
Abstract
In this study, new chalcone compounds having the chemical structure of 6-(3-aryl-2-propenoyl)-2(3H)-benzoxazolones (1–8) were synthesised and were characterised by 1H-NMR, 13 C-NMR, and HRMS spectra. Cytotoxic and carbonic anhydrase (CA) inhibitory effects of the compounds were investigated. Cytotoxicity results pointed out that compound 4, 6-[3-(4-trifluoromethylphenyl)-2-propenoyl]-3H-benzoxazol-2-one, showed the highest cytotoxicity (CC50) and potency-selectivity expression (PSE) value, and thus can be considered as a lead compound of this study. According to the CA inhibitory results, IC50 values of the compounds 1–8 towards hCA I were in the range of 29.74–69.57 µM, while they were in the range of 18.14 – 48.46 µM towards hCA II isoenzyme. Ki values of the compounds 1–8 towards hCA I were in the range of 28.37 ± 6.63–70.58 ± 6.67 µM towards hCA I isoenzyme and they were in the range of 10.85 ± 2.14 – 37.96 ± 2.36 µM towards hCA II isoenzyme.
Introduction
Cancer is the second cause of death in the world after the cardiovascular system diseases. According to The World Health Organisation (WHO) report, 13.1 million people will die because of cancer by 2030Citation1. Anticancer drugs in clinics have several adverse effects such as nausea, vomiting, hair loss, and pain, in addition to low selectivity and drug resistance problemsCitation2. Their associated limitations and adverse effects are still prompting the researchers to develop more potent, selective, and safer anticancer drug candidates. Chemotherapeutic drugs commonly used for cancer treatment in clinics are alkylating anticancer agents. These compounds interact with amino and hydroxyl groups which are available nucleic acids and proteins and lead to unwanted side effects in other tissues except neoplasmsCitation3.
α,β-unsaturated ketones are bioactive moieties having alkylation ability. They have an affinity for thiolsCitation4,Citation5 while they are either inert or far less reactive towards amino and hydroxyl groups which are available nucleic acidsCitation6,Citation7. It was reported that before the cell division, level of glutathione, which is a thiol compound, increasesCitation8. That is why it can be supposed that compounds which are thiol selective alkylators may perform selective toxicity against tumour tissuesCitation9–11 and these types of compounds may have advantages over available anticancer drugs in the market.
Chalcones are 1,3-diaryl-2-propen-1-ones, which consist of two aromatic rings connected by a three-carbons chain including α,β-unsaturated carbonyl systemCitation12. Depending on substitution on the aryl ring, chalcones have a wide range of biological activities such as antiinflammatoryCitation13,Citation14, antimicrobialCitation15,Citation16, antioxidantCitation17, cytotoxic/anticancerCitation18,Citation19, chemopreventiveCitation20, topoisomerase inhibitingCitation4, carbonic anhydrase (CA) inhibitingCitation21,Citation22 and acetylcholine esterase inhibiting activitiesCitation23.
Benzoxazolones are considered as “privileged scaffolds” in the design of pharmacological probesCitation24. Benzoxazolones have high flexibility for chemical modifications allowing changes to the characteristics of side-chains on a rigid platformCitation25. As a result, benzoxazolones exhibit various biological activities such as anti-HIVCitation26, anticancerCitation27,Citation28, analgesicCitation29, antiinflammatoryCitation30, antimicrobialCitation31, and antioxidantCitation32 activities. The functionalisation of the nitrogen atom at the third position of the benzoxazolone moiety is of interest since the electronic characteristics of this atom can be decisive for the biological activityCitation24.
Some chalcones bearing 2(3H)-benzoxazolone were reported with strong cytotoxic activityCitation33,Citation34. However, there is a very limited number of studies about them. One of them is Ivanova and co-workers’ study. They reported antileukemic effects of benzoxazolone derived chalcones on BV-173 cell-line by inducing apoptotic cell deathCitation33. The fact that these compounds showed selective and strong anticancer activity pointed out the importance of benzoxazolone bearing chalcone molecules in designing new anticancer candidate molecules.
CA is a metalloenzyme that catalyses the interconversion between CO2 and bicarbonate. CAs play an important role in many physiological and pathological processes such as biosynthetic reactions (such as gluconeogenesis, lipogenesis, and ureagenesis), respiration and transport of CO2/bicarbonate, electrolyte secretion in a variety of tissues/organs, calcification, and tumorigenicityCitation35,Citation36.
There are eight CA families which are genetically different such as the α-, β-, γ-, δ-, ζ- η-, θ-, and the recently reported ι-CAsCitation37. α-CAs are available in human and it has 15 subtypes. Three of them (CA VIII, X, and XI) are non-catalytic as do not possess a zinc ion in their active site. The other 12 CA isozymes have differences in terms of catalytic activity, location, and cell distributions. According to the location, these are cytosolic isozymes (CA I, II, III, VII, and XIII), membrane-bounded isozymes (CA IV, IX, XII, and XIV), secreted salivary isozyme (CA VI), and mitochondrial isozyme (CA V)Citation38–40.
Activation or inhibition of several CAs is a potential strategy for diagnosis and/or treatment of several diseases such as glaucoma, epilepsy, several neurological diseases, and obesity. Antiglaucomal agents (CA II, IV, and XII), diuretics (CA II, IV, XII, and XIV), antiepileptics (CA VII, and XIV) inhibit several isoenzymes of CA. Some chalcone compounds have been reported as attractive scaffolds for the development of new CA inhibitorsCitation21,Citation41.
CA inhibitors commonly include sulfonamideCitation35,Citation42 or phenolCitation22,Citation35,Citation41 functional group. However, there are several studies reporting that chalcone compounds without these functional groups showed potent CA inhibiting effectCitation43–46. In addition, there was no study about CA inhibiting potency of benzoxazolone or its chalcone derivatives.
In this study, it was aimed to synthesise some chalcone compounds having the chemical structure of 6-(3-aryl-2-propenoyl)-2(3H)-benzoxazolones (1–8) in which α,β-unsaturated ketone moiety is available to evaluate their cytotoxicity (towards both tumour cell lines and non-tumour cells) and inhibition profile towards CA hCAI and II with the expectation to find out a new candidate molecule/s which may direct further designs.
Experimental
Chemistry
The nuclear magnetic resonance (NMR) spectra (1H-NMR, and 13C-NMR) were recorded on a Bruker AVANCE III 400 MHz (Bruker, Karlsruhe, Germany) spectrometer [400 MHz (1H) and 100 MHz (13C)]. Chemical shifts are given as δ values in ppm against tetramethylsilane as the internal standard and J values were expressed in Hz. Mass spectra of the compounds were taken using a liquid chromatography ion trap-time of flight tandem mass spectrometer (Shimadzu, Kyoto, Japan) equipped with an electrospray ionisation (ESI) source, operating in both positive and negative ionisation mode. Shimadzu’s LCMS Solution software was used for data analysis. Melting points were determined using an Electrothermal 9100 instrument (IA9100, Bibby Scientific Limited, Staffordshire, UK) and are uncorrected. Reactions were monitored by thin-layer chromatography (TLC) using silica gel 60 HF254 (Merck KGaA, Darmstadt, Germany).
Synthesis of 6-acetyl-2(3H)-benzoxazolone
Dimethylformamide (13 ml, 172 mmol) was slowly added on aluminum chloride (80 g, 600 mmol). The mixture was heated at 45 °C for 5 min. 2(3H)-benzoxazolone (8.1 g, 60 mmol) and acetyl chloride (6,4 ml, 90 mmol) were added into the solution of aluminum chloride (Scheme 1). Then the reaction mixture was heated at 80 °C for 3 h and poured on ice water (200 ml) with HCl (30 ml, 37%). The precipitated crude product was collected by filtration and it was air-dried and crystallised from ethanol. (Yield: 77%, m.p: 231–234 °C, brown crystals)Citation27.
Scheme 1. Synthesis of the compounds 1–8. Ar: Phenyl (1); 4-methylphenyl (2); 4-Methoxyphenyl (3); 4-trifluoromethylphenyl (4); 3-hyrdoxyphenyl (5); 4-isopropylphenyl (6); 4-dimethylaminophenyl (7); 4-benzyloxyphenyl (8).
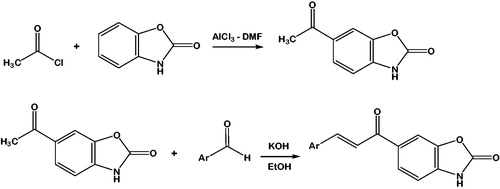
Synthesis of chalcone compounds 1, 2, 3, 4, and 6
To the mixture of 6-acetyl-2(3H)-benzoxazolone (ketone, 5.6 mmol) and a suitable aldehyde [benzahdehyde (1), 4-methylbenzaldehyde (2), 4-methoxybenzaldehyde (3), 4-trifluorobenzaldehyde (4), 4-isopropylbenzaldehyde (6)] in ethanol (5 ml) in 1:1 mol ratios, aqueous solution of KOH (10%, 5 ml) was added (Scheme 1). Reaction content was stirred at room temperature for 24 h. Reactions were followed by TLC. When the reaction finished, the content of the reaction flask was poured on ice-water (100 ml) and neutralised by HCl (37%). The precipitated solid product was filtered and washed with cold waterCitation27. The crude compounds were purified by crystallisation from a suitable solvent (Acetonitrile: ethanol for compounds 1–4; acetonitrile: methanol for compound 6)41.
6-(3-Phenyl-2-propenoyl)-3H-benzoxazol-2-one (1)
Yield 77%. Mp: 230–232 °C. 1H-NMR (DMSO-d6) δ (ppm) 12.03(bs, 1H, NH), 8.11(d, 1H, arom. H, J = 1.2 Hz), 8.07(dd,1H, arom. H, J1 = 8.2 Hz, J2 = 1.6 Hz), 7.99(d,1H, Ar-CH=, J = 15.5 Hz), 7.89–7.91(m, 2H, arom. H), 7.74(d, 1H, =CHCO, J = 15.5 Hz), 7.47(d, 1H, arom. H, J = 1.2 Hz), 7.45(d, 2H, arom. H, J = 2.5 Hz), 7.24(d, 1H, arom. H, J = 8.2 Hz). 13C-NMR (DMSO-d6) δ (ppm) 187.8, 154.9, 144.2, 143.9, 135.5, 135.2, 132.3, 131.0, 129.38, 129.37, 129.36, 126.2, 122.3, 110.0. HRMS (ESI-MS) m/z calculated [M + H]+ 266.0812; measured 266.0803.
6-[3-(4-Metyhlphenyl)-2-propenoyl]-3H-benzoxazol-2-one (2)
Yield 83%. Mp: 258–260 °C. 1H-NMR (DMSO-d6) δ (ppm) 8.08 (d, 1H, arom. H, J = 1.5 Hz), 8.05 (dd,1H, arom. H, J1= 8.1 Hz, J2 = 1.5 Hz), 7.93 (d, 1H, Ar-CH=, J = 15.5 Hz), 7.79 (d, 2H, arom. H, J = 8.0 Hz), 7.70 (d, 1H, =CHCO, J = 15.5 Hz), 7.27 (d, 2H, arom. H, J = 8.0 Hz), 7.22 (d, 1H, arom. H, J = 8.1 Hz), 2.35 (s, 3H, CH3). 13C-NMR (DMSO-d6) δ (ppm) 187.7, 155.2, 144.2, 144.0, 141.1, 135.9, 132.5, 132.3, 129.98, 129.97, 129.42, 129.41, 126.1, 121.2, 110.0, 109.8, 21,6. HRMS (ESI-MS) m/z calculated [M + H]+ 280.0968; measured 280.0967.
6-[3-(4-Methoxyphenyl)-2-propenoyl]-3H-benzoxazol-2-one (3)
Yield 58%. Mp: 210–213 °C. 1H-NMR (DMSO-d6) δ (ppm) 8.08 (d, 1H, arom. H, J = 1.4 Hz), 8.04 (dd, 1H, arom. H, J1 = 8.2 Hz, J2 =1.4 Hz), 7.86 (d, 2H, arom. H, J = 8.8 Hz), 7.84 (d, 1H, Ar-CH =, J = 15.5 Hz), 7.71 (d, 1H, =CHCO, J = 15.5 Hz), 7.22 (d,1H, arom. H, J = 8.2 Hz), 7.01 (d,2H, arom. H, J = 8.8 Hz), 3.82 (s, 3H, =OCH3). 13C-NMR (DMSO-d6) δ (ppm) 187.6, 161.8, 155.0, 144.2, 143.9, 135.3, 132.6, 131.3, 127.9, 126.0, 119.7, 114.8, 110.0, 109.9, 55.9. HRMS (ESI-MS) m/z calculated [M + H]+ 296.0917; measured 296.0918.
6-[3-(4-Trifluoromethylphenyl)-2-propenoyl]-3H-benzoxazol-2-one (4)
Yield 80%. Mp: 257–259 °C. 1H-NMR (DMSO-d6) δ (ppm) 7.24 (d, 1H, arom. H, J = 8.2 Hz), 7.78 (d,1H, =CHCO, J = 15.7 Hz), 7.80 (d, 2H, arom. H, J = 8.2 Hz), 8.06 (d, 1H, arom. H, J = 1.6 Hz), 8.09 (d, 1H, Ar-CH =, J = 15.7 Hz), 8.12–8.14 (m, 3H, arom. H). 13C-NMR (DMSO-d6) δ (ppm) 187.7, 154.9, 143.9, 142.2, 139.2, 135.7, 131.9, 130.5, 129.9, 126.2, 124.9, 122.7, 119.1, 110.1. HRMS (ESI-MS) m/z calculated [M + H]+ 334.0686; measured 334.0687.
6-[3-(4-Isopropylphenyl)-2-propenoyl]-3H-benzoxazol-2-one (6)
Yield 33%. Mp: 220–222 °C. 1H-NMR (DMSO-d6) δ (ppm) 12.08 (bs, 1H, NH), 8.10 (d, 1H, arom. H, J = 1.4 Hz), 8.06 (dd, 1H, arom. H, J1=8.2 Hz, J2 =1.4 Hz), 7.93 (d, 1H, Ar-CH=, J = 15.5 Hz), 7.82 (d, 2H, arom. H, J = 8.2 Hz), 7.72 (1H, d, =CHCO, J = 15.5 Hz) 7.34 (d, 2H, arom. H, J = 8.2 Hz), 7.24 (d, 1H, arom. H, J = 8.2 Hz), 2.98–2.89 (m, 1H,CH), 1.22 (d, 6H, CH3, J = 6.9 Hz). 13C-NMR (DMSO-d6) δ (ppm) 187.8, 154.9, 151.9, 144.3, 143.9, 135.3, 132.9, 132.4, 129.55, 129.54, 127.35, 126.12, 121.3, 110.0, 33.9, 24.1. HRMS (ESI-MS) m/z calculated [M + H]+ 308.1281; measured 308.1286.
Synthesis of the compounds 5, 7, and 8
To the mixture of 6-acetyl-2(3H)-benzoxazolone (ketone, 5.6 mmol) and a suitable aldehyde [3-hydroxybenzaldehyde (5), 4-dimethylaminobenzaldehyde (7), 4-benzyloxybenzaldehyde (8)] in 1:1 mol ratios in ethanol (2 ml), aqueous solution of KOH (10%, 2 ml) was added. Then the mixture was irradiated by microwave (50–80 °C, 25–60 W) for 15 min (compound 7), 20 min (compound 8) and 30 min (compound 5). Reactions were followed by TLC. When the reaction finished, the content of the reaction flask was poured on ice-water (100 ml) and neutralised by HCl (37%). The solid precipitated was filtered and washed with cold water. The crude compounds were purified by crystallisation from acetonitrile.
The chemical structures of the compounds were confirmed by 1H-NMR, 13C-NMR, and HRMS.
6-[3-(3-Hydroxyphenyl)-2-propenoyl]-3H-benzoxazol-2-one (5)
Yield 44%. Mp: 277–279 °C. 1H-NMR (DMSO-d6) δ (ppm) 12.10 (bs, 1H, NH), 9.61(s, 1H, OH), 8.08 (d, 1H, arom. H, J = 1.5 Hz), 8.03 (dd, 1H, arom. H, J1 = 8.4 Hz, J2 =1.5 Hz), 7.86 (d, 1H, Ar-CH =, J = 15.4 Hz), 7.62 (d, 1H, =CHCO, J = 15.4 Hz), 7.31 (d, 1H, arom. H, J = 7.7 Hz), 7.24 (d, 1H, arom. H, J = 7.7 Hz), 7.21 (d, 2H, arom. H, J = 8.05 Hz), 6.85 (dd, 1H, arom. H, J1 = 8.05 Hz, J2 =1.5 Hz). 13C-NMR (DMSO-d6) δ (ppm) 188.1, 158.4, 155.2, 144.7, 144.2, 136.7, 135.6, 132.6, 130.5, 126.4, 120.6, 118.4, 116.0, 110.2. HRMS (ESI-MS) m/z calculated [M + H]+ 282.0761; measured 282.0746.
6-[3-(4-Dimethylaminophenyl)-2-propenoyl]-3H-benzoxazol-2-one (7)
Yield 48%. Mp: 248–250 °C. 1H-NMR (DMSO-d6) δ (ppm) 12.02 (1 H, bs, NH), 8.00 (d,1H, arom. H, J = 8.3 Hz), 7.85 (d, 1H, Ar-CH =, J = 10.4 Hz), 7.70 (d,2H, arom. H, J = 8.9 Hz), 7.67 (d, 1H, =CHCO, J = 10.4 Hz), 7.66 (s,1H, arom. H), 7.19 (d, 1H, arom. H, J = 8.3 Hz), 6.73 (d, 2H, arom. H, J = 8.9 Hz), 2.99 (s, 6H, CH3). 13 C-NMR (DMSO-d6) δ (ppm) 187.0, 155.2, 152.7, 145.7, 144.1, 135.1, 133.3, 131.5, 125.9, 122.8, 116.4, 112.4, 110.1, 110.0, 40.6. HRMS (ESI-MS) m/z calculated [M + H]+ 309.1234; measured 309.1230.
6-[3-(4-Benzyloxyphenyl)-2-propenoyl]-3H-benzoxazol-2-one (8)
Yield 63%. Mp: 243–245 °C. 1H-NMR (DMSO-d6) δ (ppm) 8.07 (s, 1H, arom. H), 8.03 (dd, 1H, arom. H, J1 = 8.2 Hz, J2 = 1.3 Hz), 7.87–7.82 (m, 3H, arom. H), 7.69 (d, 1H, arom. H, J = 15.4 Hz), 7.46–7.30 (m, 4H, arom. H), 7.20 (d, 2H, arom. H, J = 8.4 Hz), 7.08 (d, 2H, arom. H, J = 8.8 Hz), 5.20 (s, 2H, CH2). 13C-NMR (DMSO-d6) δ (ppm) 187.8, 161.1, 155.3, 144.4, 144.2, 137.4, 135.6, 132.7, 131.5, 129.2, 128.6, 128.5, 128.3, 126.2, 120.0, 115.9, 110.2, 110.1, 70.1. HRMS (ESI-MS) m/z calculated [M + H]+ 372.1230; measured 372.1218.
Biological activity
Cytotoxicity test
Materials
The following chemicals and reagents were obtained from the indicated companies: Dulbecco’s modified Eagle’s medium (DMEM) from GIBCO BRL (Grand Island, NY); foetal bovine serum (FBS), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), doxorubicin (DXR), and dimethyl sulphoxide (DMSO) from Wako Pure Chem. Ind. (Osaka, Japan); and culture plastic dishes and plates (96-well) were purchased from Becton Dickinson (Franklin Lakes, NJ).
Cell culture
Human normal oral mesenchymal cells, gingival fibroblast (HGF), and periodontal ligament fibroblast (HPLF) established from the first premolar tooth extracted from the lower jaw of a 12-year-old girlCitation47 and human OSCC cell line HSC-2 (derived from tongue), purchased from Riken Cell Bank (Tsukuba, Japan), were cultured at 37 °C in DMEM supplemented with 10% heat-inactivated FBS, 100 units/ml penicillin G, and 100 µg/ml streptomycin sulphate under a humidified 5% CO2 atmosphere. HGF and HPLF cells at 10–18 population doubling levels were used in this study.
Assay for cytotoxic activity
Cells were inoculated at 2.5 × 103 cells/0.1 ml in a 96-microwell plate (Becton Dickinson Labware, Franklin Lakes, NJ). After 48 h, the medium was replaced with 0.1 ml of fresh medium containing different concentrations of single test compounds. Cells were incubated further for 48 h and the relative viable cell number was then determined by the MTT methodCitation22,Citation48–54. All benzoxazolone derivatives were dissolved with DMSO at the concentration of 40 mM and stored until use. Control cells were treated with the same amounts of DMSO (0.00156, 0.03125, 0.0625, 0.125, 0.25, 0.5, and 1.0%) and the cell damage induced by DMSO was subtracted from that induced by test agents. In brief, cells were stained with MTT reagent, dissolved with DMSO, and the absorbance of the MTT-stained cell lysate was measured at 560 nm, using a microplate reader (Infinite F 50R, TECAN, Kawasaki, Japan). Control cells were treated with the same amounts of DMSO and the cell damage induced by DMSO was subtracted from that induced by test agents. The concentration of compound that reduced the viable cell number by 50% (CC50) was determined from the dose-response curve and the mean value of CC50 for each cell type was calculated from triplicate assays.
Calculation of tumour specificity
Tumour specificity (TS) was calculated using the following equation: TS = Mean CC50 against three normal oral cell types (HGF, HPLF)/Mean CC50 against four OSCC cell lines (HSC-2). Since HGF cells were derived from gingival tissue, the relative sensitivity of these cells was also compared (as mean CC50 against HGF/mean CC50 against HSC-2).
Calculation of potency-selectivity expression
Potency-selectivity expression (PSE) was calculated by the following equation: PSE = Mean CC50 against two normal oral cell types/(CC50 against four OSCC cell lines)Citation2×100 (HGF, HPLF, HSC-2) and as mean CC50 against HGF/(CC50 against HSC-2)2×100 using the pair of cell types from the same tissue (gingiva).
Carbonic anhydrase inhibition
The purification of cytosolic CA isoenzymes (CA I and CA II) was previously described with a simple one-step method by a Sepharose-4B-L tyrosine-sulphanilamide affinity chromatographyCitation42,Citation53,Citation55–58. The protein quantity in the column effluents was determined spectrophotometrically at 280 nm. Sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) was applied with a Bio-Rad Mini Gel system Mini-PROTEIN system (Bio-Rad Laboratories, Inc., Shanghai, China) after purification of both CA isoenzymes. Briefly, it was performed in acrylamide for the running (10%) and the stacking gel (3%) contained SDS (0.1%), respectively. The increase in absorbance of the reaction medium was spectrophotometrically recorded at 348 nm. Also, the quantity of protein was determined at 595 nm according to the method as described previouslyCitation55–60. Bovine serum albumin was used as a standard protein. The IC50 values were obtained from activity (%) versus compounds plots. For the calculation of Ki values, three different concentrations were used. The Lineweaver–Burk curves were drawn and calculations were realised as beforeCitation55–60.
Results and discussion
Chemistry
The compounds 1–8, 6-(3-aryl-2-propenoyl)-2(3H)-benzoxazolones, were synthesised successfully according to Scheme 1. Aryl part was changed as phenyl (1), 4-methylphenyl (2), 4-methoxyphenyl (3), 4-trifluoromethylphenyl (4), 3-hydroxyphenyl (5), 4-isopropylphenyl (6), 4-dimethylaminophenyl (7), and 4-benzyloxyphenyl (8). The compounds except 1, 3, 5 were reported for the first time in this study.
As indicated in Scheme 1, 6-acetyl-2(3H)-benzoxazolone was synthesised by Friedel–Crafts acylation first. The product was obtained in good yield and purity. Since direct acylation of 2(3H)-benzoxazolone is regioselective and always leads to a 6-acyl derivativeCitation34.
The chalcones 1–4, and 6 were synthesised by the conventional synthesis method using Claisen–Schmidt condensation reaction between 6-acetyl-2(3H)-benzoxazolone and suitable aldehyde as shown in Scheme 1. On the other hand, chalcones 5, 6, 7, and 8 were synthesised by microwave irradiation method.
The structures of the compounds were confirmed by 1H-NMR, 13C-NMR, and HRMS. In particular, analysis of 1H-NMR spectra of the compounds 1–8 revealed that all compounds (except compound 7) were both geometrically pure and were configured trans, as derived from coupling constant J: 15.4–15.7 Hz for vinyl protons, observed at 7.60–8.09 ppm. The compound 7 was configured cis with coupling constant J: 10.4 Hz for vinyl protons. The aromatic ring protons were observed at the area of 7.0–8.0 ppm, as expected. The 13 C-NMR spectra of all compounds, carbons of carbonyl groups were appeared about 187 ppm, as expected. HRMS results were also confirmed the chemical structures of the compounds.
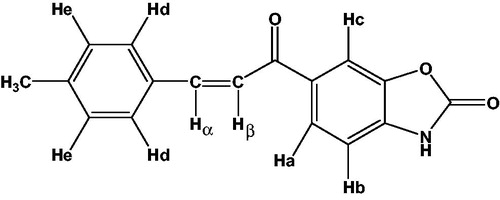
Although all the spectral results were presented in the experimental section, the details of 1H-NMR spectra of compound 2 are given in as an example.
Figure 1. The details of 1H-NMR spectra of compound 2 as represantative 1H-NMR. δ (ppm) Ha: 8.05 (dd, 1H, JHa-Hb: 8.1 Hz, JHa-Hc: 1.5 Hz), Hb: 7.22 (d, 1H, JHa-Hb: 8.1 Hz), Hc: 8.08 (d, 1H, JHa-Hc: 1.5 Hz), Hd: 7.79 (d, 2H, JHd-He: 8.0 Hz), He: 7.27 (d, 2H, JHd-He: 8.0 Hz), Hα: 7.93 (d, 1H, JHα-Hβ: 15.5 Hz), Hβ: 7.70 (d, 1H, JHα-Hβ: 15.5 Hz) CH3 protons: 2.35 (s, 3H).
Cytotoxic/anticancer activity
The cytotoxicities of the synthesised compounds, 1–8, have been investigated in vitro against oral squamous cancer cell line (HSC-2) and human normal oral cells (HGF and HPLF). The reference compounds used were DXR and 5-fluorouracil (5-FU) which are clinically in use for cancer treatment. Cytotoxicity results of compounds 1–8 were presented in . All of the compounds showed lower cytotoxicity than DXR but showed higher cytotoxicity than 5-FU. Cytotoxicities of the compounds were in the range of 4.0– 30.2 µM towards HSC-2 cell line. The compounds showed 1.3–9.4 times more cytotoxic than 5-FU. When the cytotoxicities of the compounds were considered, compound 4, 6-[3–(4-Trifluoromethylphenyl)-2-propenoyl]-3H-benzoxazol-2-one, was found as the most potent cytotoxic molecule towards HSC-2 cell line.
Table 1. Cytotoxic activities of the compounds 1–8 towards human OSCC cell lines and human oral normal cells
The first point to be considered for the compounds is whether they are tumour cytotoxins. Therefore, the compounds in the series were also evaluated against HGF and HPLF non-malignant cells and the data are presented in . Under clinical conditions, tumours are surrounded by different types of normal cells. Therefore, tumour selectivity values (TS) were calculated. TS were calculated for the compounds by dividing the average CC50 value towards normal cells into the average CC50 value towards cancer cell lines (). According to , the TS values of the compounds were greater than 1. This indicated that the compounds were tumour-selective. The cytotoxicity results pointed out that compound 2 had the highest TS value in the series.
Lead compounds should have both prominent cytotoxic potential and selective cytotoxicity for tumours. In order to identify the most promising compounds in terms of both good potencies and selectively cytotoxic, the PSE values were calculated according to equation i.e. [Average TS figure (determination of TS)/Average CC50 value (a measure of potency)Citation2] × 100. PSE values were presented in . The PSE values of the compounds were in the range of 8–159. The number of compounds in the series with a PSE value greater than 30 is six. These six compounds appear to serve as lead compounds to improve new analogues.
The substituent on the phenyl ring rather than hydrogen was found to be a generally useful modification in increasing cytotoxicity (1.5 and 5.5 times) and selectivity (1.6–2.7 times). An exception was compound 7 for cytotoxicity and selectivity. This may result from the changes in the electronic structure of the phenyl ring. When methyl substituent is introduced into the 4 position of the phenyl ring (compound 2), the cytotoxicity increased 2.2 times comparing to the non-substituted derivative (compound 1), and TS increased 2.7 times. On the other hand, when the isopropyl group was substituted at the 4 position of phenyl ring in compound 6, the increase in cytotoxicity (2.2 times) was the same as the methyl derivative (compound 2) and the increase in selectivity of compound 6 was 1.8 times. These results suggested that the substitution of an alkyl group at the 4 position of phenyl was a useful modification in both the cytotoxicity and selectivity, especially when small size alkyl group substitution had better in increasing selectivity. It suggested that 4 position of the phenyl ring may play a crucial role in the interaction of the molecule with the active site of enzyme or protein. Compound 4, wherein the trifluoromethyl group was substituted at the 4 position of the phenyl ring, supported this suggestion although the increase in selectivity in compound 4 was slightly less (1.8 times) than compound 2. Additionally to TS, cytotoxicity increased dramatically (5.5 times) in compound 4. This is noteworthy. When the electron-donating groups with resonance effect as 4-methoxy and 4-benzyloxy groups (compounds 3 and 8) were substituted at the 4 position of the phenyl ring, the increase in cytotoxicity (1.5 and 1.9 times, respectively) was lower than compounds 2 and 4 which carry 4-methyl and 4-trifluoromethyl substituents on the phenyl ring. However, when the substitution of 4 position of phenyl was dimethylamine (compound 7) cytotoxicity reduced 1.4 times comparing to the non-substituted derivative, compound 1. In addition, substituents at 4 position in compounds 3 and 8 lead to a lower increase in selectivity comparing to the compounds 2 and 4’s.
When the PSE values of the compounds were considered for the compounds having substitution at 4 position of phenyl ring, the results observed were interesting. The total electronic contributions of the substituents at 4 position of phenyl ring were as follows according to literatureCitation61; σisopropyl: −0.15, σmethyl: −0.17, σmethoxy: −0.27, σdimethylamine: −0.83 and σtrifluoromethyl: 0.54. The compounds bearing the substituent at issue were 6, 2, 3, 7, and 4. The results presented at show that the replacement of hydrogen by electron-withdrawing substituent at the 4 position of the phenyl ring increased the PSE values of the compounds (except for compound 2). It means as long as the Hammet value increased, the PSE value of the compound increased. There was a high positive correlation between σ and PSE values (r: 0.7918). It also reflects that electron-withdrawing substituents at the 4 position of phenyl had a positive effect on PSE.
Compound 4 carrying 4-trifluoromethyl substituent on phenyl ring was the most impressive compound of our design in terms of cytotoxicity and PSE value. This compound can be considered as a drug candidate for further investigations. The cytotoxicity and PSE value of the compound 4 were 5.5 and 9.9 times more potent than non-substituted compound 1, respectively. While the cytotoxicity of compound 4 was 8 times low cytotoxic than DXR, it was 9.4 times more cytotoxic than reference compound 5-FU and its PSE (about 2 times) value was higher than 5-FU.
Another issue that can contribute to cytotoxicity can be the solubility of the compounds which was reflected by Partition Coefficients (logP). The logP values of the compounds were calculated by ChemDraw program (Ultra 7.0)[logP values: 2.71 (compound 1), 3.20 (compound 2), 2.58 (compound 3), 3.63 (compound 4), 2.32 (compound 5), 3.94 (compound 6), 2.99 (compound 7), 4.31 (compound 8)]. The correlations between logP and CC50 values were investigated and it was noticed that there was a low negative correlation (r: −0.2614).
Carbonic anhydrase inhibitory effects
The human CA I and II inhibitory effects of the compounds 1–8 were reported for the first time in this study and the inhibition data are shown in . Acetazolamide (AZA) was used as a reference drug for both hCA I and II isoenzymes. The compounds 1–8 showed lower CA inhibitory effects than the reference drug, AZA. According to , IC50 values of the compounds 1–8 towards hCA I were in the range of 29.74–69.57 µM, while they were in the range of 18.14–48.46 µM towards hCA II isoenzyme. IC50 values of AZA were 16.58 and 8.37 µM towards hCA I and hCA II, respectively. According to the IC50 values, compound 4, wherein the trifluoromethyl group was substituted at the 4 position of the phenyl ring showed the best activity (29.74 µM) towards hCA I and compound 8 carrying 4-benzyloxy substituent on phenyl ring showed the best activity (18.14 µM) towards hCA II.
Table 2. Inhibitory effects of the compounds 1–8 on hCA I and hCA II isoenzymes.
According to , Ki values (inhibitory potency) of the compounds 1–8 towards hCA I were in the range of 28.37 ± 6.63 – 70.58 ± 6.67 µM towards hCA I isoenzyme and they were in the range of 10.85 ± 2.14 – 37.96 ± 2.36 µM towards hCA II isoenzyme. Ki values of AZA were 30.18 ± 7.77 µM and 4.41 ± 0.55 µM towards hCA I and hCA II, respectively. So, some compounds (compounds 3, 4, 6, and 8) had similar Ki values with AZA towards hCA I while all compounds had higher Ki values than AZA towards hCA II which suggests that they are worse inhibitor than AZA.
Conclusions
Newly synthesized 6-(3-aryl-2-propenoyl)-2(3H)-benzoxazolones, the compounds 1–8, were reported here for the first time with their cytotoxic properties and potential inhibitory effects on hCA I and II. According to the cytotoxicity results of the compounds, compound 4 was the most impressive lead compound of the study with remarkably PSE value (159) for further testing and investigations. On the other hand, according to Ki values compounds 2, 3, 4, 6, and 8 can be considered for the development of new CA I inhibitors due to similar Ki values to AZA but they are not suitable derivatives for the development of new CA II inhibitors since they had higher Ki values than AZA.
Disclosure statement
The authors declare no conflict of interest.
Additional information
Funding
Related Research Data
References
- Ferlay J, Colombet M, Soerjomataram I, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer 2019;144:1941–1953.
- Raghavendra NM, Pingili D, Kadasi S, et al. Dual or multi-targeting inhibitors: the next generation anticancer agents. Eur J Med Chem 2018;143:1277–300.
- Ashby J. Potential carcinogenicity of alkylating agents. Chem Brit 1978;14:595.
- Gul HI, Cizmecioglu M, Zencir S, et al. Cytotoxic activity of 4′-hydroxychalcone derivatives against Jurkat cells and their effects on mammalian DNA topoisomerase I. J Enzyme Inhib Med Chem 2009;24:804–7.
- Erciyas E, Erkaleli HI, Cosar G. Antimicrobial evaluation of some styryl ketone derivatives and related thiol adducts. J Pharm Sci 1994;83:545–8.
- Dimmock JR, Kumar P, Allen TM, et al. Synthesis and cytotoxic evaluation of some carbohydrazones and thiocarbohydrazones of various unsaturated ketones and related Mannich bases. Pharmazie 1997;52:182–6.
- Das U, Sharma RK, Dimmock JR. 1,5-diaryl-3-oxo-1,4-pentadienes: a case for antineoplastics with multiple targets. Curr Med Chem 2009;16:2001–20.
- Stern H. Sulfhydryl groups and cell division. Science 1956;124:1292–3.
- Dimmock JR, Raghavan SK, Bigam GE. Evaluation of Mannich bases of 2-arylidene-1,3-diketones versus murine P388 leukemia. Eur J Med Chem 1988; 23:111–7.
- Dimmock JR, Sidhu KK, Chen M, et al. Evaluation of some Mannich bases of cycloalkanones and related compounds for cytotoxic activity. Eur J Med Chem 1993;28:313–22.
- Pati HN, Das U, Sakagami H, et al. 1,3-diaryl-2-propenones and 2-benzylidene-1,3-indandiones: a quest for compounds displaying greater toxicity to neoplasms than normal cells. Archiv Der Pharmazie 2010;343:535–41.
- Orlikova B, Tasdemir D, Golais F, et al. Dietary chalcones with chemopreventive and chemotherapeutic potential. Genes Nutr 2011;6:125–47.
- Takahashi T, Takasuka N, Iigo M, et al. Isoliquiritigenin, a flavonoid from licorice, reduces prostaglandin E2 and nitric oxide, causes apoptosis, and suppresses aberrant crypt foci development. Cancer Sci 2004;95:448–53.
- Nowakowska Z. A review of anti-infective and anti-inflammatory chalcones. Eur J Med Chem 2007;42:125–37.
- Fiore C, Eisenhut M, Ragazzi E, et al. A history of the therapeutic use of liquorice in Europe. J Ethnopharmacol 2005;99:317–24.
- Kocyigit UM, Budak Y, Gürdere MB, et al. Synthesis of chalcone-imide derivatives and investigation of their anticancer and antimicrobial activities, carbonic anhydrase and acetylcholinesterase enzymes inhibition profiles. Arch Physiol Biochem 2018;124:61–8.
- Ghosh A, Mandal S, Banerji A, et al. A new chalcone from Pongamia pinnata and its antioxidant properties. Nat Prod Commun 2009;4:209–10.
- Gul HI, Yamali C, Gunesacar G, et al. Cytotoxicity, apoptosis, and QSAR studies of phenothiazine derived methoxylated chalcones as anticancer drug candidates. Med Chem Res 2018;27:2366–78.
- Yadav VR, Prasad S, Sung B, Aggarwal BB. The role of chalcones in suppression of NF-κB-mediated inflammation and cancer. Int Immunopharmacol 2011;11:295–309.
- Kim HG, Oh HJ, Ko JH, et al. Lanceoleins A-G, hydroxychalcones, from the flowers of Coreopsis lanceolata and their chemopreventive effects against human colon cancer cells. Bioorg Chem 2019;85:274–81.
- Gul HI, Yamali C, Bulbuller M, et al. Anticancer effects of new dibenzenesulfonamides by inducing apoptosis and autophagy pathways and their carbonic anhydrase inhibitory effects on hCA I, hCA II, hCA IX, hCA XII isoenzymes. Bioorg Chem 2018;78:290–7.
- Yamali C, Tugrak M, Gul HI, et al. The inhibitory effects of phenolic Mannich bases on carbonic anhydrase I and II isoenzymes. J Enzyme Inhib Med Chem 2016;31:1678–81.
- Yerdelen KO, Gul HI. Synthesis and anticholinesterase activity of fumaramide derivatives. Med Chem Res 2013;22:4920–9.
- Murty MSR, Ram KR, Rao RV, et al. Synthesis and preliminary evaluation of 2-substituted-1,3-benzoxazole and 3-(3-substituted)propyl-,3-benzoxazol-2(3H)-one derivatives as potent anticancer agents. Med Chem Res 2011;20:576–86.
- Chiarotto I, Feroci M, Orsini M, et al. Electrogenerated N-heterocyclic carbenes: N-functionalization of benzoxazolones. Tetrahedron 2009;65:3704–10.
- Deng B, Cullen MD, Zhou ZG, et al. Synthesis and anti-HIV activity of new alkenyldiarylmethane (ADAM) non-nucleoside reverse transcriptase inhibitors (NNRTIs) incorporating benzoxazolone and benzisoxazole rings. Bioorg Med Chem 2006;14:2366–74.
- Ivanova Y, Momekov G, Petrov O, et al. Cytotoxic Mannich bases of 6-(3-aryl-2-propenoyl)-2(3H)-benzoxazolones. Eur J Med Chem 2007;42:1382–7.
- Kumar D, Jacob MR, Reynolds MB, Kerwin SM. Synthesis and evaluation of anticancer benzoxazoles and benzimidazoles related to UK-1. Bioorg Med Chem 2002;10:3997–4004.
- Unlu S, Onkol T, Dundar Y, et al. Synthesis and analgesic and anti-inflammatory activity of some new (6-acyl-2-benzoxazolinone and 6-acyl-2-benzothiazolinone derivatives with acetic acid and propanoic acid residues. Arch Pharm 2003;336:353–60.
- Koksal M, Gokhan N, Kupell E, et al. Synthesis, analgesic and antiinflammatory properties of certain 5-/6-acyl-3-(4-substituted-1-piperazinylmethyl)-2-benzoxazolinones derivatives. Archiv Der Pharmazie 2005;338:117–25.
- Koksal M, Gokhan N, Erdogan H, et al. Synthesis of 3-(4-substituted benzoylmethyl)-2-benzoxazolinones and screening antimicrobial activities. Farmaco 2002;57:535–8.
- Orhan H, Doğruer DS, Cakir B, et al. The in vitro effects of new non-steroidal antiinflammatory compounds on antioxidant system of human erythrocytes. Exp Toxicol Pathol 1999;51:397–402.
- Ivanova Y, Momekov G, Petrov O. Synthesis of novel substituted 1,3-diarylpropenone derivatives and their in vitro cytotoxic activity. Lett Drug Design Discov 2009;6:353–7.
- Ivanova YB, Momekov GT, Petrov OI. New heterocyclic chalcones. Part 6. Synthesis and cytotoxic activities of 5-or 6-(3-aryl-2-propenoyl)-2(3H)-benzoxazolones. Heterocycl Commun 2013;19:23–8.
- Supuran CT. Carbonic anhydrase inhibitors and their potential in a range of therapeutic areas. Exp Opin Ther Patents 2018;28:709–12.
- Supuran CT, Carbonic anhydrase inhibitor—no donor hybrids and their pharmacological applications. In: Morbidelli L, Bonavida B, eds. Therapeutic application of nitric oxide in cancer and inflammatory sisorders. Cambridge, MA: Academic Press; 2019. pp. 229–242.
- Thacker PS, Shaikh P, Angeli A, et al. Synthesis and biological evaluation of novel 8-substituted quinoline-2-carboxamides as carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2019;34:1172–7.
- Supuran CT. Carbonic anhydrases and metabolism. Metabolites 2018;8:25.
- Supuran CT. Carbonic anhydrase activators. Future Med Chem 2018;10:561–73.
- Nocentini A, Supuran CT. Carbonic anhydrase inhibitors as antitumor/antimetastatic agents: a patent review (2008–2018). Exp Opin Ther Patents 2018;28:729–40.
- Yamali C, Gul HI, Sakagami H, Supuran CT. Synthesis and bioactivities of halogen bearing phenolic chalcones and their corresponding bis Mannich bases. J Enzyme Inhib Med Chem 2016;31:125–31.
- Yamali C, Gul HI, Ece A, et al. Synthesis, molecular modeling, and biological evaluation of 4-[5-aryl-3-(thiophen-2-yl)-4,5-dihydro-1H-pyrazol-1-yl] benzenesulfonamides toward acetylcholinesterase, carbonic anhydrase I and II enzymes. Chem Biol Drug Design 2018;91:854–66.
- Gencer N, Bilen C, Demir D, et al. In vitro inhibition effect of some chalcones on erythrocyte carbonic anhydrase I and II. Artif Cell Nanomed Biotechnol 2013;41:384–8.
- Stellenboom N. Comparison of the inhibitory potential towards carbonic anhydrase, acetylcholinesterase and butyrylcholinesterase of chalcone and chalcone epoxide. J Biochem Mol Toxicol 2019;33:e22240.
- Tutar U, Koçyiğit ÜM, Gezegen H. Evaluation of antimicrobial, antibiofilm and carbonic anhydrase inhibition profiles of 1, 3‐bis‐chalcone derivatives. J Biochem Mol Toxicol 2019;33:e22281.
- Burmaoglu S, Yilmaz AO, Polat MF, et al. Synthesis of novel tris-chalcones and determination of their inhibition profiles against some metabolic enzymes. Arch Physiol Biochem 2019;1–9. DOI: 10.1080/13813455.2019.1623265.
- Kantoh K, Ono M, Nakamura Y, et al. Hormetic and anti-radiation effects of tropolone-related compounds. In Vivo 2010;24:843–51.
- Tugrak M, Yamali C, Sakagami H, Gul HI. Synthesis of mono Mannich bases of 2-(4-hydroxybenzylidene)-2, 3-dihydroinden-1-one and evaluation of their cytotoxicities. J Enzyme Inhib Med Chem 2016;31:818–23.
- Unluer E, Gul HI, Demirtas A, et al. Synthesis and bioactivity studies of 1-aryl-3-(2-hydroxyethylthio)-1-propanones. J Enzyme Inhib Med Chem 2016;31:105–9.
- Sakagami H, Uesawa Y, Masuda Y, et al. Quantitative structure-cytotoxicity relationship of newly synthesized piperic acid esters. Anticancer Res 2017;37:6161–8.
- Gul HI, Tugrak M, Sakagami H. Synthesis of some acrylophenones with N-methylpiperazine and evaluation of their cytotoxicities. J Enzyme Inhib Med Chem 2016;31:147–51.
- Gul HI, Tugrak M, Sakagami H, et al. Synthesis and bioactivity studies on new 4-(3-(4-substitutedphenyl)-3a,4-dihydro-3H-indeno[1,2-c]pyrazol-2-yl) benzenesulfonamides. J Enzyme Inhib Med Chem 2016;31:1619–24.
- Gul HI, Mete E, Eren SE, et al. Designing, synthesis and bioactivities of 4-[3-(4-hydroxyphenyl)-5-aryl-4,5-dihydro-pyrazol-1-yl]benzenesulfonamides. J Enzyme Inhib Med Chem 2017;32:169–75.
- Yamali C, Ozgun DO, Gul HI, et al. Synthesis and structure elucidation of 1-(2,5/3,5-difluorophenyl)-3-(2,3/2,4/2,5/3,4-dimethoxyphenyl)-2-propen-1-ones as anticancer agents. Med Chem Res 2017;26:2015–23.
- Gul HI, Yamali C, Yesilyurt F, et al. Microwave-assisted synthesis and bioevaluation of new sulfonamides. J Enzyme Inhib Med Chem 2017;32:369–74.
- Ozgun DO, Yamali C, Gul HI, et al. Inhibitory effects of isatin Mannich bases on carbonic anhydrases, acetylcholinesterase, and butyrylcholinesterase. J Enzyme Inhib Med Chem 2016;31:1498–501.
- Burmaoglu S, Yilmaz AO, Taslimi P, et al. Synthesis and biological evaluation of phloroglucinol derivatives possessing α‐glycosidase, acetylcholinesterase, butyrylcholinesterase, carbonic anhydrase inhibitory activity. Arch Pharm 2018;351:1700314.
- Timur I, Kocyigit UM, Dastan T, et al. In vitro cytotoxic and in vivo antitumoral activities of some aminomethyl derivatives of 2, 4‐dihydro‐3H‐1, 2, 4‐triazole‐3‐thiones – Evaluation of their acetylcholinesterase and carbonic anhydrase enzymes inhibition profiles. J Biochem Mol Toxicol 2019;33:e22239.
- Gul HI, Tugrak M, Gul M, et al. New phenolic Mannich bases with piperazines and their bioactivities. Bioorg Chem 2019;90:103057.
- Gul HI, Mete E, Taslimi P, et al. Synthesis, carbonic anhydrase I and II inhibition studies of the 1, 3, 5-trisubstituted-pyrazolines. J Enzyme Inhib Med Chem 2017;32:189–92.
- Hansch C, Leo A. Substituent constants for correlation analysis in chemistry and biology. Hoboken, NJ: Wiley; 1979.