
Abstract
Haspin is a mitotic protein kinase required for proper cell division by modulating Aurora B kinase localisation and activity as well as histone phosphorylation. Here a series of imidazopyridazines based on the CHR-6494 and Structure Activity Relationship was established. An assessment of the inhibitory activity of the lead structures on human Haspin and several other protein kinases is presented. The lead structure was rapidly optimised using a combination of crystal structures and effective docking models, with the best inhibitors exhibiting potent inhibitory activity on Haspin with IC50 between 6 and 100 nM in vitro. The developed inhibitors displayed anti-proliferative properties against various human cancer cell lines in 2D and spheroid cultures and significantly inhibited the migration ability of osteosarcoma U-2 OS cells. Notably, we show that our lead compounds are powerful Haspin inhibitors in human cells, and did not block G2/M cell cycle transition due to improved selectivity against CDK1/CyclinB.
Graphical Abstract
Introduction
The deregulation of protein phosphorylation is directly responsible for the pathogenesis of several diseases and protein kinases are therefore considered as major drug targets. Specific targeting of kinases that are essential for unwanted cancerous cells is therefore an important step towards therapy. The search for pharmacological kinase inhibitors has become a major approach to discover new therapeutic agents. The most commonly used conventional anticancer agents such as taxanes and vinca alkaloids aim at impairing the ability of the cancerous cell to divide by targeting the microtubule cytoskeleton leading to mitotic arrest and ultimately cell deathCitation1. Nowadays, more specific ways disturbing cell division are being explored by the pharmaceutical community that involve targeting essential mitotic protein kinases whose expression or activity are found deregulated in many cancersCitation2–5.
Aurora B kinase controls many aspects of mitosis, including the correction of microtubule-kinetochore attachment errors and cytokinesisCitation6. The deregulation of its activity or expression is often associated with genomic instability and aneuploidy, commonly observed in a vast majority of solid tumours and hematological malignanciesCitation7. In 2013, more than a dozen Aurora kinase inhibitors were tested in phase I clinical trialsCitation8. Despite recent advances, the effectiveness of Aurora B kinase inhibitors has so far been limited by the development of resistance and side effectsCitation9. It is therefore of great interest to develop new strategies targeting Aurora B indirectly through its upstream natural regulators. Many other protein kinases control Aurora B activity directly or indirectly, making them potential therapeutic targetsCitation10–12. Amongst them, the protein kinase Haspin (also known as GSG2, germ cell associated 2) which has emerged as a key regulator of Aurora B functions. It has been shown to act upstream of Aurora B, regulating both its centromeric localisation and activityCitation13–17. It also plays several key roles during mitosis both in maintaining chromatin and centromeric cohesion and spindle pole structuresCitation13,Citation18–20. Haspin phosphorylates Histone H3 on threonine 3 (H3T3ph) at mitosis leading to the recruitment and clustering of Aurora B to the centromereCitation21–23. This clustering is essential for Aurora B kinase activation, which in return phosphorylates and activates Haspin, creating a positive feedback loop between the two kinasesCitation16. Haspin kinase appears essential for mitosis as its depletion or inhibition results in cell death caused by mitotic catastropheCitation24,Citation25. Additionally, Haspin kinase is overexpressed in several malignancies such as Burkitt’s lymphoma, B cell chronic lymphocytic leukaemia and pancreatic cancersCitation26–29. Hence, Haspin represents an interesting target for cancer therapy.
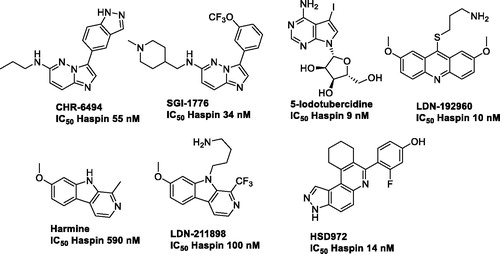
Haspin has atypical structural features that are unique to this kinase, increasing the likelihood of identifying specific inhibitors that may result in fewer off-target effectsCitation30,Citation31. There are currently a few Haspin inhibitors described in the literature (): bicyclic heterocyclic structures such as imidazopyridazine CHR-6494 from Chroma Biotech or SGI-1776Citation24,Citation32; the nucleotide like 5-iodotubercidineCitation13,Citation17 and finally methoxylated fused tricyclic derivatives containing an acridine (LDN-192960) or a β-carboline such as in harmine or LDN-211898Citation17,Citation33,Citation34. More recently, 3H-pyrazolo[4,3-f]quinolone as HSD972Citation35, and natural productsCitation29,Citation36 were additionally reported.
Figure 1. Example of Haspin inhibitors.
The heterocyclic derivative CHR-6494 is active both in vitro, on tumour cellsCitation24,Citation37 and in vivo as it reduced angiogenesis and tumour growth on nude mice xenografts of HCT-116 cells. CHR-6494 affects also cell viability and mobility of several melanoma cancer lines when used alone or in combination with the Mitogen-activated protein kinase kinase (MEK) inhibitor Trametinib which acts synergisticallyCitation37. However, the compound displays poor selectivity against other kinases but the structure offers an attractive starting point for optimisation.
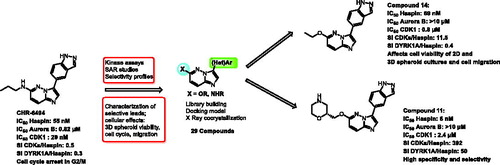
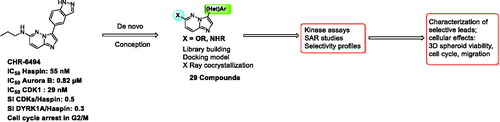
In this study, we report the development of novel imidazopyrimidine derivatives that display improved potency towards Haspin and improved selectivity (). Synthesis was fully optimised and modulations were performed in parallel with docking studies. To guide the design of new inhibitors, we solved a high-resolution crystal structure of an early derivative with Haspin. The structure confirmed the expected binding mode interacting with the hinge of the kinase in an adenosine triphosphate (ATP) competitive way. For ligand optimisation, further docking experiments were performed using this structural model.
Figure 2. Presented work.
Structure activity relationships were established and selectivity was assessed using a representative kinase panel. Several cellular studies were performed to demonstrate the specific mode of action of newly synthesised derivatives. Very promising novel leads were obtained that exhibited anti-proliferative properties against various human cancer cell lines grown in 2D and 3D spheroid cell cultures and significantly inhibited the migration ability of osteosarcoma U-2 OS cells.
Results and discussion
Chemistry
First, we developed an efficient synthesis of the CHR-6494 derivative in three steps as this compound was not commercially available at the beginning of this work. The first step consisted of a nucleophilic aromatic substitution (SNAr) in position C-6 with propylamine in N-methyl-2-pyrrolidone (NMP) under microwave irradiation which furnished compound 3 in 86% yield. Furthermore, a region-selective bromination of compound 3 in the presence of N-bromosuccinimide (NBS) led to compound 4 in quantitative yield. The displacement of this halogen was carried out by a Suzuki–Miyaura cross coupling, using 1H-indazole-5-boronic acid as partner, in presence of caesium carbonate as base in a mixture of ethanol/water as solvent under microwave irradiation to give compound 5 in the moderate yield of 28% (Scheme 1).
Scheme 1. Reagents and conditions: i) HNRR' (5.0 equiv.), NMP, M.W., 180 °C, 1 h; ii) NBS (1.0 equiv.), ACN, r.t. 1 h; iii) ArB(OH)2 or ArB(Pin)2, Na2CO3, Pd(PPh3)4 (0.1 equiv.), Dioxane/H2O (9/1), M.W., 150 °C, 1h30. iv) ROH, NaH, NMP, M.W., 180 °C, 1 h; v) ROH, NaH, THF, r.t., 1–5 h; vi) HCO2H, Et3N, Pd(OAc)2 (0.1 equiv.), Xantphos (0.2 equiv.), THF, M.W., 150 °C, 15 min.
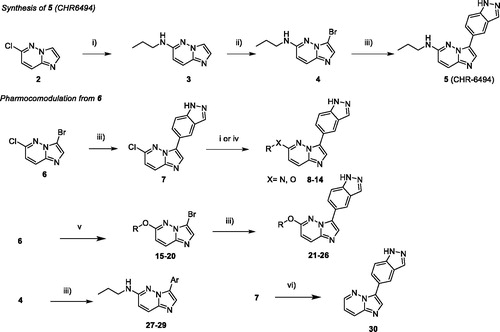
This linear synthetic pathway prompted us to develop a versatile skeleton to explore molecular diversity in C-3 and C-6 positions. We focussed our attention on a bis-halogenated platform 6, which can be regio-selectively functionalised in C-3 and C-6 positions (Scheme 1) by chlorine and bromine discriminationCitation38. However, the method suffered in our hands from long reaction times under thermal conditions, leading to purification difficulties and low yields. Fortunately, the lack of reactivity was fully circumvented using microwave activation, which improved kinetic parameters of the reaction and significantly increased the yield during both palladium cross coupling and SNAr reactionsCitation39–41.
A direct regio-selective Suzuki cross-coupling reaction was first performed with 1H-indazole-5-boronic acid under microwave irradiation resulting in a satisfactory yield of compound 7 (61%), which was efficient in comparison to the use of conventional reaction conditions (around 10%). In a second step, starting from compound 7, SNAr occurred in C-6 using various nucleophiles under microwave irradiation and afforded the attempted compounds 8–14 in moderate yields due to the presence of the indazole moiety. Several primary or secondary aliphatic amines were successfully introduced by nucleophilic substitution. An attempted reaction with amino alcohols led only to nitrogen reactions. Condensation of the alcohols required the addition of a base enhancing reactivity for completion of the reaction at a lower temperature in reaction times of only 10 min.
To pursue the building of a focussed library around this scaffold, we modified the synthetic pathway due to the presence of the indazole moiety which led to some limitations such as low solubility or reactivity. We also switched the two steps and began by the SNAr, with amines or various chiral alcohols in presence of NaH as base which furnished compounds 15–20 in a moderate to high yields ().
Table 1. Structure of synthesised derivatives and yields.
Next, the Suzuki reaction was performed using indazole boronic acid to furnish compounds 21–26 in moderate yields. Furthermore, benzimidazole and 2-aminopyrimidine as indazole isosters were introduced on bromo derivative 4 to give compounds 27 and 28 in 23% and 73% yield, respectively. We also introduced on compound 4 a naphthyl moiety instead of indazole, to generate derivative 29 with a very satisfactory yield. Without any donor/acceptor hydrogen bond system, this compound was unable to interact with the Haspin hinge region and served as a negative control for biochemical and biological analysis. To complete the construction of the library, we performed the reduction of the C5-Cl bond with a palladium catalyst and microwave irradiation to give the desired product 30 in 37% of yield ().
Kinase assays
We tested the inhibitory activity of the 18 synthesised compounds on recombinant HsHaspin activity compared to the reference compound CHR-6494 5 () with the main objective of obtaining more selective Haspin inhibitors and of being able to assess the potency of a selective inhibitor in cellular assays with reduced interference of off-targets.
Table 2. IC50 (µM) of the imidazopyridazine derivatives on various protein kinases (in bold for haspin kinase). For selected compounds, the selectivity index for each kinase vs Haspin is shown in parentheses.
As expected, compounds without any heteroatom attached to the imidazopyridazine core in C-6 (compound 30) or lacking the indazole part in C-3 (compounds 27–29), lost their activity for Haspin. It was evidence that these two elements play a crucial role maintaining potent inhibitory activity and that they define the key pharmacophore. Modulation of the propyl amine in C-6 led to variation in Haspin inhibition. Methylation of the NH group confirmed that the labile proton was not essential (compound 8). Cycloalkylamines as well as piperidine were used as substituents in C-6 without any significant changes in activity (compounds 9–11). The cyclopentyl group seemed preferred, with an improved activity for compound 9 at IC50 = 31 nM. Finally, a second hydrophilic function was introduced on the small amino alkyl side chain in compound 12 and resulted in activity that remained identical to that of other derivatives of this subfamily.
While switching the aminopropyl group for propyloxy in C-6 proved disappointing (compound 13), the ethoxyl residue was very well tolerated as derivative 14 exhibited good Haspin inhibition with an IC50 = 69 nM. The best results were obtained when the C-6 alkyloxy group was substituted by heteroalkyl cycles. With prolinol derivatives, the chiral compounds 25 (R) and 26 (S) were found to be very active with a preference for the S configuration. A real activity improvement was achieved by switching the pyrrolidine for a morpholine (compounds 21–24). The position of the heteroatom had a real impact. Interaction with the kinase active site was favoured when the two electron rich oxygen atoms, which are strong H bond acceptors, were located close to each other. This behaviour was confirmed by the IC50 of derivatives 21 and 22 (6 and 12 nM, respectively) which were more active than 23 and 24 (25 and 20 nM, respectively). Finally, the (S) derivative 21 and its enantiomer 22 are to our knowledge the most active Haspin inhibitors in the imidazopyrimidazine series ever reported and join the short list of very potent Haspin inhibitors.
The selectivity of each derivative was quantified on several other kinases. CHR-6494 (5) appeared in our hands as a pan kinase inhibitor with moderate selectivity and was surprisingly more active on CDK9 and DYRK1A than on Haspin (). For the developed imidazopyridazine library, the selectivity vs Aurora B was excellent, as the newly designed Haspin inhibitors did not inhibit this kinase at 1 µM. Noteworthy, 12 derivatives did not affect this enzyme at 10 µM (compounds 11–14, 21–25, 28, 30). CDK2 and CDK5 inhibition was fully enhanced in the case of C-6-O substituted molecules (compounds 13,14, 21–24). The highest selectivity for CDK9 and DYRK1A occurred with the morpholino containing derivatives (compounds 21–24), which exhibited a (sub)micromolar IC50 range on both kinases. The best selectivity was clearly achieved with 21 (IC50 Haspin = 6 nM) with selectivity for CDK2, 5, 9 and DYRK1A of 716, 150, 28 and 50-fold, respectively. Altogether, these results showed that our chemical series displayed increased efficacy and selectivity.
Values are IC50 expressed in µM and calculated from dose–response curves (each point on the curves was performed in triplicate). Selectivity indexes (SI; in brackets) are calculated as follows: SI = IC50 kinase X/IC50 Haspin. ND: not determined. *IC50 values obtained using the ADP-Glo methodology (see Experimental section, Supplementary Material).
Binding mode and molecular modelling studies
We first conducted an ATP competition assay with compound 12 on Haspin kinase activity (). We tested an ATP concentration range from 5 to 240 µM on compound 12 concentrations ranging from 0.001 to 10 µm. Our data clearly show the competition between compound 12 and ATP, as the calculated IC50 increased from 6 nM at 5 µM ATP to 950 nM at 240 µM ATP.
Figure 3. Binding mode of selected compounds with Haspin. (A) ATP competition assay with compound 12. (B) Crystal structure of Haspin with compound 12. The inhibitor is displayed in stick representation with yellow carbon atoms, and the key interactions with the kinase ATP binding site are shown. (C) Superimposition of the binding mode of compounds 12 (yellow carbon atoms) and 21 (grey carbon atoms) in Haspin active site. The three-letter amino acid code and residue number are labelled next to each side chain.
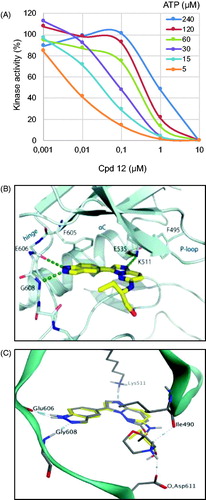
We next determined the co-crystal structure of Haspin in complex with derivative 12 to determine the binding mode of the inhibitor within the kinase (). The inhibitor adopted a planar conformation, positioning the indazole moiety for hydrogen bonds to the hinge region. This orientation resulted in the imidazopyridazine group protruding further in the pocket, interacting with the catalytic lysine Lys511. The amino alkyl side chain decoration tucked beneath the β1 and β2Citation42, and occupied the space towards the solvent exposed region of the pocket. This binding mode of compound 12 demonstrated shape complementarity to the ATP-binding pocket of the kinase, thus explaining the good potency of the inhibitor. These results also strongly suggest that our compounds behave as type I kinase inhibitors.
Derivative 21 was docked into the active site of Haspin to understand key interactions with the protein residues. Docking experiments showed that the binding mode of inhibitor 21 was conserved in the ATP active site and formed similar hydrogen bond interactions with the hinge region and catalytic lysine Lys511 to compound 12 (). Interestingly, the morpholine group made two additional hydrogen bond interactions between the protonated nitrogen and the oxygen of the backbone carbonyl of isoleucine Ile490 and the carboxyl group of the side chain of aspartic acid Asp611. These extra hydrogen bonds could explain the strong potency of inhibitor 21 compared to compound 12 or CHR-6494 5. In addition, this interaction can be seen as a salt bridge between the ammonium of the morpholine group and the carboxylate anion group of the side chain of aspartic acid Asp611. The difference of activity between CHR-6494 5 and inhibitor 21 towards DYRK1A could be explained by the lack of one hydrogen bond acceptor or a lack of one salt bridge, since the position of Asp611 in Haspin corresponds to asparagine Asn234 in DYRK1A. Moreover, the NH2 side chain of Asn234 may be exposed to the protonated nitrogen, thus forming an unfavourable interactionCitation43.
Cellular effects
Cell viability
We subsequently tested the effects of selected compounds on the cell viability of various highly proliferative cancerous cell lines () from osteosarcoma (U-2 OS), colorectal cancer (HCT116), breast cancer (HBL100) and neuroblastoma (SH-SY5Y) as well as non-cancerous retinal fibroblast RPE-1 immortalised with hTERT, cultured under a conventional monolayer (2-Dimensions) format.
Table 3. Effects on cell viability.
Cell viability was assessed after 48 h treatment with selected compounds in a dose-dependent manner (). Among cancerous cell lines, U-2 OS appeared to be the most sensitive one, responding to most compounds apart from compound 12 and our negative control 29 with EC50 ranging from 2.8 µM for derivative 14 to 8.6 µM for compound 22. Compounds were generally more efficient at inhibiting the viability of normal RPE-1 cells than the other cells of cancerous origin. None of the compounds appeared to affect the viability of SH-SY5Y cells and only derivatives 12 and 14 slightly affected HBL100 cells (EC50 of 6.6 and 9.5 µM respectively). The negative compound 29 did not impact any of the tested cell lines (all EC50 >10 µM). Amongst the selected compounds, 14 appeared the most active one, affecting all cell lines apart from SH-SY5Y. This compound displayed the strongest impact on the viability of U-2 OS and HCT116 cells with EC50 of 2.8 µM and 1.7 µM, respectively.
Cells were incubated with increasing doses of each compound for 48 h. Cell viability was determined by MTS assay and EC50 (µM) were calculated from the dose–response curves of each compound on the indicated human cell lines (each point made in triplicate).
Effects on 3D spheroids
It is known that the impact of drugs on cell division may change markedly depending upon environmental conditions. Therefore, in order to gain in physiological predictivityCitation44, the anti-proliferative activity of derivative 14 was further addressed on multicellular spheroids (), still using compounds 5 and 29 as reference and negative control respectively. As RPE-1 and SH-SY5Y cells failed to form spheroids, U-2 OS, HCT116 and HBL100 cell lines were used for this approach.
Figure 4. Effects of compounds on spheroid viability. (A) Cell viability in spheroids from HCT116, HBL100 and U-2 OS cells was measured after 2 and 10 days of treatment with DMSO, staurosporine (1 µM) or compounds 5, 14, 29 at 5 µM, at a single dose on day 0. Cell viability was expressed in percentage of the DMSO control set at 100%. Data were acquired in triplicates, results are mean ± SEM, ***p ≤ 0.001, *p ≤ 0.05 (two-tailed unpaired t-test). (B) U-2 OS spheroids were treated with 5, 2.5 or 1 µM of compounds 5, 12, 14, 21 or 22, or with 1 µM of staurosporine or DMSO, at a single dose on day 0. Cell viability was measured after 2 and 10 days and expressed in percentage of the DMSO control set at 100%. Data were acquired in triplicates; graphs represent the mean of 2 independent experiments. Results are mean ± SEM, ****p ≤ 0.0001 (two-tailed unpaired t-test).
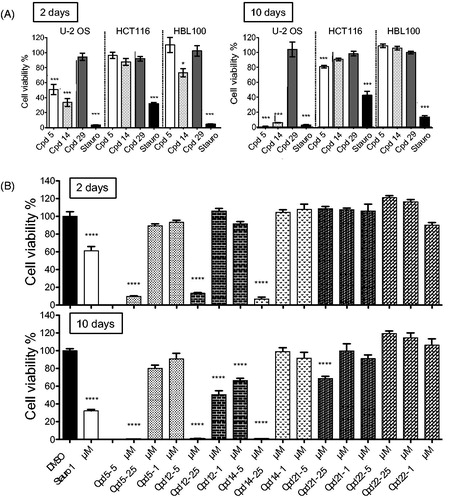
A 48-h exposure with 5 µM of compound 5 triggered a significant decrease in U-2 OS spheroid viability (49%), without any significant effect on that of HCT116 and HBL-100 cells (, left panel). Notably, derivative 14 impacted U-2 OS spheroid viability slightly more (33%) and showed an additional – rather moderate – activity on HBL-100 spheroids (73%). These results demonstrated that compound 14, compared to 5, had similar or slightly stronger effects on U-2 OS spheroid viability and confirmed our results on 2D culture cell viability. After 10-day exposure, the effect on U-2 OS spheroid viability was markedly increased with both compounds while HCT116 and HBL100 remained mostly unresponsive (, right panel). At 5 µM, cell viability was down to 1% and 5% for compounds 5 and 14, respectively.
As U-2 OS spheroids appeared the most responsive to our inhibitors, we tested our other hit compounds (14, 12, 21, 22) at three concentrations (1, 2.5 and 5 µM) in cell viability assays after 2 and 10 days (). After 2-day treatments, spheroid viability was strongly affected by compounds 5, 14 and 12 at 5 µM concentration (9.7, 6.9, 13.1% remaining viability, respectively) while compounds 21 and 22 showed no effects. Interestingly, after a 10-day treatment compound 12 appeared as the most effective inhibitor using U-2 OS cells, showing growth inhibition at 2.5 and 1 µM (50 and 66% remaining viability, respectively). For the other compounds these lower concentrations were mostly ineffective. However, a slight decrease in spheroid viability was observed for compound 21 at 5 µM concentration (69%). Taken together, our data showed that this series displayed some anti-proliferative properties without being generally cytotoxic. Results on U-2 OS spheroids clearly indicated stronger anti-proliferative effects for less selective compounds.
Effects on cellular Haspin kinase
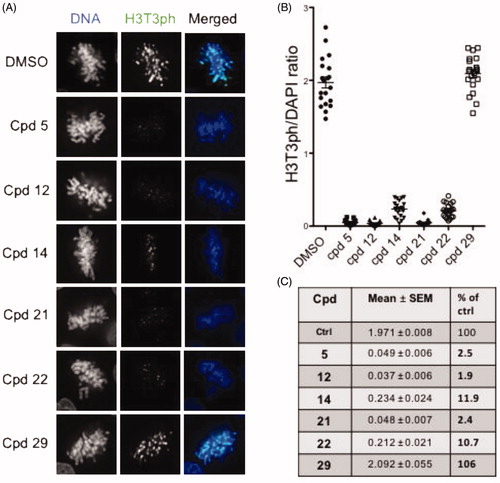
We next tested in cells the functional effects of compounds 12, 14, 21 and 22 as well as compounds 5 and 29 as reference and negative controls, respectively. We monitored and quantified Haspin activity by immunofluorescence and image analysis in U-2 OS cells following the phosphorylation of Threonine 3 of Histone H3 (H3T3ph) as a specific marker (). Cells were treated with 0.5 µM of the various compounds for 24 h. As shown in , treatment of cells with compounds 5, 12, 14, 21 and 22 greatly reduced the H3T3ph signal in selected prometaphase/metaphase cells. We noted that typical chromosome misalignment defects were observed as previously reported in Haspin depletion studiesCitation14. Quantification of the H3T3ph signal vs DAPI is represented in . Compounds 5, 21 and 12 showed the strongest effect on cellular Haspin activity with only 2.5, 2.4 and 1.9% of H3T3ph signal remaining in prometaphase cells, respectively. This effect was slightly milder with compound 22- and 14-treated cells where 10.7 and 11.9% of H3T3p signal respectively remained (). The negative compound 29 showed a value similar to that of the DMSO control. A similar experiment performed on cells treated with a lower dose of 0.05 µM of compounds for 24 h revealed that cellular Haspin activity was still inhibited by more than 80% by compounds 5, 12, 21 and 22 (Supplementary Figure S5). These results showed that all tested compounds are highly cell penetrant and inhibit endogenous Haspin kinase. A comparison of their effects showed that compounds 5, 12 and 21 were the most efficient at reducing cellular Haspin activity.
Figure 5. In-cell evaluation of Haspin inhibition. (A) immunofluorescence staining of U-2 OS cells treated for 24 h with 500 nM of each compound or 0.1% of DMSO. Haspin activity was monitored by staining of phosphorylated Histone H3 on threonine 3 (H3T3ph, green), deoxyribonucleic acid DNA was visualised by DAPI (4′,6-diamidino-2-phenylindole) staining (blue). Bar 10 µm. (B) Quantification of Haspin activity in prometaphase cells from (A); box and whiskers representation, n ≥ 30. (C) Statistical analysis of data obtained in (B). Results are mean ± SEM, p values are indicated (two-tailed unpaired t-test). Results are also shown in percentage of the DMSO control.
Effects on cell cycle
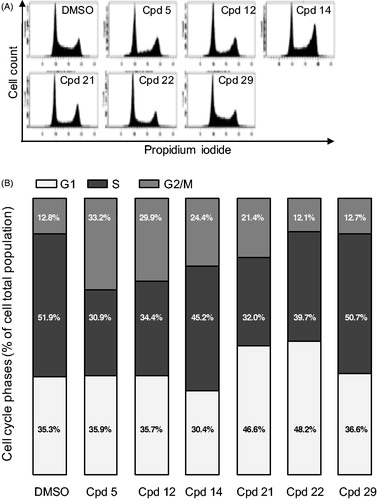
We next addressed the effect of our compounds on the cell cycle. U-2 OS cells were treated for 24 h with 2.5 µM of compounds and their cell cycle profile was analysed by flow cytometry (). A lower dose of 0.5 µM was simultaneously tested showing no effect on the cell cycle of U-2 OS cells (data not shown), concurring with the results obtained on cell viability assays (). However, flow cytometry profiles, after treatment with compounds 5, 12, 14 and 21 at 2.5 µM, showed a strong increase of cells in G2/M phase of the cell cycle (33.3, 29.9, 24.4 and 21.4%, respectively vs. 12.8% in control DMSO-treated cells, accompanied with a reduction of cells in S phase (30.9, 34.4, 45.2 and 32%, respectively vs. 51.9% in control cells). These results are consistent with impaired cell cycle phase progression and transition checkpoints which are under the control of specific protein kinases, the Cyclin-dependent kinases (CDK/cyclin)Citation45,Citation46. The S-phase reduction coupled with a G2/M increase observed for compounds 5 and 12 could be in part explained by their strong inhibition of CDK2, a CDK essential for the progression through these phases (IC50 of 91 and 110 nM, respectively, )Citation46. However, the S-phase reduction coupled with a G1 increase observed for compounds 21 and 22 cannot be explained by a CDK2 inhibition (IC50 of 4.3 and 5.3 µM, respectively). Such effects could result from the inhibition of G1-specific CDKs such as CDK4 and CDK6Citation47,Citation48, which remains to be tested.
Figure 6. Cell cycle effects of selected compounds. (A) U-2 OS cells were treated with 2.5 µM of each compound for 24 h before ethanol fixation and propidium iodide staining. DNA content was measured by flow cytometry and representative profiles are shown. (B) quantification of the proportion of cells in each phase of the cell cycle from the experiment described in (A). Results are mean percentage (n = 3 independent experiments).
While a slight delay in M phase is expected by inhibition of Haspin, the strong accumulation of cells in G2/M phases observed with compounds 5 and 12 cannot be solely linked to this inhibition but is likely the consequence of the inhibition of several kinases regulating mitotic entry. We recently reported inhibition of 5 on CDK1 (Cyclin-dependent kinase 1) during the first divisions of sea urchin embryosCitation49. We therefore tested our compounds on two obvious candidates, HsCDK1 and HsPLK1 (Polo-like_kinase 1) both required for mitotic entryCitation50 ().
Table 4. IC50 (µM) of selected compounds on HsPLK1 and HsCDK1.
Values are IC50 expressed in µM and calculated from dose–response curves (each point from the curves was performed in duplicate). SI are calculated as follows: SI = IC50 CDK1/IC50 Haspin (shown in ). NR: not relevant.
None of the tested compounds inhibited PLK1 (IC50>10 µM). However, two compounds (5 and 12) showed strong inhibition of CDK1 (IC50 of 29 and 52 nM, respectively), which could account for the accumulation of cells in G2/M phase.
Altogether, these data showed that our compounds strongly affect the cell cycle in diverse ways. Some of these effects, shared by compounds 5 and 12, can be explained by their ability to inhibit CDK1 and CDK2. The cell cycle kinase profile of these compounds could be of interest for further pharmacological development at a time when the marketing of CDK inhibitors such as Palbociclib, Abemaciclib or Ribociclib is thrivingCitation51.
Effects on cell migration
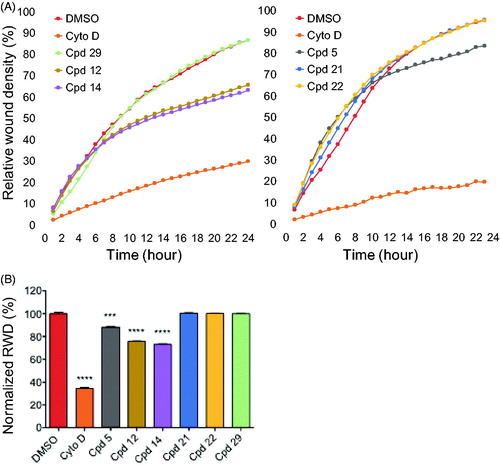
We tested the effect of our most potent inhibitors on U-2 OS cell migration in a wound healing assay over a duration of 24 h where relative wound density (RWD) was measured every hour (). The experiment was conducted in the presence of 0.5% FBS (fetal bovine serum). This condition allowed us to differentiate migrating cells from dividing cells which could interfere with the migration results. Compounds were tested at 0.6 µM alongside a DMSO control and cytochalasine D at 0.1 µg/mL as a positive control in two independent sets of experiments due to system limitations. The compound concentration was chosen as the maximum concentration that remained non-toxic to the cells for the length of the experiment in 0.5% FBS. After 24 h, cells treated with compounds 21 and 22 reached around 100% wound closure similar to the DMSO control and compound 29-treated cells (). In contrast, both compound 12- and 14-treated cells migrated significantly slower, reaching 88 and 73% of normalised wound closure after 24 h, respectively (). The latter results confirmed that the observed effects were the result of an impaired cell migration not linked to cell division. Taken together, these results indicated that both compounds 12 and 14 exhibited an inhibitory effect on cell migration, suggesting an interesting anti-angiogenic potential. They also suggested that this effect on cell migration may not be linked to Haspin inhibition itself but rather to an off-target effect of these compounds.
Figure 7. Effects of compounds on cell migration. (A) scratch wound cell migration assays were conducted on U-2 OS cells after treatment with 0.6 µM of each compound or 0.1 µg/mL cytochalasine D or 0.1% DMSO in two sets of experiments. Images were acquired every hour for 24 h 1by real-time live-cell microscopy (IncuCyte, Essen Bioscience) and images were analysed to determine the relative wound density (RWD) (IncuCyte software). Data are RWD mean ± SEM (n = 4). (B) Bar graph representation of the end point (24 h) of the experiment in A normalised against DMSO control (100%) for both sets of experiments. Results are mean percentage ± SEM; ***p ≤ 0.001, ****p ≤ 0.0001(two-tailed unpaired t-test).
Conclusion
We synthesised and assessed a novel series of imidazopyridines starting from CHR-6494, a known Haspin inhibitorCitation24. We tested the inhibitory properties of the compounds against HsHaspin and showed that the highest activity was obtained with compounds 21 and 22 (6 and 12 nM, respectively) compared to that of CHR-6494 (55 nM). These two compounds also shared the highest SI towards all the protein kinases tested. We saw a modest increase in selectivity of compounds 12 and 14 which notably did not inhibit Aurora B kinase (average SI of 24.2 and 23.6, respectively). All the compounds tested achieved in-cell Haspin inhibition, with compounds 12 and 21 being the most effective. We demonstrated that our best hits, compounds 21 and 22, were potent inhibitors of endogenous Haspin in human cells, which compared to the reference CHR-6494 or compound 12, did not strongly affect G2/M transition due to their selectivity against CDK1 and CDK2. Hence, compounds 21 and 22 can be considered interesting and selective tools that will help to dissect Haspin function from other cell cycle regulating kinases. On the other hand, the lower SI of compound 12 and 14 endow them with substantial anti-proliferative properties against human cancer spheroids as well as anti-migrating effects. These highly sought-after properties mean that they could be used as starting material for the potential development of anti-cancer therapeutics.
Supplemental Material
Download PDF (5.8 MB)Acknowledgements
The authors thank the staff at Diamond Light Source for their assistance during data collection.
Disclosure statement
The authors declare no conflict of interest.
Additional information
Funding
References
- Wilson L, Jordan MA. New microtubule/tubulin-targeted anticancer drugs and novel chemotherapeutic strategies. J Chemother 2004;16: 83–5.
- Keen N, Taylor S. Aurora-kinase inhibitors as anticancer agents. Nat Rev Cancer 2004;4:927–36.
- de Carcer G, Perez de Castro I, Malumbres M. Targeting cell cycle kinases for cancer therapy. Curr Med Chem 2007;14:969–85.
- Schmidt M, Bastians H. Mitotic drug targets and the development of novel anti-mitotic anticancer drugs. Drug Resist Updat 2007;10:162–81.
- Mountzios G, Terpos E, Dimopoulos MA. Aurora kinases as targets for cancer therapy. Cancer Treat Rev 2008;34:175–82.
- Ruchaud S, Carmena M, Earnshaw WC. Chromosomal passengers: conducting cell division. Nat Rev Mol Cell Biol 2007;8:798–812.
- Vader G, Lens SM. The Aurora kinase family in cell division and cancer. Biochim Biophys Acta 2008;1786:60–72.
- Cheung CH, Sarvagalla S, Lee JY, et al. Aurora kinase inhibitor patents and agents in clinical testing: an update (2011–2013). Expert Opin Ther Pat 2014;24:1021–38.
- Guo J, Anderson MG, Tapang P, et al. Identification of genes that confer tumor cell resistance to the aurora B kinase inhibitor, AZD1152. Pharmacogenomics J 2009;9:90–102.
- Jelluma N, Brenkman AB, van den Broek NJ, et al. Mps1 phosphorylates Borealin to control Aurora B activity and chromosome alignment. Cell 2008;132:233–46.
- Petsalaki E, Akoumianaki T, Black EJ, et al. Phosphorylation at serine 331 is required for Aurora B activation. J Cell Biol 2011;195:449–66.
- Petsalaki E, Zachos G. Clks 1, 2 and 4 prevent chromatin breakage by regulating the Aurora B-dependent abscission checkpoint. Nat Commun 2016;7:11451.
- De Antoni A, Maffini S, Knapp S, et al. A small-molecule inhibitor of Haspin alters the kinetochore functions of Aurora B. J Cell Biol 2012;199:269–84.
- Dai J, Higgins JM. Haspin: a mitotic histone kinase required for metaphase chromosome alignment. Cell Cycle 2005;4:665–8.
- Dai J, Sultan S, Taylor SS, Higgins JM. The kinase Haspin is required for mitotic histone H3 Thr 3 phosphorylation and normal metaphase chromosome alignment. Genes Dev 2005;19:472–88.
- Wang F, Ulyanova NP, van der Waal MS, et al. A positive feedback loop involving Haspin and Aurora B promotes CPC accumulation at centromeres in mitosis. Curr Biol 2011;21:1061–9.
- Wang F, Ulyanova NP, Daum JR, et al. Haspin inhibitors reveal centromeric functions of Aurora B in chromosome segregation. J Cell Biol 2012;199:251–68.
- Dai J, Sullivan BA, Higgins JM. Regulation of mitotic chromosome cohesion by Haspin and Aurora B. Dev Cell 2006;11:741–50.
- Dai J, Kateneva AV, Higgins JM. Studies of Haspin-depleted cells reveal that spindle-pole integrity in mitosis requires chromosome cohesion. J Cell Sci 2009;122:4168–76.
- Liang C, Chen Q, Yi Q, et al. A kinase-dependent role for Haspin in antagonizing Wapl and protecting mitotic centromere cohesion. EMBO Rep 2018;19:43–56.
- Kelly AE, Ghenoiu C, Xue JZ, et al. Survivin reads phosphorylated histone H3 threonine 3 to activate the mitotic kinase Aurora B. Science 2010;330:235–9.
- Wang F, Dai J, Daum JR, et al. Histone H3 Thr-3 phosphorylation by Haspin positions Aurora B at centromeres in mitosis. Science 2010;330:231–5.
- Yamagishi Y, Honda T, Tanno Y, Watanabe Y. Two histone marks establish the inner centromere and chromosome bi-orientation. Science 2010;330:239–43.
- Huertas D, Soler M, Moreto J, et al. Antitumor activity of a small-molecule inhibitor of the histone kinase Haspin. Oncogene 2012;31:1408–18.
- McKinley KL, Cheeseman IM. Large-scale analysis of CRISPR/Cas9 cell-cycle knockouts reveals the diversity of p53-dependent responses to cell-cycle defects. Dev Cell 2017;40:405–20.e2.
- Dave SS, Fu K, Wright GW, et al. Molecular diagnosis of Burkitt’s lymphoma. N Engl J Med 2006;354:2431–42.
- Rosenwald A, Alizadeh AA, Widhopf G, et al. Relation of gene expression phenotype to immunoglobulin mutation genotype in B cell chronic lymphocytic leukemia. J Exp Med 2001;194:1639–47.
- Han X, Kuang T, Ren Y, et al. Haspin knockdown can inhibit progression and development of pancreatic cancer in vitro and vivo. Exp Cell Res 2019;385:111605.
- Bastea LI, Hollant LMA, Doppler HR, et al. Sangivamycin and its derivatives inhibit Haspin-Histone H3-survivin signaling and induce pancreatic cancer cell death. Sci Rep 2019;9:16588.
- Eswaran J, Patnaik D, Filippakopoulos P, et al. Structure and functional characterization of the atypical human kinase Haspin. Proc Natl Acad Sci U S A 2009;106:20198–203.
- Villa F, Capasso P, Tortorici M, et al. Crystal structure of the catalytic domain of Haspin, an atypical kinase implicated in chromatin organization. Proc Natl Acad Sci U S A 2009;106:20204–9.
- Chen LS, Redkar S, Bearss D, et al. Pim kinase inhibitor, SGI-1776, induces apoptosis in chronic lymphocytic leukemia cells. Blood 2009;114:4150–7.
- Patnaik D, Jun X, Glicksman MA, et al. Identification of small molecule inhibitors of the mitotic kinase Haspin by high-throughput screening using a homogeneous time-resolved fluorescence resonance energy transfer assay. J Biomol Screen 2008;13:1025–34.
- Cuny GD, Robin M, Ulyanova NP, et al. Structure-activity relationship study of acridine analogs as Haspin and DYRK2 kinase inhibitors. Bioorg Med Chem Lett 2010;20:3491–4.
- Opoku-Temeng C, Dayal N, Aflaki Sooreshjani M, Sintim HO. 3H-pyrazolo[4,3-f]quinoline Haspin kinase inhibitors and anticancer properties. Bioorg Chem 2018;78:418–26.
- Kim JE, Lee SY, Jang M, et al. Coumestrol epigenetically suppresses cancer cell proliferation: coumestrol is a natural Haspin kinase inhibitor. Int J Mol Sci 2017;18:2228.
- Han L, Wang P, Sun Y, et al. Anti-melanoma activities of Haspin inhibitor CHR-6494 deployed as a single agent or in a synergistic combination with MEK inhibitor. J Cancer 2017;8:2933–43.
- Bendjeddou LZ, Loaec N, Villiers B, et al. Exploration of the imidazo[1,2-b]pyridazine scaffold as a protein kinase inhibitor. Eur J Med Chem 2017;125:696–709.
- Belaroussi R, El Hakmaoui A, Akssira M, et al. Regioselective synthesis of 2,4‐substituted pyrido[1′,2′:1,5]pyrazolo[3,4‐d]pyrimidines through sequential Pd‐catalyzed arylation and SNAr reactions. Eur J Org Chem 2016; 2016:3550–8.
- Copin C, Henry N, Buron F, Routier S. Palladium-catalyzed direct arylation of 2,6-disubstituted imidazo[2,1-b][1,3,4]thiadiazoles. Synlett 2016;27:1091–5.
- Belaroussi REH, Percina A, Chartier N, et al. Synthesis of 1,4-disubstituted pyrido[1′,2′:1,5]pyrazolo[3,4-d]pyridazines by means of SNAr and palladium-catalysed reactions. Eur J Org Chem 2015;2015:4006–17.
- Modi V, Dunbrack RL Jr. Defining a new nomenclature for the structures of active and inactive kinases. Proc Natl Acad Sci U S A 2019;116:6818–27.
- Chaikuad A, Diharce J, Schröder M, et al. An unusual binding model of the methyl 9-anilinothiazolo[5,4-f] quinazoline-2-carbimidates (EHT 1610 and EHT 5372) confers high selectivity for dual-specificity tyrosine phosphorylation-regulated kinases. J Med Chem 2016;59:10315–21.
- Edmondson R, Broglie JJ, Adcock AF, Yang L. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay Drug Dev Technol 2014;12:207–18.
- Nigg EA. Cyclin-dependent protein kinases: key regulators of the eukaryotic cell cycle. Bioessays 1995;17:471–80.
- Hochegger H, Takeda S, Hunt T. Cyclin-dependent kinases and cell-cycle transitions: does one fit all?. Nat Rev Mol Cell Biol 2008;9:910–6.
- Sherr CJ. Growth factor-regulated G1 cyclins. Stem Cells 1994;12: 47–55. discussion 55–47.
- Meyerson M, Harlow E. Identification of G1 kinase activity for cdk6, a novel cyclin D partner. Mol Cell Biol 1994;14:2077–86.
- Feizbakhsh O, Pontheaux F, Glippa V, et al. A Peak of H3T3 Phosphorylation Occurs in Synchrony with Mitosis in Sea Urchin Early Embryos. Cells 2020;9:898.
- Parrilla A, Cirillo L, Thomas Y, et al. Mitotic entry: the interplay between Cdk1, Plk1 and Bora. Cell Cycle 2016;15:3177–82.
- Carles F, Bourg S, Meyer C, Bonnet P. PKIDB: a curated, annotated and updated database of protein kinase inhibitors in clinical trials. Molecules 2018; 23:908.