
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
A novel series of pyrazole analogues including hydrazones, pyrazolo[4,3-c]-pyridazines, pyrazolo[3,4-e][1,2,4]triazine and pyrazolo[3,4-d][1,2,3]triazoles was designed, synthesised and screened for their in vitro antimicrobial and DHFR inhibition activity. Compounds bearing benzenesulphonamide moiety incorporated with 3-methyl-5-oxo-1H-pyrazol-4(5H)-ylidene) hydrazine 3a or 6-amino-7-cyano-3-methyl-5H-pyrazolo[4,3-c]pyridazine 6a revealed excellent and broad spectrum antimicrobial activity comparable to ciprofloxacin and amphotericin B as positive antibiotic and antifungal controls, respectively. Furthermore, these derivatives proved to be the most active DHFR inhibitors with IC50 values 0.11 ± 1.05 and 0.09 ± 0.91 µM, in comparison with methotrexate (IC50 = 0.14 ± 1.25 µM). The in silico studies were done to calculate the drug-likeness and toxicity risk parameters of the newly synthesised derivatives. Additionally, the high potency of the pyrazole derivatives bearing sulphonamide against DHFR was confirmed with molecular docking and might be used as an optimum lead for further modification.
Introduction
The increased microbial resistance has led to the demand for new bactericidal and fungicidal agentsCitation1. The reasons for resistance are the inaccurate diagnosis as well as misuse and widespread use of antimicrobial agentsCitation2. So, a worldwide effort to search for new generation drugs was stimulated to get new potent, resistance-free, and safer antimicrobial agentsCitation3,Citation4.
Recently, dihydrofolate reductase (DHFR) has been considered to be a universal and attractive enzyme which is present in all organisms. Its essential function is to catalyse the reduction of dihydrofolate to tetrahydrofolate within the thymidylate synthesis cycle. As a result, inhibition of DHFR causes “thymineless death”Citation5–8. Inhibitors of DHFR explored a crucial role in medicine like methotrexate that is a non-selective inhibitor and a confirmed agent used in oncology for the treatment of rheumatoid arthritis and several cancersCitation9. Thus, there are a vast number of interesting target profiles and literatures achievable with DHFR and its inhibitors.
Pyrazoles are a class of interesting heterocyclic compounds characterised by the presence of two nitrogen atoms adjacent to three carbon atoms in five-membered aromatic ring structureCitation10. Pyrazole derivatives play an imperative role in wide spectrum of biological activities including antibacterialCitation11, antifungalCitation12, anti-inflammatoryCitation13–15, analgesicCitation16, oestrogen receptor bindingCitation17, neuroprotectiveCitation18, antineoplastic agentsCitation19. Furthermore, several reports exhibited that pyrazoles I–IV and pyrazolo[3,4-d]pyrimidine V () had high bactericidal and fungicidal activity against the reference strainsCitation20–24. Additionally, the pyrazole-containing cores (VI–VII) were found to be active as antimicrobial and antimalarial through inhibition of DHFRCitation25–27 (). Regarding to their broad spectrum of biological activities, pyrazole ring has been considered as a favourable unit for addition in the field of drug discovery and therefore in the pharmaceutical industryCitation10.
Figure 1. Some reported pyrazole hybrid molecules as antimicrobial agents.
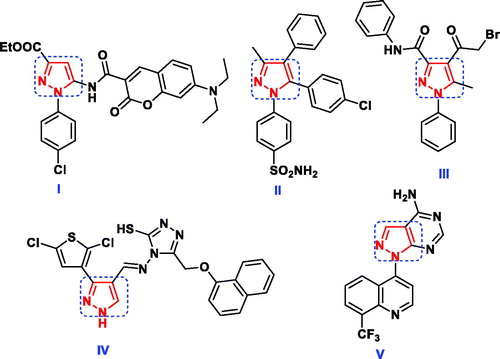
Figure 2. Potent DHFR inhibitors owning pyrazole scaffold as antimicrobial and antimalarial agents.
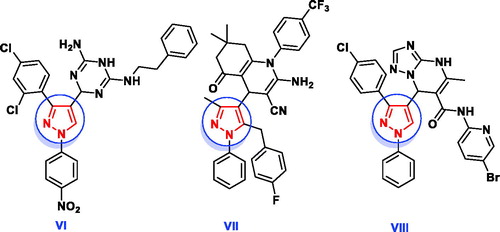
In addition, several pharmacologically active structural units, for example, pyridazines, 1,2,4-triazines and 1,2,3-triazoles are being explored to identify novel lead antimicrobial molecules (). The pyridazine derivative IX revealed promising in vitro antimicrobial activities against Gram + ve and Gram -ve bacteria in comparison with tetracyclineCitation28. Nagawade et al. reported that the pyridazine-3-carboxylic acid analogue X showed gratifying in vitro antibacterial results approaching that of corresponding reference, ciprofloxacinCitation29. Compound containing 1,2,4-triazine nucleus XI has been reported to possess better antimicrobial activity with less toxicity than ciprofloxacinCitation30. The derivative bearing 1,2,3-triazole scaffold XIIa displayed overall encouraging efficiency against all screened microbial strains except Aspergillus niger, while the other derivative XIIb exhibited better antimicrobial potency against all strains except Bacillus subtilis and Aspergillus nigerCitation31.
Figure 3. Reported antimicrobial leads containing pyridazine, 1,2,4-triazine and 1,2,3-triazole moieties.
Motivated by the aforementioned findings, and upon continuation of our research programme in the field of discovery of pyrazole bearing antimicrobial agentsCitation32–34, some novel pyrazole prototypes were designed and synthesised after exploring molecular fusion with pyridazine, triazine and triazole moieties. The synthesised compounds comprising pyrazole motif were evaluated in vitro for their antimicrobial activities against human pathogenic microbes and their inhibitory activities against DHFR enzyme. Finally, molecular docking and in silico studies were used to explain the obtained biological data.
Experimental
Chemistry
All melting points were determined on a Gallenkamp apparatus and are uncorrected. The IR spectra were measured on a Pye-UnicamSP300 instrument in potassium bromide discs. The 1H-NMR and 13C-NMR spectra were recorded on Varian Mercury VX (300 MHz) spectrometer (with operating frequencies 300.07 MHz for 1H using TMS as an internal standard and 75.45 MHz for 13C). Chemical shifts (δ) are reported in parts per million (ppm), and coupling constants (J) are reported in Hertz (Hz). NMR spectra were recorded at temperature (30–45 °C) and were referenced to the residual signals of DMSO-d6. Elemental analyses were carried out by the Micro analytical Centre of Cairo University, Giza, Egypt. The antimicrobial activities were carried out in the Medical Mycology Laboratory of the Regional Centre for Mycology and Biotechnology of Al-Azhar University, Cairo, Egypt.
General procedure for preparation of hydrazones 3a and 3b
To a solution of 1 (1 g, 10 mmol) and sodium acetate (1.64 g, 20 mmol ( in absolute ethanol (20 ml), the appropriate diazonium salt of aromatic amines 2a and 2b (10 mmol) [prepared by diazotising a solution of each of sulphanilamide and 1-naphthyl amine, respectively (10 mmol) in hydrochloric acid (1.5 ml) with a solution of sodium nitrite (0.69 g, 10 mmol) in 5 ml water in ice bath], was added portion wise with stirring at 0–5 °C. The reaction mixture was stirred for further 1 h and diluted with water. The obtained precipitate was collected by filtration, washed with H2O and recrystallised from ethanol to afford the corresponding hydrazine derivatives 3a and 3b, respectively.
4-(2-(3-Methyl-5-oxo-1,5-dihydro-4H-pyrazol-4-ylidene)hydrazineyl)benzenesulfonamide (3a)
Yield: 79%; yellow crystals; m.p. 201–203 °C; IR (KBr) ν cm−1 3472, 3346 (NH2), 3235, 3120 (2NH), 3057 (CH-arom.), 2925 (CH-aliph.), 1733 (CO), 1367, 1145 (SO2); 1H NMR (DMSO-d6) δ = 2.61 (s, 3H, CH3), 6.44 (s, 2H, NH2, exchangeable with D2O), 7.19 − 7. 22 (d, J = 7.2 Hz, 2H, Ar-H), 7.60 − 7.62 (d, J = 7.2 Hz, 2H, Ar-H), 9.15 (s, 1H, NH, exchangeable with D2O), 12.37 (s, 1H, NH-hydrazone); 13C NMR (DMSO-d6): 12.9, 117.8, 127.4, 130.3, 130.6, 146.1, 149.1, 164.5. Anal. Calcd. for C10H11N5O3S (281.29): C, 42.70; H, 3.94; N, 24.90; S, 11.40%. Found: C, 42.51; H, 3.72; N, 24.73; S, 11.75%.
5-Methyl-4–(2-(naphthalene-1-yl)hydrazineylidene)-2,4-dihydro-3H-pyrazol-3-one (3b)
Yield 80%; pale yellow crystals; m.p. 189–191 °C; IR (KBr) ν cm−1 3368, 3242 (2NH), 3071 (CH-arom.), 2948 (CH-aliph.), 1730 (CO); 1H NMR (DMSO-d6) δ = 2.48 (s, 3H, CH3), 7.34 − 7.66 (m, 6H, naphthyl-H), 8.18 − 8.21 (d, J = 8.1 Hz, 1H, naphthyl-H), 9.16 (s, 1H, NH, exchangeable with D2O), 12.66 (s, 1H, NH-hydrazone, exchangeable with D2O); 13C NMR (DMSO-d6): 14.1, 106.3, 120.2, 122.1, 122.3, 124.2, 125.5, 127.1, 129.4, 131.0, 143.1, 147.2, 182.2. Anal. Calcd. for C14H12N4O (252.27): C, 66.65; H, 4.79; N, 22.21%. Found: C, 66.88; H, 4.57; N, 22.43%.
General procedure for preparation of compounds 6a–d
A mixture of 3a (2.81 g, 10 mmol) with each of either malononitrile (4a) (0.66 g, 10 mmol) or ethyl cyanoacetate (4b) (1.13 g, 10 mmol) was fused in the presence of ammonium acetate (1.54 g, 20 mmol) at 150 °C for 30 min, then left to stand at room temperature and triturated with absolute ethanol. The solid products so formed were collected by filtration and recrystallised from ethanol/DMF to give 6a and 6b, respectively.
Similarly, a mixture of 3b (2.52 g, 10 mmol) with each of either malononitrile (4a) (0.66 g, 10 mmol) or ethyl cyanoacetate (4b) (1.13 g, 10 mmol) was fused in the presence of ammonium acetate (1.54 g, 20 mmol) at 150 °C for 30 min, then left to stand at room temperature and triturated with absolute ethanol. The solid products so formed were collected by filtration and recrystallised from ethanol/DMF to give 6c and 6d, respectively.
4-(6-Amino-7-cyano-3-methyl-5H-pyrazolo[4,3-c]pyridazin-5-yl)benzenesulphonamide (6a)
Yield: 68%; green crystals; m.p. 298–300 °C; IR (KBr) ν cm−1 3441, 3357, 3232, 3125 (2NH2), 3054 (CH-arom.), 2933 (CH-aliph.), 2205 (C≡N), 1336, 1174 (SO2); 1H NMR (DMSO-d6) δ = 2.77 (s, 3H, CH3), 6.87 (s, 2H, NH2, exchangeable with D2O), 7.28 − 7.30 (J = 7.2 Hz, d, 2H, Ar-H), 8.08 − 8.10 (J = 7.2 Hz, d, 2H, Ar-H), 8.69 (s, 2H, NH2, exchangeable with D2O); 13C NMR (DMSO-d6): 11.1, 82.1, 115.0, 116.1, 129.2, 131.9, 137.8, 140.6, 147.8, 155.3, 162.6. Anal. Calcd. for C13H11N7O2S (329.34): C, 47.41; H, 3.37; N, 29.77; S, 9.74%. Found: C, 47.62; H, 3.58; N, 29.54; S, 9.95%.
4-(7-Cyano-3-methyl-6-oxo-1,6-dihydro-5H-pyrazolo[4,3-c]pyridazin-5-yl)benzenesulphonamide (6b)
Yield: 66%; brown crystals; m.p. 294–296 °C; IR (KBr) ν cm−1 3437, 3329 (NH2), 3241 (NH), 3051 (CH-arom.), 2943 (CH-aliph.), 2207 (C≡N), 1728 (CO), 1341, 1162 (SO2); 1H NMR (DMSO-d6) δ = 2.25 (s, 3H, CH3), 6.72 (s, 2H, NH2, exchangeable with D2O), 7.77 − 7.79 (d, J = 7.2 Hz, 2H, Ar-H), 8.10 − 8.13 (d, J = 7.2 Hz, 2H, Ar-H), 9.60 (s, 1H, NH, exchangeable with D2O); 13C NMR (DMSO-d6): 10.6, 78.3, 115.6, 120.2, 130.5, 134.9, 138.7, 142.3, 148.2, 160.1, 169.7. Anal. Calcd. for C13H10N6O3S (330.32): C, 47.27; H, 3.05; N, 25.44; S, 9.71%. Found: C, 47.48; H, 3.28; N, 25.66; S, 9.92%.
6-Amino-3-methyl-5-(naphthalen-1-yl)-5H-pyrazolo[4,3-c]pyridazine-7-carbonitrile (6c)
Yield 65%; brown crystals; m.p. 288–290 °C; IR (KBr) ν cm−1 3328, 3232 (NH2), 3086 (CH-arom.), 2944 (CH-aliph.), 2215 (C≡N); 1H NMR (DMSO-d6) δ = 2.60 (s, 3H, CH3) 7.33–7.62 (m, 9H, Ar-H + NH2, exchangeable with D2O); 13C NMR (DMSO-d6): 10.1, 99.7, 107.5, 115.0, 118.3, 120.8, 121.0, 125.0, 127.7, 128.5, 129.5, 131.2, 139.7, 141.2, 149.9, 155.5, 162.6. Anal. Calcd. for C17H12N6 (300.32): C, 67.99; H, 4.03; N, 27.98%. Found: C, 67.78; H, 4.23; N, 27.75%.
3-Methyl-5-(naphthalen-1-yl)-6-oxo-5,6-dihydro-1H-pyrazolo[4,3-c]pyridazine-7-carbonitrile (6d)
Yield 61%; brown crystals; m.p. 282–284 °C; IR (KBr) ν cm−1 3386 (NH), 3073 (CH-arom.), 2964 (CH-aliph.), 2217 (C≡N), 1732 (CO); 1H NMR (DMSO-d6) δ = 2.47 (s, 3H, CH3), 7.49–7.78 (m, 6H, Ar-H), 8.24 − 8.27 (d, J = 8.1 Hz, 1H, naphthyl-H), 9.42 (s, 1H, NH, exchangeable with D2O); 13C NMR (DMSO-d6): 12.9, 99.8, 108.1, 115.3, 121.2, 121.6, 123.7, 16.6, 126.9, 127.8, 129.3, 130.8, 138.7, 142.2, 146.1, 162.0, 168.7. Anal. Calcd. for C17H11N5O (301.30): C, 67.77; H, 3.68; N, 23.24%. Found: C, 67.56; H, 3.89; N, 23.47%.
General procedure for preparation of compounds 8a and 8b
Dissolve either (2.81 g, 10 mmol) of 3a or (2.52 g, 10 mmol) of 3b in absolute ethanol (30 ml), and then add 0.5 ml of triethylamine. Each of the solutions was treated with phenylisothiocyanate (1.35 g, 10 mmol). The reaction mixtures were heated under reflux for 8 h then cooled. The so formed precipitate during heating in each case was collected by filtration and recrystallised from dioxane to give 8a and 8b, respectively.
4-(7-Methyl-4-phenyl-3-thioxo-3,4-dihydro-2H-pyrazolo[3,4-e][1,2,4]triazin-2-yl)benzenesulphonamide (8a)
Yield: 59%; light yellow crystals; m.p. 275–277 °C; IR (KBr) ν cm−1 3322, 3268 (NH2), 3080 (CH-arom.), 2953 (CH-aliph.), 1386, 1182 (SO2), 1317 (CS); 1H NMR (DMSO-d6) δ = 2.28 (s, 3H, CH3), 6.25 (s, 2H, NH2, exchangeable with D2O), 6.77–8.06 (m, 9H, Ar-H); 13C NMR (DMSO-d6): 21.6, 124.2, 127.11, 127.14, 129.4, 132.0, 134.5, 136.2, 137.9, 143.5, 151.4, 157.6, 176.1. Anal. Calcd. for C17H14N6O2S2 (398.46): C, 51.24; H, 3.54; N, 21.09; S, 16.09%. Found: C, 51.55; H, 3.75; N, 21.32; S, 16.30%.
7-Methyl-2-(naphthalen-1-yl)-4-phenyl-2,4-dihydro-3H-pyrazolo[3,4-e][1,2,4]triazine-3-thione (8b)
Yield 56%; light yellow crystals; m.p. 257–259 °C; IR (KBr) ν cm−1 3066 (CH-arom.), 2941 (CH-aliph.), 1278 (CS); 1H NMR (DMSO-d6) δ = 2.48 (s, 3H, CH3), 7.61–7.88 (m, 11H, Ar-H), 8.11 − 8.14 (d, J = 8.1 Hz, 1H, naphthyl-H); 13C NMR (DMSO-d6): 20.0, 112.1, 118.2, 123.1, 123.3, 124.9, 126.2, 127.1, 127.7, 129.0, 129.8, 133.1, 134.9, 135.0, 136.8, 137.0, 140.6, 151.0, 157.6, 174.1. Anal. Calcd. for C21H15N5S (369.44): C, 68.27; H, 4.09; N, 18.96%. Found: C, 68.48; H, 4.30; N, 18.74%
General procedure for preparation of compounds 9a and 9b
Dissolve either (2.81 g, 10 mmol) of 3a or (2.52 g, 10 mmol) of 3b in dimethylformamide (30 ml), and then add 0.5 ml of triethylamine. Each of the solutions was treated with hydroxylamine hydrochloride (0.69 g, 10 mmol). The reaction mixtures were heated under reflux for 24 h then left to cool, and poured into crushed ice and acidified with 10% HCl. The solid product obtained was collected by filtration and recrystallised from dioxane/ethanol to give 9a and 9b, respectively.
4-(6-Methylpyrazolo[3,4-d][1,2,3]triazol-2(4H)-yl)benzenesulphonamide (9a)
Yield: 63%; pale yellow crystals; m.p. > 300 °C;IR (KBr) ν cm−1 3381, 3257 (NH2), 3136 (NH), 3077 (CH-arom.), 2925 (CH-aliph.), 1385, 1131 (SO2); 1H NMR (DMSO-d6) δ = 2.89 (s, 3H, CH3), 6.31(s, 2H, NH2, exchangeable with D2O), 7.54 − 7.57 (d, J = 7.2 Hz, 2H, Ar-H), 7.64 − 7.66 (d, J = 7.2 Hz, 2H, Ar-H), 12.62 (s, 1H, NH, exchangeable with D2O); 13C NMR (DMSO-d6): 10.2, 127.8, 129.4, 134.2, 141.9, 143.9, 146.1. Anal. Calcd. for C10H10N6O2S (278.29): C, 43.16; H, 3.62; N, 30.20; S, 11.52%. Found: C, 43.37; H, 3.84; N, 30.41; S, 11.75%.
6-Methyl-2-(naphthalen-1-yl)-2,4-dihydropyrazolo[3,4-d][1,2,3]triazole (9b)
Yield: 67%; pale yellow crystals; m.p. > 300 °C; IR (KBr) ν cm−1 3292 (NH), 3086 (CH-arom.), 2944 (CH-aliph.); 1H NMR (DMSO-d6) δ = 2.72 (s, 3H, CH3) 7.40–8.20 (m, 7H, Ar-H), 12.76 (s, 1H, NH, exchangeable with D2O); 13C NMR (DMSO-d6): 10.3, 123.2, 123.5, 125.5, 125.9, 127.3, 129.8, 130.6, 132.1, 134.5, 139.1, 146.1. Anal. Calcd. for C14H11N5 (249.27): C, 67.46; H, 4.45; N, 28.10%. Found: C, 67.46; H, 4.45; N, 28.10%.
Biological activity
Antimicrobial activity sensitivity assay
The target compounds 3–9 were screened in vitro opposite to various types of bacteria, Streptococcus pneumoniae and Bacillus subtilis as examples of Gram-positive bacteria, and Pseudomonas aeruginosa and Escherichia coli as examples of Gram-negative bacteria and for their Antifungal activities against Aspergillus fumigates and Candida albicans, respectively. Solutions of concentrations (1 µg/mL) of the target compounds were used. The agar media were inoculated with different microorganism’s culture tested after 24 h of inoculation at 37 °C for bacteria and for antifungal tested after 72 h of inoculation at 28 °C. Ciprofloxacin and amphotericin B were used as standard antibacterial and antifungal drugs, respectively. The diameter of inhibition zone (mm) was measured for the biologically activity using the diffusion techniqueCitation35. The active compounds 3a, 3b, 6a, 6b, 8a and 9a were further investigated to determine their antimicrobial activity expressed in terms of minimum inhibitory concentration (MIC) using the modified agar well diffusion method that mentioned above. The different concentrations (triplicate) of each compound were tested and compared with standard drugs.
Dihydrofolate reductase (DHFR) inhibition
The newly synthesised active targets 3a, 3b, 6a, 6b, 8a and 9a were assessed for their in vitro inhibition against dihydrofolate reductase (DHFR) in confirmatory diagnostic unit, Vacsera, Egypt. Methotrexate was used as a reference drug following the previously mentioned methodCitation36. The obtained results are depicted as IC50 values of enzyme inhibition in . The assay mixture contained 50 µM Tris–HCl buffer (pH 7.4), 50 µM NADPH, 10 µl DMSO or the same volume of DMSO solution containing the test compounds to a final concentration of 10−11 to 10−5 M, and 10 µl of DHFR, in a final volume of 1.0 ml. After addition of the enzyme, the mixture was incubated at room temperature for 2.0 min, and the reaction was initiated by adding 5 µl of dihydrofolic acid, the change in absorbance (ΔOD/min) was measured by the spectrophotometer at 340 nm and 22 °C, kinetic programme (reading every 15 s for 2.5 min). Results are reported as % inhibition of enzymatic activity calculated using the following formula:
where ΔOD/min: the spectrophotometer readings 12.3: extinction coefficient for the DHFR reaction at 340 nm. V: Enzyme volume in mL (the volume of enzyme used in the assay) d: The dilution factor of the enzyme sample. mgP/mL: enzyme concentration of the original sample before dilution.
In silico calculations of molecular properties
Molecular descriptors express the pharmacokinetic, pharmacodynamic and physicochemical effects of all newly synthesised targets 3, 6, 8 and 9. The lipophilicity (milogP) and topological polar surface area (tPSA) were measured using the online Molinspiration softwareCitation37 while the drug score, drug-likeness and aqueous solubility were evaluated via the OSIRIS property explorer softwareCitation38. Furthermore, good bioavailability is more preferable for targets having TPSA of ≤140 Å2 and ≤10 rotatable bondsCitation39.
Molecular modelling study
The molecular docking a powerful tool to understanding and rationalise the obtained biological results. The interactions of the newly synthesised compounds 3a and 6a having the highest DHFR inhibitory activity were investigated with the active site of the target kinase to study their mode of binding and orientations. All the molecular modelling studies were carried out using MOE, 10.2008 softwareCitation40,Citation41. All minimisations were performed with MOE until an RMSD gradient of 0.05 kcal.mol−1 Å−1 with MMFF94x force field and the partial charges were automatically calculated. The co-crystallised structure of the dihydrofolate reductase enzyme was downloaded from Protein Data Bank website (PDB ID: 1DLS)Citation36. The enzyme structure was prepared for molecular docking using Protonate 3D protocol in MOE with the default options. The co-crystallised inhibitor was used to define the active site for molecular docking. Triangle Matcher placement method and London dG scoring function were used in the docking protocol. Docking setup was first validated by self-docking of the co-crystallised inhibitor in the enzyme active site giving a docking pose with an energy score (S) = −11.68 kcal/mol and an RMSD of 0.88 Å from the co-crystalised ligand pose (). Then, the validated molecular docking setup was used to investigate the ligand–target interactions of the newly synthesised compounds 3a and 6a in the DHFR active site to predict their binding pattern and to investigate their ability to satisfy the required structural features for binding interactions ( and ).
Results and discussion
Chemistry
The synthetic strategies approved for the synthesis of the intermediate and target compounds are outlined in Schemes 1 and 2. Thus, 3-methylpyrazol-5(4H)-one 1 was coupled smoothly with diazonium salts 2a and 2b in the presence of ethanol and sodium acetate at 0–5 °C, to afford the respective hydrazines 3a and 3b (Scheme 1). The latter products were established on the basis of their analytical and spectral data. The 1H NMR spectrum of 3a, as an example, revealed the two signals at δ 9.15 and 12.37 ppm due to two NH groups, in addition to the presence of amino, methyl and aromatic protons. While, its 13C NMR spectrum revealed the signals at δ 12.9 and 164.5 ppm assigned to the carbons of the CH3 and CO groups, (See Scheme 1 and Experimental part). Moreover, the obtained arylhydrazino derivatives have been utilised as starting materials for preparing the targeted pyrazolopyridazine ring system. Fusion of hydrazines 3a and 3b with malononitrile (4a) in the presence of ammonium acetate over melting point afforded the 6-aminopyrazolo [4,3-c]-pyridazine derivatives 6a and 6c. The structures of 6a and 6c were established via inspection of their spectral data. For example, the infra-red of 6a indicated the presence of the two NH2 and C≡N absorption band at ν = 3441, 3357, 3232, 3125 and 2205 cm−1. While, its 1H NMR revealed singlet signal at δ 2.77 ppm which was assigned for CH3 group, two signals at δ 6.87 and 8.69 due to two NH2 protons and the absence of any signals may be attributed to NH protons, beside aromatic protons in the molecule. The 13CNMR of 6a showed signals at 11.1and 115.0 for the carbons of the CH3, and C≡N groups, in addition to the sp2 carbon atoms (Scheme 1 and Experimental part). The formation of 6a and 6c from 3a and 3b is assumed to proceed through the intermediate of non-isolable adducts 5a and 5c, which cyclised into the imino structure i via loss of water molecule, which tautomerised into the end amino products 6a and 6c (Scheme 1).
Scheme 1. Synthesis of pyrazolopyridazine derivatives.
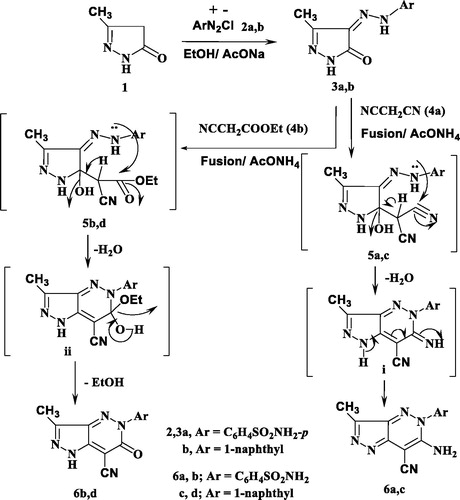
Similarly, 3a and 3b reacted with ethyl cyanoacetate (4b) in the same above reaction conditions to afford 6-oxopyrazolo[4,3-c]pyridazine derivatives 6b and 6d based on the elemental analysis and spectral data. The IR spectra of 6b and 6d indicate the presence of C≡N absorption bands at 2207, 2217 cm−1, respectively and C=O at 1728, 1732 cm−1, respectively. 1H NMR spectra of 6b and 6d showed the presence of only one signal attributed to NH at 9.60 and 9.42 ppm, respectively. Also, its 13C NMR spectra revealed the signals of amidic C=O at 169.7 and 168.7 ppm, respectively.
The structure 6b and 6d was assumed to be formed through the non-isolable adducts 5b and 5d, which underwent cyclisation into structure ii via loss of water molecule then loss of ethanol molecule to give the end reaction products the oxo derivatives 6b and 6d (Scheme 1).
The starting material 3a and 3b used in this study was proved to be a versatile for synthesis of some novel pyrazolo[3,4-e][1,2,4]triazines and pyrazolo[3,4-d]-[1,2,3]triazoles.
Thus, 3a and 3b were reacted with phenylisothiocyanate in refluxing ethanol containing a catalytic amount of triethylamine to give the pyrazolotriazine derivatives 8a and 8b. The composition and structure of products 8a and 8b were established by the results of analytical and compatible spectroscopic data (IR, 1H NMR and 13C NMR), (Scheme 2 and Experimental part).
Scheme 2. Synthesis of pyrazolotriazine and pyrazolotriazole derivatives.
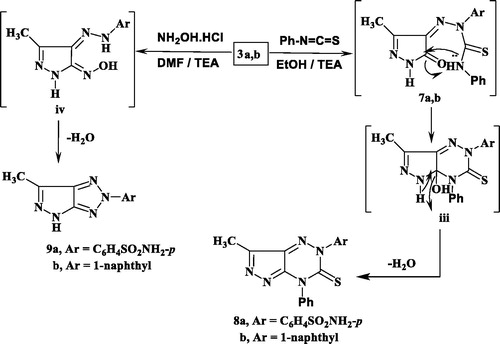
The infra-red spectra of 8a and 8b indicated the absence of the 2NH and carbonyl absorption bands and revealed the characteristic absorption band for the thiocarbonyl group. For example, the 1H NMR spectrum of 8a in DMSO-d6 revealed two singlets at δ 2.28, 6.25 ppm which were assigned for methyl and NH2 protons, respectively, and disappearance of any signal attributable to the NH proton, in addition to the presence of aromatic protons. Also, 13C NMR spectrum showed confirmatory signal of the CS group around δ 176.1 ppm. Formation of 8 was proceeded via the nucleophilic addition of 3 to phenyl isothiocyanate to give the non-isolable adduct 7 followed by cyclisation through the nucleophilic attack of N atom to the carbon of the C = O group giving structure iii which upon elimination of H2O molecule afforded the end reaction products 8a and 8b.
Further illustration of the hydrazone structure 3 came from the reaction with hydroxyl amine hydrochloride in the presence of dimethyl formamide (DMF)/triethylamine solution under reflux to give the final product pyrazolotriazole derivative 9. The structures of 9a and 9b were in consistence with their respective analytical and spectral studies (Scheme 2).
The IR spectrum of 9a, taken as a typical example, was characterised by the disappearance of absorption band due to the CO group. Its 1HNMR showed two singlet at δ 2.89, 6.31 ppm which were assigned for CH3 and NH2 protons, respectively and a singlet at δ 12.62 ppm attributable to the pyrazole-NH proton, in addition to the presence of aromatic protons (cf. Scheme 2 and Experimental Section). The carbonyl group of pyrazole moiety was also absent in 13C-NMR spectra.
Biological activity
Antimicrobial sensitivity assay
The antimicrobial activity of all synthesised compounds 3, 6, 8 and 9 was evaluated by agar well diffusion methodCitation35 using ciprofloxacin and amphotericin B as standard references for antibacterial and antifungal activity, respectively. The screening was done for the in vitro antibacterial activity against Gram-positive Streptococcus pneumoniae and Bacillus subtilis, and Gram-negative Pseudomonas aeruginosa and Escherichia coli, and for the in vitro antifungal activity against Aspergillus fumigates and Candida albicans. The measure zones of inhibition are presented in in mm. By investigation of the given data, it was observed that compounds 3a, 3b, 6a, 6b, 8a and 9a displayed excellent inhibition zone diameter ranging from 11.6 to 28.7 mm against all tested strains in comparison with the reference drugs. On the other hand, the remaining derivatives showed weak or no antimicrobial activity.
Figure 4. Antimicrobial activity of the most active compounds against different bacterial and fungal strains compared with the reference drugs, ciprofloxacin and amphotericin B, respectively.
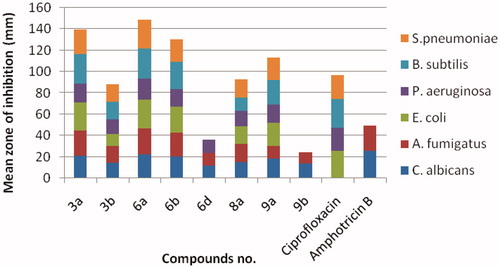
Minimum inhibitory concentration (MIC) of the most active compounds 3a, 3b, 6a, 6b, 8a and 9a was measured in vitro using twofold serial dilution techniqueCitation35. The results of MIC were recorded in . It was noticed that compound 6a among other derivatives revealed excellent and the highest MIC value all over the tested strains. Moreover, the derivatives 3a and 6b were equipotent with the reference, amphotericin B against A. fumigates (MIC = 1.95 µg/mL) and exhibited two folds decrease in the potency than the standard, ciprofloxacin against S. pneumonia (MIC = 1.95 and 0.98 µg/mL, respectively). Additionally, 6b was equipotent in the antifungal activity with reference against A. fumigates. All the remaining derivatives illustrated from moderate-to-weak antimicrobial activity.
Table 1. Minimal inhibitory concentrations (MICs) of the synthesised compounds against the tested pathogenic bacteria and fungi.a
Structure–activity relationship (SAR) study
By inspection of the previous data, it was clear that all derivatives having benzene sulphonamide moiety showed higher and remarkable antimicrobial activity than those having naphthalenyl one. Incorporation of 3-methyl-5-oxopyrazoline with hydrazinyl open chain at p-4 displayed noticeable and excellent antimicrobial activity especially against S. pneumonia, A. fumigates and C. albicans in compound 3a. Fusion of the 3-methyl-5-oxopyrazoline with pyridazine moiety giving 6-amino-7-cyano-3-methyl-5H-pyrazolo[4,3-c]pyridazine improved the potency to the highest value in 6a against almost strains in comparison with references. However, 6b revealed approximately the same excellent potency of 3a. Replacement of pyridazine with triazine or triazole moieties caused drop in the antimicrobial activity in 8a and 9a, respectively.
In vitro enzyme assay on dihydrofolate reductase (DHFR)
The inhibitory activity of the synthesised compounds 3a, 3b, 6a, 6b, 8a and 9a were examined against DHFR using reported procedureCitation36. The results of the inhibitory activity are shown as IC50 values in using methotrexate as a positive control. As expressed in , compounds 3a and 6a proved to be the highest active inhibitors with IC50 values 0.11 ± 1.05 and 0.09 ± 0.91 µM, in comparison with Methotrexate (IC50 = 0.14 ± 1.25 µM). However, the rest derivatives revealed moderate inhibitory activities via 8a and 9a (IC50 = 1.36 ± 0.12 and 1.45 ± 0.23 µM, respectively) and weak ones through 3b and 6b (IC50 = 18.30 ± 0.81 and 5.24 ± 0.37 µM, respectively).
Table 2. In vitro inhibitory activities of the screened compounds 3a, 3b, 6a, 6b, 8a and 9a against DHFR enzyme.
Correlation between the chemical structure and the inhibitory activity of the screened derivatives over DHFR enzyme revealed that the existence of benzene sulphonamide moiety in 3a, 6a, 8a and 9a led to enhanced activity, and the potency order was 6a > 3a > 8a > 9a. Moreover, the attachment of pyrazoline ring with hydrazinyl group in 3a or hybridisation with pyridazine in 6a possessed better inhibitory activity than with triazine or triazole in 8a and 9a.
In silico calculations of molecular properties
Drug-likeness parameters
A molecular property is a complex balance of various structural features which determine whether a specific molecule is similar to the known drugs and to facilitate the drug-likeness of the candidate drug. Therefore, the calculated molecular descriptors of the new compounds expressed in terms of calculating log P, molecular size, flexibility and the presence of hydrogen-donor and acceptors using Molinspiration toolCitation37 and the results are depicted in .
Table 3. Calculated molecular properties of the synthesised compounds 3, 6, 8 and 9 for assessment of the drug likeness.
By analysing the obtained data, it can be concluded that there are linear correlations between the lipophilicity and the activity of the new derivatives and all of them are fully in agreement with the Lipinski’s rule. All the tested compounds have the number of rotatable bonds in the range of 1–3, with the values ranging from 2 into 3 allowed to the most bioactive compounds 3a, 3b, 6a, 6b, 8a and 9a and therefore, obviously exhibiting small conformational flexibility. Results shown in indicated that all of the analysed compounds have Topological polar surface area (TPSA) values ˂140 Å2 except for compounds 6a and 6b have values 153.59 and 147.54, respectively, and therefore are candidate for good solubility, capacity for penetrating cell membranes and intestinal absorption. Compounds with nOHNH value (H-bond donors) less than 5 exhibited increased solubility in cellular membranes and all the test compounds have nOHNH value in the range of 0–4 which is less than 5. Compounds have nON value (H-bond acceptors) in the range of 5–9 and molecular weight in the range of 249.28–398.47. Molecular volumes of the compounds in the series increased as increasing MW and ranged from 216.61 to 317.53. The partition coefficient is vital to examine the physico-chemical properties associated with biological activity. The milogP values for all compounds under investigation are less than 5, outlining good lipophilicity (as defined by the pH-partition hypothesis) with various possible biological sites reasonable oral absorption and lower permeation across biological membranes but higher aqueous solubility especially for compounds 3a, 6a, 6b, 8a and 9a that are most bioactive compounds. Higher solubility is a good factor in drug formation and might generally be reduced in more highly hydrophobic compounds. The greater lipophilicity of these compounds may protect against ROS damage and is due in part to their smaller polar surface area which is another useful descriptor of the oral bioavailability and drug transport properties. As can be seen, compounds 3b and 6c that contains the nonpolar naphthyl substituent exhibited greater lipophilicity (milogP = 2.54 and 2.63, respectively) as compared to compound 3a and 6a (milogP = 0.07 and −0.04) with polar group (sulphonamide). Also, the same phenomena were observed in compounds 6d, 8b, 9b, that contains naphthyl substituent exhibited greater lipophilicity (milogP = 2.48, 3.94, 2.70, respectively) as compared to compounds 6b, 8a, 9a (milogP = −0.02, 1.27, 0.03) with polar group (sulphonamide). Our study revealed that compounds 6c, 6d, 8b and 9b (greater lipophilicity) are biologically either inactive or less active, but compounds 6a, 6b, 8a and 9a (least lipophilicity) are highly active against the tested bacteria and fungi, while compound 3a are highly active than 3b. Our results exhibited that there is a clear relationship between lipophilicity and antimicrobial activity. The most active compounds against all microbial strains were 3a (milogP = 0.07), 6a (milogP = −0.04), 6b (milogP = −0.02), 8a (milogP = 1.27) and 9a (milogP = 0.03). In the light of the above results, we achieved that all tested compounds satisfy the “Rule of five” and meet all criteria for good permeability and bioavailability.
Toxicity risks
Toxicity risks and physicochemical properties of the newly prepared compounds were evaluated through the methodology developed by OsirisCitation38. The data given in , displayed that all compounds are expected to be non-irritating and high risk of reproductive effects. Also, compounds 3b, 6b, 6d, 8b and 9b have shown the non-mutagenic and non-tumorigenic effects. The aqueous solubility of a compound significantly affects its absorption and distribution properties. It is well known that more than 80% of the drugs on the market have estimated solubility values greater than −4. From , it was noted that compounds 3a, 3b, 6b, 9a and 9b exhibited solubility values above −4 and they are expected to have good aqueous solubility which significantly affects their absorption and distribution characteristics. Osiris programme was used for calculating the fragment-based drug-likeness of the synthesised compounds; a positive value indicates that the designed molecule contains fragments that are frequently present in commercial drugs. The results indicated that all target compounds have drug-likeness values except 6c and 6d in the comparable zone with that of the standard drugs. The drug score combines drug likeness, miLogP, solubility, molecular weight, and toxicity risks in one handy value that may be used to judge the compound’s overall potential to qualify for a drugCitation39. A value of 0.5 or more makes the compound a promising lead for future development of safe and efficient drugs. The overall drug score values for the synthesised compounds were calculated and compared to those of the standard drugs. Compounds 3a, 6b and 9a own good drug score values ().
Table 4. Toxicity risks, solubility, drug-likeness, and drug score of the synthesised compounds.
Molecular modelling study
In the current study, the molecular docking study was performed in order to rationalise the obtained In vitro enzyme assay results on dihydrofolate reductase. So, the interactions of the highly potent synthesised compounds 3a and 6a with the active site of DHFR were explored using MOE (Molecular Operating Environment) software 10.2008Citation40,Citation41 and through downloading of the protein data bank file (PDB ID: 1DLS)Citation36 that contains the co-crystallised ligand, methotrexate. The molecular docking protocol was verified by re-docking of the original ligand in the vicinity of the active site of DHFR indicating that the docking protocol used is suitable for the intended docking study. This is shown by the score energy of −11.68 kcal/mol, the small root mean standard deviation (RMSD) between the experimental co-crystallised inhibitor pose and the docked pose of 0.88 Å and the highly noticed superimposition between them ().
Figure 5. 2D and 3D Views (A, B) of the original ligand, methotrexate re-docked in the active site of DHFR (PDB ID: 1DLS) using MOE software. 3D representation (C) of the superimposition of the docking pose (yellow) and the co-crystallised (red) of methotrexate with an RMSD of 0.88 Å.
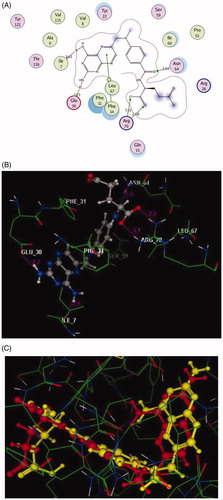
The pteridine moiety of the co-crystallised ligand, methotrexate interacts with the active site of DHFR by two different interactionsCitation42; hydrogen bonding of the two amino groups with Ile7 and Glu30 (distance: 1.93 and 1.73 Å, respectively), and arene–arene interaction with Phe34. Moreover, the amide oxygen forms H-bond acceptor with the side chain of Asn64 (distance: 2.64 Å), while the two oxygen of the carboxylic group shares the binding by two H-bond acceptors with Arg70 (distance: 2.68 and 3.12 Å, respectively). This beside many hydrophobic interactions with various amino acid residues: Ile7, Val8, Ala9, Tyr22, Arg28, Phe31, Gln35, Ser59, Ile60, Pro61, Val115, Tyr121, and Thr136, as shown in ().
The docked compounds 3a and 6a explored higher negative energy score of −12.17 and −13.48 kcal/mol indicating higher predicted binding affinity than the co-crystallised ligand. They were fit in the active site of the enzyme in a similar way via binding of the sulphonamide moiety with the side chain of Ser59 through two hydrogen bonds ( and ). The two nitrogen of the hydrazinyl moiety in 3a displayed two H-bonds, acceptor with the backbone of Ser118 and donor with side chain of Thr146 (distance: 2.72 and 1.62 Å, respectively). The oxygen of pyrazolone scaffold shared fixation through two H-bond acceptors with the side chain of Ser119 (distance: 2.85 and 3.05 Å) ().
Figure 6. 2D and 3D Views (A, B) of the compound 3a docked in the active site of DHFR (PDB ID: 1DLS) using MOE software. Dotted lines and arrows represent hydrogen bonds.
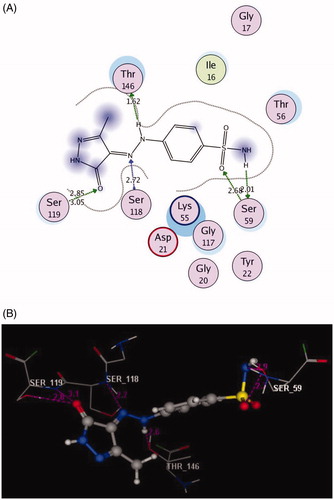
Figure 7. 2D and 3D Views (A, B) of the compound 6a docked in the active site of DHFR (PDB ID: 1DLS) using MOE software. Dotted lines and arrows represent hydrogen bonds.
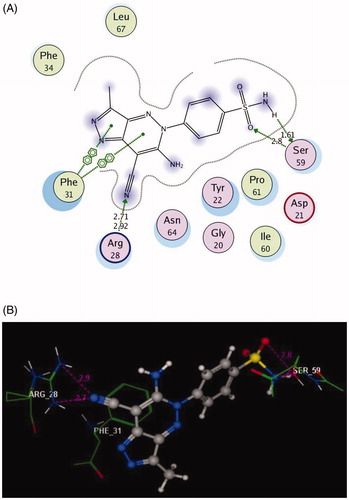
The bulky pyrazolo[4,3-c]pyridazine scaffold in the compound 6a enforced a bound conformation via two arene–arene interactions with Phe31. It could be assumed that chain elongation with –NH–N= hydrazinyl fragment between pyrazole and benzenesulfonamide in 3a, deprived pyrazole ring from binding with the essential amino acid Phe31, although the insertion of N–N chain in cyclised ring fused to pyrazole in 6a gave the chance to be near with Phe31. Furthermore, the cyano substitution on the pyazolopyridazine moiety in 6a allowed two hydrogen bond acceptors with the side chain of Arg28. (distance: 2.71 and 2.92 Å, respectively) ().
Finally, the presence of the sulphonamide substitution in the examined compounds allowed for the H-bond formation with the side chain of Ser59 that was responsible for the observed superior inhibitory activities against DHFR. Notably, the 7-cyanopyrazolo[4,3-c]pyridazine substitution demonstrated excellent binding profile leading to synergistic effect.
Conclusion
In summary, a series of pyrazole derivatives 3, 6, 8 and 9 bearing different heterocyclic systems was designed and synthesised. All synthesised analogues were examined for their in vitro antimicrobial, DHFR inhibition activity and in silico studies. The antimicrobial data revealed the ability of the compounds 3a and 6a to inhibit the growth of the screened panel of six strains with excellent MIC values in comparison with the reference drugs. In addition, the in vitro inhibitory activity against DHFR enzyme illustrated that compounds 3a and 6a were the most potent ones in comparison with methotrexate. Based on the previously obtained data from DHFR inhibition assay, the pyrazoles 3a and 6a containing sulphonamide substitution illustrated good fitting and favourable binding modes with DHFR enzyme in the docking study through formation of a critical hydrogen bond with the essential amino acid Ser59.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Naeem A, Badshah SL, Muska M, et al. The current case of quinolones: synthetic approaches and antibacterial activity. Molecules 2016;21:268.
- Welte T, Pletz MW. Antimicrobial treatment of nosocomial meticillin-resistant Staphylococcus aureus (MRSA) pneumonia: current and future options. Int J Antimicrob Agents 2010; 36:391–400.
- Frere JM. Beta-lactamases and bacterial resistance to antibiotics. Mol Microbial 1995; 16:385–95.
- Devasahayam G, Scheld WM, Hoffman PS. Newer antibacterial drugs for a new century. Expert Opin Invest Drugs 2010;19:215–34.
- Foye WO, Lemke TL, Williams DA, Principles of medicinal chemistry. 4th ed. Media (PA): Williams and Wilkins; 2005:442–456.
- Champe PC, Harvey RA, Lippincott’s illustrated reviews: biochemistry. 2nd ed. Philadelphia (PA): Lippincott Williams and Wilkins; 1994:350–356.
- Berman EM, Werbel LM. The renewed potential for folate antagonists in contemporary cancer chemotherapy. J Med Chem 1991;34:479–85.
- El-Gazzar YI, Georgey HH, El-Messery SM, et al. Synthesis, biological evaluation and molecular modeling study of new (1,2,4-triazole or 1,3,4-thiadiazole)-methylthio-derivatives of quinazolin-4(3H)-one as DHFR inhibitors. Bioorg Chem 2017; 72:282–92.
- Al-Rashood ST, Aboldahab IA, Nagi MN, et al. Synthesis, dihydrofolate reductase inhibition, antitumor testing, and molecular modeling study of some new 4(3H)-quinazolinone analogs. Bioorg Med Chem 2006;14:8608–21.
- Siliveri S, Bashaboina N, Vamaraju HB, Raj S. Design, synthesis, molecular docking, ADMET studies and biological evaluation of pyrazoline incorporated 1,2,3-triazole benzene sulphonamides. Int J Pharm Pharm Sci 2019;11:6–15.
- Karthikeyan MS, Holla BS, Kumari NS. Synthesis and antimicrobial studies on novel chloro-fluorine containing hydroxy pyrazolines. Eur J Med Chem 2007;42:30–6.
- Ragavan RV, Vijayakumar V, Kumari NS. Synthesis and antimicrobial activities of novel 1,5-diaryl pyrazoles. Eur J Med Chem 2010; 45:1173–80.
- Lee YT, Chung YK. Silver(I)-catalyzed facile synthesis of pyrazoles from propargyl N-sulfonylhydrazones. J Org Chem 2008; 73:4698–701.
- Ezawa M, Garvey DS, Janero DR, et al. Design of a heteroaryl modified 1, 5-disubstituted pyrazole cyclooxygenase-2 (COX-2) selective inhibitors. Lett Drug Design Dis 2005; 2:40–3.
- Кorobko D, J. Hadjipavlou-litina D, Logoyda L`. Antioxidant and anti-inflammatory properties of a series of new 7,8-disubstituted theophylline containing a pyrazole ring. Asian J Pharm Clin Res 2018; 11:448–50.
- Menozz G, Merello L, Fossa P, et al. Synthesis, antimicrobial activity and molecular modeling studies of halogenated 4-[1H-imidazol-1-yl(phenyl)methyl]-1,5-diphenyl-1H-pyrazoles. Bioorg Med Chem 2004;12:5465–83.
- Naoum F, Kasiotis KM, Magiatis P, Haroutounian SA. Synthesis of novel nitro-substituted triaryl pyrazole derivatives as potential estrogen receptor ligands. Molecules 2007; 12:1259–73.
- Cocconcelli G, Diodato E, Caricasole A, et al. Aryl azoles with neuroprotective activity-parallel synthesis and attempts at target identification. Bioorg Med Chem 2008; 16:2043–52.
- Manojkumar P, Ravi TK, Gopalakrishnan S. Antioxidant and antibacterial studies of arylazopyrazoles and arylhydrazonopyrazolones containing coumarin moiety. Eur J Med Chem 2009; 44:4690–4.
- Liu H, Ren Z-L, Wang W, et al. Novel coumarin-pyrazole carboxamide derivatives as potential topoisomerase II inhibitors: design, synthesis and antibacterial activity. Eur J Med Chem 2018; 157:81–7.
- Sharshira EM, Hamada N. Synthesis and antimicrobial evaluation of some pyrazole derivatives. Molecules 2012; 17:4962–71.
- Farag AM, Mayhoub AS, Barakat SE, Bayomi AH. Synthesis of new N-phenylpyrazole derivatives with potent antimicrobial activity. Bioorg. Med. Chem 2008; 16:4569–78.
- Vijesh AM, Isloor AM, Shetty P, et al. New pyrazole derivatives containing 1,2,4-triazoles and benzoxazoles as potent antimicrobial and analgesic agents. Eur J Med Chem 2013; 62:410–5.
- Holla BS, Mahalinga M, Karthikeyan MS, et al. Synthesis of some novel pyrazolo[3,4-d]pyrimidine derivatives as potential antimicrobial agents. Bioorg. Med. Chem 2006; 14:2040–7.
- Zhang T-Y, Zheng C-J, Wu J, et al. Synthesis of novel dihydrotriazine derivatives bearing 1,3-diaryl pyrazole moieties as potential antibacterial agents. Bioorg Med Chem Lett 2019; 29:1079–84.
- Sapariya NH, Vaghasiya BK, Thummar RP, et al. Synthesis, characterization, in silico molecular docking study and biological evaluation of a 5-(phenylthio) pyrazole based polyhydroquinoline core moiety. New J Chem 2017; 41:10686–94.
- Bhatt JD, Chudasama CJ, Patel KD. Microwave assisted synthesis of pyrimidines in ionic liquid and their potency as non-classical malarial antifolates. Arch Pharm Chem Life Sci 2016; 349:791–800.
- Kandile NG, Mohamed MI, Zaky H, Mohamed HM. Novel pyridazine derivatives: Synthesis and antimicrobial activity evaluation. Eur J Med Chem 2009; 44:1989–96.
- Nagawade RR, Khanna VV, Bhagwat SS, Shinde DB. Synthesis of new series of 1-Aryl-1,4-dihydro-4-oxo-6-methyl pyridazine-3-carboxylic acid as potential antibacterial agents. Eur J Med Chem 2005; 40:1325–30.
- Arshad M, Bhat AR, Hoi KK, et al. Synthesis, characterization and antibacterial screening of some novel 1,2,4-triazine derivatives. Chin Chem Lett 2017; 28:1559–65.
- Kaushik CP, Pahwa A. Convenient synthesis, antimalarial and antimicrobial potential of thioethereal 1,4-disubstituted 1,2,3-triazoles with ester functionality. Med Chem Res 2018; 27:458–69.
- Gad-Elkareem MAM, Othman I. Synthesis and antimicrobial evaluation of some new thienopyrazoles, pyrazolothienopyridines and pyrazolothienopyrimidines via Gewald reaction. Int J Pharma Sci 2017; 7:1808–16.
- Hassan AS, Askar AA, Nossier ES, et al. Antibacterial evaluation, in silico characters and molecular docking of Schiff Bases derived from 5-aminopyrazoles. Molecules 2019; 24:3130.
- Khalifa NM, El-Sayed AS, Abd El-Karim SS, et al. 1,3,4-Triarylpyrazoles containing 2-thioxoimidazolidinones and different fused systems: synthesis and antimicrobial activity. Russ J Gen Chem 2018; 88:2646–52.
- Ibrahim HS, Eldehna WM, Abdel-Aziz HA, et al. Improvement of antibacterial activity of some sulfa drugs through linkage to certain phthalazin-1(2H)-one scaffolds. Eur J Med Chem 2014; 85:480–6.
- El-Naggar M, Sallam HA, Shaban SS, et al. Design, synthesis, and molecular docking study of novel heterocycles incorporating 1,3,4-thiadiazole moiety as potential antimicrobial and anticancer agents. Molecules 2019;24:1066.
- The online Molinspiration software. Available from: https://www.molinspiration.com/
- The OSIRIS property explorer software. Available from: http://www.organic-chemistry.org/prog/peo/
- Othman IMM, Gad-Elkareem MAM, El-Naggar M, et al. Novel phthalimide based analogues: design, synthesis, biological evaluation, and molecular docking studies. J Enzy Inh Med Chem 2019; 34:1259–70.
- Molecular Operating Environment (MOE), Version 2008.10. Montreal (QC): Chemical Computing Group Inc. Available from: https://www.chemcomp.com/MOEMolecular_Operating_Environment.htm
- Elzahabi HAS, Nossier ES, Khalifa NM, et al. Anticancer evaluation and molecular modeling of multi-targeted kinase inhibitors based pyrido[2,3-d]pyrimidine scaffold. J Enzy Inh Med Chem 2018; 33:546–57.
- Lewis WS, Cody V, Galitsky N, et al. Methotrexate-resistant variants of human dihydrofolate reductase with substitutions of Leucine 22. J Bio Chem 1995; 270:5057–64.