Abstract
A series of N-cyanomethyl aromatic sulfonamides and bis-sulfonamides was prepared by reaction of arylsulfonyl halides with aminoacetonitrile. The obtained derivatives incorporated various aryl moieties, such as 4-halogeno/alkyl/aryl/nitro-substituted-phenyl, pentafluorophenyl or 2-naphthyl. Moderate inhibitory activity was detected for some compounds against the cytosolic human isoform II of the metalloenzyme carbonic anhydrase (CA, EC 4.2.1.1), hCA II, with inhibition constants of 90, 180 and 560 nM for the 4-nitrophenyl-, 4-iodophenyl- and pentafluorophenyl-N-cyanomethylsulfonamides, respectively. Other derivatives acted as weak inhibitors of isoforms hCA I (KIs of 720 nM–45 μM), hCA II (KIs of 1000–9800 nM) and hCA IX (KIs of 900–10200 nM). Thus, the N-cyanomethylsulfonamide zinc binding group is less effective than the sulfonamide, sulfamate or sulfamide ones for the design of effective CA inhibitors.
Introduction
The α-carbonic anhydrases (CAs, EC 4.2.1.1) constitute a family of metalloenzymes that catalyze the reversible hydration of CO2 to bicarbonate and a proton Citation1-5. We have been working with the molecular cloning of some of the 16 presently known human CA (hCA) isoforms Citation6-8, as well as screening for inhibitory effects of a variety of compounds on most of them, showing that such various isozymes (e.g., hCA I, II, IV, VA, VB, VII, IX, XII, XIII and XIV) constitute valid targets for the development of novel antiglaucoma, antitumor, antiobesity or anticonvulsant drugs Citation9-13. Furthermore, very recently representatives of the α- or β-CA class have been cloned and characterized in other organisms, such as Plasmodium falciparum [Citation14], Mycobacterium tuberculosis [Citation15], Cryptococcus neoformans [Citation16] or Candida spp. [Citation17], some of them being also investigated from the inhibition point of view [Citation14,Citation16], as it has been proved that these enzymes are critical for the growth or virulence of these pathogens Citation14-17. Since many of these organisms are highly pathogenic, and present different degrees of resistance to the currently available drugs targeting them, inhibition of their CAs may constitute novel approaches to fighting such diseases Citation14-17. Thus, there is a constant need for the design of novel classes of CA inhibitors (CAIs) Citation1-5, belonging to different classes of compounds, as such derivatives may lead to interesting pharmacological applications.
In addition to the classical sulfonamide zinc binding group (ZBG) for the design of CAIs Citation1-5, recently we also explored alternative ZBGs, discovering that N-substituted sulfonamides [Citation18], sulfamates and their derivatives [Citation19,Citation20], as well as sulfamides and their derivatives Citation21-23, may lead to effective inhibitors targeting several physiologically relevant isoforms.
Continuing our work in exploring different ZBGs for the design of CAIs, we report here that N-cyanomethyl aromatic sulfonamides and bis-sulfonamides show interesting inhibitory activity against the cytosolic isoforms CA I and II, and the transmembrane, tumor-associated isozyme CA IX.
Materials and methods:
General
Melting points were determined on a Büchi Melting Point 510 and are uncorrected. 1H-NMR spectra were recorded on a Bruker AC-250 spectrometer using DMSO-d6 as solvent and tetramethylsilane as internal standard. Chemical shifts are expressed in δ (ppm) downfield from tetramethylsilane, and coupling constants (J) are expressed in Hertz. Electron Ionization mass spectra (30 eV) were recorded in positive or negative mode on a Waters MicroMass ZQ. Arylsulfonyl halides, aminoacetonitrile, solvents, buffers and inorganic reagents were from Sigma-Aldrich, Milan, Italy and were used without further purification. Recombinant human CA isoforms I, II and IX were prepared as reported earlier by our group Citation9-13Citation20–23.
General procedure
N-Cyanomethylsulfonamides were prepared as previously described by our group for obtaining N-cyanosulfonamides, by reaction of arylsulfonyl halides with 2-amino acetonitrile in aqueous acetone [Citation24].
N-Cyanomethyl-4-fluorophenyl sulfonamide (1)
mp 72°C; 1H NMR (DMSO-d6, 250 MHz) δ 8.6 (t, 1H, J = 5.7 Hz), 7.9 (m, 2H), 7.4 (t, 2H, J = 9 Hz), 4.1 (d, 2H, J = 2 Hz); MS ESI+ m/z 237 (M+H)+. ESI− m/z 213 (M − H)− .
N-Cyanomethyl-4-chlorophenyl sulfonamide (2)
mp 111°C; 1H NMR (DMSO-d6, 250 MHz) δ 8.6 (t, 1H, J = 6 Hz), 7.8 (d, 2H, J = 8.5 Hz), 7.7 (d, 2H, J = 8.5 Hz), 4.1 (d, 2H, J = 3.5 Hz); MS ESI− m/z 229 (M − H)− .
N-Cyanomethyl-4-bromophenyl sulfonamide (3)
mp 126°C; 1H NMR (DMSO-d6, 250 MHz) δ 8.7 (t, 1H, J = 6 Hz), 7.8 (d, 2H, J = 8.5 Hz), 7.7 (d, 2H, J = 8.5 Hz), 4.1 (d, 2H, J = 3.2 Hz); MS ESI+m/z 299 (M+Na)+. ESI− m/z 275 (M − H)− .
N-Cyanomethyl-4-iodophenyl sulfonamide (4)
mp 147°C; 1H NMR (DMSO-d6, 250 MHz) δ 8.7 (t, 1H, J = 5.7 Hz), 8 (d, 2H, J = 7 Hz), 7.6 (d, 2H, J = 7 Hz), 4.1 (d, 2H, J = 2.5 Hz); MS ESI− m/z 321 (M − H)− .
N-Cyanomethyl-4-methylphenyl sulfonamide (5)
mp 101°C; 1H NMR (DMSO-d6, 250 MHz) δ 8.5 (s, 1H), 7.7 (d, 2H, J = 7.5 Hz), 7.4 (d, 2H, J = 7.5 Hz), 4.06 (s, 2H), 2.4 (s, 3H); MS ESI− m/z 209 (M − H)− .
N-Cyanomethyl-4-tertbutylphenyl sulfonamide (6)
mp 96°C; 1H NMR (DMSO-d6, 250 MHz) δ 8.5 (t, 1H, J = 5.9 Hz), 7.7 (d, 2H, J = 7 Hz), 7.6 (d, 2H, J = 7 Hz), 4.06 (s, 2H), 1.3 (s, 9H); MS ESI+m/z 275 (M+Na)+.
N-Cyanomethyl-4-biphenyl sulfonamide (7)
mp 95°C; 1H NMR (DMSO-d6, 250 MHz) δ 8.6 (s, 1H), 7.9–7.4 (m, 9H), 4.1 (s, 2H); MS ESI− m/z 271 (M − H)− .
N-Cyanomethyl-4-nitrophenyl sulfonamide (8)
mp 93°C; 1H NMR (DMSO-d6, 250 MHz) δ 9 (s, 1H), 8.4 (d, 2H, J = 7 Hz), 8.1 (d, 2H, J = 7 Hz), 4.2 (s, 2H); MS ESI− m/z 240 (M − H)− .
N-Cyanomethyl-pentafluorophenyl sulfonamide (9)
mp 98°C; 1H NMR (DMSO-d6, 250 MHz) δ 4.3 (s, 2H); MS ESI− m/z 285 (M − H)− .
N-Cyanomethyl-2-naphthyl sulfonamide (10)
mp 93°C; 1H NMR (DMSO-d6, 250 MHz) δ 8.68 (t, 1H, J = 6 Hz), 8.5 (s, 1H), 8.2 (m, 2H), 8.1 (d, 1H, J = 7.5 Hz), 7.8 (d, 1H, J = 7.5 Hz), 7.7 (m, 2H), 4.15 (d, 2H, J = 3.2 Hz); MS ESI+m/z 269 (M+Na)+. ESI− m/z 245 (M − H)− .
Bis-N-cyanomethyl-1,3-benzene-di-sulfonamide (11)
mp 87°C; 1H NMR (DMSO-d6, 250 MHz) δ 8.9 (s, 1H), 8.2–8.1 (m, 3H), 4.1 (s, 4H); MS ESI− m/z 313 (M − H).
CA inhibition assay
An Applied Photophysics (Oxford, UK) stopped-flow instrument was used for assaying the CA-catalysed CO2 hydration activity [Citation25]. Phenol red (at a concentration of 0.2 mM) was used as indicator, working at the absorbance maximum of 557 nm, with 10 mM Hepes (pH 7.5) as buffer, 0.1 M Na2SO4 (for maintaining constant the ionic strength), following the CA-catalyzed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7–17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor at least six traces of the initial 5–10% of the reaction were used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (1 mM) were prepared in distilled-deionized water with 10–20% (v/v) DMSO (which is not inhibitory at these concentrations) and dilutions up to 0.1 nM were done thereafter with distilled-deionized water. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E-I complex. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3, from Lineweaver-Burk plots, as reported earlier, and represent the mean from at least three different determinations Citation9-13. Human CA I and CA II were from Sigma-Aldrich (Milan, Italy) whereas recombinant hCA IX (catalytic domain) were prepared as reported earlier by us [Citation10,Citation13].
Results and discussion
N-Cyanosulfonamides of the type RSO2NHCN were shown by our group to lead to effective CAIs targeting isoforms CA II and IV [Citation24]. It is known that CAIs (of the sulfonamide, sulfamate or sulfamide type) bind to the Zn(II) ion within the enzyme active site in the deprotonated form, at the same time participating in numerous hydrogen bonds and hydrophobic interactions with amino acid residues from the cavity, such multiple interactions explaining the high affinity of many inhibitors for various CA isozymes Citation1-6Citation9–13. Considering RSO2NHCN [Citation24] compounds as lead molecules, we investigate here whether a bulkier substituent at the sulfonamide moiety than the cyano one, i.e., the cyanomethyl group, may still be used for the design of effective CAIs, which in this way would incorporate a new ZBG, the N-cyanomethylsulfonamide one. Thus, by using the same chemistry as reported earlier for the preparation of N-cyanosulfonamides [Citation24], we obtained a series of N-cyanomethylsulfonamides by reaction of arylsulfonyl halides A with aminoacetonitrile B, under Schotten-Baumann conditions, as shown in Scheme [Citation24]. A group of derivatives of type 1–11 have been prepared in this way, which incorporate various aryl moieties, such as 4-halogeno/alkyl/aryl/nitro-substituted-phenyl, pentafluorophenyl or 2-naphthyl. A bis-N-cyanomethyl-sulfonamide (compound 11) has also been obtained, as it is known that benzene-1,3-disulfonamides act as efficient CAIs, with one of the clinically used drugs (dichlorophenamide, DCP) belonging to this class of derivatives Citation1-5.
Scheme 1 Preparation of N-cyanomethylsulfonamides 1–11 by reaction of arylsulfonyl halides A with aminoacetonitrile B.
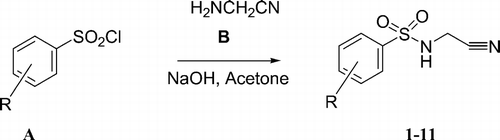
Derivatives 1–11 have been investigated as inhibitors of three physiologically relevant CA isoforms, the ubiquitous human cytosolic hCA I and II, as well as the transmembrane, human, tumor-associated hCA IX ().
Table I. Inhibition data for isozymes hCA I, II and IX with N-cyanomethylsulfonamides 1–11 prepared in the present study, and the clinically used sulfonamides acetazolamide (AAZ) and dichlorophenamide (DCP) as standard inhibitors. 

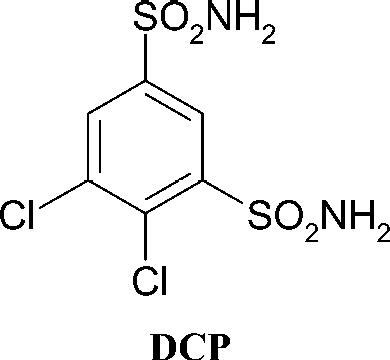
As seen from the data of , the N-cyanomethylsulfonamides 1–11 investigated here show inhibitory activity against the three investigated CA isoforms. The following structure–activity relationship correlations were observed for this group of derivatives: (i) against the slow cytosolic isozyme hCA I Citation1-4, the N-cyanomethylsulfonamides 1–11 showed weak inhibitory activity, with inhibition constants in the range 720 nM–45 μM, being thus much less effective inhibitors as compared to the unsubstituted sulfonamides used clinically acetazolamide or dichlorophenamide (which showed KIs in the range 250–1200 nM). Only the pentafluorophenyl-substituted derivative 9 showed interesting activity against this isozyme, being in fact a more efficient inhibitor than the aromatic unsubstituted sulfonamide DCP (but less effective as compared to the heterocyclic sulfonamide AAZ). Otherwise, the substitution pattern at the benzene nucleus slightly influenced activity, except for the bis-substituted derivative 11 which was the least effective CAI (KI of 45 μM). The weak inhibitory activity of these derivatives is presumably due to the rather bulky nature of the N-cyanomethylsulfonamide ZBG, which may hinder its access in the neighborhood of the Zn(II) ion of the enzyme active site for efficient binding. In fact the bis-sulfonamide 11 investigated here, which is also the most bulky one in the series of investigated compounds, was also the least effective hCA I inhibitor; (ii) against the physiologically most relevant and ubiquitous isoform hCA II, the N-cyanomethyl-sulfonamides 1–11 showed inhibition constants in the range 90–9800 nM. Thus, two derivatives, 4 and 8, may be considered as effective hCA II inhibitors, as their KIs, of 90–180 nM, are only 2.3–4.7 times higher as that of the aromatic disulfonamide DCP (KI of 38 nM), which has clinical applications as an antiglaucoma drug Citation1-4. These two compounds incorporate the 4-nitrophenyl- and the 4-iodophenyl moiety. The best hCA II inhibitor is the former derivative, in which the nitro group in the para position probably enhances the acidification of the SO2NH proton, which is beneficial for the inhibition of CA due to the fact that inhibitors generally bind the Zn(II) ion as monoanions Citation1-4. However, small variations in the moieties substituting the phenyl ring lead to a pronounced decrease of hCA II inhibitory activity, such as for example in the perfluorophenyl compound 9 (which is a medium potency inhibitor, with a KI of 560 nM) or the other derivatives 1–3, 5 and 6, which showed KIs in the range 1000–3500 nM. On the other hand, in the case of derivatives incorporating bulkier aryl groups (biphenyl, 2-naphthyl—compounds 7 and 10) as well as for the bis-sulfonamide derivative 11, the hCA II inhibitory activity is even more decreased, such compounds possessing KIs in the range 7600–9800 nM. It is thus obvious that generally the N-cyanomethylsulfonamides are less effective hCA II inhibitors as compared to the unsubstituted sulfonamides (such as for example AAZ and DCP, ), the sulfamates [Citation19,Citation20] or the sulfamides Citation21-23; (iii) due to the rather weak hCA I and II inhibitory activity in this group of derivatives and because hCA IX is rather difficult to prepare [Citation26], we investigated the inhibition of this transmembrane, tumor-associated isoform only with the most effective hCA II inhibitors detected so far (the active sites of hCA II and hCA IX are rather similar) [Citation10]. Indeed, the four compounds showed weak hCA IX inhibitory activity, with KIs in the range 900–10200 nM, being thus much less effective as compared to the unsubstituted sulfonamides (AAZ and DCP show KIs of 25–50 nM against this isoform).
In conclusion, in a series of N-cyanomethylsulfonamides incorporating substituted-phenyl or naphthyl moieties, moderate hCA II inhibitory activity has been detected for some compounds (inhibition constants of 90–560 nM for the 4-nitrophenyl-, 4-iodophenyl- and pentafluorophenyl-N-cyanomethylsulfonamides, respectively), whereas other such derivatives acted as weak inhibitors of isoforms hCA I (KIs of 720 nM–45 μM), hCA II (KIs of 1000–9800 nM) and hCA IX (KIs of 900–10200 nM). Thus, the N-cyanomethylsulfonamide zinc binding group is less effective than the sulfonamide, sulfamate or sulfamide ones for the design of effective CA inhibitors.
Acknowledgements
This research was financed in part by a grant from the 6th framework program of EU (EUROXY project).
Notes
Related Research Data
References
- Supuran CT, Scozzafava A, Conway J. Carbonic anhydrase—its inhibitors and activators, HJ Smith, C Simons, Series. CRC Press, Boca Raton, New York, London 2004; 1–363
- Pastorekova S, Parkkila S, Pastorek J, Supuran CT. Carbonic anhydrases: Current state of the art, therapeutic applications and future prospects. J Enz Inhib Med Chem 2004; 19: 199–229
- Supuran CT, Scozzafava A, Casini A. Carbonic anhydrase inhibitors. Med Res Rev 2003; 23: 146–189
- Scozzafava A, Mastrolorenzo A, Supuran CT. Modulation of carbonic anhydrase activity and its applications in therapy. Expert Opin Ther Pat 2004; 14: 667–702
- Sly WS, Hu PY. Human carbonic anhydrases and carbonic anhydrase deficiencies. Annu Rev Biochem 1995; 64: 375–401
- Lehtonen J, Shen B, Vihinen M, Casini A, Scozzafava A, Supuran CT, Parkkila AK, Saarnio J, Kivela AJ, Waheed A, Sly WS, Parkkila S. Characterization of CA XIII, a novel member of the carbonic anhydrase isozyme family. Biol Chem 2004; 279: 2719–2727
- Fujikawa-Adachi K, Nishimori I, Taguchi T, Onishi S. Human mitochondrial carbonic anhydrase VB. CDNA cloning, mRNA expression, subcellular localization, and mapping to chromosome X. J Biol Chem 1999; 274: 21228–21233
- Fujikawa-Adachi K, Nishimori I, Taguchi T, Onishi S. Human carbonic anhydrase XIV (CA14): cDNA cloning, mRNA expression, and mapping to chromosome 1. Genomics 1999; 61: 74–81
- Nishimori I, Vullo D, Innocenti A, Scozzafava A, Mastrolorenzo A, Supuran CT. Carbonic anhydrase inhibitors. The mitochondrial isozyme VB as a new target for sulfonamide and sulfamate inhibitors. J Med Chem 2005; 48: 7860–7866
- Pastorekova S, Casini A, Scozzafava A, Vullo D, Pastorek J, Supuran CT. Carbonic anhydrase inhibitors: The first selective, membrane-impermeant inhibitors targeting the tumor-associated isozyme IX. Bioorg Med Chem Lett 2004; 14: 869–873
- Vullo D, Voipio J, Innocenti A, Rivera C, Ranki H, Scozzafava A, Kaila K, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of the human cytosolic isozyme VII with aromatic and heterocyclic sulfonamides. Bioorg Med Chem Lett 2005; 15: 971–976
- Vullo D, Innocenti A, Nishimori I, Pastorek J, Scozzafava A, Pastorekova S, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of the transmembrane isozyme XII with sulfonamides—a new target for the design of antitumor and antiglaucoma drugs?. Bioorg Med Chem Lett 2005; 15: 963–969
- Cecchi A, Hulikova A, Pastorek J, Pastorekova S, Scozzafava A, Winum J-Y, Montero J-L, Supuran CT. Carbonic anhydrase inhibitors. Sulfonamides inhibit isozyme IX mediated acidification of hypoxic tumors. Fluorescent sulfonamides design as probes of membrane-bound carbonic anhydrase isozymes involvement in tumorigenesis. J Med Chem 2005; 48: 4834–4841
- Krungkrai J, Scozzafava A, Reungprapavut S, Krungkrai SR, Rattanajak R, Kamchonwongpaisan S, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of Plasmodium falciparum carbonic anhydrase with aromatic sulfonamides: Towards antimalarials with a novel mechanism of action?. Bioorg Med Chem 2005; 13: 483–489
- Suarez Covarrubias A, Larsson AM, Hogbom M, Lindberg J, Bergfors T, Bjorkelid C, Mowbray SL, Unge T, Jones TA. Structure and function of carbonic anhydrases from Mycobacterium tuberculosis. J Biol Chem 2005; 280: 18782–18789
- Klengel T, Liang WJ, Chaloupka J, Ruoff C, Schroppel K, Naglik JR, Eckert SE, Mogensen EG, Haynes K, Tuite MF, Levin LR, Buck J, Muhlschlegel FA. Fungal adenylyl cyclase integrates CO2 sensing with cAMP signaling and virulence. Curr Biol 2005; 15: 2021–2026
- Bahn YS, Cox GM, Perfect JR, Heitman J. Carbonic anhydrase and CO2 sensing during Cryptococcus neoformans growth, differentiation, and virulence. Curr Biol 2005; 15: 2013–2020
- Briganti F, Pierattelli R, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Part 37. Novel classes of carbonic anhydrase inhibitors and their interaction with the native and cobalt-substituted enzyme: Kinetic and spectroscopic investigations. Eur J Med Chem 1996; 31: 1001–1010
- Winum JY, Scozzafava A, Montero JL, Supuran CT. Sulfamates and their therapeutic potential. Med Res Rev 2005; 25: 186–228
- Bonnac L, Innocenti A, Winum JY, Casini A, Montero JL, Scozzafava A, Barragan V, Supuran CT. Carbonic anhydrase inhibitors: Aliphatic N-phosphorylated sulfamates—a novel zinc-anchoring group leading to nanomolar inhibitors. J Enz Inhib Med Chem 2004; 19: 275–278
- Casini A, Winum JY, Montero JL, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors: Inhibition of cytosolic isozymes I and II with sulfamide derivatives. Bioorg Med Chem Lett 2003; 13: 837–840
- Winum JY, Scozzafava A, Montero JL, Supuran CT. The sulfamide motif in the design of enzyme inhibitors. Expert Opin Ther Pat 2006; 16: 27–47
- Winum J-Y, Innocenti A, Nasr J, Montero J-L, Scozzafava A, Vullo D, Supuran CT. Carbonic anhydrase inhibitors: Synthesis and inhibition of cytosolic/tumor-associated carbonic anhydrase isozymes I, II, IX and XII with N-hydroxy-sulfamides—A new zinc binding function in the design of inhibitors. Biorg Med Chem Lett 2005; 15: 2353–2358
- Supuran CT, Scozzafava A, Briganti F. Carbonic anhydrase inhibitors. N-Cyanosulfonamides: a new class of high affinity isozyme II and IV inhibitors. J Enz Inhib 1999; 14: 289–306
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971; 246: 2561–2573
- Vullo D, Franchi M, Gallori E, Pastorek J, Scozzafava A, Pastorekova S, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of the tumor-associated isozyme IX with aromatic and heterocyclic sulfonamides. Bioorg Med Chem Lett 2003; 13: 1005–1009