neutrophil elastase), myeloperoxidase (MPO), defensins, and lysozyme. Secondary granules store lactoferrin and tertiary granules contain metalloproteases and gelatinase [Citation101]. Formation of the immunological synapse can stimulate degranulation, leading to the release of these compounds into the extracellular space [Citation101]. It has been shown that MPO [Citation102], defensins [Citation103] and granzyme B [Citation104] isolated from neutrophil granules have cytotoxic capacities. In addition, various released components can recruit and activate other immune cells [Citation105,Citation106]. However, in vitro studies showed that neutrophils with a degranulation defect did not have decreased ADCC ability [Citation107], suggesting that other effector functions play a more important role in tumor cell killing. Another cytotoxic effect of neutrophils is the release of neutrophil extracellular traps (NETs) [Citation108]. During NETosis, neutrophils release their nuclear components, such as DNA and histones, but also cytotoxic factors [Citation109]. NETs are then able to trap pathogens. However, currently no studies have identified a role of NETosis in tumor cell killing. Recently, a new cytotoxic mechanism of neutrophils, i.e. trogocytosis, was identified [Citation107,Citation110]. Trogocytosis is a process in which neutrophils internalize parts of the tumor cell membrane. This results in disruption of the cancer cell plasma membrane, leading to a necrotic form of cell death, referred to as trogoptosis () [Citation107]. Thus, as neutrophils have multiple cytotoxic effector functions, recruitment and stimulation of neutrophils with anti-TAA antibodies might further enhance tumor cell killing [Citation111,Citation112].
Figure 3. Neutrophil-mediated trogocytosis of tumor cells via bispecific antibody therapy.
Human neutrophils express different types of FcγRs, with either high or low affinity for IgG. Resting neutrophils express low-affinity FcγRs, i.e. FcγRIIIb (CD16b) and FcγRIIa (CD32a), while stimulation of neutrophils with G(M)-CSF induces expression of the high-affinity FcγRI (CD64) [Citation113,Citation114]. FcγRIIIb is the most abundantly expressed FcγR on neutrophils. However, it does not seem to be involved in killing of target cells, as blockade with FcγRIIIb specific antibodies did not decrease ADCC by neutrophils. Conversely, selective blockade of FcγRIIa or FcγRI reduced killing of nucleated target cells [Citation115]. Targeting of FcγRI on stimulated neutrophils significantly enhanced tumor cell cytotoxicity [Citation113]. Therefore, various BsAbs targeting FcγRI have been created [Citation116]. An overview of the BsAbs in preclinical or clinical stages is provided in .
The HEA125x197 BsAb targets FcγRI with one Fab arm, while the other Fab domain recognizes Ep-CAM [Citation117], which is a TAA that is overexpressed on virtually all carcinoma cells. In the presence of 10 µg/ml HEA125x197, IFN-γ and G-CSF-stimulated neutrophils were able to kill significantly more CiMe ovarian carcinoma cells, compared to IgG anti-Ep-CAM mAbs [Citation117].
The MDX-210 BsAb consists of 2 Fab domains targeting FcγRI on macrophages and neutrophils and HER-2 on HER-2-positive cancer cells. Cytotoxicity assays with neutrophils from G-CSF-treated patients and SK-BR-3 breast cancer cells showed that addition of 0.4 µg/ml MDX-210 enhanced neutrophil-mediated killing of the target cells [Citation118]. This effect was not observed when neutrophils from healthy donors were used, presumably because these neutrophils lack FcγRI expression. Also, a humanized version of MDX-210 (i.e. MDX-H210) was developed [Citation72]. Human neutrophils were incubated with G-CSF and IFN-γ prior to a co-culture with HER-2/neu expressing cancer cells, after which 0.1 µg/ml MDX-H210 was added, which resulted in enhanced neutrophil-mediated killing of the tumor cells. Due to these promising preclinical results, MDX-210 and MDX-H210 were tested in clinical trials. A phase I trial studied the effect of MDX-210 in 15 patients with advanced breast or ovarian cancer that overexpressed HER-2 [Citation119]. MDX-210 infusion at various doses (0.35–10.0 mg/m2) was well tolerated, and tumor reduction was observed in 2 out of 10 patients. A combination therapy of MDX-210 and filgastrin (therapeutically used G-CSF) was also tested in a phase I clinical trial to further enhance cytotoxicity by neutrophils [Citation118]. Advanced breast cancer patients were treated with G-CSF injections on 8 consecutive days, and with a single injection of MDX-210 on day 4 with different doses per cohort (0.35–100 mg/m2). The combination of G-CSF and MDX-210 was well tolerated. In addition, enhanced plasma levels of the inflammatory cytokines IL-6 and TNF-α postinfusion and infiltration of neutrophils into the tumor indicated immunologic effects of the combination treatment [Citation118]. Similar phase I clinical trials using MXD-H210 (0.35–200 mg/m2) and G-CSF to treat advanced breast cancer patients also showed limited toxicities and increased plasma levels of IL-6 and TNF-α [Citation120,Citation121]. In addition, cytotoxic capabilities of patient’s neutrophils were analyzed. Neutrophils were isolated from the blood and incubated with SK-BR-3 breast cancer cells in the presence of MDX-H210. Patient neutrophils showed enhanced cytotoxic capacities compared to neutrophils from healthy donors [Citation120]. In addition to G-CSF stimulation, IFN-γ and GM-CSF can also induce the expression of FcγRI on neutrophils. Therefore, MDX-H210 therapy was tested in combination with IFN-γ in a phase I clinical trial [Citation122]. Twenty-three patients with advanced HER-2-positive cancers (i.e. breast, prostate and lung cancers) were treated with alternating doses of IFN-γ and MDX-H210 (1–70 mg/m2) every 24 h for 3 weeks. Minimal toxicity was observed. Immunological effects of the treatment were not investigated in this study. The combination of MDX-H210 and GM-CSF was also investigated for toxicity and antitumor effects. In a pilot trial, 13 patients with HER-2-positive cancers (i.e. breast, ovarian, colorectal, lung, and renal) were treated with GM-CSF and various doses of MDX-H210 (1–20 mg/m2) [Citation123]. Again, limited toxicity and increased plasma levels of IL-6, TNF-α and G-CSF were observed in patients. However, after 3 weeks of treatment, significant levels of human anti-bispecific antibodies (HABA) were detected in 5 out of 11 patients. This trial was followed by a phase II study, combining 15 µg/m2 MDX-H210 and GM-CSF for the treatment of HER-2-positive prostate cancer [Citation124]. Clinical responses were observed in 35% of advanced prostate cancer, with a reduction of >50% in prostate-specific antigen (PSA) levels. Of note, observed responses due to treatment with MDX-210 or MDX-H210 are likely the result of different FcγRI expressing immune cells, e.g. neutrophils and macrophages. However, also various grade 3 and 4 adverse events were observed in treated patients [Citation124].
MDX-447 is an FcγRI-targeting BsAb, which consists of two humanized Fab domains, targeting both FcγRI and EGFR [Citation116]. In a phase I/II clinical trial, patients with refractory, EGFR-overexpressing tumors (including head and neck, kidney, bladder, and prostate cancers) were treated with various doses of MDX-447 alone (1–15 mg/m2), or in combination with G-CSF. Generally, the treatment was well tolerated with limited toxicity. In addition, 9 out of 36 patients experienced stable disease for 3–6 months [Citation116]. In another phase I trial, no objective responses were observed in patients treated with different doses of MDX-447 (1–40 mg/m2) [Citation125]. Taken together, FcγRI targeting on neutrophils can potentiate tumor cell killing in vitro. However, in most patients, no significant antitumor responses were observed by treatment with FcγRI targeting BsAbs, and clinical trials involving MDX-210, MDX-H210, and MDX 447 have been discontinued. There are several explanations for the disappointing results of FcγRI BsAb. First, generally patients who had been heavily pretreated with chemo-, radio- or hormonal therapy to treat advanced solid cancer were enrolled in trials. Second, the relative short half-life of BsAb may have precluded effective antitumor effects. For instance, treatment with bispecific T-cell engagers (BiTEs) that consists of two different single-chain variable fragments (scFvs) necessitates administration with continuous intravenous infusion [Citation126]. Third, FcγRI BsAbs were often given in combination with G-CSF or GM-CSF, which recruits immature neutrophils from the bone marrow. With hindsight, this was an unlucky choice, as FcγRI is downregulated during neutrophil maturation, and immature neutrophils were likely less able to induce tumor cell killing through FcγRI [Citation127].
4.2. Neutrophil FcαRI targeting
Similar to macrophages, neutrophils also express FcαRI, which can be used as an alternative target to stimulate neutrophil-mediated ADCC [Citation127–131]. In fact, activation through FcαRI is more potent than activation via FcγR on neutrophils. For example, FcαRI cross-linking by IgA, but not FcγR cross-linking by IgG, induces the release of leukotriene B4 (LTB4), which is a potent neutrophil chemoattractant [Citation132]. In a 3D culture of SK-BR-3 tumor cells in collagen, FcαRI targeting with an FcαRIxHER-2 BsAb induced neutrophil migration toward tumor colonies, which was not observed in the presence of an FcγRIxHER-2 BsAb [Citation15]. In addition, neutrophil migration was enhanced in the presence of endothelial (HUVEC) cells. Possibly, pro-inflammatory cytokines released by neutrophils, i.e. IL-1β and TNFα, stimulated HUVEC cells to produce the neutrophil chemokine IL-8 [Citation15]. FcαRI targeting on neutrophils was shown to induce various cytotoxic functions, such as phagocytosis, respiratory burst, degranulation, ADCC, and NETosis [Citation133,Citation134]. In addition, induction of autophagy in tumor cells as well as necrosis was observed in the presence of neutrophils and FcαRI BsAbs [Citation135].
Increased tumor cell killing by neutrophils after targeting FcαRI compared to FcγR targeting was observed for multiple TAA in vitro [Citation136–143]. In vitro, addition of 1 µg/ml FcαRIxHER-2 BsAbs enhanced SK-BR-3 tumor cell lysis by IFN-γ or G-CSF stimulated neutrophils, compared to FcγRIxHER-2 BsAbs (i.e. MDX-210) [Citation127]. Since G-CSF mobilizes immature neutrophils from the bone marrow, the effect of FcαRI and FcγRI targeting on bone marrow-derived neutrophils was investigated. Only FcαRIxHER-2 BsAbs were able to induce tumor cell killing by immature neutrophils, whereas FcγRIxHER-2 BsAbs were ineffective [Citation127].
FcαRIxCD20 BsAbs consisting of two Fab domains have been described, targeting FcαRI on neutrophils and CD20 on B cell lymphomas [Citation139]. Human neutrophils were able to effectively kill multiple B cell lymphoma cell lines (i.e. BALL, BJAB, RAJI, CESS, RM-1, and ARH-77) in vitro when FcαRIxCD20 BsAbs were present, but not in the presence of IgG anti-CD20 mAbs (all at 2 µg/ml). In addition, neutrophils isolated from G-CSF or GM-CSF treated patients killed significantly more tumor cells compared to neutrophils from healthy donors [Citation139]. Another BsAb targeting FcαRI and CD20, consists of one Fab arm targeting FcαRI, while the Fc-region contains an anti-CD20 domain [Citation79]. In vitro, this BsAb effectively promoted killing of CD20+ B cell lymphoma Raji cells by human neutrophils at antibody concentrations as low as 0.1 µg/ml, while the control antibody rituximab (IgG) was unable to induce ADCC [Citation79].
As described above, we recently developed anti-FcαRI BsAbs, which contained an fully active IgG1 Fc part [Citation82]. Co-incubation of human neutrophils with A431 tumor cells resulted in enhanced tumor cell killing in the presence of 10 µg/ml FcαRIxEGFR BsAbs, compared to IgG α-EGFR or IgA α-EGFR mAbs, although IgA α-EGFR mediated killing was slightly higher at lower doses (0.1 µg/ml). Live-cell imaging showed that FcαRIxEGFR BsAbs and IgA α-EGFR mAbs induced swarming of neutrophils toward A431 cancer cells. FcαRIxEGFR BsAbs also induced degranulation of neutrophils, as measured by increased lactoferrin as well as release of IL-8 and neutrophil chemoattractants [Citation82]. Similar experiments were performed with FcαRIxgp75 BsAbs and murine B16F10gp75 tumor cells. FcαRIxgp75 BsAbs mediated enhanced killing compared to IgG α-gp75 mAbs at antibody concentrations of 10 µg/ml. In addition, FcαRIxgp75 BsAbs induced degranulation. In vivo, treatment with 100 µg FcαRIxgp75 BsAbs significantly reduced growth of subcutaneous B16F10gp75 tumors, compared to IgG α-gp75 mAbs, which was partly dependent on neutrophils, as specific depletion with anti-GR-1 mAbs reduced the efficacy of therapy [Citation82]. Thus, as FcαRI induces potent effector functions by neutrophils and induces potent antitumor responses, FcαRI BsAbs represent promising molecules for tumor targeting.
4.3. Blocking the CD47-SIRPα axis on neutrophils
Neutrophils also express the inhibitory receptor SIRPα. Therefore, antibodies targeting either CD47 or SIRPα may enhance neutrophil responses. In vitro, SK-BR-3 breast cancer cells opsonized with trastuzumab (IgG anti-HER2 mAbs) were more potently killed by human neutrophils, in the presence of anti-CD47 mAbs [Citation144]. Similar results were obtained with anti-SIRPα mAbs in ADCC assays with trastuzumab-opsonized SK-BR-3 breast cancer cells and human neutrophils [Citation98]. In addition, anti-SIRPα mAbs also enhanced elimination of Burkitt’s lymphoma cells in the presence of rituximab (IgG anti-CD20 mAbs) by human neutrophils. The efficacy of treatment with anti-SIRPα mAbs (i.e. KWAR23) was also tested in vivo [Citation98]. Transgenic mice, in which the extracellular domain of murine SIRPα was replaced with human SIRPα, were injected with Burkitt’s lymphoma cells and subsequently treated with rituximab or KWAR23 alone, or combined. Neither treatment with rituximab nor KWAR23 affected tumor growth. However, combination therapy strongly decreased tumor growth, which was reduced after specific depletion of macrophages or neutrophils, indicating that both cell types played a role in tumor killing during combination therapy [Citation98].
Additionally, the effect of CD47xEGFR-IgG1 BsAbs on neutrophil effector functions was investigated. EGFR+CD47+ cancer cells were labeled with DiD and incubated with human neutrophils and CD47xEGFR-IgG1 BsAbs, after which trogocytic activity of neutrophils was determined [Citation90]. Addition of 1 µg/ml EGFRxCD47-IgG1 BsAbs enhanced trogocytosis of tumor cells by neutrophils in an EGFR-dependent manner in multiple EGFR+CD47+ cancer cell lines (FaDu, A431, DLD1, Pae1, SKOV3, and OvCAR3). Increase in trogocytosis subsequently resulted in rapid tumor cell killing [Citation90], supporting that CD47-SIRPα blockade will also be beneficial to induce more potent antitumor responses by neutrophils.
5. Conclusion
Antibody therapy has become a powerful therapeutic weapon for patients with cancer. BsAbs that link two targets, i.e. tumor cells and immune effector cells, can be potential tools to further enhance tumor cell elimination. Various BsAbs have been generated, which are currently tested in preclinical and clinical trials. Myeloid cells, including macrophages and neutrophils, are interesting targets for BsAb therapy, as they naturally possess different cytotoxic properties that can be utilized to eliminate cancer cells. In addition, TANs and especially TAMs are already abundantly present in the TME, obviating the need for recruitment as in situ stimulation may lead to potent tumor cell killing. Moreover, anti-FcαRI BsAbs will induce recruitment of more neutrophils. Combining BsAb therapy with checkpoint blockade will likely further enhance antitumor responses, and in general, BsAbs will preferentially bind to cells expressing both targets, e.g. CD47 and PD-L1, thereby promoting specific elimination of cancer cells and not normal cells. However, in some cases, both markers are expressed on either malignant or healthy cells, e.g. CD47 and CD19 on malignant and normal B cells. Consequently, healthy cells may also be depleted as a result of BsAb treatment. Nonetheless, as a result of the higher specificity of BsAb binding, presumably lower antibody doses are needed to achieve opsonization of tumor cells and thereby clinical responses. Taken together, recruiting myeloid cells with BsAbs represents a promising novel therapy for the treatment of cancer.
6. Expert opinion
Myeloid cells are currently overlooked as potential effector cells, as their presence in tumors is generally linked with poor prognosis of cancer patients. Whereas it is true that myeloid cells can have pro-tumor and immunosuppressive properties, they also have intrinsic cytotoxic properties, which can be utilized in the presence of (bispecific) antibody therapy. As such, we anticipate that macrophages and neutrophils are promising effector cells in antibody-based immunotherapy of cancer. Targeting dendritic cells with BsAb may additionally represent a promising approach to induce adaptive antitumor responses. Despite significant progress to target myeloid cells, no drugs have yet been approved. This can be due to various reasons such as high turnover of myeloid cells and myeloid cell heterogeneity and plasticity [Citation145]. Single-cell RNA sequencing is a powerful technique to gain a better understanding of tumor associated myeloid cells and how to effectively target specific subsets, as this may help to characterize cell diversity and identify heterogeneous phenotypic states. For instance, recently C1QC+ TAMs and SPP1+ TAMs were identified in colorectal cancer, which did not show similarities with either M1 or M2 dichotomous phenotypes [Citation146]. It was observed that C1QC+ TAMs were able to phagocytose and present antigen, while SPP1+ TAMs played a role in angiogenesis, which benefits the tumor. Hence, specific TAMs subsets could be targeted which may lead to improved anti-cancer therapy.
In general, macrophages are particularly effective in eliminating tumor cells through phagocytosis in the presence of antibodies. They express several FcγR, and selective targeting of activating receptors (i.e. FcγRI, FcγRIIa or FcγRIIIa) by BsAb will bypass potential inhibition via the inhibitory FcgRIIb. Macrophages also express FcαRI, although the ability for ADCP is slightly less in the presence of IgA anti-TAA mAbs or anti-FcαRI BsAb [Citation13]. Most tumors already contain a major population of macrophages, which makes them ideal candidates for BsAb therapy strategies as macrophages will be directly activated and kill tumor cells without the need for recruitment. Until now, the use of checkpoint inhibitors has shown promising results. Studies have shown that therapeutic blockade of PD-1/PD-L1 reduced tumor growth in immunodeficient mice by increasing phagocytosis of TAMs against tumor cells.This suggests that anti-PD-1/PD-L-1 blockade in combination with BsAbs might enhance macrophage-mediated antitumor immunity [Citation145].
Similarly, neutrophils are abundantly present in the blood and bone marrow and can be easily expanded by G-CSF therapy, without manipulation or the need for complicated ex vivo culture. This is an advantage compared to other forms of immunotherapy. For example, it is challenging to generate sufficient numbers of activated clinical NK cells or (CAR) T cells [Citation147]. Furthermore, neutrophils kill via non-apoptotic mechanisms, which could be of benefit if tumor cells have gained mutations in apoptotic pathways [Citation111]. Neutrophils are most potently targeted via FcαRI, as this will result in enhanced killing as well as neutrophil migration. Moreover, FcαRI targeting will result in release of pro-inflammatory cytokines, which is not observed after cross-linking of FcγR [Citation15] (van Gool and van Egmond, personal communication). Since the production of clinically grade IgA mAbs with sufficient half-life is difficult, we propose to use BsAbs for FcαRI targeting. Moreover, an IgG1 anti-FcαRI BsAb with a fully functional Fc tail may represent the most optimal format as this will combine activation of macrophages and neutrophils. Additional targeting of the CD47-SIRPα pathway also is an interesting option. Because CD47 is expressed on tumor cells and the inhibitory receptor SIRPα on both macrophages and neutrophils [Citation13,Citation144], targeting either CD47 or SIRPα would lead to stronger ADCP by macrophages and trogocytosis by neutrophils. Thus, we propose that IgG1 anti-FcαRI BsAb therapy combined with blockage of the CD47-SIRPα axis represents the most optimal way for the recruitment of myeloid cells in cancer therapy. However, IL-17 inhibition might be also an interesting strategy, because it has been shown that IL-17 stimulates expansion and polarization into metastasis promoting neutrophils in breast cancer. Furthermore, it has been shown in preclinical studies that anti-PD1 inhibitor with anti-CXCR2 (targeting neutrophils) has been effective in reducing tumor growth [Citation148]. Thus, reprogramming or targeting these myeloid cells by using various molecules or checkpoints in combination with BsAbs may be a promising tool to harness innate antitumor immunity.
Article highlights
The tumor microenvironment influences the state of myeloid cells, polarizing them into cytotoxic effector cells or immunosuppressive regulators.
Macrophages have the ability to eliminate tumor cells via antibody-dependent cellular phagocytosis.
Neutrophils possess various cytotoxic mechanisms, including phagocytosis, degranulation, NET release, and trogocytosis.
Bispecific antibodies (BsAbs) targeting FcγRs or FcαRI may be used to enhance tumor cell killing and to promote pro-inflammatory responses, hereby overcoming the immunosuppressive tumor microenvironment.
Combinations of BsAbs with other molecules, e.g. checkpoint inhibitors or cytokines, may further enhance the antitumor immune response.
Abbreviations
ADCC: antibody-dependent cellular cytotoxicity, ADCP: antibody-dependent cellular phagocytosis, BsAb: bispecific antibodies, DC: dendritic cell, EGFR: endothelial growth factor receptor, Ep-CAM: epithelial cell adhesion molecule, Fab: fragment antigen-binding, FcR: Fc receptor, G-CSF: granulocyte colony-stimulation factor, GM-CSF: granulocyte macrophage-colony stimulator factor, IL: interleukin, IFN-γ: interferon-gamma, LLC: Lewis lung cancer, mAb: monoclonal antibody, MPO: myeloperoxidase, MUC-1: human epithelial mucin, NET: neutrophil extracellular trap, NSCLC: non-small cell lung cancer, SIRPα: signal regulatory protein alpha, TAA: tumor-associated antigen, TAM: tumor-associated macrophages, TAN: tumor-associated neutrophils, Tg: transgenic, TME: tumor microenvironment, TNF-α: tumor necrosis factor-alpha.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Sung H, Ferlay J, Siegel RL. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021 May;71(3):209–249.
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011 Mar 4;144(5):646–674.
- Dunn GP, Bruce AT, Ikeda H. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002 Nov;3(11):991–998.
- He X, Xu C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020 Aug;30(8):660–669.
- Yamamoto K, Venida A, Yano J. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature. 2020 May;581(7806):100–105.
- Weber R, Groth C, Lasser S. IL-6 as a major regulator of MDSC activity and possible target for cancer immunotherapy. Cell Immunol. 2021 Jan;359:104254.
- Horii M, Regulatory MT. B cells and T cell regulation in cancer. J Mol Biol. 2021 Jan 8;433(1):166685.
- Umansky V, Adema GJ, Baran J. Interactions among myeloid regulatory cells in cancer. Cancer Immunol Immunother. 2019 Apr;68(4):645–660.
- Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science. 2013 Dec 20;342(6165):1432–1433.
- Braster R, O’Toole T, van Egmond M. Myeloid cells as effector cells for monoclonal antibody therapy of cancer. Methods. 2014 Jan 1;65(1):28–37.
- Shuptrine CW, Surana R, Weiner LM. Monoclonal antibodies for the treatment of cancer. Semin Cancer Biol. 2012 Feb;22(1):3–13.
- Zahavi D, Weiner L. Monoclonal antibodies in cancer therapy. Antibodies (Basel). 2020 Jul 20;9:3.
- Gul N, van Egmond M. Antibody-dependent phagocytosis of tumor cells by macrophages: a potent effector mechanism of monoclonal antibody therapy of cancer. Cancer Res. 2015 Dec 1;75(23):5008–5013.
- Goldberg BS, Ackerman ME. Antibody-mediated complement activation in pathology and protection. Immunol Cell Biol. 2020 Apr;98(4):305–317.
- Otten MA, Bakema JE, Tuk CW. Enhanced FcalphaRI-mediated neutrophil migration towards tumour colonies in the presence of endothelial cells. Eur J Immunol. 2012 Jul;42(7):1815–1821.
- Bispecific Antibodies SH. Antibody-drug conjugates for cancer therapy: technological considerations. Biomolecules. 2020 Feb 26;10:3.
- Sedykh SE, Prinz VV, Buneva VN. Bispecific antibodies: design, therapy, perspectives. Drug Des Devel Ther. 2018;12:195–208.
- Brinkmann U, Kontermann RE. The making of bispecific antibodies. MAbs. 2017 Feb/Mar;9(2):182–212.
- Labrijn AF, Janmaat ML, Reichert JM. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discov. 2019 Aug;18(8):585–608.
- Thakur A, Huang M, Lum LG. Bispecific antibody based therapeutics: strengths and challenges. Blood Rev. 2018 Jul;32(4):339–347.
- Loffler A, Kufer P, Lutterbuse R. A recombinant bispecific single-chain antibody, CD19 x CD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes. Blood. 2000 Mar 15;95(6):2098–2103.
- Dreier T, Lorenczewski G, Brandl C. Extremely potent, rapid and costimulation-independent cytotoxic T-cell response against lymphoma cells catalyzed by a single-chain bispecific antibody. Int J Cancer. 2002 Aug 20;100(6):690–697.
- Kawamoto H, Minato N. Myeloid cells. Int J Biochem Cell Biol. 2004 Aug;36(8):1374–1379.
- Mantovani A, Marchesi F, Jaillon S. Tumor-associated myeloid cells: diversity and therapeutic targeting. Cell Mol Immunol. 2021 Mar;18(3):566–578.
- Kim J, Bae JS. Tumor-associated macrophages and neutrophils in tumor microenvironment. Mediators Inflamm. 2016;2016:6058147.
- Dysthe M, Parihar R. Myeloid-derived suppressor cells in the tumor microenvironment. Adv Exp Med Biol. 2020;1224:117–140.
- Wellenstein MD, de Visser KE. Cancer-cell-intrinsic mechanisms shaping the tumor immune landscape. Immunity. 2018 Mar 20;48(3):399–416.
- Salmaninejad A, Valilou SF, Soltani A. Tumor-associated macrophages: role in cancer development and therapeutic implications. Cell Oncol (Dordr). 2019 Oct;42(5):591–608.
- Franklin RA, Liao W, Sarkar A. The cellular and molecular origin of tumor-associated macrophages. Science. 2014 May 23;344(6186):921–925.
- Beltraminelli T, De Palma M. Biology and therapeutic targeting of tumour-associated macrophages. J Pathol. 2020 Apr;250(5):573–592.
- Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010 Mar 19;140(6):883–899.
- Mantovani A, Marchesi F, Malesci A. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017 Jul;14(7):399–416.
- Martinez FO, Sica A, Mantovani A. Macrophage activation and polarization. Front Biosci. 2008. 1. Jan(13):453–461.
- Mantovani A, Sica A, Sozzani S. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004 Dec;25(12):677–686.
- Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008 Dec;8(12):958–969.
- Vesely MD, Kershaw MH, Schreiber RD. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29(1):235–271.
- Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010 Apr 2;141(1):39–51.
- De Palma M, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell. 2013 Mar 18;23(3):277–286.
- Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol. 2002 Mar;196(3):254–265.
- Ni C, Yang L, Xu Q. CD68- and CD163-positive tumor infiltrating macrophages in non-metastatic breast cancer: a retrospective study and meta-analysis. J Cancer. 2019;10(19):4463–4472.
- DeNardo DG, Brennan DJ, Rexhepaj E. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011 Jun;1(1):54–67.
- Zhang QW, Liu L, Gong CY. Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PLoS One. 2012;7(12):e50946.
- Komohara Y, Fujiwara Y, Ohnishi K. Tumor-associated macrophages: potential therapeutic targets for anti-cancer therapy. Adv Drug Deliv Rev. 2016 Apr 1;99(Pt B):180–185.
- Mantovani A, Cassatella MA, Costantini C. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. 2011 Jul 25;11(8):519–531.
- Mocsai A. Diverse novel functions of neutrophils in immunity, inflammation, and beyond. J Exp Med. 2013 Jul 1;210(7):1283–1299.
- Ley K, Hoffman HM, Kubes P. Neutrophils: new insights and open questions. Sci Immunol. 2018 Dec 7;3(30):30.
- Gruijs M, Sewnath CAN, Egmond MV. Therapeutic exploitation of neutrophils to fight cancer. Semin Immunol. 2021 Dec;15:101581.
- Coffelt SB, Wellenstein MD, de Visser KE. Neutrophils in cancer: neutral no more. Nat Rev Cancer. 2016 Jul;16(7):431–446.
- Grecian R, Whyte MKB, Walmsley SR. The role of neutrophils in cancer. Br Med Bull. 2018 Dec 1;128(1):5–14.
- Burn GL, Foti A, Marsman G. The Neutrophil. Immunity. 2021 Jul 13;54(7):1377–1391.
- Jaillon S, Ponzetta A, Di Mitri D. Neutrophil diversity and plasticity in tumour progression and therapy. Nat Rev Cancer. 2020 Sep;20(9):485–503.
- Fridlender ZG, Sun J, Kim S. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. 2009 Sep 8;16(3):183–194.
- Templeton AJ, McNamara MG, Seruga B. Prognostic role of neutrophil-to-lymphocyte ratio in solid tumors: a systematic review and meta-analysis. J Natl Cancer Inst. 2014 Jun;106(6):dju124.
- Guthrie GJ, Charles KA, Roxburgh CS. The systemic inflammation-based neutrophil-lymphocyte ratio: experience in patients with cancer. Crit Rev Oncol Hematol. 2013 Oct;88(1):218–230.
- Di Carlo E, Forni G, Lollini P. The intriguing role of polymorphonuclear neutrophils in antitumor reactions. Blood. 2001 Jan 15;97(2):339–345.
- Li X, Liu R, Su X. Harnessing tumor-associated macrophages as aids for cancer immunotherapy. Mol Cancer. 2019 Dec 5;18(1):177.
- Oflazoglu E, Stone IJ, Brown L. Macrophages and Fc-receptor interactions contribute to the antitumour activities of the anti-CD40 antibody SGN-40. Br J Cancer. 2009 Jan 13;100(1):113–117.
- Oflazoglu E, Stone IJ, Gordon KA. Macrophages contribute to the antitumor activity of the anti-CD30 antibody SGN-30. Blood. 2007 Dec 15;110(13):4370–4372.
- Bologna L, Gotti E, Da Roit F. Ofatumumab is more efficient than rituximab in lysing B chronic lymphocytic leukemia cells in whole blood and in combination with chemotherapy. J Immunol. 2013 Jan 1;190(1):231–239.
- Akewanlop C, Watanabe M, Singh B. Phagocytosis of breast cancer cells mediated by anti-MUC-1 monoclonal antibody, DF3, and its bispecific antibody. Cancer Res. 2001 May 15;61(10):4061–4065.
- Ashraf SQ, Umana P, Mossner E. Humanised IgG1 antibody variants targeting membrane-bound carcinoembryonic antigen by antibody-dependent cellular cytotoxicity and phagocytosis. Br J Cancer. 2009 Nov 17;101(10):1758–1768.
- Huls G, Heijnen IA, Cuomo E. Antitumor immune effector mechanisms recruited by phage display-derived fully human IgG1 and IgA1 monoclonal antibodies. Cancer Res. 1999 Nov 15;59(22):5778–5784.
- Lefebvre ML, Krause SW, Salcedo M. Ex vivo-activated human macrophages kill chronic lymphocytic leukemia cells in the presence of rituximab: mechanism of antibody-dependent cellular cytotoxicity and impact of human serum. J Immunother. 2006 Jul-Aug;29(4):388–397.
- Rafiq S, Butchar JP, Cheney C. Comparative assessment of clinically utilized CD20-directed antibodies in chronic lymphocytic leukemia cells reveals divergent NK cell, monocyte, and macrophage properties. J Immunol. 2013 Mar 15;190(6):2702–2711.
- Watanabe M, Wallace PK, Keler T. Antibody dependent cellular phagocytosis (ADCP) and antibody dependent cellular cytotoxicity (ADCC) of breast cancer cells mediated by bispecific antibody, MDX-210. Breast Cancer Res Treat. 1999 Feb;53(3):199–207.
- Gruijs M, Braster R, Overdijk MB. Epidermal growth factor receptor as target for perioperative elimination of circulating colorectal cancer cells. J Oncol. 2022;2022:3577928.
- Richards DM, Endres RG. The mechanism of phagocytosis: two stages of engulfment. Biophys J. 2014 Oct 7;107(7):1542–1553.
- Hogarth PM, Pietersz GA. Fc receptor-targeted therapies for the treatment of inflammation, cancer and beyond. Nat Rev Drug Discov. 2012 Mar 30;11(4):311–331.
- Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. 2008 Jan;8(1):34–47.
- Clynes RA, Towers TL, Presta LG. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 2000 Apr;6(4):443–446.
- Sundarapandiyan K, Keler T, Behnke D. Bispecific antibody-mediated destruction of Hodgkin’s lymphoma cells. J Immunol Methods. 2001 Feb 1;248(1–2):113–123.
- Keler T, Graziano RF, Mandal A. Bispecific antibody-dependent cellular cytotoxicity of HER2/neu-overexpressing tumor cells by Fc gamma receptor type I-expressing effector cells. Cancer Res. 1997 Sep 15;57(18):4008–4014.
- Keler T, Wallace PK, Vitale LA. Differential effect of cytokine treatment on Fc alpha receptor I- and Fc gamma receptor I-mediated tumor cytotoxicity by monocyte-derived macrophages. J Immunol. 2000 Jun 1;164(11):5746–5752.
- Grugan KD, Dorn K, Jarantow SW. Fc-mediated activity of EGFR x c-Met bispecific antibody JNJ-61186372 enhanced killing of lung cancer cells. MAbs. 2017 Jan;9(1):114–126.
- Vijayaraghavan S, Lipfert L, Chevalier K. Amivantamab (JNJ-61186372), an Fc enhanced EGFR/cMet bispecific antibody, induces receptor downmodulation and antitumor activity by monocyte/macrophage trogocytosis. Mol Cancer Ther. 2020 Oct;19(10):2044–2056.
- Aleyd E, Heineke MH, van Egmond M. The era of the immunoglobulin A Fc receptor FcalphaRI; its function and potential as target in disease. Immunol Rev. 2015 Nov;268(1):123–138.
- Reljic R. In search of the elusive mouse macrophage Fc-alpha receptor. Immunol Lett. 2006 Sep 15;107(1):80–81.
- Launay P, Grossetete B, Arcos-Fajardo M. Fcalpha receptor (CD89) mediates the development of immunoglobulin A (IgA) nephropathy (Berger’s disease). Evidence for pathogenic soluble receptor-Iga complexes in patients and CD89 transgenic mice. J Exp Med. 2000 Jun 5;191(11):1999–2009.
- Li B, Xu L, Pi C. CD89-mediated recruitment of macrophages via a bispecific antibody enhances anti-tumor efficacy. Oncoimmunology. 2017;7(1):e1380142.
- van Egmond M, Hanneke van Vuuren AJ, JG VDW. The human Fc receptor for IgA (Fc alpha RI, CD89) on transgenic peritoneal macrophages triggers phagocytosis and tumor cell lysis. Immunol Lett. 1999 May 3;68(1):7–83.
- van Egmond M, van Vuuren AJ, Morton HC. Human immunoglobulin A receptor (FcalphaRI, CD89) function in transgenic mice requires both FcR gamma chain and CR3 (CD11b/CD18). Blood. 1999 Jun 15;93(12):4387–4394.
- Heemskerk N, Gruijs M, Temming AR. Augmented antibody-based anticancer therapeutics boost neutrophil cytotoxicity. J Clin Invest. 2021 Mar 15;131(6). DOI:10.1172/JCI134680. .
- Kelton W, Mehta N, Charab W. IgGA: a “cross-isotype” engineered human Fc antibody domain that displays both IgG-like and IgA-like effector functions. Chem Biol. 2014 Dec 18;21(12):1603–1609.
- Borrok MJ, Luheshi NM, Beyaz N. Enhancement of antibody-dependent cell-mediated cytotoxicity by endowing IgG with FcalphaRI (CD89) binding. MAbs. 2015;7(4):743–751.
- Li B, Xu L, Tao F. Simultaneous exposure to FcgammaR and FcalphaR on monocytes and macrophages enhances antitumor activity in vivo. Oncotarget. 2017 Jun 13;8(24):39356–39366.
- Barclay AN, van den Berg TK. The interaction between signal regulatory protein alpha (SIRPalpha) and CD47: structure, function, and therapeutic target. Annu Rev Immunol. 2014;32(1):25–50.
- Chao MP, Alizadeh AA, Tang C. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell. 2010 Sep 3;142(5):699–713.
- Chen S, Lai SWT, Brown CE. Harnessing and enhancing macrophage phagocytosis for cancer therapy. Front Immunol. 2021;12:635173.
- Chao MP, Tang C, Pachynski RK. Extranodal dissemination of non-Hodgkin lymphoma requires CD47 and is inhibited by anti-CD47 antibody therapy. Blood. 2011 Nov 3;118(18):4890–4901.
- Hendriks M, Ploeg EM, Koopmans I. Bispecific antibody approach for EGFR-directed blockade of the CD47-SIRPalpha “don’t eat me” immune checkpoint promotes neutrophil-mediated trogoptosis and enhances antigen cross-presentation. Oncoimmunology. 2020 Sep 29; 9(1):1824323.
- Seshacharyulu P, Ponnusamy MP, Haridas D. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012 Jan;16(1):15–31.
- Buatois V, Johnson Z, Salgado-Pires S. Preclinical development of a bispecific antibody that safely and effectively targets CD19 and CD47 for the treatment of B-Cell lymphoma and leukemia. Mol Cancer Ther. 2018 Aug;17(8):1739–1751.
- Hatterer E, Barba L, Noraz N. Co-engaging CD47 and CD19 with a bispecific antibody abrogates B-cell receptor/CD19 association leading to impaired B-cell proliferation. MAbs. 2019 Feb/Mar;11(2):322–334.
- Chen SH, Dominik PK, Stanfield J. Dual checkpoint blockade of CD47 and PD-L1 using an affinity-tuned bispecific antibody maximizes antitumor immunity. J Immunother Cancer. 2021 Oct;9(10).
- Wang Y, Ni H, Zhou S. Tumor-selective blockade of CD47 signaling with a CD47/PD-L1 bispecific antibody for enhanced anti-tumor activity and limited toxicity. Cancer Immunol Immunother. 2020 Feb;70(2):365–376.
- Law CL, Gordon KA, Toki BE. Lymphocyte activation antigen CD70 expressed by renal cell carcinoma is a potential therapeutic target for anti-CD70 antibody-drug conjugates. Cancer Res. 2006 Feb 15;66(4):2328–2337.
- McEarchern JA, Oflazoglu E, Francisco L. Engineered anti-CD70 antibody with multiple effector functions exhibits in vitro and in vivo antitumor activities. Blood. 2007 Feb 1;109(3):1185–1192.
- Ring NG, Herndler-Brandstetter D, Weiskopf K. Anti-SIRPalpha antibody immunotherapy enhances neutrophil and macrophage antitumor activity. Proc Natl Acad Sci U S A. 2017 Dec 5;114(49):E10578–E10585.
- Hubert P, Heitzmann A, Viel S. Antibody-dependent cell cytotoxicity synapses form in mice during tumor-specific antibody immunotherapy. Cancer Res. 2011 Aug 1;71(15):5134–5143.
- van Spriel AB, Leusen JH, van Egmond M. Mac-1 (CD11b/CD18) is essential for Fc receptor-mediated neutrophil cytotoxicity and immunologic synapse formation. Blood. 2001 Apr 15;97(8):2478–2486.
- Faurschou M, Borregaard N. Neutrophil granules and secretory vesicles in inflammation. Microbes Infect. 2003 Nov;5(14):1317–1327.
- Clark RA, Olsson I, Klebanoff SJ. Cytotoxicity for tumor cells of cationic proteins from human neutrophil granules. J Cell Biol. 1976 Sep;70(3):719–723.
- Lichtenstein A. Mechanism of mammalian cell lysis mediated by peptide defensins. Evidence for an initial alteration of the plasma membrane. J Clin Invest. 1991 Jul;88(1):93–100.
- Martin A, Seignez C, Racoeur C. Tumor-derived granzyme B-expressing neutrophils acquire antitumor potential after lipid A treatment. Oncotarget. 2018 Jun 19;9(47):28364–28378.
- Cowland JB, Borregaard N. Granulopoiesis and granules of human neutrophils. Immunol Rev. 2016 Sep;273(1):11–28.
- Nemeth T, Mocsai A. Feedback amplification of neutrophil function. Trends Immunol. 2016 Jun;37(6):412–424.
- Matlung HL, Babes L, Zhao XW. Neutrophils kill antibody-opsonized cancer cells by trogoptosis. Cell Rep. 2018 Jun 26; 23(13):3946–3959e6.
- Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. 2018 Feb;18(2):134–147.
- Brinkmann V, Reichard U, Goosmann C. Neutrophil extracellular traps kill bacteria. Science. 2004 Mar 5;303(5663):1532–1535.
- Valgardsdottir R, Cattaneo I, Klein C. Human neutrophils mediate trogocytosis rather than phagocytosis of CLL B cells opsonized with anti-CD20 antibodies. Blood. 2017 May 11;129(19):2636–2644.
- Heemskerk N, van Egmond M. Monoclonal antibody-mediated killing of tumour cells by neutrophils. Eur J Clin Invest. 2018 Nov;48(Suppl 2):e12962.
- van Egmond M, Bakema JE. Neutrophils as effector cells for antibody-based immunotherapy of cancer. Semin Cancer Biol. 2013 Jun;23(3):190–199.
- Valerius T, Repp R, de Wit TP. Involvement of the high-affinity receptor for IgG (Fc gamma RI; CD64) in enhanced tumor cell cytotoxicity of neutrophils during granulocyte colony-stimulating factor therapy. Blood. 1993 Aug 1;82(3):931–939.
- Futosi K, Fodor S, Mocsai A. Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int Immunopharmacol. 2013 Nov;17(3):638–650.
- Valerius T, Wurflein D, Stockmeyer B. Activated neutrophils as effector cells for bispecific antibodies. Cancer Immunol Immunother. 1997 Nov-Dec;45(3–4):5–142.
- Curnow RT. Clinical experience with CD64-directed immunotherapy. An overview. Cancer Immunol Immunother. 1997 Nov-Dec;45(3–4):210–215.
- Schweizer C, Strauss G, Lindner M. Efficient carcinoma cell killing by activated polymorphonuclear neutrophils targeted with an Ep-CAMxCD64 (HEA125x197) bispecific antibody. Cancer Immunol Immunother. 2002 Dec;51(11–12):621–629.
- Repp R, Valerius T, Wieland G. G-CSF-stimulated PMN in immunotherapy of breast cancer with a bispecific antibody to Fc gamma RI and to HER-2/neu (MDX-210). J Hematother. 1995Oct;4(5):415–421.
- Valone FH, Kaufman PA, Guyre PM. Phase Ia/Ib trial of bispecific antibody MDX-210 in patients with advanced breast or ovarian cancer that overexpresses the proto-oncogene HER-2/neu. J Clin Oncol. 1995 Sep;13(9):2281–2292.
- Repp R, van Ojik HH, Valerius T. Phase I clinical trial of the bispecific antibody MDX-H210 (anti-FcgammaRI x anti-HER-2/neu) in combination with Filgrastim (G-CSF) for treatment of advanced breast cancer. Br J Cancer. 2003 Dec 15;89(12):2234–2243.
- Pullarkat V, Deo Y, Link J. A phase I study of a HER2/neu bispecific antibody with granulocyte-colony-stimulating factor in patients with metastatic breast cancer that overexpresses HER2/neu. Cancer Immunol Immunother. 1999 Apr;48(1):9–21.
- Lewis LD, Beelen AP, Cole BF. The pharmacokinetics of the bispecific antibody MDX-H210 when combined with interferon gamma-1b in a multiple-dose phase I study in patients with advanced cancer. Cancer Chemother Pharmacol. 2002 May;49(5):375–384.
- Posey JA, Raspet R, Verma U. A pilot trial of GM-CSF and MDX-H210 in patients with erbB-2-positive advanced malignancies. J Immunother. 1999 Jul;22(4):371–379.
- James ND, Atherton PJ, Jones J. A phase II study of the bispecific antibody MDX-H210 (anti-HER2 x CD64) with GM-CSF in HER2+ advanced prostate cancer. Br J Cancer. 2001 Jul 20;85(2):152–156.
- Fury MG, Lipton A, Smith KM. A phase-I trial of the epidermal growth factor receptor directed bispecific antibody MDX-447 without and with recombinant human granulocyte-colony stimulating factor in patients with advanced solid tumors. Cancer Immunol Immunother. 2008 Feb;57(2):155–163.
- Goebeler ME, Bargou RC. T cell-engaging therapies - BiTEs and beyond. Nat Rev Clin Oncol. 2020 Jul;17(7):418–434.
- Otten MA, Rudolph E, Dechant M. Immature neutrophils mediate tumor cell killing via IgA but not IgG Fc receptors. J Immunol. 2005 May 1;174(9):5472–5480.
- Mmj VG, van Egmond M. IgA and FcalphaRI: versatile players in homeostasis, infection, and autoimmunity. Immunotargets Ther. 2020;9:351–372.
- Bakema JE, van Egmond M, Immunoglobulin A. A next generation of therapeutic antibodies?. MAbs. 2011 Jul-Aug;3(4):352–361.
- Monteiro RC, van de Winkel JGJ. IgA Fc receptors. Annu Rev Immunol. 2003;21(1):177–204.
- Valerius T, Stockmeyer B, van Spriel AB. FcalphaRI (CD89) as a novel trigger molecule for bispecific antibody therapy. Blood. 1997 Dec 1;90(11):4485–4492.
- van der Steen L, Tuk CW, Bakema JE. Immunoglobulin A: fc(alpha)RI interactions induce neutrophil migration through release of leukotriene B4. Gastroenterology. 2009 Dec;137(6):2018–29 e1–3.
- Bakema JE, van Egmond M. The human immunoglobulin A Fc receptor FcalphaRI: a multifaceted regulator of mucosal immunity. Mucosal Immunol. 2011 Nov;4(6):612–624.
- Aleyd E, van Hout MW, Ganzevles SH. IgA enhances NETosis and release of neutrophil extracellular traps by polymorphonuclear cells via Fcalpha receptor I. J Immunol. 2014 Mar 1;192(5):2374–2383.
- Bakema JE, Ganzevles SH, Fluitsma DM. Targeting FcalphaRI on polymorphonuclear cells induces tumor cell killing through autophagy. J Immunol. 2011 Jul 15;187(2):726–732.
- Brandsma AM, Bondza S, Evers M. Potent Fc receptor signaling by IgA leads to superior killing of cancer cells by neutrophils compared to IgG. Front Immunol. 2019;10:704.
- Rouwendal GJ, van der Lee MM, Meyer S. A comparison of anti-HER2 IgA and IgG1 in vivo efficacy is facilitated by high N-glycan sialylation of the IgA. MAbs. 2016;8(1):74–86.
- van Egmond M, van Spriel AB, Vermeulen H. Enhancement of polymorphonuclear cell-mediated tumor cell killing on simultaneous engagement of fcgammaRI (CD64) and fcalphaRI (CD89). Cancer Res. 2001 May 15;61(10):4055–4060.
- Stockmeyer B, Dechant M, van Egmond M. Triggering Fc alpha-receptor I (CD89) recruits neutrophils as effector cells for CD20-directed antibody therapy. J Immunol. 2000 Nov 15;165(10):5954–5961.
- Dechant M, Vidarsson G, Stockmeyer B. Chimeric IgA antibodies against HLA class II effectively trigger lymphoma cell killing. Blood. 2002 Dec 15;100(13):4574–4580.
- Lohse S, Derer S, Beyer T. Recombinant dimeric IgA antibodies against the epidermal growth factor receptor mediate effective tumor cell killing. J Immunol. 2011 Mar 15;186(6):8–3770.
- Stadick H, Stockmeyer B, Kuhn R. Epidermal growth factor receptor and g250: useful target antigens for antibody mediated cellular cytotoxicity against renal cell carcinoma?. J Urol. 2002 Feb;167(2 Pt 1):12–707.
- Pascal V, Laffleur B, Debin A, Pascal V, Laffleur B, Debin A . Anti-CD20 IgA can protect mice against lymphoma development: evaluation of the direct impact of IgA and cytotoxic effector recruitment on CD20 target cells. Haematologica. 2012 Nov; 97(11):1686–1694.
- Zhao XW, van Beek EM, Schornagel K. CD47-signal regulatory protein-alpha (SIRPalpha) interactions form a barrier for antibody-mediated tumor cell destruction. Proc Natl Acad Sci U S A. 2011 Nov 8;108(45):18342–18347.
- Nakamura K, Smyth MJ. Myeloid immunosuppression and immune checkpoints in the tumor microenvironment. Cell Mol Immunol. 2020 Jan;17(1):1–12.
- Zhang L, Li Z, Skrzypczynska KM. Single-cell analyses inform mechanisms of myeloid-targeted therapies in colon cancer. Cell. 2020 Apr 16; 181(2):442–459e29.
- Choi JW, Lee ES, Kim SY. Cytotoxic effects of ex vivo-expanded natural killer cell-enriched lymphocytes (MYJ1633) against liver cancer. BMC Cancer. 2019 Aug 19;19(1):817.
- Wu M, Ma M, Tan Z. Neutrophil: a new player in metastatic cancers. Front Immunol. 2020;11:565165.