
Abstract
Phosphoinositide 3-kinase Delta (PI3Kδ) plays a key role in B-cell signal transduction and inhibition of PI3Kδ was confirmed to have clinical benefit in certain types of activation of B-cell malignancies. Herein, we reported a novel series of 4-pyrrolidineoxy or 4-piperidineamino substituted quinazolines, showing potent PI3Kδ inhibitory activities. Among these compounds, 12d, 14b and 14c demonstrated higher potency against PI3Kδ with the half maximal inhibitory concentration (IC50) values of 4.5, 3.0, and 3.9 nM, respectively, which were comparable to idelalisib (IC50 = 2.7 nM). The further PI3K isoforms selectivity evaluation showed that compounds 12d, 14b and 14c have excellent PI3Kδ selectivity over PI3Kα, PI3Kβ, and PI3Kγ. Moreover, compounds 12d, 14b and 14c also displayed different anti-proliferative profiles against a panel of four human B cell lines including Ramos, Raji, RPMI-8226, and SU-DHL-6. The molecular docking simulation indicated several key hydrogen bonding interactions were formed. This study suggests the introduction of pyrrolidineoxy or piperidineamino groups into the 4-position of quinazoline leads to new potent and selective PI3Kδ inhibitors.
Graphical Abstract
1. Introduction
Phosphoinositide 3-kinases (PI3Ks) play a pivotal role in multiple cellular functions including cell growth, development, migration, angiogenesis, and survivalCitation1. Upon stimulation of cytokine signaling, PI3Ks function as an intracellular secondary messenger transforming phosphatidylinositol 4,5-bisphosphate (PIP2) into phosphatidylinositol 3,4,5-trisphosphate (PIP3) via phosphorylation catalysation, thereafter activation of the downstream signal transducer (Akt, mTOR) and subsequent activator of transcriptionCitation2. There are four PI3K isoforms validated, including PI3Kα, PI3Kβ, PI3Kγ, and PI3Kδ. PI3Kα and PI3Kβ are ubiquitously expressed in multiple cells whereas PI3Kδ and PI3Kγ are identified as predominant expression in hematopoietic cellsCitation3. In particular, PI3Kδ is found responsible for the B-cell receptor (BCR) signaling downstream transduction and constitutive studies show activation of BCR signaling pathway is a hallmark of B-cell malignancies such as chronic lymphocytic leukemia (CLL), follicular lymphoma (FL), mantle cell lymphoma (MCL), small lymphocytic lymphoma (SLL), diffuse large B-cell lymphoma (DLBCL), and indolent non-Hodgkin’s lymphoma (iNHL)Citation4. Therefore, PI3Kδ is thought as suitable target for the potential treatment of certain B-cell malignancies, as well as immunologic disorders (due to its specific role in controlling immune cell function)Citation5. Notably, small molecules selective PI3Kδ inhibitor idelalisib (Compound 1) has been recently approved by Food and Drug Administration (FDA) for treatment of CLL, FL, and SLL, which solidify the therapeutic concept of PI3Kδ inhibitor ()Citation6,Citation7.
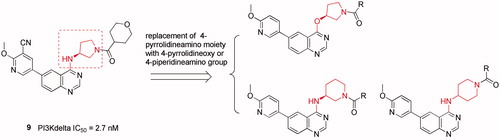
Despite the first-in-class approved, potent and oral selective PI3Kδ inhibitor, idelalisib was tagged with black-box warning and demonstrated struggling with severe adverse events in the later clinical validationCitation8. Therefore, there is an urgent need to develop second generation PI3Kδ inhibitor with lower toxicity and fewer side effects. Duvelisib (Compound 2), another potent PI3Kδ inhibitor, shared chemical similarity to idelalisib, however, this was recently terminated in the phase III clinical trials due to underneath efficiency. Many other analogues derived from the chemical structure of idelalisib were recently reported and showed strong PI3Kδ efficacy and selectivity, for instances Compounds 3 (PI3Kδ: half maximal inhibitory concentration (IC50) = 2.2 nM), 4 (PI3Kδ: IC50 = 1.0 nM), and 5 (PI3Kδ: IC50 = 4.6 nM)Citation9–11. Nevertheless, our drug discovery efforts are engaged into the development of PI3Kδ inhibitors with novel and distinct chemotypes. Recently, we reported a new series of potent PI3Kδ inhibitors, chemically featured by a quinazoline scaffold and a 6-benzamide moiety such as Compound 7 (PI3Kδ: IC50 = 17 nM)Citation12, derived from the Novartis’s patented Compound 6 (PI3Kδ: IC50 = 9 nM) with potent PI3Kδ inhibition and selectivityCitation13–15. A subsequent structural modification was carried out and a series of 4-anilinequinazolines was synthesised, exemplified by Compound 8 (PI3Kδ: IC50 = 9.3 nM) showing improved PI3Kδ inhibitionCitation16. Later, further structural investigation by replacing the 4-aniline with a 4-pyrrolidineamino moiety led to a series of potent and selective PI3Kδ inhibitors, such as Compound 9 (PI3Kδ: IC50 = 2.7 nM), showing equivalence to idelalisib in our examination ()Citation17. Encouraged by these fantastic findings, we decided to develop a new series of quinazoline based PI3Kδ inhibitors by introducing functionalised pyrrolidineoxy or piperidineamino group at the 4-position of quinazoline instead of the pyrrolidineamino moiety. Herein, we disclose the synthesis, biological evaluation of this series of 4-pyrrolidineoxy and 4-piperidineamino substituted quinazolines as potent and selective PI3Kδ inhibitors ().
2. Results and discussion
2.1. Chemistry
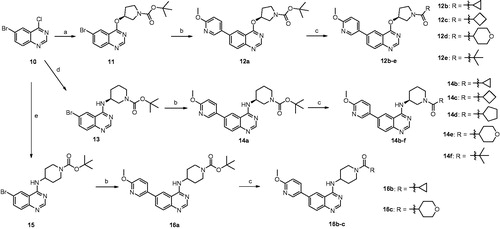
The 4-pyrrolidineoxy and 4-piperidineamino substituted quinazoline derivatives were synthesised according to the synthetic routes outlined in Scheme 1. Treatment of 6-bromo-4-chloroquinazoline (Compound 10) with (S)-1-Boc-3-hydroxypyrrolidine in the presence of sodium hydride (NaH) gave (S)-4-pyrrolidineoxyqui-nazoline (Compound 11) in 70% yield, which was subsequently reacted with 6-methoxy-3-pyridinylboronic acid using Suzuki coupling condition to generate Compound 12aCitation18,Citation19. Compound 12a reacted with TFA at room temperature to get rid of the tert-butyloxycarbonyl protecting group (Boc group) and then was acylated with diverse acids to afford Compounds 12(b–e). Compounds 14(a–f) and 16(a–c) were prepared by employing the similar synthetic proceduresCitation20. Compound 10 was treated with (S)-1-Boc-3-aminopiperidine or 1-Boc-4-aminopiperidine to give intermediate Compounds 13 and 15, respectively, which in turn underwent Suzuki coupling reaction, deprotection, and condensation to produce Compounds 14(a–f) and 16(a–c) successfully (Scheme 1).
Scheme 1. Reagents and conditions: (a) (S)-1-Boc-3-hydroxypyrrolidine, anhydrous THF, NaH, rt, overnight, 70%; (b) 6-methoxy-3-pyridinylboronic acid, Na2CO3, PdCl2 (dppf), DME/H2O, reflux, 4 h, 65–81%; (c) (i)TFA, CH2Cl2, rt, 2 h, 23–91%; (ii) diverse acids, DMF, HATU, DIPEA, rt, 12 h, 23–91%; (d) (S)-1-Boc-3-aminopiperidine, DMF, DIPEA, 90 °C, 6 h, 90%; (e) 1-Boc-4-aminopiperidine, DMF, DIPEA, 90 °C, 6 h, 52%.
2.2. PI3Kδ inhibitory activity for the title compounds
All the newly synthesised compounds were assessed for their PI3Kδ inhibitory activities and idelalisib was employed as the positive control. The 4-pyrrolidineoxy substituted quinazoline analogs were firstly examined and the results are shown in . It was found all the 4-pyrrolidineoxy substituted quinazoline analogues displayed significant PI3Kδ inhibitory activities under the concentration of 100 nM. The initial Compound 12a bearing a (S)-4-(1-Boc-pyrrolidin-3-yl)oxy side chain showed an inhibitory ratio of 79% at the concentration of 100 nM, while replacement of the tert-butoxy group with cyclopropyl group (Compound 12b: 90%) afforded enhanced PI3Kδ inhibitory activity, showing an IC50 value of 9.3 nM. Switch of the cyclopropyl group (Compound 12b) to cyclobutyl (Compound 12c: IC50 = 6.1 nM) and tetrahydro-2H-pyran-4-yl (Compound 12d: IC50 = 4.9 nM) groups led to higher PI3Kδ inhibitory activity, whereas replacement with the branched tert-butyl (Compound 12e: 53%) resulted in weak PI3Kδ potency. In the 4-pyrrolidineoxy subseries, Compound 12d bearing tetrahydro-2H-pyran-4-yl side chain afforded the most potent PI3Kδ inhibitory activity, which was approximately equivalent to control drug idelalisib (IC50 = 2.7 nM; ).
Table 1. PI3Kδ inhibitory activity of 4-pyrrolidineoxy substituted quinazolinesTable Footnotea.
Subsequently, the 4-piperidineamino substituted quinazoline analogues were evaluated and the data are shown in . The initial (S)-4-(1-Boc-piperidin-3-yl)amino Compound 14a showed weak PI3Kδ inhibitory activity with an inhibitory ratio of 51% at the concentration of 100 nM. However, replacement of tert-butoxy group with diverse cyclic aliphatic substituents afforded highly improved PI3Kδ inhibitory activity. Analogue of Compound 14b bearing a cyclopropyl group gave an IC50 value of 3 nM, and analogue of Compound 14c tailed with a cyclobutyl group showed almost comparable potency, with an IC50 value of 3.9 nM, whereas analogues of Compound 14d with a cyclopentyl terminal and Compound 14e containing a tetrahydro-2H-pyran-4-yl tail showed a little less potent PI3Kδ inhibition than that of Compound 14b, with IC50 values of 8.7 and 5.2 nM, respectively. Again, an attempt to shift the cyclic group to non-cyclic alkyl group such as tert-butyl (Compound 14f: 54%) resulted in PI3Kδ inhibition largely reduced. Otherwise, an exploration of changing the (S)-4-(piperidin-3-yl)amino side chain into 4-(piperidin-4-yl)amino group was also conducted, and three analogues were synthesised. However, unfortunately, Compound 16a bearing a Boc group (Compound 16a: 72%) and Compound 16b with a cyclopropyl group (Compound 16 b: 70%) showed moderate PI3Kδ inhibition, while Compound 16c incorporated with tetrahydro-2H-pyran-4-yl group(Compound 16c: 48%) produced unsatisfactorily weak potency. This suggested the spatial orientation of the tailed acyl substituents was critical for PI3Kδ inhibition, which was consistent to the structure-activity relationship of our previously reported 6-aryl substituted 4-anilinequinazoline series. In this preliminary PI3Kδ inhibition evaluation, three compounds 12d, 14b, and 14c showed IC50 values beneath 5 nM, being approximately comparable to idelalisib, which were picked out for further evaluation.
Table 2. PI3Kδ inhibitory activity of 4-piperidineamino substituted quinazolinesTable Footnotea.
2.3. Isoform selectivity of the new PI3Kδ inhibitors
Based on the above preliminary PI3Kδ inhibitory activity results, Compounds 12d, 14b, and 14c were subsequently evaluated for their selectivity among PI3Kα, PI3Kβ, and PI3Kγ. As shown in , all three compounds 12d, 14b, and 14c showed much lower potency against other three PI3K isoforms than that of PI3Kδ, although they displayed moderate PI3Kα inhibition. Compound 12d with an IC50 value of 4.5 nM against PI3Kδ demonstrated 11-fold, 131-fold, and 103-fold selectivity over PI3Kα, PI3Kβ and PI3Kγ, respectively, whereas Compounds 14 b and 14c displayed the similar PI3Kδ selectivity which were 12- and 15-fold over PI3Kα, 105- and 96-fold over PI3Kβ, and 34- and 31-fold over PI3Kγ, respectively. Moreover, it was noted that selectivity of compound 12d against the PI3Kβ and PI3Kγ isoforms was much higher than idelalisib, although the poor selectivity against PI3Kα was observed ().
Table 3. Isoform selectivity of compounds against PI3K (p110α, p110β, p110γ, and p110δ)
2.4. In vitro anti-proliferative assays of the new PI3Kδ inhibitors
Furthermore, Compounds 12d, 14b, and 14c were tested for their anti-proliferative activities against four human B cell lines including Ramos, Raji, RPMI-8226, and SU-DHL-6with idelalisib and SAHA as reference compounds. As shown in , Compound 12d exhibited most potent anti-proliferation against RPMI-8226 (IC50 = 44 nM) among these four cell lines, whereas Compound 14b showed significantly potent anti-proliferative activity against Ramos, Raji, and SU-DHL-6, but moderate anti-proliferation against RPMI-8226 and Compound 14c also showed strong anti-proliferativity against SU-DHL-6 with an IC50 value of 1.49 nM. It was found that the reference PI3Kδ inhibitor idelalisib displayed markedly anti-proliferative activity against SU-DHL-6, whereas another reference drug SAHA (vorinostat) afforded significantly anti-proliferation against Ramos, Raji, and RPMI-8226. In a word, three Compounds 12d, 14b, and 14c as well as idelalisib were observed showing different anti-proliferative profiles in the four human B cell lines ().
Table 4. Anti-proliferative activities of new compounds in vitro
2.5. Molecular modeling study
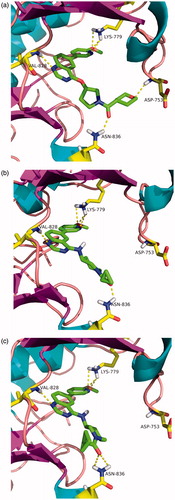
To further understand the potent PI3Kδ inhibition, molecular docking simulations of Compounds 12d, 14b, and 14c within human PI3Kδ enzyme were performed. As shown in , the docked pose of each Compound (12d, 14b and 14c) ma es the similarly favorable interactions with the PI3Kδ binding pocket of structure 2WXP as expected, namely, three key hydrogen bonds with the hinge residue, the quinazoline scaffold with Val828, the methoxypyridyl moiety with Lys779, as well as the carbonyl group with Asn836. Moreove r, it was observed that, although, the oxygen of the tetrahydro-2H-pyran-4-yl group in Compound 20a formed an additional hydrogen bond with Asp753, it seemed to show little contribution for improving the inhibitory activity in this case ().
Figure 1. Representative structures for previously reported potent PI3Kδ inhibitors.
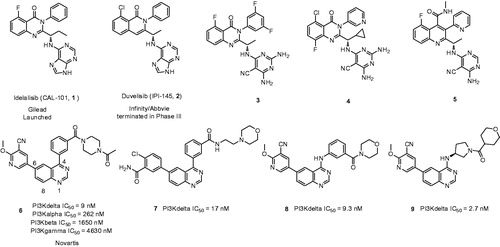
Figure 2. Design of the 4-pyrrolidineoxy and 4-piperidineamino substituted quinazolines as PI3Kδ inhibitors.
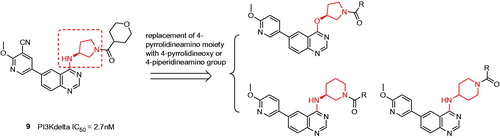
Figure 3. Molecular docking studies of Compounds 12d (a), 14b (b) as well as 14c (c) into the site of PI3Kδ (PDB code: 2WXP). Compound is shown as sticks. Hydrogen bonds within 2.5 Å are shown as yellow dashed lines.
3. Conclusion
In summary, we have synthesised and evaluated a novel series of quinazoline derivatives by introducing a functionalised 4-pyrrolidineoxy or 4-piperidineamino groups as potent PI3Kδ inhibitors. The structure-activity relationship (SAR) was discussed and many derivatives showed nanomolar PI3Kδ inhibitory activities, particularly, Compounds 12d, 14b, and 14c demonstrating preferably potent PI3Kδ inhibitory activities with IC50 values of 4.5, 3, and 3.9 nM, respectively, approximately comparable to idelalisib (IC50 = 2.7 nM). Moreover, Compounds 12d, 14b, and 14c showed excellent PI3Kδ isoform selectivity over PI3Kα, PI3Kβ, and PI3Kγ. These three compounds also displayed different anti-proliferative profiles against a panel of four human B cell lines. The molecular docking study indicated several key hydrogen bonding interactions formations, which may explain their higher PI3Kδ. This study suggests the introduction of pyrrolidineoxy or piperidineamino groups into the 4-position of quinazoline leads to new potent and selective PI3Kδ inhibitors
Disclosure statement
The authors declare no conflict of interest.
Additional information
Funding
Related Research Data
References
- Liu P, Cheng H, Roberts TM, Zhao J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov 2009;8:627–44.
- Ciraolo E, Morello F, Hirsch E. Present and future of PI3K pathway inhibition in cancer: perspectives and limitations. Curr Med Chem 2011;18:2674–85.
- Wei M, Wang X, Song Z, et al. Targeting PI3Kδ: emerging therapy for chronic lymphocytic leukemia and beyond. Med Res Rev 2015;35:720–52.
- Davids MS, Brown JR. Targeting the B cell receptor pathway in chronic lymphocytic leukemia. Leuk Lymphoma 2012; 53:2362–70.
- Fruman DA, Rommel C. PI3Kδ inhibitors in cancer: rationale and serendipity merge in the clinic. Cancer Discov 2011; 1:562–72.
- Fruman DA, Cantley LC. Idelalisib-a PI3Kδ inhibitor for B-cell cancers. N Engl J Med 2014;370:1061–2.
- Norman P. Selective PI3Kδ inhibitors, a review of the patent literature. Expert Opin Ther Pat 2011;21:1773–90.
- Markham A. Idelalisib: first global approval. Drugs 2014; 74:1701–7.
- Patel L, Chandrasekhar J, Evarts J, et al. 2,4,6-triaminopyrimidine as a novel hinge binder in a series of PI3Kδ selective inhibitors. J Med Chem 2016;59:3532–48.
- Patel L, Chandrasekhar J, Evarts J, et al. Discovery of orally efficacious phosphoinositide 3-kinase δ inhibitors with improved metabolic stability. J Med Chem 2016;59:9228–42.
- de Turiso FG, Hao X, Shin Y, et al. Discovery and in vivo evaluation of the potent and selective PI3Kδ inhibitors 2-((1S)-1-((6-Amino-5-cyano-4-pyrimidinyl)amino)ethyl)-6-fluoro- N-methyl-3-(2-pyridinyl)-4-quinolinecarboxamide (AM-0687) and 2-((1S)-1-((6-Amino-5-cyano-4-pyrimidinyl)amino)ethyl)-5-fluoro-N-methyl-3-(2-pyridinyl)-4-quinolinecarboxamide (AM-1430)). J Med Chem 2016;59:7252–67.
- Xin M, Hei YY, Zhang H, et al. Discovery of 6-benzamide containing 4-phenylquinazoline derivatives as novel PI3Kδ inhibitors. Lett Drug Des Dis 2017;14:167–74.
- Furet P, Hebach C, Hogenauer et al. Quinazoline derivatives as PI3K modulators. Patent WO2013027711; 2013.
- Hoegenauer K, Soldermann N, Stauffer F, et al. Discovery and pharmacological characterization of novel quinazoline-based PI3K delta-selective inhibitors. ACS Med Chem Lett 2016;7:762–7.
- Hoegenauer K, Soldermann N, Hebach C, et al. Discovery of novel pyrrolidineoxy-substituted heteroaromatics as potent and selective PI3K delta inhibitors with improved physicochemical properties. Bioorg Med Chem Lett 2016; 26:5657–62.
- Xin M, Hei YY, Zhang H, et al. Design and synthesis of novel 6-aryl substituted 4-anilinequinazoline derivatives as potential PI3Kδ inhibitors. Bioorg Med Chem Lett. 2017;27:1972–7.
- Xin M, Duan W, Feng Y, et al. Bioorg Med Chem 2018; Available from: https://doi.org/10.1016/j.bmc.2018.03.002.
- Hei YY, Xin M, Zhang H, et al. Synthesis and antitumor activity evaluation of 4,6-disubstituted quinazoline derivatives as novel PI3K inhibitors. Bioorg Med Chem Lett 2016; 26:4408–13.
- Zhang H, Xin M, Xie XX, et al. Synthesis and antitumor activity evaluation of PI3K inhibitors containing 3-substituted quinazolin-4(3H)-one moiety. Bioorg Med Chem 2015;23:7765–76.
- Xin M, Wen J, Tang F, et al. The discovery of novel N-(2-pyrimidinylamino) benzamide derivatives as potent hedgehog signaling pathway inhibitors. Bioorg Med Chem Lett 2013; 23:6777–83.