
ABSTRACT
Introduction: Direct targeting of Bcl-2 members for therapeutic purposes in cancer has become a clinical reality with the FDA approval of ABT-199/Venetoclax. Other highly specific BH3-mimetics are in pre-clinical development. Understanding the functional interactions among the Bcl-2 family is of prime importance to fully exploit their potential. NOXA is considered a rather weak BH3-only member but it has unexplored potential in various settings, which are of relevance in cancer. NOXA is best known as a selective inhibitor of MCL1, itself overexpressed in many cancers, and this protein pair forms an important rheostat in many forms of cell stress.
Areas covered: We summarize the distinct pathways that induce NOXA RNA and protein, and how this may be exploited in solid and hematopoietic cancers, with a focus on multiple myeloma and chronic lymphocytic leukemia.
Expert opinion: The therapeutic potential to induce NOXA is not yet fully explored nor exploited, and we suggest 1) areas that require further fundamental investigation, including replicative stress and epigenetics, 2) areas where translation to therapeutic application seems more imminent (ER stress, ROS, inhibition of NOXA degradation) 3) a complementary approach to inducing NOXA by direct targeting of MCL1 via the novel BH3 mimetic S63845 and similar compounds.
KEYWORDS:
1. Introduction
1.1. The BCL-2 family of proteins
Regulated cell death or apoptosis is a process whereby superfluous, damaged or pathological cells are removed via ordered proteolytic breakdown. As a result of diverse stress signals, cytoplasmic cysteine proteases called caspases become activated, which trigger the actual execution of apoptosis. The most prominent executioner is caspase-3 [Citation1]. Two upstream routes to apoptosis are known; the extrinsic or death receptor pathway and the intrinsic or mitochondrial pathway. The intrinsic pathway is predominant in most cell types [Citation2], and is controlled at the mitochondrion as central sensor organelle of cell stress.
Stress signals culminate in shifts in expression levels and/or interactions in a group of proteins, which collectively form the Bcl-2 family. The Bcl-2 family consists of three subgroups: the pro-survival Bcl-2-like (BCL2, BCLXL, MCL1, BCL2A1, BCLB, and BCLW), the pro-apoptotic multi-domain BAX and BAK1 (to which also the more obscure member Bok is assigned), and the single domain BH3-only proteins (BIM, PUMA/BBC3, BID, BAD, BIK, BMF, NOXA, HRK). Of note, there are additional BCL-2(like) proteins which are less well studied or have predominantly non-apoptotic functions, and these members, or splice variants with opposing functions of e.g. MCL1 and BCLXL that are generally expressed at low levels will not be discussed further in the present review [Citation3–Citation5]. Changes in the balance between Bcl-2-like and BH3-only members occur upon pro-survival or stress/apoptotic stimuli. This can lead to activation and multimerization of BAX and/or BAK1, which causes pore formation in the mitochondrial outer membrane. Through these pores, inter-membrane proteins leak into the cytoplasm, most prominently cytochrome C, which becomes a co-factor for caspase-9 activation once bound by the adapter protein APAF-1 in the cytoplasm [Citation6]. Caspase-9 and its co-factors form the multimeric apoptosome, which constitutes the generator of active caspase-3. Although minor mitochondrial damage or membrane permeabilization and subsequent caspase-9 activation can still be quenched by inhibitory fail-safe mechanisms, massive pore formation by BAX/BAK1, cytochrome C release and apoptosome formation can be considered as the point of no return for apoptosis [Citation7]. It follows that, in order to promote or prevent apoptosis for therapeutic purposes, the interactions among the Bcl-2 family are crucial determinants.
1.2. NOXA is a weak BH3-only member, or is it perhaps not?
Structurally, the Bcl-2 family is quite uniform despite the fact that individual members can have opposing pro-death or pro-survival functions. The Bcl-2-like and BAX-like members share homology domains (numbered BH1-4) and have a similar 3D structure. The most numerous subgroup of Bcl-2 proteins is formed by the BH3-only proteins, which only share the 10–12 amino acid BH3 domain, and have no (common) ordered structure. However, upon binding with a Bcl-2-like member, the BH3-only domain is inserted into a hydrophobic groove of their binding partner [Citation8]. Initially, it was unclear what the distinction among the pro- and anti-apoptotic Bcl-2 members was, as overexpression of any BH3-only protein can trigger apoptosis (in fact that is a prominent criterion to be included in the family). Conversely, all Bcl-2-like members can protect against various apoptotic stimuli when overexpressed [Citation9–Citation11].
In several seminal studies, the group of David Huang uncovered that the BH3-only proteins have vastly different interactions and binding properties. Some, such as BIM and BID are promiscuous and bind to all the pro-survival Bcl-2 members, while BAD and NOXA have a much more restricted binding [Citation12,Citation13]. In particular, BAD interacts with BCL-2, BCLXL and BCLW, while NOXA binds only to MCL1 and BCL2A1. This division of BH3-only proteins in strong and weak interactors is reflected by their capacity to trigger apoptosis; BID and BIM are generally strong apoptosis activators, since they bind all pro-survival Bcl-2 members. NOXA in contrast, is considered a ‘weak’ or indirect activator as it can only bind and neutralize the pro-survival members MCL1 and BCL2A1, thereby preventing them from keeping BAX and BAK1 in check. These (non-mutually exclusive) modes of direct versus indirect control of BAX and BAK1 have been an area of quite some controversy [Citation14–Citation18], and not all details have been elucidated yet. Currently, there seems consensus that BIM, BID and most probably PUMA [Citation19] can directly activate BAX or BAK1 in a hit-and-run mechanism which will trigger apoptosis, whereas the other BH3-only proteins cannot ().
Figure 1. Functional interactions among Bcl-2 family members.
Inhibitory interactions among pro-survival members (dark grey boxes), and strong (dark grey ovals) versus weak (light grey ovals) BH3-only members. Stimulatory (arrow) and inhibitory (blockline) interactions among pro-survival Bcl-2 members, BAX/BAK1, and strong BH3-only members. NOXA is tentatively represented as activator of BAK and BAX based on Refs. 20–23.Figure compiled and adapted from information and similar schemes represented in Refs [Citation8,Citation9,Citation18,Citation20].
![Figure 1. Functional interactions among Bcl-2 family members.Inhibitory interactions among pro-survival members (dark grey boxes), and strong (dark grey ovals) versus weak (light grey ovals) BH3-only members. Stimulatory (arrow) and inhibitory (blockline) interactions among pro-survival Bcl-2 members, BAX/BAK1, and strong BH3-only members. NOXA is tentatively represented as activator of BAK and BAX based on Refs. 20–23.Figure compiled and adapted from information and similar schemes represented in Refs [Citation8,Citation9,Citation18,Citation20].](/cms/asset/83f5cab4-f964-4fd9-bbe8-ca7ea3246fa5/iett_a_1349754_f0001_b.gif)
Still, In addition to BIM, BID and PUMA, there are convincing reports that BMF and NOXA can also directly bind BAX and potentially also BAK1, depending on whether BIM and BID are present, and whether other anti-apoptotic proteins such as BCLXL are inhibited [Citation21]. The potential of NOXA as direct activator is weaker than that of BIM or BIM, and also seems to critically depend on whether full-length NOXA protein [Citation22] or a BH3-peptide is applied [Citation23]. Genetic data support that NOXA can indeed bind directly to BAX, and thereby induce cell death in BIM/BID/PUMA triple knock-out cells [Citation20]. Biochemically, BAX binding capacity was uncovered using full-length, in vitro translated NOXA. Combined these data suggests that, if valid under physiological circumstances, the interaction of NOXA with BAX/BAK is of significantly lower affinity than that of BIM or BID. Nevertheless, these studies hint that perhaps the dust has not yet settled in the arena of Bcl-2 family interactions (). Moreover, they implicate that under certain conditions, the role of NOXA can be decisive in triggering apoptosis.
Therefore, there is a rationale to explore in detail whether any hidden potential of NOXA can be unleashed to combat cancer. Before we go deeper into the properties of NOXA and the pathways it may be involved in that can be targeted for therapeutic purposes, we will first address the important interaction with its binding partner MCL-1, followed by NOXA gene and mRNA expression under various forms of cell stress. It will become clear that NOXA gene and protein regulation is complex and can be activated by a multitude of signals involved in DNA damage, cell division/proliferation and cell stress. We will then summarize recent developments regarding drugs that directly target pro-survival Bcl-2 members, most prominent the so-called BH3 mimetics, before we merge these sections in a conclusion and expert opinion section.
2. Protein regulation of MCL1 and NOXA
Under what circumstances can NOXA have a significant impact on apoptosis? It follows from its established interactions that this occurs in cells that depend on their survival on either MCL1 or BCL2A1. BCL2A1 function has been mostly described in the context of lymphocytes after antigen or CD40 stimulation, and, due to a paucity of good reagents for both mouse A1 and human BCL2A1, detailed insight into its importance or contribution to apoptosis is still incomplete [Citation24]. With the recent description of modified BH3 peptide reagents that are highly specific for BCL2A1 over other pro-survival Bcl-2 members [Citation25], insight into the functional contribution of BCL2A1 may soon improve. In this light, the recent description that a peptide based on human NOXA is able to covalently attach to BCL2A1 via cysteine bonds, with great selectivity over MCL-1, may allow further probing into the role of BCL2A1 but also of NOXA, in cancer cells but also under specific conditions such as UV light and ROS generation[Citation26].
For MCL1 there is ample evidence for its importance in many cell types and under various circumstances. MCL1 is widely expressed in hematopoietic and other tissues, and is regulated by a variety of transcriptional, post-transcriptional and – translational processes. The consensus model is that upstream signals control phosphorylation of various residues and thereby proteasomal degradation. Four E3 ubiquitin-ligases (MULE [Citation27], SCFβ-TrCP [Citation28], SCFFbw7 [Citation29], and Trim17 [Citation30]) and a deubiquitinase (USP9X [Citation31]), that respectively mediate and oppose MCL1 ubiquitination, have been identified. Moreover, MCL1 is overexpressed in a variety of cancers[Citation32]. NOXA binds and sequesters MCL1, thereby lowering the cell’s capacity to counteract direct activator BH3 only members. In addition, it has been shown that NOXA binding actively decrease the half-life of MCL1 actively[Citation13]. One way MCL1 protein levels are regulated, is via ubiquitination and proteasomal targeting mediated by the E3 ligase MULE, which also contains a BH3-only domain able to bind MCL1[Citation27]. Recent findings characterizing the conformational dynamic change of a MCL1 region following NOXA or BIM binding respectively[Citation33] explain the fact that, NOXA hands over MCL1 to the ubiquitination system [Citation34,Citation35] in contrast to BIM that inhibits its ubiquitination. Indeed, the conformation of a dynamic Q221R222N223 motif of the BH3 domain of MCL1 controls MCL1 ubiquitination. While NOXABH3 binding modifies this motif toward a helical conformation favoring MCL1 ubiquitination, BIM binding biases it toward a non-helical conformation inhibiting MCL1 ubiquitination[Citation33] ( 2).
Figure 2. Protein regulation of NOXA and MCL1, and drugs that target these aspects.
Schematic overview of current knowledge of protein regulation of NOXA and MCL1 via the (de)ubiquitination machinery. Drugs with proven or clearly expected clinical potential are indicated by boxes at the steps they target. See main text for details. Ub = Ubiquitin, K11, K48 denote lysine11 or Lys48 linkage in ubiquitin chains.
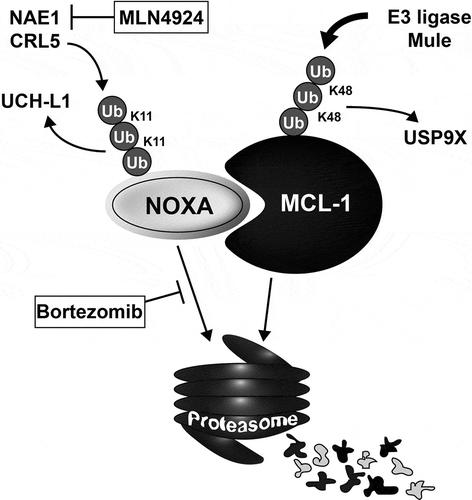
In addition to being a potential chaperone for MCL1, there are various reports that NOXA itself is also regulated via post-translational control, specifically by the (de)-ubiquitination cycling system [Citation36]. The possibility that NOXA is ubiquitinated at lysine residues was investigated and this culminated with the surprising finding that it is degraded via the 26S proteasome, but in a ubiquitin-independent way [Citation37,Citation38]. In contrast, the de-ubiquitinating enzyme UCH-L1 was postulated to protect NOXA from proteasomal degradation by removing Lys(48)-linked polyubiquitin chains [Citation39]. Epigenetic silencing of UCH-L1 in various cancer types would then determine whether NOXA was controlled by this system or not. However, this explanation did not clarify which E3 ligase is responsible for ubiquitination of Noxa. Recently, a possible answer was provided in a study showing that the Cullin-Ring-ligase-5 (CLR5) E3 ligase can ubiquitinate NOXA by Lysine-11 ubiquitin ligation, instead of by the canonical Lysine-48 ligation, resulting in the proteasomal targeting of NOXA (). NOXA levels, and the responsible neddylating enzyme UBE2F correlated with survival in lung cancer patients, providing in in vivo support for these findings [Citation40].
In agreement with the notion that the CLR5 E3 ligase controls NOXA protein half-life, an inhibitor of the proximal regulator of the neddylation system (NEDD8-activating enzyme, NAE) called MLN4924 is able to kill various cancer cells by induction of NOXA [Citation41–Citation43]. MLN4924 or pevonedistat is currently in phase I-II for various cancers.
Thus, it now seems plausible that NOXA itself, in addition to its best studied binding partner MCL1, is under tight post-translational control. This protein pair, which has a large impact on cell survival, is thus in a prime position to function as a rheostat under various forms of cell stress. In the following sections we will summarize conditions of cell stress where a role for NOXA has been either clearly shown, or was suggested but where the mechanism is still largely unclear.
3. NOXA transcriptional and protein regulation by various forms of cell stress
3.1. Chromosomal locus
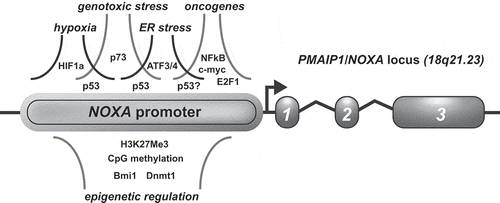
The NOXA transcript which encodes NOXA was first identified as a phorbol-12-myristate-13-acetate (PMA) responsive gene in the Jurkat acute T-cell leukemia cell line and in human peripheral blood cells, and consequently, the gene was named PMA-induced protein 1 (PMAIP1)[Citation44]. The human PMAIP locus resides at the cytogenetic band 18q21.23, and the gene contains three exons of which exon 2 is not encoded in the NOXA protein that spans only 54 amino acid residues. Two unstable splice variants of 136 and 70 amino acids have been described, which have no apparent role in the regulation of apoptosis. Apparently, the first reported PMAIP transcript originated from one of these non-functional transcripts [Citation45], therefore, we maintain here the designation NOXA. The NOXA promoter region extents over approximately 4 kb, and contains several enhancer regions located upstream and downstream of the actual gene locus. These regulatory regions contain binding sites for over 40 different transcription factors and co-activators suggesting that NOXA can be differentially regulated by many signals and conditions (). This explains the marked tissue/cell type-specific regulation of NOXA expression (reviewed by Ploner et al. [Citation46]. Here, we discuss the different modalities of NOXA regulation and its consequences for health and disease.
Figure 3. Signaling and cell stress pathways that impact on NOXA gene expression.
Schematic depiction of the NOXA locus and promoter region. Transcription factors and cell stress pathways implicated in the induction of NOXA mRNA expression are shown above. Epigenetic factors and regulators are shown below. Arrow indicates transcription start site (TSS). See text for detailed description. H3K27Me3 = Histone H3 lysine27 trimethylation.
3.2. DNA damage and genotoxic stress
Originally, NOXA was described to be regulated by p53, as it was found that adenoviral delivery of p53 into p53-deficient mouse embryonic fibroblasts (MEFs) resulted in the rapid induction of NOXA mRNA. NOXA is part of the p53-dependent transcriptional response following genotoxic stress, shown by induction of NOXA mRNA upon γ-irradiation. However, the functional consequences of DNA damage-induced NOXA expression were shown to be cell type-specific, as NOXA-deficient thymocytes underwent apoptosis upon γ-irradiation as efficiently as wildtype thymocytes, despite induction of NOXA expression. In contrast, MEFs from NOXA-deficient mice were more resistant to γ-irradiation and DNA damaging agents such as adriamycin and etoposide than wildtype MEFs [Citation47–Citation49]. The mechanistic basis for these cell type and cell state specific effects remains elusive. Importantly, it has been shown that Puma deficiency conferred significantly more resistance to DNA damage-induced apoptosis of thymocytes and untransformed fibroblasts, whereas the expression of the oncogenic E1A protein sensitized cells to NOXA-induced apoptosis. The mechanism of the differential involvement of NOXA and PUMA under these conditions remains to be studied [Citation50]. In addition, not only the cellular context but also the nature of the genotoxic stressor is of importance in determining the involvement of the different BH3-only family members in DNA damage-induced apoptosis. Ultraviolet (UV) radiation-induced cell death is p53-dependent, and in this context, loss of NOXA suppressed UV radiation-induced apoptosis of keratinocytes, whereas loss of PUMA provided no protection [Citation51]. In addition, anti-mitotic drugs such as microtubule-targeting agents cause mitotic arrest, which when unresolved leads to DNA damage and p53 activation. It was recently demonstrated that NOXA is the rate-limiting BH3-only protein in mitotic arrest-associated cell death [Citation52], but whether this requires an intact p53 pathway remains to be confirmed. In agreement, in a genome-wide siRNA screen in HeLa cervical carcinoma cells treated with the anti-mitotic drug taxol, NOXA was identified a critical initiator of mitotic cell death [Citation53].
The NOXA promoter contains a consensus p53-binding motif just proximal to the transcription start site (TSS) that is important for promoter activity. However, additional studies have demonstrated that p73, which is a member of the p53 family of proteins, is also able to bind the p53 consensus site and provoke transactivation [Citation54]. Indeed, p73 was implicated in the p53-independent NOXA induction following genotoxic stress and in adenoviral E1A-mediated apoptosis [Citation55]. There are several other indications that genotoxic stress can induce NOXA in a p53-independent fashion, for instance by cisplatin treatment [Citation56,Citation57].
In conclusion, the function of NOXA in DNA damage-induced cell death generally seems subordinate to that of PUMA, yet the known data imply that the regulation of NOXA expression may represent a potential therapeutic target in defined situations.
3.3. Epigenetic regulation
It has become clear that NOXA expression is also dynamically regulated at the epigenetic and chromatin level. For instance, the polycomb group gene Bmi1 repressed NOXA expression in memory CD4+ T cells by inducing trimethylation of histone 3 on Lysine 27 (H3K27Me3), and by recruitment of the DNA methyltransferase Dnmt1, resulting in CpG methylation. Accordingly, enhanced cell death of Bmi1−/- memory T cells was observed [Citation58]. Chemical compounds that alter DNA and chromatin marks, such as histone deacetylase (HDAC) inhibitors and DNA methyltransferase inhibitors, constitute a novel class of epigenetic drugs with promising activities toward cancer and immune-mediated diseases [Citation59]. Interestingly, the HDAC inhibitor SNX-275 (Entinostat) was active in acute myeloid leukemia (AML) blasts, by transcriptional upregulation of BIM (BCL2L11) and NOXA [Citation60]. Other HDAC inhibitors such as Kendine 92 and suberoylanilide hydroxamic acid (SAHA, also known as vorinostat), induced apoptosis of chronic lymphocytic leukemia (CLL) cells by increasing the expression of PUMA, BIM and NOXA [Citation61].
3.4. Hypoxia
Next to genotoxic stress, various other cellular stressors result in the induction of NOXA expression. Hypoxia is a well-known source of cellular stress related to insufficient oxygen supply to tissues and cells, and is a characteristic feature of most tumors caused by inadequate vascularization and blood supply. The adaptive response to hypoxia involves altered metabolism and proliferative capacity and ultimately cell death in case of extreme or prolonged hypoxia. The hypoxia inducible factor – alpha subunit (Hif1α) transcription factor plays a pivotal role in hypoxic adaptation [Citation62]. Hypoxic conditions were shown to induce NOXA expression in several different tissues and tumors in a p53-independent fashion, by Hif1α-dependent transactivation and binding to a hypoxia-response element in the NOXA promoter [Citation63]. Induction of expression seems to be an early response to hypoxia and results in the reactive oxygen species (ROS)-dependent release of cytochrome C and cell death, which could have therapeutic potential [Citation63]. Hypoxia is intimately connected to activation of the mammalian target of rapamycin (mTOR) signaling and the unfolded protein response (UPR) stress response [Citation64], which are both implicated in the induction of NOXA expression, see also below.
3.5. ER stress
The endoplasmatic reticulum (ER) plays an essential role in the regulation of cellular homeostasis by controlling protein folding assembly and transport. Many cellular stress conditions trigger the accumulation of improperly folded proteins resulting in ER stress. To handle ER stress, the unfolded protein response (UPR) takes place as a cytoprotective response that switches to apoptosis if the ER stress cannot be resolved. Selective BH3-only proteins such as BIM, NOXA and perhaps to a lesser extent PUMA [Citation65] have been identified as the main players responsible for the cell death by ER stress. The role of BIM in ER stress induced apoptosis has been unequivocally demonstrated previously. BIM is subject to C/EBP-homologous protein (CHOP)-mediated transcriptional induction as well as post-translational control [Citation66]. Our purpose here is to focus on the subsequently uncovered critical role of NOXA. Indeed, knockdown of NOXA strongly reduced cell death induced by ER stress which can be elicited by numerous drugs such as the ER-associated protein degradation (ERAD) inhibitor fenretinide [Citation67], the SERCA inhibitor thapsigargin [Citation68], the ‘BH3 mimetic like’ such as Gossypol, Apogossypol, HA14-1, S1 [Citation69,Citation70], oncolytic virus [Citation71], pemetrexed [Citation72], or the heat shock response inducer aurin [Citation73]. Of note, the induction of NOXA by certain ‘BH3 mimetic-like’ compounds allows to distinguish them from the true BH3 mimetics that do not exhibit this property [Citation74]. The UPR signaling involves three adaptive pathways that operate in parallel namely IRE1, PERK and ATF6. While crosstalk between the UPR branches is of functional importance, only the PERK signaling leads to the induction of Noxa. Indeed, PERK activation mediates the phosphorylation of eIF2α resulting in attenuation of global translation and in parallel to the specific translational activation of ATF4, one of the main transcriptional regulators of the UPR response. Of interest, knockdown of ATF4 strongly reduced NOXA expression suggesting that ATF4 functions as a transcriptional activator of NOXA in tumor cells under ER stress [Citation68]. On the other hand, it was shown that both ATF3 and ATF4 are induced by ER stress and that only ATF3 is able to directly bind to the NOXA promoter [Citation75] and indirectly recruits ATF4 to it. Altogether, regardless of the cellular model and the drugs involved, the transcription of NOXA in an ATF4/ATF3-dependent manner plays a major role in the apoptosis induced under ER stress. This implies that induction of ER stress by any of the means mentioned above, or by novel therapeutic compounds, is in fact an indirect way of unleashing the apoptotic potential of NOXA.
3.6. Proteasome inhibition
Bortezomib is a highly selective and reversible inhibitor of the 26S proteasome, which was initially described as an inhibitor of the NF-κB pathway. Bortezomib is approved for treating multiple myeloma (MM) and mantle cell lymphoma (MCL), two life-threatening hematological malignancies with a dismal clinical outcome. In both diseases, Bortezomib is used as initial therapy or for relapsed/refractory patients. While its mechanism of action is not yet fully unraveled, there is consensus regarding its multifactor biological consequences, which include ER stress and apoptosis induction, ROS production and alteration of NF-κB activity. Among these processes, the ER stress due to the inhibition of the degradation of ubiquitinated proteins is considered to be the central mechanism triggering cell death [Citation76]. The engagement of the intrinsic mitochondrial pathway by Bortezomib was consistently characterized by the induction of NOXA in numerous malignant cell types i.e. hematopoietic cells (MM, MCL, CLL) as well as solid tumor cells (melanoma, carcinoma cells) [Citation67,Citation68,Citation75,Citation77,Citation78]. In all malignant cells except in CLL cells [Citation38], the rapid induction of NOXA protein after proteasome inhibitor exposure was preceded by an up-regulation of NOXA mRNA. Knockdown of NOXA invariably protected malignant cells from Bortezomib-induced cell death, underlining the critical and common role of NOXA in Bortezomib-induced apoptosis, although this protection may remain partial. Two other common characteristics of NOXA induction are its independence of the basal level of NOXA and of the p53 status. Indeed, the rapid induction of NOXA protein takes place even if it is undetectable before exposure to Bortezomib [Citation77,Citation79], suggesting that NOXA itself is under strict control by the proteasome. Finally, although the activation of p53 pathway maybe dispensable for NOXA induction, the fold increase of NOXA mRNA was superior in p53 wild-type cells compared to p53 mutant cells, when comparing the same cell type [Citation79]. Regardless of the cellular model studied, the PERK/eIf2a/ATF4 pathway appears as one of the main mechanisms responsible for NOXA induction in response to Bortezomib. The expression of the non-phosphorylated form of eIf2α in MEF cells triggers resistance to Bortezomib accompanied by the absence of the induction of ATF4 and NOXA [Citation67]. In agreement with the implication of the PERK/ATF4 pathway, the low basal levels of ATF3 and ATF4 in MM patients are associated with weak efficiency of Bortezomib/dexamethasone treatment via the reduced induction of NOXA and CHOP [Citation80]. The induction of NOXA through the activation of the PERK pathway was also found with carfilzomib, a second-generation proteasome inhibitor [Citation81,Citation82].
Besides the implication of the ATF3/ATF4 in the induction of NOXA by proteasome inhibitors, several other mechanisms have been proposed according to the cell type studied. In MM, the KLF9 gene was identified as significantly over-expressed in patients that responded to Bortezomib compared to non-responder patients. KLF9 was shown to directly interact with the PMAIP1 promoter in a Bortezomib-dependent manner [Citation83]. Furthermore, the ectopic expression of KLF9 was sufficient to induce NOXA and apoptosis in the absence of Bortezomib, indicating that KLF9 levels beyond a certain threshold are able to induce apoptosis through NOXA expression. In MCL, NOXA induction was first shown to be dependent on ROS generation [Citation79], as, ROS scavengers blocked NOXA upregulation in response to Bortezomib. More recently, NOXA induction was shown to be dependent on PRDM1 [Citation84]. PRDM1, also known as Blimp-1, is a transcriptional repressor required for terminal B cell differentiation. Knockdown of PRDM1 blocks both basal and Bortezomib-induced NOXA expression. Because PRDM1 is a repressor protein, it has been proposed that it could suppress a player whose absence may allow NOXA induction. Thus, although ATF3 and ATF4 play essential roles in the induction of NOXA by proteasome inhibitors, the role of other, cell type specific, transcription factors cannot be ruled out for optimal transcription of NOXA in response to proteasome inhibitors.
3.7. Metabolic stress – glucose and nutrients
As mentioned above, various stress response pathways regulate MCL1 protein half-life . A prominent pathway is via PI3K/AKT/GSK3β, which has been implicated in various cell types and conditions, ranging from cytokine- to glucose deprivation [Citation85–Citation87]. Apart from its contribution as BH3 protein and chaperone for MCL1 degradation, a unique role for NOXA in glucose- or nutrient-deprivation has been difficult to identify mechanistically [Citation88]. An intriguing possibility is that NOXA may alter cellular metabolism by shunting glucose to the pentose phosphate pathway, which was proposed to be regulated by Ser13 phosphorylation [Citation89]. Such a switch of function of NOXA from apoptotic to metabolic regulator is somewhat reminiscent of the dual role of BAD in apoptosis and glucose metabolism in endocrine tissues [Citation90], but firm proof has yet to be provided.
We and others have investigated a potential role of the ATP/AMP-sensing kinase AMPK in MCL1 phosphorylation [Citation88,Citation91]. A direct functional interaction could not be found, yet there is the intriguing action of the AMPK activator AICAR that induces cell death in an AMPK-independent fashion, which required BIM/NOXA [Citation91]. The picture that emerges is that the NOXA/MCL1 protein pair can be considered a rheostat that senses cellular metabolism via glucose and other nutrient levels in various cell types. The sensors or kinases that would couple glucose, nutrient or ATP levels to MCL1 or NOXA phosphorylation remain to be identified. Thus, although cellular metabolism is clearly linked to apoptosis, therapeutic exploitation of this potential via targeting of MCL1/NOXA requires further mechanistic investigation.
3.8. Autophagy
Autophagy is a degradative process whereby cells recycle their organelles and large protein complexes. In most cells this is a steady state flux that can be increased in times of stress, such as nutrient shortage. Autophagy is positively and negatively controlled by AMPK and MTOR, respectively. Cellular constituents are engulfed in membrane compartments that subsequently fuse with lysosomes to degrade the cargo. In recent years, the genes and proteins involved in this complex process have been largely elucidated [Citation92]. In general, it has been proposed that ‘too much’ autophagy will lead to cell death, either in a caspase-dependent or – independent fashion. Furthermore, cancer cells require active autophagy in order to cope with their intrinsically stressed state [Citation93], which implicates that targeting autophagy would be therapeutically beneficial. The (reciprocal) link between autophagy and cell death has been studied for many years, but the basic aspects are still unclear and sometimes quite controversial, for a summary of open questions, see [Citation94]. An often studied connection is the central autophagy regulator Beclin, which contains a BH3(like) domain allowing interaction with BCL2, BCLXL and MCL1. The affinity of the Beclin-BCLXL interaction is reported to be in the micro-molar range [Citation95,Citation96], which is substantially lower than the nano-molar affinities of ‘regular’ BH3 proteins, implying that additional interactions or protein modifications are required for Beclin in order to compete with other BH3 proteins, and thereby shift the apoptotic balance.
In relation to autophagic cell death, two types of NOXA involvement have been suggested. The first involves endosomal TLR3, or cytoplasmic dsRNA recognition by MDA5, which induces NOXA protein in melanoma cells by an as yet unidentified mechanism [Citation97]. Second, oncogenic, inducible Ras activity in cell lines was reported to result in autophagic cell death, by ERK-dependent upregulation of NOXA protein. NOXA-dependent autophagy was associated with displacement and degradation of MCL1 from Beclin-1 [Citation98], suggesting that NOXA itself may compete with Beclin to liberate MCL1 and trigger lethal autophagy. Additionally, various reports mention NOXA in connection with autophagy, but generally, these also contain features of ER- or proteasomal stress. As described above, it is known that NOXA plays a role under these conditions, making it difficult to adequately assess the role of NOXA in the autophagic aspects of cell death. In summary, several basic aspects of autophagic cell death are still unclear, and a specific role for NOXA has been described in restricted cases, which does not yet suggest modes of therapeutic application.
4. BCL-2 members as therapeutic target – BH3 mimetics and beyond
The preceding sections have focused mostly on NOXA and highlighted situations where its induction may play a decisive role in cell death induction. In addition, the impressive progress on direct therapeutic targeting of pro-survival Bcl-2 members in cancer warrants a summary of these developments, which we will subsequently merge from the perspective of NOXA biology.
In 2005, the first ‘true’ anti-Bcl2 family compound was reported, now dubbed BH3 mimetics. Abbott developed a potent inhibitor (ABT-737) of BCL2, BCLXL and BCLW which kills susceptible cells in a BAX/BAK1 dependent fashion [Citation99]. An orally administrable variant, ABT-263 or Navitoclax, had clinical efficacy in CLL and other B-cell malignancies [Citation100], but inhibition of BCLXL in platelets led to thrombocytopenia and dose limiting toxicity [Citation101], which halted further development. Subsequently, a search for compounds that target single Bcl-2 family members, has led to important clinical as well as pre-clinical success. Conversion of ABT-263 into a Bcl-2-specific compound resulted in ABT-199, which efficiently kills BCL2-dependent cancer cells such as CLL but spares platelets [Citation102]. The success story of ABT-199 or Venetoclax is now well-known, and FDA approval for previously treated CLL with 17p deletion was granted in April 2016. Venetoclax shows impressive clinical efficacy with minimal side-effects in CLL [Citation103].
This success sparked further development that resulted in BCLXL- as well as MCL1-specific compounds – we will mention here the most well-studied. WEHI-539 and the more recently developed A-1155463 inhibit BCLXL with subnanomolar affinity [Citation104,Citation105]. Though potent BCLXL inhibition obviously has the drawback that it will induce (transient) thrombocytopenia, this may be a manageable hazard in cancers that strongly depend on BCLXL. MCL1-specific inhibitors have also been developed [Citation106,Citation107]. The initial compounds such as A-1210477 were specific though not very potent, but very recently the high affinity MCL1 inhibitor S63845 was described [Citation108]. Given the severe effects of even single allele ablation of MCL1 in mice [Citation109–Citation112], the predominant notion in the field was that inhibiting MCL1 would be toxic for non-malignant tissues. The data with S63845 contradict this and suggest there is a clear therapeutic window. In hindsight, this may not be too surprising as many tumors in fact express MCL1 at levels far above normal cells. Interestingly, S63845 causes increased MCL1 protein stability, indicating it behaves like BIM rather than NOXA[Citation108].
Additional MCL1 inhibitors are being developed by other companies, notably AMG 176 which is in Phase 1 for MM (http://www.amgenpipeline.com/pipeline/) and optimization of A-1210477 [Citation107] can also be expected. Another novel development is engineering of covalent compounds that target the MCL1 BH3 groove and/or other reactive cysteine or lysine residues [Citation113,Citation114]. While a detailed description and comparison of the data obtained with these novel MCL1 inhibitors is beyond the scope of this review, it seems clear that covalent targeting may greatly enhance specificity. On the other hand, introducing reactive groups into compounds may also enhance binding to non-target proteins. Future research in cellular and animal or human systems will have to determine pharmacological properties and clinical potential of these novel compounds. In addition, their availability will allow to discriminate whether they act as ‘pseudo’ NOXA or rather like BIM to target MCL1 or BCL2A1 [Citation26]. Finally, these highly specific and cell-permeable reagents can also be used to quantitatively profile each cancer cell type for its dependence on pro-survival BCL2 members [Citation115], an approach that was previously only possible with relatively high concentrations of peptides and permeabilized cells [Citation116].
5. NOXA induction in cancer
5.1. Melanoma and solid cancers
Induction of NOXA gene expression and protein function have been linked to cell death in various hematopoietic and solid cancers. We will briefly summarize data on several stress pathways leading to NOXA involvement in solid cancers, before we discuss in more details two hematopoietic cancers where NOXA expression and function can be clearly linked to existing and novel therapeutic applications.
Especially for melanoma, many reports have described various inducers and pathways of cell stress leading to NOXA gene and-or protein induction and cell death. These pathways can be considered exemplary for other solid cancers and have been recently reviewed [Citation117]. The most significant pathways have been mentioned in the preceding sections: proteasome inhibition, ER stress, and generation of reactive oxygen species (ROS) by various types of drugs, have been broadly recognized as inducers of NOXA. In addition, inhibitors that target the epigenome may de-repress NOXA gene transcription [Citation118].
5.2. Multiple myeloma
MM is a plasma-cell malignancy that is heterogeneous, and can be classified into four main subtypes of MM patients defined as hyperdiploid (HY), CCND1 t(11;14), MAF t(16;14), or t(14.20), and MMSET t(4;14). In this pathology, the combined profile of BCL2, BCLXL, and MCL1 is sufficient to distinguish these 4 molecular subgroups of MM [Citation119]. NOXA is also very heterogeneously expressed in the different subgroups of myeloma patients. The CCND1 and MMSET subgroups have high levels of Noxa, while the HY and MAF subgroups express very heterogeneous but consistently lower levels of NOXA[Citation119]. A unique feature of the CCND1 subgroup is the high level of BCL2 and the lowest levels of MCL1. Thus, we can hypothesize that in this subgroup the anti-apoptotic role of MCL1 is mainly neutralized by the high expression of Noxa, explaining that this subgroup is mainly BCL2 dependent. In agreement with this hypothesis, the majority of MM patients responsive to the BCL2 antagonist Venetoclax (ABT-199) are found in the BCL2high MCL1low and NOXAhigh CCND1 subgroup. In myeloma cell lines, a very heterogeneous expression of NOXA is reported, which is not related to p53 status [Citation120]. Indeed, some cell lines have undetectable Noxa, either at mRNA or protein level, while other myeloma cell lines express very high levels of endogenous NOXA [Citation120]. Since the vast majority of MM cells are known to be strongly MCL1 dependent for survival [Citation121,Citation122], NOXA is expected to play a major role in the induction of cell death through the neutralization of MCL1 [Citation77,Citation123,Citation124]. In accordance, S63845 is effective in MM cells that depend on MCL1 [Citation108].
5.3. Chronic lymphocytic leukemia
CLL is a genetically and clinically heterogeneous malignancy of mature B cells. A hallmark of chronic lymphocytic leukemia (CLL) is a dichotomy in survival and proliferation between the leukemic cells in peripheral blood (PB) and the lymph node (LN) compartments. PB CLL cells are in cell cycle arrest and sensitive to pro-apoptotic signals, whereas CLL cells in the LN proliferate and upregulate proteins that mediate resistance to apoptosis [Citation125,Citation126]. Analysis of apoptosis genes in CLL compared to normal LN B cell populations demonstrated not only the well-known overexpression of BCL2 in CLL, but also overexpression of BH3-only members BMF [Citation127] and NOXA[Citation128]. The functional significance of increased BMF has not yet been elucidated. A major difference between the LN and PB compartments is a change in the NOXA/MCL1 balance [Citation129], which was clearly linked to apoptosis sensitivity in general, and to Bortezomib in particular. These observations have several implications for (novel) therapeutic options, which will be discussed in relation to the recent clinical success of the BTK inhibitor ibrutinib [Citation130], and Venetoclax [Citation103].
The first aspect is the cause of the intrinsic high levels of NOXA in CLL. Although NOXA was originally described as a p53-responsive gene, its expression in CLL is not mediated via p53, as it is not induced further by chemotherapeutics, and also occurs in p53 deficient patient samples [Citation128]. Another option is that increased expression of NOXA is a reflection of CLL biology, since CLL is intimately linked to continuous B cell receptor (BCR) signaling [Citation131], and NOXA is induced in normal B cells by BCR triggering [Citation132]. This could imply that CLL cells originate from a phase where BCR triggering lowers the apoptosis threshold by expression of NOXA, but apoptosis is then prevented by (pre)-malignant transformation. This notion is supported by a murine model of CLL [Citation133]. Arguing against it is the fact that in LN CLL cells, where BCR triggering is presumed to be ongoing [Citation126], NOXA expression is low, whereas the reverse is true for the PB compartment. This suggests that NOXA overexpression in the PB reflects another form of cell stress.
Second, NOXA expression is high and indeed, PB CLL cells are very susceptible to Bortezomib in vitro [Citation129], yet this does not translate into clinical efficacy of Bortezomib, for reasons that are not completely clear [Citation134–Citation136]. Apart from a proposed effect of dietary flavonoids present in CLL patient plasma (and apparently not, or less effective in MM), an explanation might be that the required level of ER stress required to undergo apoptosis is not attained in CLL in vivo, while this threshold is reached in MM. This is consistent with the fact that MM is a highly secretory tumor type prone to ER stress, whereas CLL is not.
Third, the fluctuating balance in NOXA/MCL1 between different compartments in relation to micro-environmental stimuli in CLL would predict that this is a relevant determinant in clinical responses/resistance to (novel) drugs. This indeed appears to be the case. It is well known that MCL1 is not targeted by the BH3 mimetics ABT-737 and Venetoclax, and that MCL1 expression is a resistance determinant for these drugs in many cancer types and also CLL [Citation137,Citation138]. Conversely, lowering MCL1 levels in CLL by for instance cyclin-dependent kinase inhibition triggers cell death [Citation139]. Clinical testing of cyclin–dependent kinase inhibitor flavopiridol in CLL however was quite toxic [Citation140], especially compared to current, more efficient alternatives. Lastly, responses to the BTK inhibitor ibrutinib, which forces CLL cells out of the LN by blocking adhesion [Citation141,Citation142], are accompanied by a reduction in MCL1 levels in recent LN emigrants. Resistance to ibrutinib was accompanied by a recurrence of MCL1 or BCLXL expression [Citation143].
6. Conclusions – NOXA as drug or MCL1 as target
The preceding sections allow several conclusions. The first is that NOXA, despite its modest status as a weak BH3-only member, may have considerable therapeutic potential, which has not yet been fully explored nor exploited. The second is that some of the multifaceted means to induce NOXA gene transcription and/or protein expression need further mechanistic investigation, while others seem to be closer to therapeutic application. Thirdly, due to its very close association with MCL1, conditions that induce NOXA will inhibit MCL1 and thereby induce apoptosis directly, or in synergy with other stimuli or compounds. Indeed, there are many reports of the synergy between NOXA induction and sensitivity for BH3 mimetics (ABT-737 or Venetoclax) in conditions where cells are resistant to the latter.
7. Expert opinion – assessing stress signaling to NOXA as key to new applications
Currently, we have an incomplete understanding of the therapeutic potential of NOXA as an inducer of cell death in cancer. The key to uncover this potential is a better understanding of the various, cancer cell type specific stress pathways. Many of these have been reported to connect in some manner with NOXA gene and/protein induction, and thereby to increased cell death susceptibility. However, a mechanistic basis for some of these instances is not yet established firmly enough, in order that subsequent steps to therapeutic exploitation may be taken.
We tentatively propose a more or less hierarchical list of cell stress pathways that have been connected with induction of NOXA (or with reduction in MCL1), in order of increasing potential of therapeutic exploitation in cancer. We base this ordering on mechanistic insight of the induction or involvement of NOXA combined with availability of specific compounds or drugs (). Evaluating the currently available insight leads to a division in four areas. We hereby exclude the clear link between chemotherapeutics, DNA damage, p53 activation and potential NOXA gene induction, as this pathway is in fact superseded by the more potent induction of PUMA along the same signaling path.
Table 1. Ordering of cell stress pathways that affect Noxa level or function, with potential drugs and stage of clinical development indicated.
First, stress pathways where mechanistic insights into the steps that lead to NOXA involvement, or specific drugs are lacking. Autophagy and metabolic stress are examples of this category. Although there are compounds available with therapeutic potential such as AICAR [Citation91], their specificity and/or a link with the NOXA/MCL1 rheostat is currently underexplored.
The second category contains instances where NOXA gene induction or de-repression has been documented, such as replicative stress, HDAC or DNA methyltransferase inhibition. For most of these instances drugs with clear therapeutic potential are available, but specific proof that the NOXA gene induction is direct rather than indirect is currently lacking. In addition, a general feature of ROS production by cells seems to be NOXA gene induction, leading to cell death. This has been reported for various compounds in diverse cancer cell types [Citation56,Citation79,Citation144,Citation145]. Although NOXA involvement is frequently demonstrated by siRNA approaches, how ROS activate NOXA transcription is not yet fully clear.
Third, clearly promising areas for clinical exploration are the drugs that result in NOXA protein accumulation. Clear examples are inducers of ER stress, inhibitors of the neddylation machinery and the proteasome (see references in ). The latter is clinically targeted by Bortezomib in MM and MCL but other cancers like melanoma, AML and CLL might be good examples where clinical trials may lead to new applications.
Fourth is the very recent development, directly leading to novel promising strategies by targeting MCL1 directly, exemplified by the novel BH3 mimetic S63845 [Citation108]. Clearly, single administration and combination therapy with a) chemotherapy, b) other targeted compounds such as kinase inhibitors, c) other BH3 mimetics such as Venetoclax, will be evaluated in the coming years which will undoubtedly lead to novel applications.
Concluding remark: The field of NOXA biology is active and still expanding, with clear expectations of both novel mechanistic insight and clinical applications.
Article highlights
Functional interactions among Bcl-2 proteins are of crucial importance in determining cancer cell survival
Direct targeting of pro-survival Bcl-2 proteins is now clinically achievable for various cancers
The therapeutic potential of NOXA is not yet fully explored nor exploited
Clinical evaluation of inducers of ER-stress that induce Noxa, as well as inhibitors of NOXA protein degradation seems promising
The therapeutic potential of NOXA can also be exploited in the context of direct targeting of MCL1 by specific and potent novel reagents
This box summarizes key points contained in the article.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Acknowledgments
We wish to thank the reviewers whose comments and suggestions have helped considerably to improve the manuscript.
Additional information
Funding
References
- Boatright KM, Salvesen GS. Mechanisms of caspase activation. Curr Opin Cell Biol. 2003;15(6):725–731.
- Strasser A, Cory S, Adams JM. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. Embo J. 2011;30(18):3667–3683.
- Gross A, Katz SG. Non-apoptotic functions of BCL-2 family proteins. Cell Death Differ. 2017.
- Otsu K, Murakawa T, Yamaguchi O. BCL2L13 is a mammalian homolog of the yeast mitophagy receptor Atg32. Autophagy. 2015;11(10):1932–1933.
- Schweers RL, Zhang J, Randall MS, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A. 2007;104(49):19500–19505.
- Li P, Nijhawan D, Budihardjo I, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91(4):479–489.
- Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11(9):621–632.
- Czabotar PE, Lessene G, Strasser A, et al. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15(1):49–63.
- Rooswinkel RW, Van De Kooij B, De Vries E, et al. Antiapoptotic potency of Bcl-2 proteins primarily relies on their stability, not binding selectivity. Blood. 2014;123(18):2806–2815.
- Campbell KJ, Bath ML, Turner ML, et al. Elevated Mcl-1 perturbs lymphopoiesis, promotes transformation of hematopoietic stem/progenitor cells, and enhances drug resistance. Blood. 2010;116(17):3197–3207.
- Strasser A, Harris AW, Cory S. bcl-2 transgene inhibits T cell death and perturbs thymic self-censorship. Cell. 1991;67(5):889–899.
- Chen L, Willis SN, Wei A, et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17(3):393–403.
- Willis SN, Chen L, Dewson G, et al. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19(11):1294–1305.
- Villunger A, Labi V, Bouillet P, et al. Can the analysis of BH3-only protein knockout mice clarify the issue of ‘direct versus indirect’ activation of Bax and Bak? Cell Death Differ. 2011;18(10):1545–1546.
- Cheng EH, Wei MC, Weiler S, et al. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell. 2001;8(3):705–711.
- Ren D, Tu HC, Kim H, et al. BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science. 2010;330(6009):1390–1393.
- Willis SN, Fletcher JI, Kaufmann T, et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315(5813):856–859.
- Doerflinger M, Glab JA, Puthalakath H. BH3-only proteins: a 20-year stock-take. Febs J. 2015;282(6):1006–1016.
- Gallenne T, Gautier F, Oliver L, et al. Bax activation by the BH3-only protein Puma promotes cell dependence on antiapoptotic Bcl-2 family members. J Cell Biol. 2009;185(2):279–290.
- Chen HC, Kanai M, Inoue-Yamauchi A, et al. An interconnected hierarchical model of cell death regulation by the BCL-2 family. Nat Cell Biol. 2015;17(10):1270–1281.
- Du H, Wolf J, Schafer B, et al. BH3 domains other than Bim and Bid can directly activate Bax/Bak. J Biol Chem. 2011;286(1):491–501. .
- Dai H, Smith A, Meng XW, et al. Transient binding of an activator BH3 domain to the Bak BH3-binding groove initiates Bak oligomerization. J Cell Biol. 2011;194(1):39–48.
- Moldoveanu T, Grace CR, Llambi F, et al. BID-induced structural changes in BAK promote apoptosis. Nat Struct Mol Biol. 2013;20(5):589–597.
- Vogler M. BCL2A1: the underdog in the BCL2 family. Cell Death Differ. 2012;19(1):67–74.
- Huhn AJ, Guerra RM, Harvey EP, et al. Selective covalent targeting of anti-apoptotic BFL-1 by cysteine-reactive stapled peptide inhibitors. Cell Chem Biol. 2016;23(9):1123–1134.
- Barile E, Marconi GD, De SK, et al. hBfl-1/hNOXA interaction studies provide new insights on the role of Bfl-1 in cancer cell resistance and for the design of novel anticancer agents. ACS Chem Biol. 2017;12(2):444–455.
- Zhong Q, Gao W, Du F, et al. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of mcl-1 and regulates apoptosis. Cell. 2005;121(7):1085–1095.
- Ding Q, He X, Hsu JM, et al. Degradation of Mcl-1 by beta-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol Cell Biol. 2007;27(11):4006–4017.
- Inuzuka H, Fukushima H, Shaik S, et al. Mcl-1 ubiquitination and destruction. Oncotarget. 2011;2(3):239–244.
- Magiera MM, Mora S, Mojsa B, et al. Trim17-mediated ubiquitination and degradation of Mcl-1 initiate apoptosis in neurons. Cell Death Differ. 2013;20(2):281–292.
- Schwickart M, Huang X, Lill JR, et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature. 2010;463(7277):103–107.
- Warr MR, Shore GC. Unique biology of Mcl-1: therapeutic opportunities in cancer. Curr Mol Med. 2008;8(2):138–147.
- Song T, Wang Z, Ji F, et al. Deactivation of Mcl-1 by dual-function small-molecule inhibitors targeting the Bcl-2 homology 3 domain and facilitating Mcl-1 ubiquitination. Angewandte Chemie (International Ed in English). 2016;55(46):14250–14256.
- Gomez-Bougie P, Menoret E, Juin P, et al. Noxa controls Mule-dependent Mcl-1 ubiquitination through the regulation of the Mcl-1/USP9X interaction. Biochem Biophys Res Commun. 2011;413(3):460–464.
- Stankiewicz AR, Livingstone AM, Mohseni N, et al. Regulation of heat-induced apoptosis by Mcl-1 degradation and its inhibition by Hsp70. Cell Death Differ. 2009;16(4):638–647.
- Pal A, Young MA, Donato NJ. Emerging potential of therapeutic targeting of ubiquitin-specific proteases in the treatment of cancer. Cancer Res. 2014;74(18):4955–4966.
- Craxton A, Butterworth M, Harper N, et al. NOXA, a sensor of proteasome integrity, is degraded by 26S proteasomes by an ubiquitin-independent pathway that is blocked by MCL-1. Cell Death Differ. 2012;19(9):1424–1434.
- Baou M, Kohlhaas SL, Butterworth M, et al. Role of NOXA and its ubiquitination in proteasome inhibitor-induced apoptosis in chronic lymphocytic leukemia cells. Haematologica. 2010;95(9):1510–1518.
- Brinkmann K, Zigrino P, Witt A, et al. Ubiquitin C-terminal hydrolase-L1 potentiates cancer chemosensitivity by stabilizing NOXA. Cell Rep. 2013;3(3):881–891.
- Zhou W, Xu J, Li H, et al. Neddylation E2 UBE2F promotes the survival of lung cancer cells by activating CRL5 to degrade NOXA via the K11 Linkage. Clin Cancer Res. 2017;23(4):1104–1116.
- Wei D, Li H, Yu J, et al. Radiosensitization of human pancreatic cancer cells by MLN4924, an investigational NEDD8-activating enzyme inhibitor. Cancer Res. 2012;72(1):282–293.
- Wang Y, Luo Z, Pan Y, et al. Targeting protein neddylation with an NEDD8-activating enzyme inhibitor MLN4924 induced apoptosis or senescence in human lymphoma cells. Cancer Biol Ther. 2015;16(3):420–429.
- Godbersen JC, Humphries LA, Danilova OV, et al. The Nedd8-activating enzyme inhibitor MLN4924 thwarts microenvironment-driven NF-kappaB activation and induces apoptosis in chronic lymphocytic leukemia B cells. Clin Cancer Res. 2014;20(6):1576–1589.
- Hijikata M, Kato N, Sato T, et al. Molecular cloning and characterization of a cDNA for a novel phorbol-12-myristate-13-acetate-responsive gene that is highly expressed in an adult T-cell leukemia cell line. J Virol. 1990;64(10):4632–4639.
- Wang Z, Sun Y. Identification and characterization of two splicing variants of human Noxa. Anticancer Res. 2008;28(3a:1667–1674.
- Ploner C, Kofler R, Noxa: VA. At the tip of the balance between life and death. Oncogene. 2008;27(Suppl 1):S84–S92.
- Fei P, Bernhard EJ, El-Deiry WS. Tissue-specific induction of p53 targets in vivo. Cancer Res. 2002;62(24):7316–7327.
- Oda E, Ohki R, Murasawa H, et al. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;288(5468):1053–1058.
- Villunger A, Michalak EM, Coultas L, et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302(5647):1036–1038.
- Shibue T, Suzuki S, Okamoto H, et al. Differential contribution of Puma and Noxa in dual regulation of p53-mediated apoptotic pathways. Embo J. 2006;25(20):4952–4962.
- Naik E, Michalak EM, Villunger A, et al. Ultraviolet radiation triggers apoptosis of fibroblasts and skin keratinocytes mainly via the BH3-only protein Noxa. J Cell Biol. 2007;176(4):415–424. .
- Haschka MD, Soratroi C, Kirschnek S, et al. The NOXA-MCL1-BIM axis defines lifespan on extended mitotic arrest. Nat Commun. 2015;6:6891.
- Diaz-Martinez LA, Karamysheva ZN, Warrington R, et al. Genome-wide siRNA screen reveals coupling between mitotic apoptosis and adaptation. Embo J. 2014;33(17):1960–1976.
- Grande L, Bretones G, Rosa-Garrido M, et al. Transcription factors Sp1 and p73 control the expression of the proapoptotic protein NOXA in the response of testicular embryonal carcinoma cells to cisplatin. J Biol Chem. 2012;287(32):26495–26505.
- Flinterman M, Guelen L, Ezzati-Nik S, et al. E1A activates transcription of p73 and Noxa to induce apoptosis. J Biol Chem. 2005;280(7):5945–5959.
- Tonino SH, Van Laar J, van Oers MH, et al. ROS-mediated upregulation of Noxa overcomes chemoresistance in chronic lymphocytic leukemia. Oncogene. 2011;30(6):701–713.
- Sheridan C, Brumatti G, Elgendy M, et al. An ERK-dependent pathway to Noxa expression regulates apoptosis by platinum-based chemotherapeutic drugs. Oncogene. 2010;29(49):6428–6441.
- Yamashita M, Kuwahara M, Suzuki A, et al. Bmi1 regulates memory CD4 T cell survival via repression of the Noxa gene. J Exp Med. 2008;205(5):1109–1120.
- Jones PA, Issa JP, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet. 2016;17(10):630–641.
- Zhou L, Ruvolo VR, McQueen T, et al. HDAC inhibition by SNDX-275 (Entinostat) restores expression of silenced leukemia-associated transcription factors Nur77 and Nor1 and of key pro-apoptotic proteins in AML. Leukemia. 2013;27(6):1358–1368.
- Perez-Perarnau A, Coll-Mulet L, Rubio-Patino C, et al. Analysis of apoptosis regulatory genes altered by histone deacetylase inhibitors in chronic lymphocytic leukemia cells. Epigenetics. 2011;6(10):1228–1235.
- Carmeliet P, Dor Y, Herbert JM, et al. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998;394(6692):485–490.
- Kim JY, Ahn HJ, Ryu JH, et al. BH3-only protein Noxa is a mediator of hypoxic cell death induced by hypoxia-inducible factor 1alpha. J Exp Med. 2004;199(1):113–124.
- Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer. 2008;8(11):851–864.
- Toth A, Nickson P, Mandl A, et al. Endoplasmic reticulum stress as a novel therapeutic target in heart diseases. Cardiovasc Hematol Disord Drug Targets. 2007;7(3):205–218.
- Puthalakath H, O’Reilly LA, Gunn P, et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129(7):1337–1349.
- Fribley AM, Evenchik B, Zeng Q, et al. Proteasome inhibitor PS-341 induces apoptosis in cisplatin-resistant squamous cell carcinoma cells by induction of Noxa. J Biol Chem. 2006;281(42):31440–31447.
- Armstrong JL, Flockhart R, Veal GJ, et al. Regulation of endoplasmic reticulum stress-induced cell death by ATF4 in neuroectodermal tumor cells. J Biol Chem. 2010;285(9):6091–6100.
- Albershardt TC, Salerni BL, Soderquist RS, et al. Multiple BH3 mimetics antagonize antiapoptotic MCL1 protein by inducing the endoplasmic reticulum stress response and up-regulating BH3-only protein NOXA. J Biol Chem. 2011;286(28):24882–24895.
- Soderquist RS, Danilov AV, Eastman A. Gossypol increases expression of the pro-apoptotic BH3-only protein NOXA through a novel mechanism involving phospholipase A2, cytoplasmic calcium, and endoplasmic reticulum stress. J Biol Chem. 2014;289(23):16190–16199.
- Kelly KR, Espitia CM, Mahalingam D, et al. Reovirus therapy stimulates endoplasmic reticular stress, NOXA induction, and augments bortezomib-mediated apoptosis in multiple myeloma. Oncogene. 2012;31(25):3023–3038.
- Yan J, Zhong N, Liu G, et al. Usp9x- and Noxa-mediated Mcl-1 downregulation contributes to pemetrexed-induced apoptosis in human non-small-cell lung cancer cells. Cell Death Dis. 2014;5:e1316.
- Davis AL, Qiao S, Lesson JL, et al. The quinone methide aurin is a heat shock response inducer that causes proteotoxic stress and Noxa-dependent apoptosis in malignant melanoma cells. J Biol Chem. 2015;290(3):1623–1638.
- Soderquist RS, Eastman A. BCL2 inhibitors as anticancer drugs: a plethora of misleading BH3 mimetics. Mol Cancer Ther. 2016;15(9):2011–2017. .
- Wang Q, Mora-Jensen H, Weniger MA, et al. ERAD inhibitors integrate ER stress with an epigenetic mechanism to activate BH3-only protein NOXA in cancer cells. Proc Natl Acad Sci U S A. 2009;106(7):2200–2205.
- Obeng EA, Carlson LM, Gutman DM, et al. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107(12):4907–4916. .
- Gomez-Bougie P, Wuilleme-Toumi S, Menoret E, et al. Noxa up-regulation and Mcl-1 cleavage are associated to apoptosis induction by bortezomib in multiple myeloma. Cancer Res. 2007;67(11):5418–5424.
- Qin JZ, Ziffra J, Stennett L, et al. Proteasome inhibitors trigger NOXA-mediated apoptosis in melanoma and myeloma cells. Cancer Res. 2005;65(14):6282–6293.
- Perez-Galan P, Roue G, Villamor N, et al. The proteasome inhibitor bortezomib induces apoptosis in mantle-cell lymphoma through generation of ROS and Noxa activation independent of p53 status. Blood. 2006;107(1):257–264.
- Narita T, Ri M, Masaki A, et al. Lower expression of activating transcription factors 3 and 4 correlates with shorter progression-free survival in multiple myeloma patients receiving bortezomib plus dexamethasone therapy. Blood Cancer J. 2015;5:e373.
- Parlati F, Lee SJ, Aujay M, et al. Carfilzomib can induce tumor cell death through selective inhibition of the chymotrypsin-like activity of the proteasome. Blood. 2009;114(16):3439–3447.
- Lamothe B, Wierda WG, Keating MJ, et al. Carfilzomib triggers cell death in chronic lymphocytic leukemia by inducing proapoptotic and endoplasmic reticulum stress responses. Clin Cancer Res. 2016;22(18):4712–4726.
- Mannava S, Zhuang D, Nair JR, et al. KLF9 is a novel transcriptional regulator of bortezomib- and LBH589-induced apoptosis in multiple myeloma cells. Blood. 2012;119(6):1450–1458.
- Desai S, Maurin M, Smith MA, et al. PRDM1 is required for mantle cell lymphoma response to bortezomib. Mol Cancer Res. 2010;8(6):907–918.
- Maurer U, Charvet C, Wagman AS, et al. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell. 2006;21(6):749–760.
- Alves NL, Derks IA, Berk E, et al. The Noxa/Mcl-1 axis regulates susceptibility to apoptosis under glucose limitation in dividing T cells. Immunity. 2006;24(6):703–716.
- Zhao Y, Altman BJ, Coloff JL, et al. Glycogen synthase kinase 3alpha and 3beta mediate a glucose-sensitive antiapoptotic signaling pathway to stabilize Mcl-1. Mol Cell Biol. 2007;27(12):4328–4339.
- Wensveen FM, Alves NL, Derks IA, et al. Apoptosis induced by overall metabolic stress converges on the Bcl-2 family proteins Noxa and Mcl-1. Apoptosis. 2011;16(7):708–721.
- Lowman XH, McDonnell MA, Kosloske A, et al. The proapoptotic function of Noxa in human leukemia cells is regulated by the kinase Cdk5 and by glucose. Mol Cell. 2010;40(5):823–833.
- Gimenez-Cassina A, Danial NN. Noxa: a sweet twist to survival and more. Molecular Cell. 2010;40(5):687–688.
- Santidrian AF, Gonzalez-Girones DM, Iglesias-Serret D, et al. AICAR induces apoptosis independently of AMPK and p53 through up-regulation of the BH3-only proteins BIM and NOXA in chronic lymphocytic leukemia cells. Blood. 2010;116(16):3023–3032.
- Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12(9):814–822.
- Galluzzi L, Pietrocola F, Bravo-San Pedro JM, et al. Autophagy in malignant transformation and cancer progression. Embo J. 2015;34(7):856–880.
- Lindqvist LM, Simon AK, Baehrecke EH. Current questions and possible controversies in autophagy. Cell Death Disc. 2015;1:15036.
- Feng W, Huang S, Wu H, et al. Molecular basis of Bcl-xL’s target recognition versatility revealed by the structure of Bcl-xL in complex with the BH3 domain of Beclin-1. J Mol Biol. 2007;372(1):223–235.
- Oberstein A, Jeffrey PD, Shi Y. Crystal structure of the Bcl-XL-Beclin 1 peptide complex: beclin 1 is a novel BH3-only protein. J Biol Chem. 2007;282(17):13123–13132.
- Tormo D, Checinska A, Alonso-Curbelo D, et al. Targeted activation of innate immunity for therapeutic induction of autophagy and apoptosis in melanoma cells. Cancer Cell. 2009;16(2):103–114.
- Elgendy M, Sheridan C, Brumatti G, et al. Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Molecular Cell. 2011;42(1):23–35.
- Oltersdorf T, Elmore SW, Shoemaker AR, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435(7042):677–681.
- Delbridge AR, Grabow S, Strasser A, et al. Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nat Rev Cancer. 2016;16(2):99–109.
- Mason KD, Carpinelli MR, Fletcher JI, et al. Programmed anuclear cell death delimits platelet life span. Cell. 2007;128(6):1173–1186.
- Souers AJ, Leverson JD, Boghaert ER, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19(2):202–208.
- Roberts AW, Davids MS, Pagel JM, et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374(4):311–322.
- Lessene G, Czabotar PE, Sleebs BE, et al. Structure-guided design of a selective BCL-X(L) inhibitor. Nat Chem Biol. 2013;9(6):390–397.
- Tao ZF, Hasvold L, Wang L, et al. Discovery of a potent and selective BCL-XL inhibitor with in vivo activity. ACS Med Chem Lett. 2014;5(10):1088–1093.
- Bruncko M, Wang L, Sheppard GS, et al. Structure-guided design of a series of MCL-1 inhibitors with high affinity and selectivity. J Med Chem. 2015;58(5):2180–2194.
- Leverson JD, Zhang H, Chen J, et al. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax). Cell Death Dis. 2015;6:e1590.
- Kotschy A, Szlavik Z, Murray J, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016;538(7626):477–482.
- Pierson W, Cauwe B, Policheni A, et al. Antiapoptotic Mcl-1 is critical for the survival and niche-filling capacity of Foxp3(+) regulatory T cells. Nat Immunol. 2013;14(9):959–965.
- Fu NY, Rios AC, Pal B, et al. EGF-mediated induction of Mcl-1 at the switch to lactation is essential for alveolar cell survival. Nat Cell Biol. 2015;17(4):365–375.
- Peperzak V, Vikstrom I, Walker J, et al. Mcl-1 is essential for the survival of plasma cells. Nat Immunol. 2013.
- Vikstrom I, Carotta S, Luthje K, et al. Mcl-1 is essential for germinal center formation and B cell memory. Science. 2010;330(6007):1095–1099.
- Akcay G, Belmonte MA, Aquila B, et al. Inhibition of Mcl-1 through covalent modification of a noncatalytic lysine side chain. Nat Chem Biol. 2016;12(11):931–936.
- Lee S, Wales TE, Escudero S, et al. Allosteric inhibition of antiapoptotic MCL-1. Nat Struct Mol Biol. 2016;23(6):600–607.
- Peperzak V, Slinger E, Ter Burg J, et al. Functional disparities among BCL-2 members in tonsillar and leukemic B-cell subsets assessed by BH3-mimetic profiling. Cell Death Differ. 2017;24(1):111–119.
- Del Gaizo Moore V, Letai A. BH3 profiling–measuring integrated function of the mitochondrial apoptotic pathway to predict cell fate decisions. Cancer Lett. 2013;332(2):202–205.
- Albert MC, Brinkmann K, Kashkar H. Noxa and cancer therapy: tuning up the mitochondrial death machinery in response to chemotherapy. Mol Cell Oncol. 2014;1(1):e29906.
- Bolomsky A, Schlangen K, Schreiner W, et al. Targeting of BMI-1 with PTC-209 shows potent anti-myeloma activity and impairs the tumour microenvironment. J Hematol Oncol. 2016;9:17.
- Gomez-Bougie P, Amiot M. Apoptotic machinery diversity in multiple myeloma molecular subtypes. Front Immunol. 2013;4:467.
- Bodet L, Menoret E, Descamps G, et al. BH3-only protein Bik is involved in both apoptosis induction and sensitivity to oxidative stress in multiple myeloma. Br J Cancer. 2010;103(12):1808–1814.
- Derenne S, Monia B, Dean NM, et al. Antisense strategy shows that Mcl-1 rather than Bcl-2 or Bcl-x(L) is an essential survival protein of human myeloma cells. Blood. 2002;100(1):194–199.
- Gong JN, Khong T, Segal D, et al. Hierarchy for targeting pro-survival BCL2 family proteins in multiple myeloma: pivotal role of MCL1. Blood. 2016.
- Ponder KG, Matulis SM, Hitosugi S, et al. Dual inhibition of Mcl-1 by the combination of carfilzomib and TG02 in multiple myeloma. Cancer Biol Ther. 2016;17(7):769–777.
- Kiziltepe T, Hideshima T, Catley L, et al. 5-Azacytidine, a DNA methyltransferase inhibitor, induces ATR-mediated DNA double-strand break responses, apoptosis, and synergistic cytotoxicity with doxorubicin and bortezomib against multiple myeloma cells. Mol Cancer Ther. 2007;6(6):1718–1727.
- Tromp JM, Tonino SH, Elias JA, et al. Dichotomy in NF-kappaB signaling and chemoresistance in immunoglobulin variable heavy-chain-mutated versus unmutated CLL cells upon CD40/TLR9 triggering. Oncogene. 2010;29(36):5071–5082.
- Herishanu Y, Perez-Galan P, Liu D, et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood. 2011;117(2):563–574.
- Morales AA, Olsson A, Celsing F, et al. Expression and transcriptional regulation of functionally distinct Bmf isoforms in B-chronic lymphocytic leukemia cells. Leukemia. 2004;18(1):41–47.
- Mackus WJ, Kater AP, Grummels A, et al. Chronic lymphocytic leukemia cells display p53-dependent drug-induced Puma upregulation. Leukemia. 2005;19(3):427–434.
- Smit LA, Hallaert DY, Spijker R, et al. Differential Noxa/Mcl-1 balance in peripheral versus lymph node chronic lymphocitic leukemia cells correlates with survival capacity. Blood. 2007;109(4):1660–1668.
- Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369(1):32–42.
- E. ten Hacken, Burger JA. Molecular pathways: targeting the microenvironment in chronic lymphocytic leukemia–focus on the B-cell receptor. Clin Cancer Res. 2014;20(3):548–556.
- Wensveen FM, Derks IA, Van Gisbergen KP, et al. BH3-only protein Noxa regulates apoptosis in activated B cells and controls high-affinity antibody formation. Blood. 2012;119(6):1440–1449.
- Slinger E, Wensveen FM, Guikema JE, et al. Chronic lymphocytic leukemia development is accelerated in mice with deficiency of the pro-apoptotic regulator NOXA. Haematologica. 2016;101(9):e374–7.
- Liu FT, Agrawal SG, Movasaghi Z, et al. Dietary flavonoids inhibit the anticancer effects of the proteasome inhibitor bortezomib. Blood. 2008;112(9):3835–3846.
- Wickremasinghe RG. Why is CLL refractory to bortezomib? Blood. 2008;112(9):3540–3541.
- Faderl S, Rai K, Gribben J, et al. Phase II study of single-agent bortezomib for the treatment of patients with fludarabine-refractory B-cell chronic lymphocytic leukemia. Cancer. 2006;107(5):916–924.
- Tromp JM, Geest CR, Breij EC, et al. Tipping the Noxa/Mcl-1 balance overcomes ABT-737 resistance in chronic lymphocytic leukemia. Clin Cancer Res. 2012;18(2):487–498.
- Thijssen R, Slinger E, Weller K, et al. Resistance to ABT-199 induced by microenvironmental signals in chronic lymphocytic leukemia can be counteracted by CD20 antibodies or kinase inhibitors. Haematologica. 2015.
- Hallaert DY, Spijker R, Jak M, et al. Crosstalk among Bcl-2 family members in B-CLL: seliciclib acts via the Mcl-1/Noxa axis and gradual exhaustion of Bcl-2 protection. Cell Death Differ. 2007;14(11):1958–1967.
- Wiernik PH. Alvocidib (flavopiridol) for the treatment of chronic lymphocytic leukemia. Expert Opin Investig Drugs. 2016;25(6):729–734.
- De Rooij MF, Kuil A, Geest CR, et al. The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood. 2012;119(11):2590–2594.
- Spaargaren M, De Rooij MF, Kater AP, et al. BTK inhibitors in chronic lymphocytic leukemia: a glimpse to the future. Oncogene. 2015;34(19):2426–2436.
- Eldering E, Peperzak V, ter Burg H, et al. Bcl-2 members as drug target and biomarkers for response to Ibrutinib and Venetoclax in CLL. Blood. 2016;128(22):2043.
- Ohshima-Hosoyama S, Davare MA, Hosoyama T, et al. Bortezomib stabilizes NOXA and triggers ROS-associated apoptosis in medulloblastoma. J Neurooncol. 2011;105(3):475–483.
- Tessoulin B, Descamps G, Moreau P, et al. PRIMA-1Met induces myeloma cell death independent of p53 by impairing the GSH/ROS balance. Blood. 2014;124(10):1626–1636.