Abstract
Homology models of cytochrome P450 RA1 (CYP26A1) were constructed using three human P450 structures, CYP2C8, CYP2C9 and CYP3A4 as templates for the model building. Using MOE software the lowest energy CYP26A1 model was then assessed for stereochemical quality and side chain environment. Further active site optimisation of the CYP26A1 model built using the CYP3A4 template was performed by molecular dynamics to generate a final CYP26A1 model. The natural substrate, all-trans-retinoic acid (atRA), and inhibitor R115866, were docked into the model allowing further validation of the active site architecture. Using the docking studies structurally and functionally important residues were identified with subsequent characterisation of secondary structure. Multiple hydrophobic interactions, including the side chains of TRP112, PHE299, PHE222, PHE84, PHE374 and PRO371, are important for binding of atRA and R115866. Additional hydrogen bonding interactions were noted as follows: atRA – C = O of the atRA carboxylate group and ARG86; R115866 – benzothiazole nitrogen and the backbone NH of SER115.
Introduction
Retinoic acid (RA) regulates genes involved in cell proliferation, differentiation and apoptosis [Citation1], and exerts activity by binding to transcription-regulatory factors in the cell nucleus known as RAR (retinoic acid receptor) and RXR (retinoid X receptor), each having subtypes α, β and δ [Citation2]. RAR receptors are activated by all-trans- (atRA) and 9-cis- (9-cis-RA) isomers of RA, whereas RXR receptors are only activated by 9-cis-RA. Upon RA binding the activated receptor transcriptionally regulates its target genes by binding to specific response elements in promoter regions of RA target genes () [Citation3,Citation4].
Figure 1 Alcohol dehydrogenases (ADH) and short-chain dehydrogenase/reductase catalyse the oxidation of retinol to retinaldehyde, which is subsequently oxidised by aldehyde dehydrogenases (ALDH) to retinoic acid. Retinoic acid, all-trans (atRA) and 9-cis (9cRA) isomers are further metabolised by cytochrome P450 enzymes (including CYP26A1, B1 and C1) to inactive polar metabolites resulting in retinoid excretion.
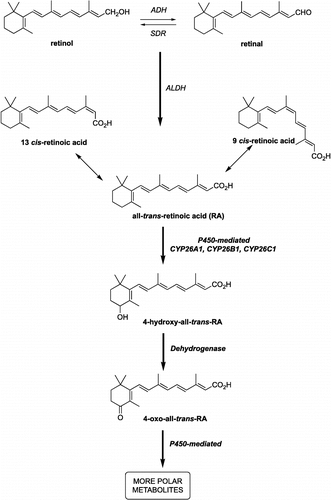
Retinoic acid is synthesised from vitamin A (retinol), which is oxidised through retinal by dehydrogenases in the cytoplasm of target cells, in low yields, to all-trans-retinoic acid (). atRA is subsequently metabolised by human liver and intestine cytochrome P450s to the inactive 4-hydroxy-RA and thence by dehydrogenases to the partially active 4-keto-RA and inactive polar metabolites () [Citation5].
The P450s responsible for 4-hydroxylation of atRA in the human liver are CYP2C8 as a major contributor as well as 3A7, 3A5, 3A4, 2C9 and 1A1 [Citation6]. However, in living tissues, atRA administration induces another RA-metabolising enzyme, CYP26 [Citation7], which recognises only atRA as its substrate, and the expression of this isozyme can be induced by atRA both in vitro and in vivo [Citation8]. Three members of the CYP26 family have now been identified: CYP26A1 [Citation7] and CYP26B1 [Citation9], which metabolise atRA in the embryo and adult and, more recently [Citation10], CYP26C1 that may have a role in the metabolism/isomerisation of both all-trans and 9-cis isomers of RA.
Retinoids in general have been used for some time in the treatment of psoriasis, cystic acne, cutaneous malignancies due to hyperkeratinisation as well as in the treatment of photo-damaged skin [Citation11,Citation12]. Retinoic acid has been used in a number of clinical situations, especially oncology and dermatology, atRA has also been shown to improve the efficacy of other treatments such as radiation, cisplatin and interferon therapies [Citation13,Citation14].
Although the amino acid sequence of CYP26A1 has been characterised no crystal structure is available. A homology model for human CYP26A1 would provide valuable information regarding active site architecture and allow docking studies to analyse ligand binding interactions, the information obtained from these studies would assist with rationale drug design of retinoid mimetics and CYP26A1 inhibitors. Importantly, crystal structures for three human cytochrome P450 enzymes, CYP2C8 [Citation15], CYP2C9 [Citation16] and CYP3A4 [Citation17] have recently become available allowing the use of a human crystal template for the homology building.
Materials and methods
Computational approaches
All molecular modelling studies were performed on a RM Innovator Pentium IV 2.4 GHz running either Linux Fedora Core 3 or Windows XP using Molecular Operating Environment (MOE) 2004.03 [Citation18] and FlexX module in SYBYL 7.0 [Citation19] molecular modelling software.
Molecular dynamics simulations were performed with GROMACS 3.2 [Citation20,Citation21] and the Gromacs force field in a NVT (canonical) environment. Individual ligand/protein complexes obtained from the docking results were soaked in a triclinic water box and minimised using a steepest descent algorithm to remove unfavorable van der Waals contacts. The system was then equilibrated via a 20 ps MD simulation at 300°K with restrained ligand/protein complex atoms. Finally, a 800 ps simulation was performed at 300°K with a time step of 2 fs and hydrogen atoms constrained with a LINCS algorithm. Visualisation of the dynamics trajectories was performed with the VMD software package, version 1.8.3 [Citation22].
All the minimisations were performed with MOE until RMSD gradient of 0.05 Kcal mol− 1Å− 1 with the forcefield specified and the partial charges were automatically calculated.
Homology searching
The protein sequence of human CYP26A1 was obtained from the ExPASy server (O43174) [Citation23]. Homologous proteins with known crystal structures were found by performing a PSI-BLAST search (comparison matrix, BLOSUM62; E-threshold, 10), using the ExPASy server, aligning the query sequence (CYP26A1) against sequences in the Protein Data Bank (PDB) [Citation24]. The alignment parameters and thresholds used for screening candidate homologues were used with their default values and BLOSUM62 comparison matrix.
Multiple sequence and structure alignment
MOE-Align, which implements a modified version of the alignment methodology originally introduced by Needleman and Wunsch [Citation25], was employed. The sequence alignment was performed with an alignment constraint between the haem cysteine residue of the query sequence (CYP26A1) and the corresponding haem cysteine residue of the templates. All the default settings in the MOE-Align panel were used for the sequence alignment.
D Model Building
Homology models were built using MOE-Homology using a single template approach with CHARMM22 forcefield. The three human P450 crystal structures CYP2C8, CYP2C9 and CY3A4 were selected as the templates for constructing CYP26A1 models. Ten intermediate models were generated and the final model was taken as the Cartesian average of all the intermediate models. The haem was positioned using the same coordinates as in the template and the complex model was energy minimised.
Model Validation
Stereochemical quality of the polypeptide backbone and side chains was evaluated using Ramachandran plots obtained from the RAMPAGE server [Citation26]. Amino acid environment was evaluated using Verify3D [Citation27] and Errat plots[Citation28]. Verify 3D assesses environment of the sidechain based on the solvent accessibility of the sidechain and the fraction of the sidechain covered by polar atoms. Errat assesses the distribution of different types of atoms with respect to one another in the protein model. Validation data for the templates CYP2C8, CYP2C9 and CYP3A4 was used as the baseline to assess the respective models.
Docking
Ligands were docked within the active site of the homology model using the FlexX docking programme of SYBYL, performed with the default values. The active site was defined by all the amino acid residues within a 6.5 Å distance from TRP112, VAL116, THR304, VAL370 and GLY373, including the haem in a heteroatom file.
The output of FlexX docking was visualised in MOE and the scoring.svl script [Citation29] was used to identify interaction types between ligand and protein.
Results and discussion
Comparative modelling methods use structural templates that have the highest sequence homology with the target protein. Homologous proteins were identified by scanning the protein sequence of CYP26A1 [Citation30], obtained from the ExPASy server [Citation23], against 3D structures deposited in the Protein Data Bank (PDB) [Citation24] using PSI-BLAST [Citation31]. The search returned amino acid sequences of different P450s isolated from different species, including the three human P450s recently deposited in the PDB. The percent sequence identity, chain length and E-value for the homologous P450 sequences are shown in . CYP3A4 (1TQN), CYP2C8 (1PQ2) and CYP2C9 (1R90) were promising templates with percent sequence identity ranging from 24-26% and good E-values.
Table I. P450 sequences identified by PSI-BLAST search using CYP26A1 as the query sequence.
All three sequences have a similar chain length to the query sequence CYP26A1 and are high resolution structures. Structural alignment of the CYP26A1 with the three human P450 templates was performed using ClustalW [Citation32] on the EBI server [Citation33].
Alignment allowed the assignment of substrate recognition sites and P450 binding motifs (ETLR, PERF and haem binding domains) ().
Figure 2 CYP26A1 sequence and ClustalW (1.82) alignment with CYP2C9, CYP2C8 and CYP3A4. The colour coding of amino acid type: red, small + hydrophobic (AVFPMILWY); blue, acid (DE); magenta, basic (RHK); green, hydroxyl + amine + basic (STYHCNGQ). Substrate recognition sites (SRS) and conserved P450 domains (ETLR, PERF and Haem binding domain) are indicated. “*” means that the residues are identical, “:” means that conserved substitutions have been observed, “.” means that semi-conserved substitutions are observed.

Models, based on each template using a single template alignment approach, were generated using Molecular Operating Environment (MOE) [Citation18] software as described in the Experimental section. To evaluate the quality of the modelled structures, the lowest energy model generated from each template was subjected to a number of checks. Stereochemical quality was assessed using Ramachandran plots [Citation34], using the Cambridge RAMPAGE server [Citation26], and amino acid environment was assessed using Verify3D [Citation27] and Errat [Citation28], using the UCLA-DOE server [Citation35] ().
Table II. Validation results for the lowest energy CYP26A1 models (produced from the three different templates) and the 3D structural templates.
Validation results would suggest that all three models performed equally well in terms of mainchain stereochemistry and amino acid environment, therefore substrate and ligand docking was required for further validation of the active site architecture.
Ligand Docking
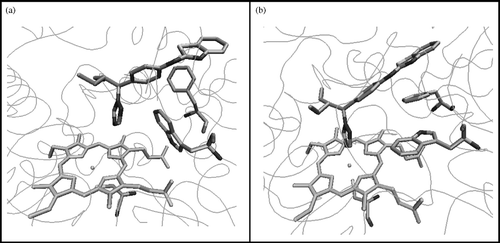
atRA was found to dock satisfactorily in the active site of all three models, with atRA orientated in a position that favours 4-hydroxylation at C-4. However, only the CYP26A1 model built using the CYP3A4 template was able to accommodate the inhibitor ligand (S)-R115866 [Citation8] in an orientation that would allow coordination between the nitrogen of the triazole heterocycle and the haem iron transition metal. However, docking studies had indicated that TRP112 was partially obstructing coordination between the triazole nitrogen of the ligand R115866 and the transition metal resulting in a distance of 5.50 Å (). Docking studies do not take into account protein flexibility therefore molecular dynamics were performed on the active site containing (S)-R115866 resulting in an optimised active site architecture. This resulted in the TRP112 residue positioned in a more favourable conformation with respect to ligand binding () with the triazole nitrogen perpendicular to the haem iron at a distance of 2.6 Å.
Figure 3 CYP26A1 model active site (a) before and (b) after active site optimisation with the (S)-R115866 bound inhibitor.
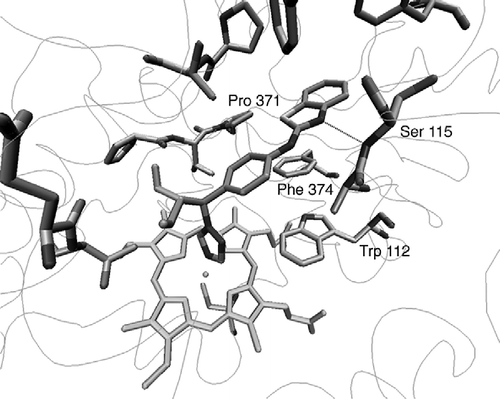
Furthermore, (S)-R115688 establishes a hydrogen bond between the benzothiazole nitrogen and the NH of SER115, as well as several hydrophobic interactions with the side chains of TRP112, PHE374, PHE84, PHE299, VAL116, PRO371 and other residues ().
Figure 4 Interactions between (S)-R115866 and the CYP26A1 active site.
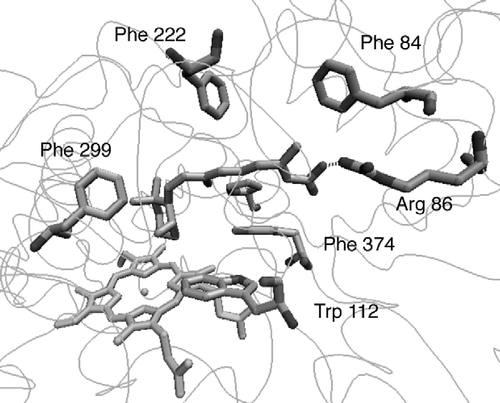
The natural substrate atRA was docked with the optimised active site. shows the putative active site of CYP26A1 containing the bound substrate orientated for 4-hydroxylation with the C4 atom positioned above the haem iron at a distance of 5.3 Å, this distance would accommodate a water molecule between the 4-position of atRA and the haem iron. atRA interacts with amino acid residues at the active site by multiple hydrophobic interactions, including the side chains of TRP112, PHE299, PHE222, PHE84, PHE374 and PRO371. Hydrogen bonding interactions between the carbonyl group of atRA and ARG86 hold the molecule within the hydrophobic tunnel.
Figure 5 atRA docked in CYP26A1 active site.
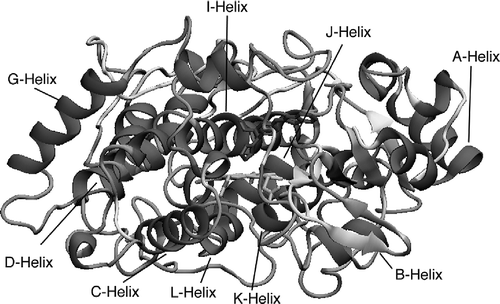
The secondary structure of the CYP26A1 model was determined using Swiss-PdbViewer 3.7 [Citation36] with identification of α–helices, β-sheets, coils and loops as shown in . The secondary structure is derived from the template structure CYP3A4 therefore further comparison with the template allows identification of specific helices and β-sheets. Studies have shown that the active site region (Figure 2) of the P450s is conserved across the entire family [Citation37] and the middle section of the I-helix (Figure 2) is also well conserved despite low overall sequence homology [Citation38]. However, although similarities do exist across the family, there are flexible regions in each P450 which have significantly different amino acid composition, such as the B'-helix, which may or may not be present, and F-G loop regions.
Table III. 3. Comparison of CYP26A1 model and CYP3A4 template secondary structure.
A good overlap between the model and template structure was found in particular for the A-, E- G-, I-, J-, J'- and K-helices. The model differs from the template in that the B'-, F- and H-helices are not identified in the model. As seen in , the large I-helix lies above the haem and is connected to the J-helix through a loop. The active site cysteine 442 lies below the haem within a coil and it is also worth noting that all the residues involved in the binding of atRA and (S)-R115688, are located in the substrate recognition sites (SRSs)().
Figure 6 3D Structure of the CYP26A1 model. α-Helices are in red, β-sheets are in yellow, loops are cyan and coils are grey.
Conclusions
The CYP26A1 model has been validated for stereochemical and amino acid environment quality using appropriate programmes, with further validation of active site architecture achieved by docking studies with the natural substrate and an inhibitor to determine the key residues involved in the ligand binding. A drug design program of CYP26A1 inhibitors using this CYP26A1 model is now ongoing based on the results obtained from the work presented here.
Acknowledgements
For MSG we would like to acknowledge the Embassy of the Arab Republic of Egypt for the award of a PhD scholarship, and for SWY we would like to acknowledge the ORS Awards Scheme for a United Kingdom Scholarship.
References
- The Retinoids: Biology Chemistry and Medicine. MB Sporn, AB Roberts, DS Goodman. Academic Press, New York 1984; 424
- Sonneveld E, Van den Brink CE, Van der Leede BJ, Schulkes RK, Petkovich M, Van der Burg B, Van der Saag PT. Human retinoic acid (RA) 4-hydroxylase (CYP26) is highly specific for all-trans-RA and can be induced through RA receptors in human breast and carcinoma cells. Cell Growth Differ 1998; 9: 629–637
- Johnson A, Chandraratna RAS. Novel retinoids with receptor selectivity and functional selectivity. Br J Dermatol (Suppl) 1999; 140: 12–17
- Torma H, Rollman O, Vahlquist A. The vitamin A metabolism and expression of retinoid-binding proteins differ in HaCaT cells and normal human keratinocytes. Arch Dermatol Res 1999; 291: 339–345
- Smith HJ, Nicholls PJ, Simons C, LeLain R. Inhibitors of steroidogenesis as agents for the treatment of hormone-dependent cancers. Exp Opin Ther Patents 2001; 11: 789–824
- Marill J, Cresteil T, Lanotte M, Chabot GG. Identification of human cytochrome P450s involved in the formation of all-trans-retinoic acid principal metabolites. Mol Pharmacol 2000; 58: 1341–1348
- Ray WJ, Bain G, Yao M, Gottlieb DI. CYP26 a novel mammalian cytochrome P450 is induced by retinoic acid and defines a new family. J Biol Chem 1997; 272: 18702–18708
- Stoppie P, Borgers M, Borghgraef P, Dillen L, Goossens J, Sanz G, Szel H, Van Hove C, Van Nyen G, Nobels G, Vanden Bossche H, Venet M, Willemsens G, Van Wauwe J. R115866 inhibits all-trans-retinoic acid metabolism and exerts retinoidal effects in rodent. J Pharmacol Exp Ther 2000; 293: 304–312
- MacLean G, Abu-Abed S, Dolle P, Tahayato A, Chambon P, Petkovich M. Cloning of a novel retinoic-acid metabolizing cytochrome P450 Cyp26B1 and comparative expression analysis with Cyp26A1 during early murine development. Mech Develop 2001; 107: 195–201
- Taimi M, Helvig C, Wisniewski J, Ramshaw H, White J, Amad M, Korczak B, Petkovich M. A novel human cytochrome P450 CYP26C1 involved in metabolism of 9-cis and all-trans isomers of retinoic acid. J Biol Chem 2004; 279: 77–81
- Ahmad N, Mukhtar H. Cytochrome P450: A target for drug development for skin diseases. J Investig Dermatol 2004; 123: 417–425
- Brecher AR, Orlow SJ. Oral retinoid therapy for dermatologic conditions in children and adolescents. J Am Acad Dermatol 2003; 49: 171–182
- Weiss GR, Liu PY, Alberts DS, Peng YM, Fisher E, Xu MJ, Scudder SA, Baker LH, Moore DF, Lippman SM. 13-cis-Retinoic acid or all-trans-retinoic acid plus inteferon-alpha in recurrent cervical cancer: A Southwest Oncology Group phase II randomized trial. Gynecol Oncol 1998; 71: 386–390
- Pettersson F, Colston KW, Dalgleish AG. Retinoic acid enhances the cytotoxic effects of gemcitabine and cisplatin in pancreatic adenocarcinoma cells. Pancreas 2001; 23: 273–279
- Schoch GA, Yano JK, Wester MR, Griffin KJ, Stout CD, Johnson EF. Structure of human microsomal cytochrome P450 2C8 Evidence for a peripheral fatty acid binding site. J Biol Chem 2004; 279: 9497–9503
- Williams PA, Cosme J, Ward A, Angove HC, Matak-Vinkovic D, Jhoti H. Crystal structure of human cytochrome P450 2C9 with bound warfarin. Nature 2003; 424: 464–468
- Yano JK, Wester MR, Schoch GA, Griffin KJ, Stout D, Johnson EF. The structure of human microsomal cytochrome P450 3A4 determined by X-ray crystallography to 2.05-Å resolution. J Biol Chem 2004; 279: 38091–38094
- Molecular Operating Environment. (MOE) Chemical Computing Group Inc Montreal Quebec Canada http://www.chemcomp.com 2004.03.
- Tripos SYBYL 7.0, Tripos Inc. South Hanley Rd St Louis Missouri 63144 USA http://www.tripos.com 1699.
- Berendsen HJC, van der Spoel D, van Drunen R. GROMACS: A message-passing parallel molecular dynamics implementation. Comp Phys Comm 1995; 91: 43–56
- Lindahl E, Hess B, van der Spoel D. GROMACS 3.0: A package for molecular simulation and trajectory analysis. J Mol Mod 2001; 7: 306–317
- Humphrey W, Dalke A, Schulten K. VMD - Visual Molecular Dynamics. J Mol Graph 1996; 14: 33–38
- The ExPASy (Expert Protein Analysis System)., proteomics server of the Swiss Institute of Bioinformatics (SIB) http://ca.expasy.org
- R CSB Protein Data Bank (PDB)., http://www.rcsb.org/pdb
- Needleman SB, Wunsch CD. A general method applicable to search for similarities in amino acid sequence of 2 proteins. J Mol Biol 1970; 48: 443–453
- RAMPAGE Server., http://ravenbioccam.ac.uk/rampage.php
- Bowie JU, Luthy R, Eisenberg D. A method to identify protein sequences that fold into a known three-dimensional structure. Science 1991; 253: 164–170
- Colovos C, Yeates TO. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Science 1993; 2: 1511–1519
- Code “scoring svl” obtained from SVL Exchange website., http://svl.chemcomp.com Chemical Computing Group Inc Montreal Canada.
- White JA, Beckett-Jones B, Guo Y-D, Dilworth FJ, Bonasoro J, Jones G, Petkovich M. cDNA cloning of human retinoic acid-metabolizing enzyme (hP450RAI) identifies a novel family of cytochromes P450. J Biol Chem 1997; 272: 18538–18541
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res 1997; 25: 3389–3402
- Higgins D, Thompson J, Gibson T, Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting position-specific gap penalties and weight matrix choice. Nucleic Acids Res 1994; 22: 4673–4680
- ClustalW WWW Service at the European Bioinformatics Institute., http://www.ebi.ac.uk/clustalw
- Lovell SS, Davis IW, Arendall, III WB, e Bakker PIW, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Cα geometry: Φ, Ψ and Cβ deviation. Proteins: Structure Function & Genetics 2002; 50: 437–450
- UCLA-DOE Institute for Genomics & Proteomics Server., http://www.doe -mbiucla.edu/Services.
- Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modelling. Electrophoresis 1997; 18: 2714–2723, (http://wwwexpasyorg/spdbv/)
- Hasemann CA, Kurumbail SS, Boddupalli SS, Peterson JA, Deisenhofer J. Structure and function of cytochrome P450: A comparative analysis of three crystal structures. Structure 1995; 41–62
- Podust LM, Poulos TL, Waterman MR. Crystal structure of cytochrome P450 14(-sterol demethylase (CYP51) from Mycobacterium tuberculosis in complex with azole inhibitors. PNAS 2001; 98: 3068–3073