
Abstract
With the aim of discovering novel IDO1 inhibitors, a combined similarity search and molecular docking approach was employed to the discovery of 32 hit compounds. Testing the screened hit compounds has led to several novel submicromolar inhibitors. Especially for compounds LVS-019 with cyanopyridine scaffold, showed good IDO1 inhibitory activity. To discover more compounds with similar structures to LVS-019, a shape-based model was then generated on the basis of it and the second-round virtual screening was carried out leading to 23 derivatives. Molecular docking studies suggested a possible binding mode of LVS-019, which provides a good starting point for the development of cyanopyridine scaffold compounds as potent IDO1 inhibitor. To improve potency of these hits, we further designed and synthesised another 14 derivatives of LVS-019. Among these compounds, LBJ-10 showed improved potency compared to the hits and displayed comparable potency to the control GDC-0919 analogue. LBJ-10 can serve as ideal leads for further modifications as IDO1 inhibitors for cancer treatment.
Introduction
Indoleamine 2,3-dioxygenase 1 (IDO1) is a haeme-containing enzyme that catalyses the first and rate limiting step of L-tryptophan conversion to N-kynurenine along the kynurenine pathwayCitation1,Citation2. IDO1 is a monomeric enzyme, which degrades different indoleamine substrates, including L-Trp, D-Trp, and tryptamine, and is ubiquitously expressed in many tissuesCitation3. The expression of IDO1 could be induced by interferon-γ, lipopolysaccharide, and tumour necrosis factorCitation4–6. Thus, within the immune system IDO1 gets highly up-regulated in response to inflammatory signals under pathophysiological conditions. Over-expression of IDO1 corrects with poor prognosis in different types of cancers, including melanoma, pancreatic, ovarian, colorectal, and othersCitation7. It is believed that IDO1 could subvert the immunomodulatory mechanism and promote tolerance local to the cancerCitation7,Citation8. Recently, multiple studies, including si-RNA, IDO1 null mice, and small-molecule inhibitors, have appeared validating IDO1 as a therapeutic target for cancer immunotherapyCitation9–14. In addition, increased IDO1 expression correlates with different tumour progression parameters and shorter patient survival has also been discoveredCitation15. IDO1 inhibitors have a synergistic effect when used in combination with anti-PD1 mAbsCitation16 and some other anticancer drugs, such as cyclophosphamide, doxorubicin, paclitaxel, and cisplatinCitation17. All this evidence confirms that IDO1 inhibition might enhance the efficacy of pharmacological cancer therapy and is a potential breakthrough approach to cancer therapy. As such, the enzyme IDO1 has emerged as a promising therapeutic target, prompting a number of potent IDO1 inhibitors have been identified recently. For the past few years, we have witnessed many efforts been focussed on discovering potential IDO1 inhibitors and hundreds of compounds have been reported according to the scientific and patent literature. However, to the best of our knowledge, only five small-molecule compounds are currently undergoing an extensive development programme (either alone or in combination) and no drug has been approved by FDA up to nowCitation18. Among these IDO1 inhibitors in clinical, 1-methyl-D-tryptophan (D-1-MT, indoximod, 1, ) is developed by NewLink Genetics, and currently is undergoing evaluation for multiple human clinical trialsCitation12,Citation19–22. NewLink Genetics developed another IDO1 inhibitor NLG-919 (also named GDC-0919, 2) which currently is in phase I clinical trials in subjects with recurrent advanced solid tumoursCitation23,Citation24. Incyte Corporation developed a clinical candidate hydroxyamidine INCB024360 (named Epacadostat, 3), which was in phase II/III clinical trials, used as a monotherapy as well as in combination use with various antibodies, for the treatment of advanced or metastatic cancersCitation25. Recently, five latest IDO1 inhibitors have also entered clinical trials within the last few months. Pfizer Inc./iTeos Therapeutics SA reported PF-06840003 (4), which is under phase I clinical trials for the treatment of patients with grade IV glioblastoma or grade III anaplastic gliomaCitation26,Citation27. Bristol-Myers Squibb Corporation reported the other latest IDO1 inhibitor BMS-986205 (ONO-7701, 5), which is in phase I/II clinical trials in combination with nivolumab in patients with advanced cancersCitation28. JiangSu Hengrui Medicine Corporation developed a small-molecule IDO1/TDO dual inhibitor SHR-9146 (NCT03208959, HTI-1090) with favourable preclinical oral bioavailability and safety profiles, which is under phase I clinical trials in subjects with advanced solid tumours. A novel prodrug of indoximod (named NLG-802 or NCT03164603) with enhanced pharmacokinetic properties was also entered into Phase I study recently by NewLink Genetics. The structures of SHR-9146 and NLG-802 have not been disclosed until now.
Figure 1. The structures of clinical IDO1 inhibitors (1–5) and hit compound (6) of INCB024360.
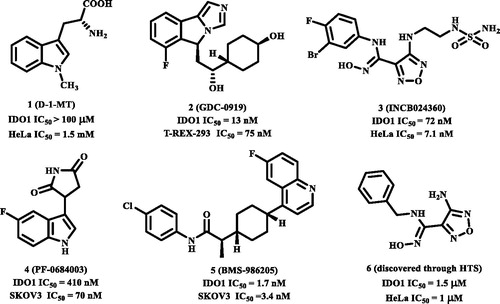
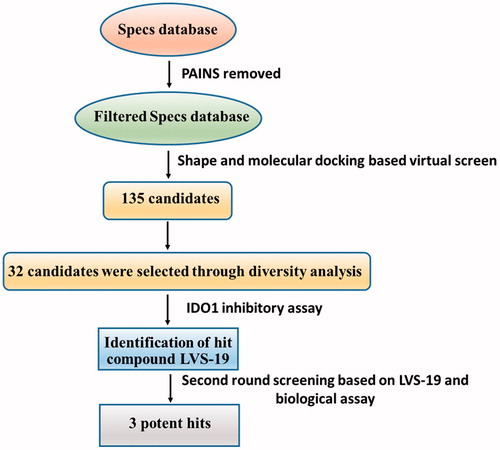
With only these IDO1 inhibitors under clinical trials and no drug has been approved, discovering novel IDO1 inhibitors suitable for development into clinical candidates is still in great demand. Further exploring these IDO1 inhibitors, we found that many of them with new structural scaffolds were discovered by structure-based design, high-throughput screening, and natural product derivatisationCitation29. Notably, the lead compound 4-amino-1,2,5-oxadiazole-3-carboximidamide (6) of currently most promising IDO1 inhibitor INCB024360 was discovered through high-throughput screening (HTS) of Incyte’s corporate collectionCitation13. It indicated that HTS is an efficient method to discover novel IDO1 inhibitors. As a complementary approach to experimental HTS, virtual screening (VS) recently has drawn lots of attention for saving time and expensesCitation30. Similarity search and molecular docking are two most commonly used methods in the discovery of small-molecule inhibitors. However, independent VS methods are facing the insufficient accuracy of scoring function which remains the bottleneck of VSCitation31,Citation32. Thus, in this study, using the combined similarity search and molecular docking approaches, we developed a VS strategy to discover new inhibitors targeting IDO1. The selected molecules were further evaluated by biochemical analysis, and the predicted binding modes of potent inhibitors were analysed. Among the potent novel molecules, a cyanopyridine scaffold named LVS-19 attracted us the most. We then selected LVS-19 to generate the rapid overlay of chemical structures (ROCS) model. With strictly chemical similarity analysis, a second-round shape-based screening was performed. Twenty-three derivatives LVS-33∼LVS-56 were finally retained and purchased from Specs database. They were also evaluated for their IDO1 inhibitory activity. A schematic of the overall VS protocol is presented in . In addition, to improve potency of these hit compounds, another 14 derivatives (LBJ-01∼LBJ-14) of LVS-019 were designed and synthesised through rational drug designation.
Figure 2. Shows the computational protocol that was applied to IDO1 inhibitors. Full details of each step are discussed in the following paragraphs.
Materials and methods
Virtual screening
Computational calculation
The Specs database file (version 01/2012) was downloaded from the official Web site (www.specs.net) and these compounds were prepared with the module of Search Conformations in MOE2016.09Citation33 to provide an average of 140 conformations. These structures were washed, i.e. all inorganic compounds were removed and all ionisable groups were set to coordinated with neutral pH conditions. Energetically minimised conformations were generated using the MMFF94x force fieldCitation34.
Creation of the ROCS model and the virtual screens based on it
The shape-based pharmacophore model in VS was generated by employing ROCSCitation35. The algorithm is based on the idea that the molecular shape of compounds is similar if the molecules overlay well and any volume mismatch results from shape dissimilarity. VSs were then performed based on the query models of INCB024360, GDC-0919, Amg-1, and LVS-019. The parameters for the ROCS run were set as follows: rankby = combo and besthits =1. The screening score for a particular database compounds was set to the maximum combo score between the database compound and model compounds.
Molecular docking
Native-docking
The crystallographic coordinates of human IDO1-inhibitors (PDB code 2D0T, 4PK5, 5EK2, 5EK3, 5EK4, and 5ETW) were used to conduct native-dockingCitation36–38 (When we have finished our works, some other crystallographic coordinates are available. We added the newly reported PDB information of these structures in the Supporting Information.) These ligands were docked back into their corresponding protein structures using Gold5.1 (CCDCs software GOLD version 5.1)Citation39,Citation40, CDOCKER (DS 4.0)Citation41, alpha PMI (Principal Moments of Inertia, MOE 2016.09)Citation42, AutoDockCitation43, and Glide (Schrödinger 2009)Citation44. To define the binding pocket of IDO1 cocrystal structure, residues around the native ligand (radius set as 8.0 Å) were selected for molecular docking. All other parameters are set as default. The docking results were evaluated through comparison of the best docked poses and the real cocrystallised pose to measure docking reliability. The best docking pose was defined as that top-scoring poses. The docking software was evaluated with root mean square deviation (RMSD) value.
Cross-docking
The complexes used in native-docking were also selected to perform cross-docking evaluation. After superposing the crystallographic complexes, the native ligands were extracted, merged in a unique database and docked into each protein structure with five docking combination protocols used in native-docking. For each of the five docking protocols and six PDB structures, a database of five ligands was docked to each of the six protein structures (the self-ligand was excluded for the cross-docking computation to follow a strict cross-docking procedure). Ten poses were generated for each of the ligand–protein couples and the RMSD values between the top-scoring pose and the crystallographic conformation of the ligand were computed. The evaluation of each protein-docking protocol couple was performed considering the RMSD of the top-scoring poses of each of the five native ligands docked on the same protein with the same protocol.
Screen with docking
GOLD was a powerful genetic algorithm (GA) method for conformation search and docking. In the present study, the binding site was defined by carefully analysing the protein–ligand interactions between IDO1 and its inhibitor. Residues around the original ligand (radius 8.0 Å) were also included in the active site. All water molecules have been removed from the protein structure. Docking studies were performed using the standard default settings with 10 GA runs on each molecule. For each of the GA runs, a maximum of 125,000 operations were performed. The binding energy of the ligands with protein was predicted using the Goldscore by choosing in the appropriate scoring function goldscore.p450_pdb.params file from those that have been supplied with the GOLD installationCitation45. All other parameters were kept at their default values.
Biological experiments
Enzyme-based IDO1 activity assay
The recombinant human IDO1 was expressed and purified as previously reportedCitation38. The IDO1 activity assay was also carried out as previously describedCitation38. In brief, the IDO1 activity of test compounds was measured in the assay buffer (100 µL) containing 50 mM potassium phosphate buffer (pH = 6.5), 25 mM neutralised ascorbic acid (10 µL), 10 µM methylene blue (10 µL), 100 µg/mL catalase (10 µL), 200 µM L-tryptophan (10 µL), purified IDO1 enzyme (10 µL, 25 µg/mL). The test compounds (10 µL, in DMSO, 0.5% final concentration) were added in 96-well plates. The reaction was carried out at 37 °C for 1 h. To convert N-formylkynurenine to kynurenine, 10 µL of NaOH (1M) was added and incubated at 60 °C for 30 min. Then the mixture was cooled at 0 °C and centrifuged at 10,000 rpm for 10 min. Lastly, 100 µL supernatant form each well was transferred to a new 96-well plate and mixed at equal volume with 2% (w/v) p-dimethylaminobenzaldehyde (p-DMAB, Ehrlich's reagent) in acetic acid. The yellow colour generated from the reaction with kynurenine was measured at 480 nm. The percentage of inhibition is reported as (100 − (A/B × 100))/100, where A is the IDO1 activity in the presence of the test compound, and B is the IDO1 activity in the absence of the test compound.
Cell-based IDO1 activity assay
HeLa cells were seeded in 96-well culture plates at a density of 5 × 104 per well. On the next day, human IFN-γ (20 ng/well) and compounds in a total volume of 200 µL culture medium containing 15 µg/mL of L-tryptophan were added to the cells. After incubation for 24 h, 140 µL of supernatant was mixed with 10 µL of 6.1 N trichloroacetic acid and the mixture was incubated for 30 min at 60 °C. The reaction mixture was then centrifuged for 20 min at 4000 rpm at 0 °C to remove sediments. About 100 µL of the supernatant was mixed with 100 µL of 2% (w/v) p-DMAB in acetic acid and measured at 480 nm. The final DMSO concentration was 0.5%. The data were analysed using Graphpad Prism 6.
Measurement of IDO1 absorbance spectra
Absorbance spectra (250–480 nm) were measured immediately after addition of compound LBJ-10 (20 µM in 0.5% DMSO) to IDO1 (5 µM) in phosphate buffer (pH 6.5) using a Tecan Safire2 microplate reader.
Determination of the inhibition type
The standard assay mixture (1 ml) contained 50 mM potassium phosphate buffer (pH = 6.5), 25 mM neutralised ascorbic acid, 10 µM methylene blue, 100 µg/mL catalase, 50 µM L-Trp, DMSO (0.5%), and eight different concentrations of IDO1. In addition, 1 µM LBJ-10 solution was added into the mixture. The reaction, at 37 °C, was started by the addition of the substrate and terminated after 60 min by adding 200 µL of 30% (w/v) trichloroacetic acid and further incubated at 50 °C for 30 min to hydrolyse N-formylkynurenine to produce kynurenine. After centrifugation at 12,000 rpm for 15 min at 20 °C, kynurenine in the supernatant (1 mL) was measured by a HPLC system with a reversed phase column (4.6 mm × 15 cm) of GL Sciences Inc. The mobile phase was 10 mM ammonium acetate containing 10% (w/v) methanol, and kynurenine was detected by absorbance at 360 nm. All determinations were carried out in duplicate. The data presented are average values. Based on the results of the activity assay, a function of rate ([V]) against enzyme amount ([E]) was proposed.
Water solubility evaluation
The aqueous solubility was determined on a Gemini Profiler instrument (pION) by the “goldstandard” Avdeef–Bucher potentiometric titration methodCitation46,Citation47.
Cell viability assay
Growth inhibition was determined by the MTT colorimetric assay. HeLa cells were plated in 96-well plates at a density of 10,000 cells/mL and allowed to attach overnight (16 h). Cells were plated in 96-well plates at a density of 10,000 cells/mL and allowed to attach overnight (16 h). Cells were then exposure to 50 µM of test compounds in triplicate for 48 h at 37 °C under a humidified atmosphere containing 5% CO2. MTT (50 µg) was added and the cells were incubated for another 4 h. Medium/MTT solutions were removed carefully by aspiration, the MTT formazan crystals were dissolved in 100 µL of DMSO, and absorbance was determined on a plate reader at 560 nm.
Chemistry
General procedures
All chemicals and reagents were purchased from commercial sources. Organic solutions were concentrated in a rotary evaporator (Bu¨chi Rotavapor) below 55 °C under reduced pressure. Silica gel thin-layer chromatography was performed on precoated plates GF-254 and visualised under UV light. Melting points were determined with a Melt-Temp II apparatus. 1H NMR and 13C NMR spectra were recorded on a Bruker AV-300 instrument using deuterated solvents with tetramethylsilane as internal standard. Chemical shifts are given in ppm (δ). The multiplicities are denoted as follows: s, singlet; d, doublet; t, triplet; q, quartette; m, multiplet; br s, broad singlet. IR spectra were recorded on a Nicolet iS10 Avatar FT-IR spectrometer using KBr film. ESI-mass and high-resolution mass spectra (HRMS) were recorded on a Water Q-Tofmicro mass spectrometer. Analytical results are within 0.40% of the theoretical values. Purity of the compounds was analysed by HPLC (Agilent Technologies 1260 Infinity) using 80:20 MeOH/H2O as the mobile phase with a flow rate of 0.8 ml/min on a C18 column. All tested compounds exhibited greater than 95% purity unless otherwise noted.
Synthesis
(E)-4-Ethoxy-1,1,1-trifluorobut-3-en-2-one (16)
To the ethoxyethene (7.2 g, 0.1 mol, 1 eq) in pyridine (11.9 g, 0.15 mol, 1.5 eq) was added trifluoroacetic anhydride by dripping slowly at 0 °C. Then stirring for 12 h at room temperature. The mixture was adjusted to neutral with 2 N HCl. The organic was washed with water and brine successively, dried over anhydrous sodium sulphate, and concentrated in vacuum to obtain yellow oily liquid (10.1 g, 60%). HRMS m/z [M + H]+ calculated for C6H8F3O2: 169.0471, found: 169.0477.
2-Hydroxy-6-(trifluoromethyl)nicotinonitrile (17)
Intermediate 16 (8.4 g, 0.05 mol, 1 eq) was dissolved in 50 ml of ethanol, and nitrile acetamide (3.5 g, 0.05 mol, 1 eq) was added at 0 °C. Then sodium ethoxide (25%, 6.8 g, 0.1 mol, 2 eq) was slowly added. The mixture was stirred under reflux for 12 h and acidified with 2 N HCl to pH =3–4. Subsequently, the solvent was concentrated in vacuum and diluted with ethyl acetate. The solution was washed with water and brine, dried over anhydrous sodium sulphate, and concentrated in vacuum to obtained the product as a yellow solid (6.1 g, 65%). HRMS m/z [M + H]+ calculated for C7H4F3N2O: 189.0270, found: 189.0270.
2-Chloro-6-(trifluoromethyl)nicotinonitrile (18a)
Compound 17 (3.0 g, 15.96 mmol, 1 eq) was added to the bottle, and phosphorus oxychloride (17.0 g, 111.69 mmol, 7 eq) was added. The mixture was refluxed for 2.5 h. Removing excess phosphorus oxychloride under reduced pressure, then poured the residue to the ice water and washed with sodium bicarbonate. The mixture was then extracted twice with AcOEt, washed five times with a saturated solution of NaCl, dried over Na2SO4 and concentrated. The crude product was purified by flash column chromatograph on silica gel (PE/EA =10:1) to give 18a in 45% yield as a milk white powder. HRMS m/z [M + H]+ calculated for C7H3ClF3N2: 206.9931, found: 206.9933.
N-(4-Fluorophenyl)-2-hydroxyacetamide (21a)
To a solution of 4-fluoroaniline (11.1 g, 0.1 mol, 1 eq) at room temperature was added glycolic acid (8.4 g, 0.11 mol, 1.1 eq). The mixture was then stirred at 115 °C for 5.5 h and quenched at room temperature by addition of a saturated solution of sodium bicarbonate then obtain precipitate 21a (14.4 g, 85%). HRMS m/z [M + H]+ calculated for C8H9FNO2: 170.0612, found: 170.0614.
N-(4-Fluorophenyl)-2-mercaptoacetamide (21b)
To a solution of 4-fluoroaniline (11.1 g, 0.1 mol, 1 eq) at room temperature was added mercaptoacetic acid (10.1 g, 0.11 mol, 1.1 eq). The mixture was then stirred at 125 °C for 6 h and quenched at room temperature by addition of a saturated solution of sodium bicarbonate then obtain precipitate 21 b (16.3 g, 88%). HRMS m/z [M + H]+ calculated for C8H9FNOS: 186.0383, found: 186.0382.
2-Mercapto-N-phenylacetamide (21c)
To a solution aniline (9.3 g, 0.1 mol, 1 eq) at room temperature was added mercaptoacetic acid (10.1 g, 0.11 mol, 1.1 eq). The mixture was then stirred at 110 °C for 6 h and quenched at room temperature by addition of a saturated solution of sodium bicarbonate then obtain precipitate 21c (14.0 g, 84%). HRMS m/z [M + H]+ calculated for C8H10NOS: 168.0478, found: 168.0472.
N-(2-Fluorophenyl)-2-mercaptoacetamide (21d)
To a solution 2-fluoroaniline (11.1 g, 0.1 mol, 1 eq) at room temperature was added mercaptoacetic acid (10.1 g, 0.11 mol, 1.1 eq). The mixture was then stirred at 120 °C for 6 h and quenched at room temperature by addition of a saturated solution of sodium bicarbonate then obtain precipitate 21d (16.1 g, 87%). HRMS m/z [M + H]+ calculated for C8H9FNOS: 186.0383, found: 186.0386.
N-(3-Chloro-4-fluorophenyl)-2-mercaptoacetamide (21e)
To a solution 3-chloro-4-fluoroaniline (14.5 g, 0.1 mol, 1 eq) at room temperature was added mercaptoacetic acid (10.1 g, 0.11 mol, 1.1 eq). The mixture was then stirred at 150 °C for 6 h and quenched at room temperature by addition of a saturated solution of sodium bicarbonate then obtain precipitate 21e (19.3 g, 88%). HRMS m/z [M + H]+ calculated for C8H8ClFNOS: 219.9994, found: 220.0000.
2-Mercapto-N-(p-tolyl)acetamide (21f)
To a solution 4-methylaniline (10.7 g, 0.1 mol, 1 eq) at room temperature was added mercaptoacetic acid (10.1 g, 0.11 mol, 1.1 eq). The mixture was then stirred at 105 °C for 6 h and quenched at room temperature by addition of a saturated solution of sodium bicarbonate then obtain precipitate 21f (15.4 g, 85%). HRMS m/z [M + H]+ calculated for C9H12NOS: 182.0634, found: 182.0636.
2-Mercapto-N-(4-methoxyphenyl)acetamide (21g)
To a solution 4-methoxyaniline (12.3 g, 0.1 mol, 1 eq) at room temperature was added mercaptoacetic acid (10.1 g, 0.11 mol, 1.1 eq). The mixture was then stirred at 100 °C for 6 h and quenched at room temperature by addition of a saturated solution of sodium bicarbonate then obtain precipitate 21 g (17.5 g, 89%). HRMS m/z [M + H]+ calculated for C9H12NO2S: 198.0583, found: 198.0586.
2-((3-Cyanopyridin-2-yl)oxy)-N-(4-fluorophenyl)acetamide (LBJ-01)
To a solution of 2-chloro-3-nitrile pyridine (1.4 g, 10 mmol, 1 eq) and 21a (2.5 g, 15 mmol, 1.5 eq) in anhydrous DMF at room temperature was added K2CO3 (1.4, 10 mmol, 1 eq). The mixture was then stirred at 20 °C for 15 min, the reaction was quenched with water then the product was obtained by filtration and the precipitate was purified by silica gel chromatography (PE/EA, v/v, 5:1) to give the title product as a white solid (1.2 g, 43%). m.p. 234–236 °C. HRMS m/z [M + Na]+ calculated for C14H10FN3OSNa: 294.0655, found: 294.0659. 1H NMR (300 MHz, DMSO-d6) δ: 10.27 (s, 1H, -NH-), 8.42 (d, J = 3.3 Hz, 1H, -ArH), 8.33 (d, J = 6.73 Hz, 1H, -ArH), 7.58 (s, 2H, -ArH), 7.24–7.12 (m, 3H, -ArH), 5.09 (s, 2H, -CH2-) ppm. 13C NMR (75 MHz, DMSO-d6) δ: 165.6, 162.5, 158.0 (1JCF = 240.0 Hz), 151.5, 144.2, 134.9, 134.8, 120.9 (3JCF = 8.3 Hz), 117.8, 115.4 (2JCF = 22.5 Hz), 95.7, 64.7 ppm.
2-((2-Cyanophenyl)thio)-N-(4-fluorophenyl)acetamide (LBJ-02)
The title compound was prepared from 2-chlorobenzonitrile and 21b following the synthesis route of LBJ-01. Yellow solid (1.1 g, 40%). m.p. 215–217 °C. HRMS m/z [M + H]+ calculated for C15H12FN2OS: 287.0654, found: 287.0668. 1H NMR (300 MHz, DMSO-d6) δ: 9.44 (s, 1H, -NH-), 8.10 (d, J = 9.0 Hz, 1H, -ArH), 7.89 (d, J = 6.0 Hz, 1H, -ArH), 7.73–7.68 (m, 2H, -ArH), 7.51 (t, J = 7.5 Hz, 1H, -ArH), 7.42 (t, J = 7.5 Hz, 1H, -ArH), 7.29 (s, 2H, -CH2-), 7.16 (t, J = 9.0 Hz, 2H, -ArH) ppm. 13C NMR (75 MHz, DMSO-d6) δ: 164.9, 162.5, 158.0 (1JCF = 238.6 Hz), 148.0, 144.3, 135.2 121.0 (3JCF = 7.5 Hz), 116.5, 116.4, 115.2 (2JCF = 21.8 Hz), 114.5, 109.6, 35.1 ppm.
2-((3-Cyanopyridin-2-yl)thio)-N-(4-fluorophenyl)acetamide (LBJ-03)
The title compound was prepared from 2-chloro-3-cyanopyridine and 21b following the synthesis route of LBJ-01. Yellow solid (1.3 g, 45%). m.p. 198–201 °C. HRMS m/z [M + H]+ calculated for C14H11FN3OS: 288.0601, found: 288.0605. 1H NMR (300 MHz, DMSO-d6) δ: 10.41 (s, 1H, -NH-), 8.71–8.69 (m, 1H, -ArH), 8.29–8.26 (m, 1H, -ArH), 7.63–7.58 (m, 2H, -ArH), 7.38–7.33 (m, 1H, -ArH), 7.17 (t, J = 8.9 Hz, 2H, -ArH), 4.26 (s, 2H, -CH2-) ppm.
2-((4-Cyanopyridin-3-yl)thio)-N-(4-fluorophenyl)acetamide (LBJ-04)
The title compound was prepared from 3-chloro-4-cyanopyridine and 21b following the synthesis route of LBJ-01. Yellow solid (1.2 g, 43%). m.p. 206–209 °C. HRMS m/z [M + H]+ calculated for C14H11FN3OS: 288.0601, found: 288.0603. 1H NMR (300 MHz, DMSO-d6) δ: 10.37 (s, 1H, -NH-), 8.69–8.67 (m, 1H, -ArH), 8.27–8.24 (m, 1H, -ArH), 7.61–7.56 (m, 2H, -ArH), 7.36–7.31 (m, 1H, -ArH), 7.18–7.12 (m, 2H, -ArH), 4.24 (s, 2H, -CH2-) ppm.
2-((3-Cyanopyridin-4-yl)thio)-N-(4-fluorophenyl)acetamide (LBJ-05)
The title compound was prepared from 4-chloro-3-cyanopyridine and 21b following the synthesis route of LBJ-01. White solid (1.3 g, 44%). m.p. 232–234 °C. HRMS m/z [M + H]+ calculated for C14H11FN3OS: 288.0601, found: 288.0603. 1H NMR (300 MHz, DMSO-d6) δ: 10.48 (s, 1H, -NH-), 8.86 (s, 1H, -ArH), 8.67 (d, J = 6.0 Hz, 1H, -ArH), 7.62–7.56 (m, 3H, -ArH), 7.21–7.13 (m, 2H, -ArH), 4.23 (s, 2H, -CH2-) ppm.
2-((2-Cyanopyridin-3-yl)thio)-N-(4-fluorophenyl)acetamide (LBJ-06)
The title compound was prepared from 3-chloro-2-cyanopyridine and 21b following the synthesis route of LBJ-01. Yellow solid (1.3 g, 45%). m.p. 211–213 °C. HRMS m/z [M + H]+ calculated for C14H11FN3OS: 288.0601, found: 288.0605. 1H NMR (300 MHz, DMSO-d6) δ: 9.63 (s, 1H, -NH-), 8.70–8.68 (m, 1H, -ArH), 8.46–8.43 (m, 1H, -ArH), 7.74–7.69 (m, 2H, -ArH), 7.57–7.53 (m, 1H, -ArH), 7.18 (t, J = 9.0 Hz, 2H, -ArH), 6.94 (s, 2H, -CH2-) ppm. 13C NMR (75 MHz, DMSO-d6) δ: 163.5, 158.0 (1JCF = 240.0 Hz), 147.1, 146.8, 146.6, 135.1, 132.2, 131.7, 123.1 (3JCF = 7.5 Hz), 122.5, 115.0 (2JCF = 22.5 Hz), 42.8 ppm.
2-((3-Cyano-6-methylpyridin-2-yl)thio)-N-(4-fluorophenyl)acetamide (LBJ-07)
The title compound was prepared from 2-chloro-3-cyanopyridine and 21b following the synthesis route of LBJ-01. Yellow solid (1.4 g, 46%). m.p. 238–240 °C. HRMS m/z [M + H]+ calculated for C15H13FN3OS: 302.0758, found: 302.0758. 1H NMR (300 MHz, DMSO-d6) δ: 10.39 (s, 1H, -NH-), 8.11 (d, J = 9.0 Hz, 1H, -ArH), 7.61–7.57 (m, 2H, -ArH), 7.20–7.13 (m, 3H, -ArH), 4.17 (s, 2H, -CH2-), 2.5 (s, 3H, -CH3) ppm.
2-((3-Cyano-6-(trifluoromethyl)pyridin-2-yl)thio)-N-(4-fluorophenyl)acetamide (LBJ-08)
The title compound was prepared from 18a and 21b following the synthesis route of LBJ-01. Yellow solid (1.5 g, 43%). m.p. 201–204 °C. HRMS m/z [M + H]+ calculated for C15H10F4N3OS: 356.0475, found: 356.0477. 1H NMR (300 MHz, DMSO-d6) δ: 10.42 (s, 1H, -NH-), 8.56 (d, J = 6.0 Hz, 1H, -ArH), 7.82 (d, J = 9.0 Hz, 1H, -ArH), 7.58–7.53 (m, 2H, -ArH), 7.15 (t, J = 9.0 Hz, 2H, -ArH), 4.24 (s, 2H, -CH2-) ppm. 13C NMR (75 MHz, DMSO-d6) δ: 164.0, 158.6 (1JCF = 238.5 Hz), 149.0, 137.4, 135.4, 132.0, 128.0, 123.9, 122.9 (3JCF = 7.5 Hz), 122.6, 114.9 (2JCF = 22.5 Hz), 97.47, 30.65 ppm.
2-((3-Cyano-6-(trifluoromethyl)pyridin-2-yl)thio)-N-(2-fluorophenyl)acetamide (LBJ-09)
The title compound was prepared from 18a and 21d following the synthesis route of LBJ-01. White solid (1.6 g, 45%). m.p. 208–211 °C. HRMS m/z [M + H]+ calculated for C15H10F4N3OS: 356.0475, found: 356.0473. 1H NMR (300 MHz, DMSO-d6) δ: 10.14 (s, 1H, -NH-), 8.56 (d, J = 9.0 Hz, 1H, -ArH), 7.83 (d, J = 9.0 Hz, 2H, -ArH), 7.30–7.23 (m, 1H, -ArH), 7.16–7.212 (m, 2H, -ArH), 4.33 (s, 2H, -CH2-) ppm.
N-(3-chloro-4-fluorophenyl)-2-((3-cyano-6-(trifluoromethyl)pyridin-2-yl)thio) acetamide (LBJ-10)
The title compound was prepared from 18a and 21e following the synthesis route of LBJ-01. White solid (1.9 g, 48%). m.p. 227–229 °C. HRMS m/z [M + H]+ calculated for C15H9ClF4N3OS: 390.0085, found: 390.0077. 1H NMR (300 MHz, DMSO-d6) δ: 10.58 (s, 1H, -NH-), 8.56 (d, J = 9.0 Hz, 1H, -ArH), 7.87–7.81 (m, 2H, -ArH), 7.46–7.35 (m, 2H, -ArH), 4.25 (s, 2H, -CH2-) ppm. 13C NMR (75 MHz, DMSO-d6) δ: 163.6, 158.4 (1JCF = 238.5 Hz), 147.5, 145.5, 142.7, 137.5, 135.0, 133.5, 123.3 (3JCF = 8.0 Hz), 116.4, 115.1 (2JCF = 22.0 Hz), 101.5, 54.25 ppm.
2-((3-Cyano-6-(trifluoromethyl)pyridin-2-yl)thio)-N-phenylacetamide (LBJ-11)
The title compound was prepared from 18a and 21c following the synthesis route of LBJ-01. Yellow solid (0.75 g, 23%). m.p. 230–233 °C. HRMS m/z [M + H]+ calculated for C15H11F3N3OS: 338.0569, found: 338.0579. 1H NMR (300 MHz, DMSO-d6) δ: 10.34 (s, 1H, -NH-), 8.55 (d, J = 9.0 Hz, 1H, -ArH), 7.81 (d, J = 9.0 Hz, 1H, -ArH), 7.54 (d, J = 6.0 Hz, 2H, -ArH), 7.30 (t, J = 9.0 Hz, 2H, -ArH), 7.05 (t, J = 7.5 Hz, 1H, -ArH), 4.25 (s, 2H, -CH2-) ppm. 13C NMR (75 MHz, DMSO-d6) δ: 164.90, 162.54, 144.32, 138.84, 128.65, 123.38, 119.24, 116.49, 116.45, 116.41, 114.54, 35.25 ppm.
2-((3-Cyano-6-(trifluoromethyl)pyridin-2-yl)thio)-N-(p-tolyl)acetamide (LBJ-12)
The title compound was prepared from 18a and 21f following the synthesis route of LBJ-01. Yellow solid (2.30 g, 62%). m.p. 212–214 °C. HRMS m/z [M + H]+ calculated for C16H13F3N3OS: 352.0726, found: 352.0733. 1H NMR (300 MHz, DMSO-d6) δ: 10.24 (s, 1H, -NH-), 8.57–8.54 (m, 1H, -ArH), 7.81 (d, J = 9.0 Hz, 1H, -ArH), 7.41 (t, J = 4.5 Hz, 2H, -ArH), 7.10 (d, J = 6.0 Hz, 2H, -ArH), 4.24 (s, 2H, -CH2-), 2.25 (s, 3H, -CH3) ppm. 13C NMR (75 MHz, DMSO-d6) δ: 164.63, 162.57, 148.03, 144.29, 136.33, 132.31, 129.02, 119.26, 116.42, 116.38, 116.34, 114.53, 109.63, 102.88, 35.22, 20.38 ppm.
2-((3-Cyano-6-(trifluoromethyl)pyridin-2-yl)thio)-N-(p-tolyl)acetamide (LBJ-13)
The title compound was prepared from 18a and 21g following the synthesis route of LBJ-01. Yellow solid (1.5 g, 42%). m.p. 217–219 °C. HRMS m/z [M + H]+ calculated for C16H13F3N3O2S: 368.0675, found: 368.0681. 1H NMR (300 MHz, DMSO-d6) δ: 10.20 (s, 1H, -NH-), 8.55 (d, J = 9.0 Hz, 1H, -ArH), 7.82 (d, J = 6.0 Hz, 1H, -ArH), 7.46–7.44 (m, 2H, -ArH), 6.89–6.86 (m, 2H, -ArH), 4.22 (s, 2H, -CH2-), 3.72 (s, 3H, -CH3) ppm. 13C NMR (75 MHz, DMSO-d6) δ: 164.38, 155.34, 144.29, 131.97, 120.83, 116.45, 116.38, 114.54, 113.81, 55.12, 54.78, 35.13, 27.31 ppm.
N-(3-Chloro-4-fluorophenyl)-2-((6-(trifluoromethyl)pyridin-2-yl)thio)acetamide (LBJ-14)
The title compound was prepared from 2-chloro-6-(trifluoromethyl)pyridine and 21e following the synthesis route of LBJ-01. White solid (1.8 g, 52%). m.p. 86–88 °C. HRMS m/z [M-H]– calculated for C14H8ClF4N2OS 363.0023, found 363.0017; 1H NMR (300 MHz, DMSO-d6) δ: 10.50 (s, 1H, -NH-), 7.95–7.86 (m, 2H, Ar-H), 7.70 (d, J = 9.0 Hz, 1H, Ar-H), 7.59 (d, J = 9.0 Hz, 1H, Ar-H), 7.45–7.37 (dd, J = 15.0 Hz, J = 9.0 Hz, 2H, Ar-H), 4.11 (s, 2H, -CH2-) ppm.
Results and discussion
Shape-based virtual screen
ROCS is a fast shape comparison application which developed based on the principle that molecules have similar shape if their volumes overlay well and any volume mismatch is a measure of dissimilarityCitation48. Besides shape description ROCS includes a colour force field allowing for basic inclusion of chemical information in the screening processCitation49,Citation50. ROCS colour force field can be used to measure chemical features complementarity and refine shape-based superpositions based on chemical similarity. The colour force field is composed of SMARTS rules that determine chemical centres plus rules to determine how centres interact.
The combination of shape matching (shape Tanimoto) and functional group matching (colour Tanimoto) expressed as Tanimoto combo score. The 3D similarity was ranked by the Tanimoto ComboScore method. Both shape and colour score vary from 0 to 1 (besthits = 1), the combo score as the sum varies between 0 and 2Citation50. In the chemical force field, a molecule is described by the spatial arrangement of six types: hydrogen-bond donors, hydrogen-bond acceptors, hydrophobes, anions, cations, and rings. Recently, several studies suggested that the bioactive conformation of a reference molecule is not needed for the enrichment of a shape-based screeningCitation51. These findings encouraged us to employ this complementary methodology (complementary to the shape of the target interaction site) as a screening model for the identification of novel scaffolds that possess similar structures with positive IDO1 inhibitors.
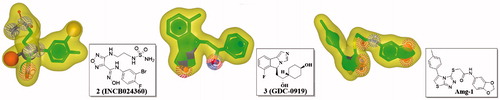
In this study, three representative compounds with diverse scaffolds, GDC-0919, INCB024360, and Amg-1, were selected as the reference molecule to generate the ROCS models. The 3D conformers of GDC-0919 and Amg-1 were obtained through the crystal structures (PDB code: 5EK4 and 4PK5, respectively). As for INCB024360, a low energy 3D conformer was generated using the MMFF94x forcefield (MOE 2016.08). The molecular shapes of these three compounds were depicted in yellow shadow (). Then, the shaped queries of these three compounds were set to screen the Specs databased, containing approximately two hundred thousand molecules. First, over 180,000 compounds in Specs database were kept after removing PAINS (Pan Assay Interference Compounds structures) using the PAINS-remover online web serverCitation52. Then, the multiple conformations of different query molecules were calculated using OMEGA with a maximum of 300 conformationsCitation53. The top five hundred compounds for each query were carried forward for docking. There were some overlapped molecules obtained from these three queries, but we did not pick them out in this step.
Figure 3. ROCS shape query derived from a low energy 3D conformation generated in MOE2016.09 of INCB024360 (2), GDC-0919 (3) and Amg-1. The green spheres illustrate ROCS ring features, the red spheres illustrate hydrogen-bond acceptors, and the blue spheres illustrate hydrogen-bond donors.
Docking results
Comparison of different crystal structures of IDO1-inhibitors
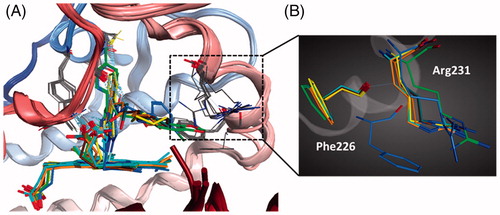
Up to date, nine crystal structures of IDO1 have been deposited in the RSCB Protein Data Bank (PDB). Among them, six structures with crystallographic resolutions higher than 3.0 Å were utilised in this work. These structures are all IDO1 with its potent inhibitors, including 4-phenylimidazole (4-PI, 8, PDB code 2D0T)Citation36, Amg-1 (9, PDB code 4PK5)Citation37, 10 (PDB code 5EK2), 11 (PDB code 5EK3), 12 (PDB code 5EK4), and 13 (PDB code 5ETW)Citation38. To compare the backbone conformation, 2D0T was used as the reference and the other structure was superposed onto it, and the RMSD was calculated (varies from 0.36 to 0.93 RMS deviation for the α-carbon atoms, ). The low RMSD indicated that these IDO1 structures showed a high similarity. However, there is a significant movement of several residues, such as Phe226 and Arg231, in the catalytic pocket of IDO1-inhibitors, indicated that the pocket of IDO1 is flexible ().
Figure 4. (A) The superimposition of six IDO1 crystal structures. (B) The significant movement of two representative residues (Phe226 and Arg231) in the catalytic pocket.
Re-docking the crystal structure using different software
In order to evaluate the accuracy of different docking programmes, including Gold 5.1 (The Cambridge Crystallographic Data Centre, UK), CDOCKET (BIOVIA, USA), Alpha PMI (Montreal, Canada), AutoDock (La Jolla, USA), and Glide (Los Angeles, USA), AutoDock, and Glide, we first performed the re-docking experiments. As shown in , among these docking software, GOLD5.1 software package with GoldScore scoring function possessed six high accuracy predictions (RMSD <1.0 Å). It indicated that Gold5.1 showed higher reproducibility than other software. For further validated the docking result, we carried the cross-docking experiment. Consistent with the result of native-docking, docking using the Gold5.1 had the smallest average RMSD (). Hence, Gold5.1 was selected and employed to filter the molecular library for discovering the putative IDO1 inhibitors by VS. As for the selection of the IDO1 crystal structures, we noticed that the IDO1 pocket was very flexible and thus the rigid receptor docking will more than likely fail in finding the diverse scaffold against IDO1. Thus, we employed multiple crystal structures on constructing an ensemble docking to deal with protein flexibility. All of the six crystal structures were employed as docking template to identify structurally diverse IDO1 inhibitors.
Figure 5. RMSD of ligands in cross-docking. The mean RMSD for the top-scoring pose of all the native ligands docked to the same protein are reported in the heat-map. The RMSD for each protein and docking protocol is represented by a colorimetric scale, going from purple to red from 0 to higher RMSD values.
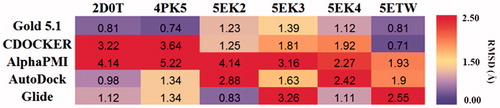
Table 1. Information of all eight of the IDO1 PDB cognate ligands.
The results of molecular docking
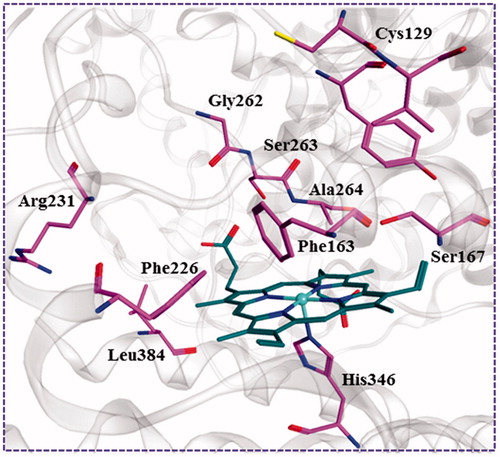
After the previous shape-based VS, the remaining compounds were submitted docking simulation using the genetic optimisation for ligand docking (Gold5.1) software package targeting multi-crystal IDO1 structures. The binding energy of the ligands with protein was predicted using GOLD score implemented in GOLD. The total GOLD score, which is represented as “Fitness,” was calculated from the contribution of hydrogen bonds and van der Waals interactions between protein and ligand as well as the contribution of intramolecular hydrogen bonds and intramolecular strain in the ligand. The binding site was defined as being any volume within 8 Å of the ligand in the crystal pose, and the residues Arg231, Leu384, Phe226, Gly262, Ser263, Ala264, Phe163, Ser167, Tyr126, Cys129, and His346 were found to be interacting residues as shown in . All compounds were each docked to the six selected IDO1 structures, and the top 1% compounds were retained based on the Goldscore Fitness in each structure. Because this number included a large degree of similar compounds, with respect to scaffold and chemical class, a diversity analysis was undertaken using the program Diverse Subset (MOE 2016.09). Compounds with high GOLD fitness score usually could form hydrogen-bond interactions with Ala264, Ser167, and Ser263 and a coordinate bond with Fe2+ ion of haeme. In addition, as shown in the interaction between the native ligand and IDO1, the formed hydrophobic interactions with the hydrophobic resides Try126, Cys129, and Phe163 of IDO1 could improve the binding affinity. Top 10% of the molecules were recruited and further visually examined. In the process of visual inspection, we removed the reported compounds against IDO1 and ruled out the structures with the unreasonable binding modes discreetly (potential IDO1 inhibitors should possess functional group which could interact with the active site haeme of IDO1 and occupy the hydrophobic A pocket). Finally, 32 compounds were purchased form Specs for in vitro testing (Figure S1). Among these compounds LVS-019 was discovered as the most potent IDO1 inhibitors.
Figure 6. The key residues in binding site of IDO1. The binding pattern was generated form the cocrystal structure (PDB code: 4PK5) depicted using MOE 2016.09. The carbon atoms of haeme and the key residues in the active site of IDO1 were coloured in green and purple, respectively.
Second round of screening based on compound LVS-019
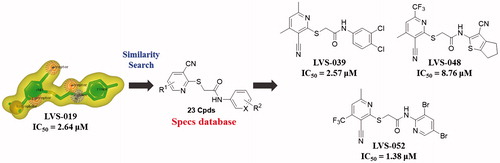
As we knew, it is equivocal to determine the activity of a chemical scaffold judging by one compound due to the potential experimental error. In order to avoid the deviation in the subsequent molecular design and confirm the activity of screening hits, a further chemical similarity search based on LVS-019 was performed. About 500 Compounds with similar molecular shape of LVS-019 were screened out. The structural similarity of these molecules was then analysed using Functional-Class Fingerprints_6 (FCFP_6) molecular fingerprintCitation54. Compounds with a Tanimoto coefficient over 0.5 were recognised as similar structures to LVS-019. After such screening, 48 derivatives of LVS-019 were retained and processed to dock by Gold5.1 to rule out the structures with the unreasonable binding modes. Finally, 23 commercial available derivatives of LVS-019 were retained and purchased to bioevaluation (Figure S2).
Biological test
Inhibitory activities of screened compounds against purified recombinant IDO1 enzyme
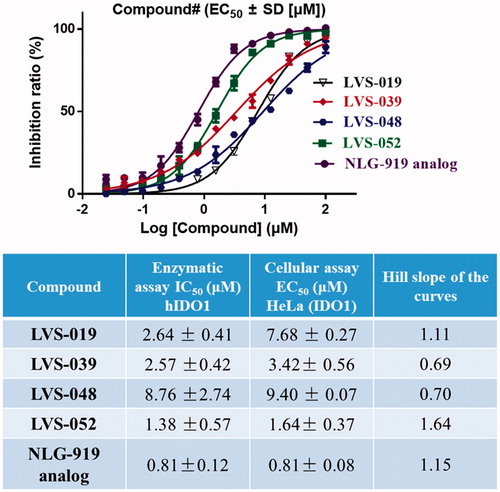
The potencies of the screened compounds in inhibiting the human IDO1 enzyme activity were initially evaluated by measuring the changes in absorbance values of the product produced from kynurenine and 4-dimethylaminobenzaldehyde at 480 nmCitation55. To ensure that our compounds did not interfere with biological assays, representative compounds and water (Figure S3) with the addition of different concentrations of kynurenine in the absence of IDO1 protein. After p-dimethylaminobenzaldehyde (p-DMAB) was added, the kynurenine concentrations were determined by measuring the absorbance at OD 480 nm. The results showed that both of the test compounds did not interfere with the signal detection. Initial enzyme-based IDO1 inhibitory activity assay was conducted for 32 compounds obtained from VS at concentrations of 10 µM using L-1-MT and GDC-0919 analogue (racemic mixture of GDC-0919) as a positive control. The results are reported in Table S1. Based on the assay, five compounds (LVS-001, LVS-002, LVS-018, LVS-019, and LVS-032) revealed potential inhibitory activity to IDO1 with inhibition rates more than 50% at 10 µM. Then, we measured the dose response behaviours of these compounds, the IC50 values of these compounds are shown in . Compound LVS-019 with cyanopyridine scaffolds had the highest activity with IC50 value of 2.64 µM. Based on the promising scaffold of LVS-019, further 23 derivatives were discovered and evaluated. The structures and the IC50 values of these 23 derivatives are shown in Figures S2 and S4, and Table S2. Among these derivatives, several compounds (LVS-039, LVS-048, and LVS-052) showed similar potency compared with LVS-019 (). Comparing these cyanopyridine scaffold compounds with the control, L-1-MT and GDC-0919 analogue, had IC50 values of 42 µM and 0.81 µM, respectively. The results indicated that these compounds were more potent inhibitors than L-1-MT and was slightly weaker than GDC-0919 analogue. It should be mentioned that these hit molecules have not been optimised yet, and there existed a possibility for improving the binding affinity of each compound after careful molecular optimisation. Hence, the discovery of the new cyanopyridine scaffold derivatives with relatively good potency was encouraging.
Figure 7. Five IDO1 inhibitors were found by VS from the database. The IDO1 inhibitory activity was obtained through enzymatic assay. The IC50 values were expressed as the mean of at least three independent determinations and are within ±15%.
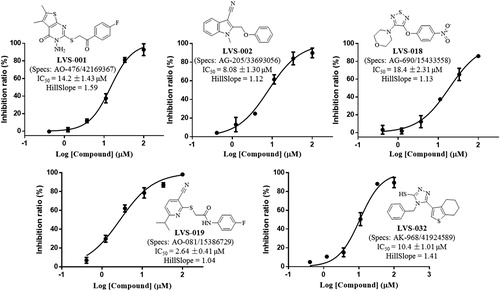
Figure 8. 23 Derivatives were discovered based on the scaffold of LVS-019. Among these compounds, LVS-039, LVS-048, and LVS-052 showed similar potency compared with LVS-019. The IC50 values of other derivatives are shown in Table S2. The IC50 values were expressed as the mean of at least three independent determinations.
Determination of IDO1 inhibitory activity in the HeLa cell-based method
The HeLa-based cellular assay is informative for drug development, as it evaluates not only the IDO1 inhibitory effect of the compounds but also their capacity to permeate the cell, their inhibition of tryptophan and kynurenine transport and their potential cytotoxicityCitation56. The representative compounds (LVS-019, LVS-039, LVS-048, and LVS-052) were also assayed for their ability to inhibit tryptophan degradation and kynurenine production in the HeLa kynurenine assay. shows the dose response curves for IFN-γ stimulated HeLa cells (IDO1 high-expressed) incubated with LVS-019, LVS-039, LVS-048, and LVS-052. The Hill slope of these dose response curves did not deviate from 1, indicated that these compounds may be specific inhibitors of IDO1. These four compounds displayed cellular IDO1 inhibitory activity in the HeLa cell line with EC50 value in the range of 1.64–9.40 µM. More importantly, a good correlation between the recombinant enzyme-based and cellular-based assays confirmed that these cyanopyridine scaffold compounds can be developed to potent IDO1 inhibitors. In addition, since the inhibition of tryptophan degradation could simply be an effect of cytotoxicity, thus the determination of cell viability was necessary when reporting cellular IC50 values. In the initial work, all of the screened compounds were determined for their cell viability using the classical MTT assy. The results are shown in Figures S5 and S6, most of these compounds displayed negligible level of toxicity against HeLa cells at 50 µM.
Figure 9. Dose-dependent inhibition of compounds LVS-019, LVS-039, LVS-048, LVS-052, and GDC-0919 analogue on cellular IDO1 inhibitory activity in the HeLa cell line. The table showed the enzymatic IC50, cellular IC50 and Hill slope of the curves of compounds LVS-019, LVS-039, LVS-048, LVS-052, and GDC-0919 analogue. The data were evaluated by Prism 6. The data are expressed as the mean ± SD calculated from three independent determinations.
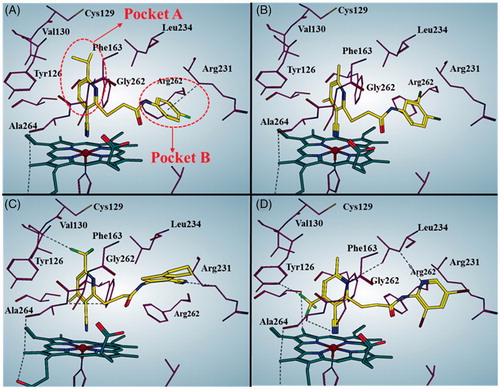
Predicted binding patters of potent inhibitors with cyanopyridine scaffold
shows the predicted binding mode of representative compounds LVS-019, LVS-039, LVS-048, and LVS-052 to the top-scored protein structures (PDB code: 4PK5). The general molecular orientation and the spatial location of the chemical features of these inhibitors were similar to that of native ligand Amg-1. The nitrile group in pyrimidine ring of these compounds directly binds to the iron-haeme and pyrimidine ring is sitting at the pocket A. Furthermore, it is predicted that the ring B occupied the pocket B which is composed of residues Arg231, Phe262, and Leu234. More importantly, the ring B of these compounds could form π–π interaction with the Phe226. These four potent compounds were more effective than the other compounds, supported that the halogen or nitrile substitution at the B ring could form an additional electrostatic interaction of Arg231 which is necessary for improving the IDO1 inhibitory activity.
Figure 10. Predicted binding modes for compound LVS-019 (A), LVS-039 (B), LVS-048 (C), and LVS-052 (D) with IDO1. The pictures were generated using MOE 2016.09. The black dotted lines indicate interactions between ligand and receptor, including hydrogen bond, halogen bond, and H…π interactions.
Design, synthesis, and preliminarily evaluation of derivatives of LVS-019
Though these four screened compounds have been confirmed that possess potential IDO1 inhibitory activity and stable binding properties. However, there are still several defects for these compounds being promising lead compounds to further drug discovery, such as the moderate IDO1 inhibitory activity at micromole level and low water solubility (18 µg/mL). Thus, we designed several novel LVS-019 derivatives (LBJ-01∼LBJ-13) with simplified structures and discussed the preliminary structure–activity relationship (SAR) of these series of compounds to find promising lead compound. In addition, to investigate the importance of the cyano group for IDO1 binding, we also designed and synthesised the compound LBJ-14 which lacks the group.
The synthesis routes used to prepare derivatives LBJ-01∼LBJ-13 are shown in Scheme 1. The key intermediate 17 was synthesised starting from ethyl vinyl ether 15 in three steps by a modification of the previously report procedureCitation57. Trifluoromethyl acetylation of 15 followed by condensation with cyanoacetamide provided pyridine 17, which was subsequently converted to 2-chloropyridine by POCl3. Intermediate 21 was prepared through the neat acylation of substituted phenylamine (20) with glycolic acid (19a) or mercaptoacetic acid (19b). Finally, the target compounds (LBJ-01∼LBJ-13) were obtained by nucleophilic aromatic substitution using the intermediates 17a–17b with 20 under weak base condition.
Scheme 1. Reagents and conditions: (i) (CF3CO)2O, pyridine, CH2Cl2, 0 °C to 25 °C, 12 h, 60%; (ii) NCCH2CONH2, EtONa, EtOH, 0 °C to reflux, 12 h, 65%; (iii) POCl3, reflux, 2.5 h, 45%; (iv) 100–150 °C, 2–6 h, 84–89%; (v) K2CO3, DMF, 20 °C, 15–45 min, 33–48%. For the detailed R groups of compounds LBJ-01–LBJ-14, see .
These 14 synthesised molecules were evaluated for their IDO1 inhibitory activity using an enzyme-based method. Encouragingly, compound LBJ-08 (IC50 = 1.87 ± 0.83 µM) and LBJ-10 (IC50 = 0.84 ± 0.05 µM) showed improved IDO1 inhibitory activity than LVS-019 (IC50 = 2.64 ± 0.94 µM) (, Figure S7). The SAR studies demonstrated that the remove of the isopropyl group (LBJ-03) would reduce IDO1 inhibitory activity. LBJ-1 with oxygen atom at the linker showed worse IDO1 activity compared with LBJ-3, indicated that the sulphur atom at the linker also has positive effect for the IDO1 inhibitory activity. Changing the 3-cyanopyridine scaffold (LBJ-03) to benzene (LBJ-02) or other substituted pyridine with nitrile group at different positions (LBJ-04∼LBJ-06) failed to show stronger inhibition efficiency. The 5-trifluoromethyl substituted cyanopyridine compound (LBJ-08) showed stronger IDO1 inhibitory activity than the unsubstituted compound LBJ-03. Based on LBJ-08, further modification focussed on the B ring, a series of different anilines substituted compounds (LBJ-09∼LBJ-13) was synthesised to explore the SAR, and most of the assayed compounds were less potent than LBJ-08 except for LBJ-10 which showed improved potency to that of LBJ-08. Compound LBJ-14 was much weaker than compound LBJ-10, indicated the necessity of the presence of cyano group in these series of compounds. With IDO1 inhibitory activity at the nanomole level, compound LBJ-10 could be a promising IDO1 inhibitor for further study. We also determined the solubility of these compounds using the “goldstandard” Avdeef–Bucher potentiometric titration method. To our disappointment, except for compound LBJ-03 with four times more soluble (75 µg/mL) than LVS-019, other compounds were on the same level with it. Thus, we would further focus on improving the drug-like properties of these series of compounds in the near future.
Table 2. Inhibitory activity of the cyanopyridine scaffold derivatives against purified human IDO1 enzyme.
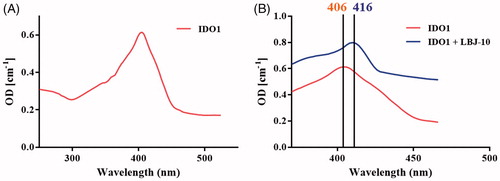
Spectroscopic and reversibility analysis of LBJ-10 binding
A UV–visible spectra study was performed to evaluate the binding of the representative compound LBJ-10 (). The absorption spectrum of the haeme group is highly sensitive to the changes in the polarity of haeme surrounding upon the ligand or substrate binding, leading to the change of spectra properties of the haemeCitation38. The UV–visible spectra of ferric IDO1 was measured in the presence and absence of compound LBJ-10. In the absence of compound LBJ-10, the absorption spectrum of ferric IDO1 exhibited a Soret peak at 406 nm. When in the presence of compound LBJ-10, the spectra shifted to 416 nm which demonstrated that compound LBJ-10 binds to ID O1 and coordinates with the iron of the haeme group.
Figure 11. Spectroscopic analysis of LBJ-10 binding. UV spectra of ferric IDO1 without (A) and with 20 μΜ concentration of compound LBJ-10 (B). The Soret’s ratio (A404/A280) of purified IDO1 was 2.15.
We also carried the additional experiment to determine the reversibility of compound LBJ-10. Detailed kinetics analysis was performed on LBJ-10 based on plotting a function of the rate (V) against the enzyme amount ([E]) ()Citation58,Citation59. The result showed that the representative LBJ-10 was categorised as reversible inhibitor (). More specific kinetic and crystal structure experiments would be carried to verify the inhibitory mode and binding mode of these series of compounds.
Figure 12. Inhibition type of LBJ-10. (A) Determination of the inhibition type. Reaction rate [V] is plotted against enzyme amount [E]. Curves with different slopes represent reversible and irreversible inhibitions, respectively. (B) Inhibition types of LBJ-10. Plot of reaction rate [V] against enzyme amount [E]. The concentrations of L-tryptophan and the inhibitor were 50 and 1 μM, respectively.
![Figure 12. Inhibition type of LBJ-10. (A) Determination of the inhibition type. Reaction rate [V] is plotted against enzyme amount [E]. Curves with different slopes represent reversible and irreversible inhibitions, respectively. (B) Inhibition types of LBJ-10. Plot of reaction rate [V] against enzyme amount [E]. The concentrations of L-tryptophan and the inhibitor were 50 and 1 μM, respectively.](/cms/asset/c64c4714-00ee-4752-b0d6-4df8af755672/ienz_a_1480614_f0012_c.jpg)
Conclusions
With the combined strategy of similarity search and molecular docking study, a diverse range of scaffolds of IDO1 inhibitors were identified from the Specs database. The shape-based screening model was developed using the ROCS method, based on the reported representative IDO1 inhibitors INCB024360, GDC-0919, and Amg-1. Subsequently, optimal docking process was further employed to screen the compounds and 32 hit compounds were finally discovered. Testing the screened hit compounds has led to several submicromolar inhibitors with novel scaffolds. Especially for compounds LVS-019 with cyanopyridine scaffold, showed the best IDO1 inhibitory activity. To discover more compounds with similar structures to LVS-019, a ROCS model was then generated on the basis of LVS-019 and then the second-round VS was carried out. Then the hits obtained by similarity search were further evaluated by GOLD, and 23 compounds were purchased to test in vitro leading to the discovery of another three potent compounds (LVS-039, LVS-048, and LVS-052). To improve potency of these hit compounds, we designed and synthesised another 14 derivatives of LVS-019 through rational drug designation. As we expected, compound LBJ-10 showed improvement potency compared to the hit compound and displayed comparable potency to the control GDC-0919 analogue. In this direction, the in silico approach described can also be used to screen existing database to identify derivatives with desired activity. Further studies on the molecular optimisation of the other hits to provide some other potent IDO1 inhibitors with novel scaffolds are currently underway.
supporting_Information.doc
Download MS Word (2 MB)Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Takikawa O, Yoshida R, Kido R. Tryptophan degradation in mice initiated by indoleamine 2,3-Dioxygenase. J Biol Chem 1986;261:3648–53.
- Paul S, Roy A, Deka SJ, et al. Nitrobenzofurazan derivatives of N'-hydroxyamidines as potent inhibitors of indoleamine-2,3-dioxygenase 1. Eur J Med Chem 2016;121:364–75.
- Hayaishi O. Properties and function of indoleamine 2,3-dioxygenase. J Biochem 1976;79:13P–21P.
- Löb S, Königsrainer A, Zieker D, et al. IDO1 and IDO2 are expressed in human tumors: levo- but not dextro-1-methyl tryptophan inhibits tryptophan catabolism. Cancer Immunol Immunother 2009;58:153–7.
- Croitoru-Lamoury J, Lamoury FMJ, Caristo M, et al. Interferon-γ regulates the proliferation and differentiation of mesenchymal stem cells via activation of indoleamine 2,3 dioxygenase (IDO). PLoS One 2011;6:e14698.
- Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol 2004;4:762–74.
- Uyttenhove C, Pilotte L, Theate I, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med 2003;9:1269–74.
- Munn DH, Mellor AL. IDO and tolerance to tumors. Trends Mol Med 2004;10:15–8.
- Zheng X, Koropatnick J, Li M, et al. Reinstalling antitumor immunity by inhibiting tumor-derived immunosuppressive molecule IDO through RNA interference. J Immunol 2006;177:5639–46.
- Muller AJ, DuHadaway JB, Chang MY, et al. Non-hematopoietic expression of IDO is integrally required for inflammatory tumor promotion. Cancer Immunol Immunother 2010;59:1655–63.
- Crosignani S, Bingham P, Bottemanne P, et al. Discovery of a novel and selective indoleamine 2,3-dioxygenase (IDO-1) inhibitor 3-(5-fluoro-1H-indol-3-yl)pyrrolidine-2,5-dione (EOS200271/PF-06840003) and its characterization as a potential clinical candidate. J Med Chem 2017;60:9617–29.
- Hou DY, Muller AJ, Sharma MD, et al. Inhibition of indoleamine 2,3-dioxygenase in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with antitumor responses. Cancer Res 2007;67:792–801.
- Yue EW, Douty B, Wayland B, et al. Discovery of potent competitive inhibitors of indoleamine 2,3-dioxygenase with in vivo pharmacodynamic activity and efficacy in a mouse melanoma model. J Med Chem 2009;52:7364–7.
- Liu X, Newton RC, Friedman SM, et al. Indoleamine 2,3-dioxygenase, an emerging target for anti-cancer therapy. Curr Cancer Drug Targets 2009;9:938–52.
- Godin-Ethier J, Hanafi LA, Piccirillo CA, et al. Indoleamine 2,3-dioxygenase expression in human cancers: clinical and immunologic perspectives. Clin Cancer Res 2011;17:6985–91.
- Spranger S, Horton B, Koblish HK, et al. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. J Immunother Cancer 2014;2:3.
- Muller AJ, Duhadaway JB, Donover PS, et al. Inhibition of indoleamine 2,3-dioxygenase, an immune-regulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat Med 2005;11:312–9.
- Pan L, Zheng Q, Chen Y, et al. Design, synthesis and biological evaluation of novel naphthoquinone derivatives as IDO1 inhibitors Design, synthesis and biological evaluation of novel naphthoquinone derivatives as IDO1 inhibitors. Eur J Med Chem 2018;157:423–36.
- Sheridan C. IDO inhibitors move center stage in immuno-oncology. Nat Biotechnol 2015;33:321–2.
- Available from: <http://clinicaltrials.gov/show/NCT01191216> [last accessed 27 Feb 2017].
- Available from: <http://clinicaltrials.gov/show/NCT00567931> [last accessed 27 Feb 2017].
- Cady SG, Sono M. 1-Methyl-DL-tryptophan, beta-(3-benzofuranyl)-DL-alanine (the oxygen analog of tryptophan), and beta-[3-benzo(b)thienyl]-DL-alanine (the sulfur analog of tryptophan) are competitive inhibitors for indoleamine 2,3-dioxygenase. Arch Biochem Biophys 1991;291:326–33.
- Available from: https://integrity.thomsonpharma.com/integrity/xmlxsl/pk_qcksrch.show_records?sessionID=1&history=&query=GDC0919&abbreviation=PRO&language=en.
- Mautino MR, Jaipuri FA, Waldo J, et al. NLG919, a novel indoleamine-2,3-dioxygenase (IDO)-pathway inhibitor drug candidate for cancer therapy. Cancer Res 2013;73:491.
- Yue EW, Sparks R, Polam P, et al. INCB24360 (Epacadostat), a highly potent and selective indoleamine-2,3-dioxygenase 1 (IDO1) inhibitor for immuno-oncology. ACS Med Chem Lett 2017;8:486–91.
- Tumang J, Gomes B, Wythes M, et al. PF-06840003: a highly selective IDO-1 inhibitor that shows good in vivo efficacy in combination with immune checkpoint inhibitors. 107th Annu Meet Am Assoc Cancer Res (AACR); 2016 April 16–20; New Orleans.
- Balog A. Discovery of a selective inhibitor of indoleamine-2,3-dioxygenase for use in the therapy of cancer. 253rd Am Chem Soc (ACS) Natl Meet; 2017 April 2–6; San Francisco.
- Molinier-Frenkel V, Castellano F. Immunosuppressive enzymes in the tumor microenvironment. FEBS Lett 2017;591:3135–57.
- Qian S, Zhang M, Chen QL, et al. IDO as a drug target for cancer immunotherapy: recent developments in IDO inhibitors discovery. RSC Adv 2016;6:7575–81.
- Zhang Y, Yang S, Jiao Y, et al. An integrated virtual screening approach for VEGFR-2 inhibitors. J Chem Inf Model 2013;53:3163–77.
- Drwal MN, Griffith R. Combination of ligand- and structure-based methods in virtual screening. Drug Discov Today Technol 2013;10:e395–401.
- Tian S, Sun H, Li Y, et al. Development and evaluation of an integrated virtual screening strategy by combining molecular docking and pharmacophore searching based on multiple protein structures. J Chem Inf Model 2013;53:2743–56.
- Molecular Operating Environment (MOE). Montreal, QC: Chemical Computing Group Inc.; 2013.
- Liem SY, Popelier PL. The hydration of serine: multipole moments versus point charges. Phys Chem Chem Phys 2014;16:4122–34.
- Sun HP, Xu XL, Wu XW, et al. Discovery and design of tricyclic scaffolds as protein kinase CK2 (CK2) inhibitors through a combination of shape-based virtual screening and structure-based molecular modification. J Chem Inf Model 2013;53:2093–102.
- Sugimoto H, Oda S, Otsuki T, et al. Crystal structure of human indoleamine 2,3-dioxygenase: catalytic mechanism of O2 incorporation by a heme-containing dioxygenase. Proc Natl Acad Sci USA 2006;103:2611–6.
- Tojo S, Kohno T, Tanaka T, et al. Crystal structures and structure activity relationships of imidazothiazole derivatives as IDO1 inhibitors. ACS Med Chem Lett 2014;5:1119–23.
- Peng YH, Ueng SH, Tseng CT, et al. Important hydrogen bond networks in indoleamine 2,3-dioxygenase 1 (IDO1) inhibitor design revealed by crystal structures of imidazoleisoindole derivatives with IDO1. J Med Chem 2016;59:282–93.
- GOLD, version 5.1. Cambridge: CCDC. Available from: www.ccdc.cam.ac.uk; September 6, 2011.
- Jones G, Willett P, Glen RC, et al. Development and validation of a genetic algorithm for flexible docking. J Mol Biol 1997;267:727–48.
- Wu G, Robertson DH, Brooks CL, et al. Detailed analysis of grid-based molecular docking: a case study of CDOCKER-A CHARMm-based MD docking algorithm. J Comput Chem 2003;24:1549–62.
- Labute P. The generalized born/volume integral implicit solvent model: estimation of the free energy of hydration using London dispersion instead of atomic surface area. J Comput Chem 2008;29:1693–8.
- Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 2009;30:2785–91.
- Glide, version 6.6. New York, NY: Schrödinger, LLC; 2015.
- Kirton SB, Murray CW, Verdonk ML, et al. Prediction of binding modes for ligands in the cytochromes P450 and other heme-containing proteins. Proteins 2005;58:836–44.
- Li X, Bian JL, Wang N, et al. Novel naphtho[2,1-d]oxazole-4,5-diones as NQO1 substrates with improved aqueous solubility: design, synthesis, and in vivo antitumor evaluation. Bioorg Med Chem 2016;24:1006–13.
- Avdeef A, Tsinman O. Miniaturized rotating disk intrinsic dissolution rate measurement: effects of buffer capacity in comparisons to traditional wood’s apparatus. Pharm Res 2008;25:2613–27.
- Sun HP, Jia JM, Jiang F, et al. Identification and optimization of novel Hsp90 inhibitors with tetrahydropyrido[4,3-d]pyrimidines core through shape-based screening. Eur J Med Chem 2014;79:399–412. −
- Fuchs JE, Spitzer GM, Javed A, et al. Minor groove binders and drugs targeting proteins cover complementary regions in chemical shape space. J Chem Inf Model 2011;51:2223–32.
- Hawkins PCD, Skillman AG, Nicholls A. Comparison of shape-matching and docking as virtual screening tools. J Med Chem 2007;50:74–82.
- Kirchmair J, Distinto S, Markt P, et al. How to optimize shape-based virtual screening: choosing the right query and including chemical information. J Chem Inf Model 2009;49:678–92.
- Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem 2010;53:2719–40.
- OMEGA, 2.4.6. Santa Fe, NM: OpenEye Scientific Software; 2012.
- Xue L, Godden JW, Stahura FL, et al. Design and evaluation of a molecular fingerprint involving the transformation of property descriptor values into a binary classification scheme. J Chem Inf Comput Sci 2003;43:1151–7.
- Röhrig UF, Majjigapu SR, Chambon M, et al. Detailed analysis and follow-up studies of a high-throughput screening for indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors. Eur J Med Chem 2014;84:284–301.
- Röhrig UF, Majjigapu SR, Vogel P, et al. Challenges in the discovery of indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors. J Med Chem 2015;58:9421–37.
- Lang RW, Wenk PF. Synthesis of selectively trifluoromethylated pyridine derivatives as potential antihypertensives. Helv Chim Acta 1988;71:596–601.
- Yu CJ, Zheng MF, Kuang CX, et al. Oren-gedoku-to and its constituents with therapeutic potential in Alzheimer’s disease inhibit indoleamine 2, 3-dioxygenase activity in vitro. J Alzheimers Dis 2010;22:257–66.
- Yang S, Li X, Hu F, et al. Discovery of tryptanthrin derivatives as potent inhibitors of indoleamine 2,3-dioxygenase with therapeutic activity in Lewis Lung Cancer (LLC) tumor-bearing mice. J Med Chem 2013;56:8321–31.