
ABSTRACT
Introduction
Early biomarker discovery studies have praised the value of their emerging results, predicting an unprecedented impact on health care. Biomarkers are expected to provide tests with increased specificity and sensitivity compared to existing measures, improve the decision-making process, and accelerate the development of therapies. For rare disorders, like Duchenne Muscular Dystrophy (DMD) such biomarkers can assist the development of therapies, therefore also helping to find a cure for the disease.
Area covered
State-of-the-art technologies have been used to identify blood biomarkers for DMD and efforts have been coordinated to develop and promote translation of biomarkers for clinical practice. Biomarker translation to clinical practice is however, adjoined by challenges related to the complexity of the disease, involving numerous biological processes, and the limited sample resources. This review highlights the current progress on the development of biomarkers, describing the proteomics technologies used, the most promising findings and the challenges encountered.
Expert opinion
Strategies for effective use of samples combined with orthogonal proteomics methods for protein quantification are essential for translating biomarkers to the patient’s bed side. Progress is achieved only if strong evidence is provided that the biomarker constitutes a reliable indicator of the patient’s health status for a specific context of use.
1. Introduction
Molecular biomarkers are measurable properties or characteristics eg. proteins, that are specific, accurate, and sensitive indicators of disease states. In clinical setup, biomarkers are routinely analyzed in biological samples, and development of novel biomarkers holds promise to radically change the way diseases are diagnosed, treated, and cured. Blood, for example, mirrors pathological changes that occur in the body, as it accesses different organs and accommodates molecular markers originating from different tissues [Citation1,Citation2]. Thus, blood samples constitute a valuable source of information for clinical management of disorders [Citation1,Citation3]. This review focuses on the current knowledge regarding discovery and validation of protein biomarkers for DMD, in blood, and highlights the challenges encountered in translating such markers to clinical applications.
2. The value of biomarkers
Numerous efforts have explored the promised land of biomarkers searching for new and/or better biomarkers, in a quest for health care improvement and development of novel therapies. Biomarkers are expected to significantly contribute to precision medicine by providing not only improved diagnostics but also new indicators for disease prediction [Citation3]. Similarly, they are anticipated to provide information regarding the individual variation in terms of eg. response to treatment, to improve the decision-making process for selection of therapies and allow adaptation of clinical interventions to the patient’s need [Citation4,Citation5]. In addition, biomarkers benefit the patient not only by improving management of the diseases with already existing treatments but also by facilitating the development of novel therapies. For incurable disorders, biomarker aid preclinical research and clinical trials, by ensuring appropriate study design and patient recruitment, as well as evaluation of treatment outcome and toxic effects. Considering the many aspects biomarkers cover and the numerous applications they are used for, classification and standardization of definitions are essential. Biomarkers are classified in: (1) diagnostic, (2) pharmacodynamic/response, (3) disease progression monitoring, (4) prognostic, (5) predictive, and (6) susceptibility/risk [Citation6,Citation7]. This classification captures the disparity between biomarkers, ensures unambiguous communication, and facilitates translation of research results to clinical practice [Citation7].
3. Duchenne Muscular Dystrophy
One of the most severe forms of muscular dystrophy is Duchenne muscular dystrophy (DMD) affecting 1:3,500 to 1:5,000 boys [Citation8–Citation11]. Affected individuals experience a progressive disease, in which loss of muscle mass and, consequently function, occurs over a period of many years with, fatal outcome [Citation12,Citation13]. The disease causes initially, delay of motor milestones and as the disease progresses, loss of motor function, resulting in a complete loss of ambulation at age between 8 and 14 years [Citation11], severe dilated cardiomyopathy and respiratory insufficiency [Citation14]. The median life expectancy estimated across 12 countries, is between 19.6 and 20.4 years, for patients that have not received respiratory support and between 28.9 and 30.6 years for patients using respiratory aid [Citation15].
DMD is an X-linked chromosomal-inherited recessive disease, caused by mutations in the DMD gene. The dystrophin gene encodes for a large protein, of 427 kDa, with a key role in muscle contraction and function [Citation16–Citation18]. Dystrophin provides a flexible connection between the actin filaments and the sarcolemma through four major functional domains: the actin-binding N-terminal domain interacting with γ-actin filaments, the central rod domain acting as a linker, the cysteine-rich domain interacting with the β-dystroglycan at the sarcolemma, and the C-terminal domain. DMD is caused by mutations, which disrupt the reading-frame, with premature translation termination and alteration or absence of the actin or the β-dystroglycan binding sites, as a consequence. The absence of a functional dystrophin protein alters the composition and structure of the dystrophin-glycoprotein complex (DGC) with consequences on structural stability of muscle fibers, signaling, and regeneration of muscle fibers subsequent injury. In DMD muscles, the DGC’s role as a link between the cytoskeleton and the extracellular matrix is compromised resulting in increased fragility of the sarcolemma during intensive contractile activity [Citation19]. The increased membrane fragility, alters its permeability to otherwise impermeable molecules with implication on calcium homeostasis, as Ca2+ ions enter through membrane breaks. The reoccurring muscle fiber damage solicits recruitment of inflammatory cells for removal of damaged fibers and subsequent muscle fiber regeneration [Citation20,Citation21]. Restoration of damaged fibers is not fully compensated due to alteration in muscle satellite cells proliferation and differentiation [Citation22,Citation23]. Thus, as the disease progresses, patients experience increasing muscle fiber degradation, inflammation, fibrosis, and fat infiltration. As a consequence of membrane breaks and/or increase deregulation of vesicle trafficking, muscle-specific proteins are most likely released into the blood stream [Citation24–Citation26]. In addition, inflammatory cells also contribute to accumulation of pro-inflammatory molecules and cytokines in blood [Citation27].
The assessed diagnostic tests for clinical management of muscular dystrophies include a wide range of physical tests, blood and biopsy tests, performed by a multi-disciplinary team including but not limited to experts in neuromuscular medicine, pediatrics, genetics. The 6-minute walk test (6MWT), 10 meter walk test (10MWT), Northern Star Ambulatory Assessment (NSAA), and performance of upper limb (PUL) are commonly used measurement tools in motor dysfunctional children. Physical tests very often rely on the patient’s capability to collaborate with the clinicians, follow instructions, and ability to perform and complete the entire test. In addition, the tests are influenced by other factors than the disease eg. patient’s motivation, physical exercise, age, psychological-disorder, and/or attention deficiency. These aspects have consequences on the sensitivity and accuracy of the functional tests. Beside physical tests, skeletal muscle composition in terms of Magnetic Resonance Imaging (MRI) measured fat fraction is currently used as an exploratory biomarkers for DMD [Citation28,Citation29]. The fat fraction in muscles correlates with functional outcomes [Citation30], physical tests, and predict deterioration of patient mobility [Citation29,Citation31,Citation32]. MRI itself is noninvasive and feasible, even in young children. However, imaging is not part of the routine and long-term clinical follow-up, due to the lack of standardized protocols for image acquisition and data analysis, high cost, and long duration of the scans [Citation33].
4. Protein biomarkers for DMD
International efforts have explored blood components eg. proteins, miRNA to identify biomarker candidates for DMD. Proteomics studies have reported several muscle-specific proteins as well as proteins involved in energy metabolism, fibrosis, and inflammation as potential biomarker candidates [Citation34–Citation37] (). Similarly, circulating microRNAs eg. miR-29 c-3 [Citation38], miR-1 and miR-206 [Citation39,Citation40] have been shown to be potential biomarkers for early detection of DMD or monitoring disease progression and treatment outcome [Citation41,Citation42]. In addition, amino acid eg. arginine, glutamine, and histidine [Citation43] as well as metabolites eg. creatine has been identified as potential biomarker candidates [Citation44,Citation45]. Additional studies of miRNA and metabolites in longitudinally collected samples can provide essential evidence to promote their use as blood biomarkers in a clinical setup [Citation46].
Table 1. List of DMD biomarkers identified. The biomarkers are ranked based on the number of publications providing evidence for association of the biomarker with DMD. Biomarkers identified in both human and animal models are listed first, followed by biomarkers identified only in DMD patients and subsequently in only DMD animal models.
4.1. Proteomic technologies and biomarker discovery and validation
State-of-the-art technologies within the proteomics field have contributed to the discovery and validation of biomarkers for the most common diseases eg. cancer, metabolic, and cardiovascular disorders [Citation61–Citation63]. This development is also beneficial for biomarker discovery in the context of rare disorders which comprises chronic, incurable diseases [Citation46,Citation64]. The initial studies exploring serum composition from patients with DMD focus mainly on diagnostic biomarkers and resulted in the discovery of creatine kinase [Citation65,Citation66] through enzymatic assays. In addition, muscle-specific enolase [Citation67,Citation68] and carbonic anhydrase III (CA3) [Citation69,Citation70] are two additional serum biomarkers identified using enzymatic or radio-immunoassays. From the initial studies, CK measurements have been translated to the clinical practice [Citation71].
In time, biomarker discovery has transitioned from single target analysis to multiplex measurements using both mass spectrometry-based methods and affinity-based methods. Proteins associated with the disease are identified through protein profiling of healthy and dystrophic tissues in mouse and canine models [Citation34,Citation72–Citation75] and further explored in samples collected from patients. The unprecedented development of high-throughput technologies has resulted in discovery of blood biomarkers through systematic protein profiling of a subset of the proteins associated with DMD or untargeted protein analysis [Citation36,Citation76,Citation77]. Protein profiling using mass spectrometry has benefit from the increased resolution and throughput and resulted in the discovery of serum biomarkers eg. factor XIIIA in mdx mice [Citation76] using MALDI-MS, myomesin-3 (MYOM3) in DMD patients using a tandem mass spectrometry approach LC/MS-MS [Citation77] and fibronectin (FN1) detected by MS/MS [Citation37]. However, serum and plasma comprise complex mixtures of proteins at concentration within a wide range, with albumin at concentration of 35–50 mg/ml being one of the most abundant proteins and IL-6 at a concentration of 0–5 pg/ml [Citation2]. This complexity has consequences on the detection of relevant targets. The presence of high abundant peptides causes decreased ionization of rare peptides with consequences on the detection of least abundant proteins [Citation78], restricting the detection limit to above 50 ng/ml [Citation79]. The complexity of serum and plasma samples can be diminished through depletion of the most abundant proteins eg. albumin and IgG [Citation80] or the 12 most abundant proteins [Citation77] or enrichment of low abundant proteins [Citation37]. However, depletion of such proteins may alter the abundance of other proteins and increase the standard error of the analysis [Citation81,Citation82]. To tackle with the complexity, samples are also fractionated based on hydrophobicity, charge, affinity, or size of the proteins [Citation37,Citation76,Citation77,Citation80]. The methods of choice in biomarker discovery studies vary resulting in discrepancies with respect to which biomarkers are detected. However, it should be noted that results obtained by mass spectrometry are confirmed with additional immune-based methods eg. ELISA and Western Blot in the case of eg. FN1 and MYOM3, respectively, [Citation37,Citation77].
The increasing availability of validated antibodies for protein profiling [Citation83] enabled discovery of potential biomarkers using a suspension bead array platform [Citation36,Citation84]. This technology allows antibody-mediated capturing of protein targets to unique color coded beads and detection of the captured molecule through a covalently bound fluorescent label [Citation36,Citation85]. Besides a high multiplexing capacity, that allows analysis of as many as 500 analytes simultaneously, this technological platform uses only microliter amount of sample and has a sensitivity of pg/ml range for analysis of proteins in complex samples [Citation86]. In addition, this technology also offers the possibility to mimic the ELISA concept, by detecting the captured target with a second antibody, making it attractive for the development of sensitive and accurate biomarker quantification assay for translation to clinical use.
Being indispensable for proteomics studies, antibodies are both praised and condemned, and their performance continuously scrutinized in particular since, antibody specificity is context and application dependent [Citation87]. Academic efforts like the The Human Protein Atlas project [Citation83] and commercial producers ensure development and access to large numbers of antibodies, validated according to developed guidelines [Citation88,Citation89]. In particular, the guidelines for enhanced validation of antibodies designed by the International Working Group for Antibody Validation (IWGAV), an ad-hoc formed group of leading researchers, promotes rigorous validation of such reagents and has been adopted by researchers and leading antibody producers [Citation89–Citation91]. Beside antibodies development of short single-stranded oligonucleotides as affinity reagents and the SomaLogic technology enabled researchers to analyze more than 1,000 targets in cross-sectional studies in the context of DMD [Citation92–Citation96]. The assay relies on the capture of the target in its native conformation to immobilized protein-specific Slow Off-rate Modified DNA aptamers (SOMAmers). The SOMAmer-target complex is isolated from the sample and subsequent dissociation of the complex, the SOMAmer quantified. Quantification is achieved through hybridization of the SOMAmers to an array using the SomaScan platform. The platform has a high multiplexing capacity that allows analysis of 5,000 targets simultaneously. Although the high-throughput screening methods are useful biomarker discovery tools, confirmation of results with more targeted assays eg. ELISA is necessary.
4.2. Biomarkers for DMD
One of the muscle-specific proteins released into the blood stream of DMD patients is creatine kinase (CK). CK is elevated in DMD patients and declines as the diseases progresses [Citation36,Citation84,Citation97,Citation98]. Its increased activity has also been used as an indicator of muscle damage in clinical practice [Citation71]. As a biomarker, serum CK is influenced by other factors than the disease, as shown in both DMD patients and mouse model studies [Citation99–Citation101]. Alone CK is not specific enough to be used as a diagnostic or prognostic biomarker [Citation102]. However, CK has been used for new born screening to facilitate early detection of muscle injury diseases as DMD [Citation103–Citation105]. Beside CK, several proteomics studies performed in the context of DMD, reported elevated blood levels of other muscle-specific proteins eg. carbonic anhydrase 3 (CA3), malate dehydrogenase 2 (MDH2), myosin light chain 3 (MYL3), cardiac muscle troponin I (TNNI3), titin (TTN) and proteins involved in energy metabolism eg. beta enolase, fructose-bisphosphate aldolase, electron transfer flavoprotein A, fibrosis eg. fibronectin and inflammation eg. interleukin [Citation46,Citation64,Citation106] ().
The majority of the identified biomarker candidates are muscle-specific proteins elevated in DMD patients in comparison to controls. Over time, these biomarkers have declining abundance trajectories, as the disease progresses [Citation107] most likely correlating with the decreasing muscle mass. The serum and/or plasma levels of CA3, MDH2, MYOM3, myosin, and troponins in DMD have been confirmed in several studies as potential disease progression monitoring biomarkers [Citation34,Citation36,Citation68,Citation70,Citation84,Citation92,Citation108]. In addition, serum MDH2 has also been identified as a predictor of loss of ambulation and associated with response to steroid treatment in a longitudinal study [Citation84] and MYOM3 as a therapy responsive biomarker in antisense oligonucleotide-mediated exon-skipping treated mdx mice [Citation77]. Beside proteins that are ubiquitously expressed in skeletal muscle, proteins specifically expressed in eg. heart provides information about cardiac status. TNNI3 and Interleukin 1 Receptor-Like 1 Protein are associated with cardiac degeneration and potential biomarkers for cardiac injury in both patients and DMD models [Citation95,Citation109,Citation110]. The use of membrane sealant in dystrophic dogs to ameliorate cardiac injury resulted in decrease of TNNI3 [Citation111]. Matrix metalloproteinase-9 (MMP9) which is involved in the degradation of the DGC protein has been shown to increase in DMD patients as the disease progresses [Citation112,Citation113]. Similar studies did not corroborate these findings raising questions regarding the differences between the cohorts analyzed in terms of eg. age, mutations, baseline levels in healthy individuals, etc. [Citation114–Citation116]. Besides muscle-specific proteins, other targets are identified as biomarker candidates. Leptin (LEP) which is produced mostly by adipocyte is identified as a serum biomarker associated with severity of metabolic syndrome [Citation117]. Furthermore, LEP together with the C reactive protein (CRP) are also inflammatory markers [Citation118]. Several reports show that the degree of inflammation is evaluated by plasma haptoglobin levels [Citation119,Citation120]. Elevated serum levels of haptoglobin are also identified in dystrophic mdx-4cv mice [Citation80]. Beside inflammation, DMD disease progression is also associated with increasing fibrosis. Proteins like IL-6, Interleukin 10 and INF-γ and Interleukin 13 are suitable for monitoring fibrosis at different disease stages [Citation92,Citation94,Citation118,Citation121]. Biomarkers associated with muscle function, inflammation, and fibrosis may recapitulate disease progression but additional biomarkers are required for the development of therapies. Cystatin C (CST3) which is a clinical biomarker for kidney injury and altered glomerular filtration rate [Citation122], has also been studied in the context of DMD [Citation123]. The results indicate that serum CST3 can be used to monitor nephrotoxicity of therapies in eg. clinical trials [Citation123]. As a toxicity biomarker, CST3 has the advantage of being independent of age (above 1,5 years), ambulatory capacity, and type of steroid treatment [Citation123].
The identified biomarkers are often associated with one or few specific clinical parameters eg. functional, cardiac status, indicating that no single biomarker can be expected to describe the disease progression. As the disease progresses the abundance of several blood biomarkers, in particular muscle-specific proteins, follow a declining trajectory that eventually plateaus in late teens, 1–2 years subsequent loss of ambulation [Citation84,Citation124]. This variation pattern makes such biomarkers more informative during the early stages of the disease in contrast to the late stages. This is also evident for biomarkers associated with cardiomyopathy which are more informative as cardiac fibrosis progresses eg. ST1 [Citation95]. This indicates that disease progression monitoring over time, for example, would benefit from monitoring more than one biomarker. To increase the accuracy of biomarker’s several studies advocate the shift from single biomarkers to biomarker ‘panels’ or ‘signatures’, a prerequisite for precision medicine [Citation3,Citation125]. Several studies support the use of biomarker panels to describe pathological changes as DMD is characterized by multiple processes decreasing muscle function, increasing fibrosis, and inflammation [Citation36,Citation94,Citation107].
4.3. Biomarker validation
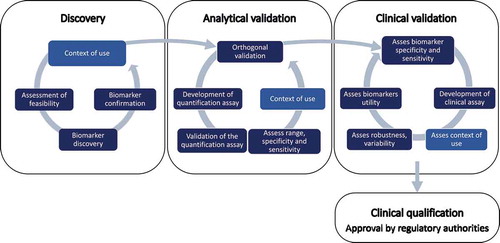
Several identified biomarkers, have been analyzed in different patient cohorts and confirmed by several research groups. However, none has been approved by regulatory authorities like the Food and Drug Administration or the European Medicines Agency. The general consensus that biomarkers are difficult to translate from research to practical use is also valid for DMD [Citation126,Citation127]. The challenges encountered relate to providing evidence that clearly defines how a biomarker is used in a specific context with the purpose of improving patient health care. The intended context of use varies and determines the burden of proof required to support translation of biomarkers to clinical practice [Citation6]. For example, a higher false discovery rate is acceptable for a biomarker used in the context of newborn screening, in contrast to a biomarker used to measure the effect of therapy in non-ambulant patients eg. steroid treatment that does not cure the disease. The biomarker development process () is often described as a linear process comprising: (1) discovery, (2) analytical validation, (3) clinical validation, and (4) qualification of biomarker but the linearity is not entirely sustained. Analysis of three cross-sectional studies performed with the same technology reported an overlap of only 12% of the biomarkers analyzed, discriminating between DMD patients and controls in all datasets [Citation107]. This indicates the necessity to confirm discovery studies in several cohorts. To discard irreproducible results early in the biomarker development process, orthogonal analytical validation strategies that use independent proteomics methods to confirm the value of biomarkers should be employed. Furthermore, the biomarker’s context of use defined during the discovery phase is not necessarily confirmed in the validation or qualification phase. The trajectory of muscle function associated biomarkers indicates that they are potential disease progression biomarkers that decrease as the disease progresses [Citation84]. However, this raises the question whether such biomarkers are useful disease progression biomarkers throughout the course of the disorder, as their abundance may decrease below the limit of detection of the quantification assay. If this is the case more sensitive assays are needed or new biomarker candidates have to be developed. Similarly, in mdx mice interleukin 13 has been reported as a disease progression marker useful during the late stages of the disease whereas IL-6, INF-γ, and IL-10 during earlier stages of DMD [Citation121]. Maintaining a circular process with feedback mechanisms ensures that biomarkers are continuously assessed, their context of use evaluated and refined in each phase, and necessary action prioritized.
Establishment of the DMD diagnosis is well developed but approval of novel therapies is hindered by the lack of validated biomarkers for monitoring treatment outcome and disease progression. Collection and access to longitudinal samples are necessary to enable testing of already discovered biomarkers for new context of use and validation of disease progression biomarkers. Therapies that restore expression of a functional dystrophin protein are developed using different strategies (1) exon skipping, (2) stop codon readthrough, (3) gene addition, (4) genome editing, or (5) myoblast transplantation. Currently, the available therapies that restore expression of functional dystrophin provide treatment for only a limited number of patients (14% for Eteplirsen approved by FDA and 13% for Ataluren conditionally approved by EMA) [Citation128,Citation129]. To facilitate the development of new therapies biomarkers suitable for clinical trials remains to be developed. Biomarkers are required also for pre-clinical studies that test novel therapies in DMD model organisms. For a more effective development of therapies biomarkers should be usable in both clinical and pre-clinical studies both patients and model organisms.
Figure 1. Biomarker development for DMD requires (1) discovery, (2) analytical validation, (3) clinical validation, and (4) qualification of biomarker. The discovery phase comprises formulation of the context of use, assessment of feasibility in terms of eg. samples resources, selection of appropriate analytical method, etc., discovery of the biomarker and confirmation in additional samples collections to assess reproducibility in several cohorts. The analytical validation phase includes orthogonal validation of the biomarker using different analytical methods, development of biomarker quantification assays for targeted analysis and validation of the biomarker quantification assay, to evaluate biomarker sensitivity, specificity, dynamic range, and variation using the developed quantification assay and refine the context of use. Clinical validation requires testing the specificity and sensitivity of the biomarker in clinical setup, development of clinical assays, assess the context of use, robustness and variability, and its utility in clinical practice.
4.4. Challenges
The scarcity of high-quality samples, the limited knowledge about disease phenotype and patient variability, makes muscular dystrophies a challenging research area. Within rare disorders, the number of samples mirrors the low prevalence of the disorders and the difficulty in retrieving samples from patients. Clinicians, care providers, and parents encounter difficulties in motivating collection of samples for other than diagnostics and/or treatment purposes. However, collection of samples and patient information has been standardized and procedures harmonized through several national and international efforts eg. EuroBioBank [Citation130] (http://www.eurobiobank.org/index.html), RD-Connect (http://rd-connect.eu/about/) [Citation131] Orphanet (https://www.orpha.net/consor/cgi-bin/index.php?lng=EN) [Citation132]. The importance of sample collection and access to such resources has been acknowledged and platforms are designed for easy access, selection, and sharing of biological samples as well as integration of omics results with phenotypic information [Citation132]. Another essential aspect is the quality of samples as blood components are subjected to alterations during retrieval, handling, and storage. Serum is prepared without additives through removal of the fibrin clot and blood cells, whereas plasma is prepared by addition of anticoagulants like EDTA, heparin, or sodium citrate. The preparation procedures influence the sample composition in terms of the total protein concentration, the volume, presence of additives, and stability [Citation133]. To overcome this limitation and increase the statistical power of the studies, strategies have been designed for multi-cohort and concurrent serum and plasma analysis [Citation36]. Biomarkers with concordant results in sample collections collected at different clinical sites and both serum and plasma represent most likely robust biomarkers not influenced by variation in sample collection and pre-analytical preparation procedures.
Another challenging aspect is the heterogeneity of the disease. Patients with severe DMD forms may loose ambulatory capacity at the age of 8 years whereas patients with milder DMD forms after the age 14 years. The patient variation in muscle function between the age of 8–14 can obscure correlation between biomarkers and functional tests like 10MWT, 6MWT, etc. In the light of these challenges studies are required to increase our understanding of the disease pathology and the different phenotypes on both cellular and tissue level.
The outcome of several clinical trials shows that tested therapies are promising but not always effective. This makes it difficult to develop pharmacodynamic biomarkers. For example, in a study comprising samples from two independent clinical trials, serum MMP9 was not found to be a predictive biomarker for treatment response with drisapersen [Citation113]. The limited efficacy of such a treatment, might not be measurable regardless of the specificity or sensitivity of a biomarker measurements.
5. Translation of biomarkers beyond DMD
Muscular dystrophies represent a large group of genetic rare disorders caused by different genetic mutations, with progressive muscle weakness symptom as the common denominator. Several muscular dystrophies share phenotypic characteristics related to deterioration of muscle function, age of onset, severity of muscle wasting symptoms, loss of mobility, short life expectancy in severe cases, and mild to severe cardiac and respiratory impairment [Citation134]. Among them the most common muscular dystrophies besides Duchenne DMD are limb-girdle muscular dystrophy (LGMD), facioscapulohumeral muscular dystrophies (FSHD) and myotonic dystrophy 1 (DM1) with a prevalence of 0.8–6.9, 3.2–4.6 and 1.7–4.2 per 100,000, respectively [Citation135–Citation139]. In addition, Becker muscular dystrophy (BMD) has similarities with DMD as it is caused by in-frame mutations in the dystrophin gene [Citation140,Citation141]. Patients affected by these muscular dystrophies also exhibit elevated serum levels of muscle proteins like CK and CA3. CA3 is a severity marker in DMD and BMD but also a serum biomarker candidate for FSHD, LGMD, congenital, and myotonic dystrophy [Citation70,Citation142]. Serum ratio of lactate dehydrogenase (LDH) isoforms in patients affected by DMD, FSHD, or LGMD has been reported to be disease-specific [Citation143]. These reports support the hypothesis that muscular dystrophies are likely to share muscle wasting biomarkers in blood.
6. Conclusion
Available proteomics technologies, sample resources, under the umbrella of international collaborations promote discovery of biomarkers. The unmet need for translating discovered biomarkers to practical tools, however, still remains unachieved in spite of the impressive number of biomarkers identified. Biomarker associated with muscle function, inflammation, fibrosis, and cardiac status, that can potentially describe different disease states are within reach. Valorization of these findings will require large efforts to be invested in analytical validation and clinical qualification of biomarkers. As DMD diagnostics has been developed during the past decades, biomarkers for monitoring disease progression and pharmacodynamic/response to treatment, are in critical demand. Such biomarkers can facilitate the development of effective therapies to cure DMD. Concerted action from clinicians, researchers, pharma companies, and patient organizations is required to make the necessary progress.
7. Expert opinion
In spite of the constraints related to the sample accessibility and disease heterogeneity, biomarker development within DMD has benefit from a number of key aspects. Several networks eg. TREAT-NMD and RD-Connect have contributed to the development of a strong stakeholder communities and promoted new partnerships and collaborative projects. Majority of the reports during the past few years originate from the involvement of multi-disciplinary research teams rather than single laboratories scattered around the world [Citation34,Citation84,Citation92,Citation107,Citation113,Citation144]. Exchange of knowledge and experience between experts contribute to the development of common goals, well-defined research objectives, selection appropriate study design and evaluation of research outcome from different perspectives eg. medical, biological, statistical, and/or analytical. In addition, patient organizations eg. Parent Project Muscular Dystrophy (www.parentprojectmd.org), Duchenne World Organization (www.worldduchenne.org), have become instrumental in promoting patients benefit and shaped the research environment [Citation145,Citation146]. The organizations have advocated not only for an active involvement of patients in clinical trials and research but also for increased sharing of resources and data according to the FAIR guidelines [Citation147,Citation148].
The efforts invested in the establishing patient registries and biobanks have provided invaluable data and biological material for research while still preserving the patients’ legal rights [Citation130,Citation132,Citation149]. Harmonized protocols for data and sample collection, handling, and storage, enable multi-cohort comparisons and meta-analysis. These type of analysis are imperative for rare disease studies in which the statistical power of the study is often low due to the limited number of cases affected. One aspect that can propel the development of biomarkers is a more active collaboration between academic research groups and pharmaceutical companies. In particular, accessibility to samples from pre-clinical and clinical trials, even if collected during clinical trials failing to prove a beneficial effect of the tested drug, can be used for research studies eg. evaluation of discovered disease progression biomarkers.
There is a general consensus that the development of biomarkers other than diagnostic is required within DMD. While biomarkers for monitoring disease progression are beneficial for clinical management of the disease, biomarkers for monitoring treatment outcome can aid the development of therapies, with positive impact on patients’ life quality and life expectancy. In addition, development of biomarkers for newborn screening are also imperative for early diagnostics and early therapeutic interventions. Evidently, several biomarkers are required to match the different requirements. In addition, majority of the most promising biomarkers have declining trajectories as the disease progresses making them more suitable to measure disease state changes during the early phase of the disease. Efforts should be invested in identifying biomarker signatures rather than single biomarkers, taking into consideration the individual variability and phenotype, enabling transition to precision medicine.
Article highlights
Proteomics technologies have enabled discovery of numerous proteins as biomarker candidates but their validation and translation to practical use, still requires additional experimental evidence that supports or undermines their value as biomarkers.
Biomarker development for Duchenne Muscular Dystrophy is associated with challenges associated with the limited number of patients and high phenotypic variation.
Involvement of several biological processed in Duchenne Muscular Dystrophy pathology and the phenotypic variation indicates that more than one biomarker is required to monitor disease progression and efficacy of treatment. Such biomarker panels are fundamental for the development of precision medicine.
Proteins associated with muscle function, inflammation, fibrosis, and cardiac status are reported as potential biomarkers. Among the most promising biomarkers 19 are confirmed using more than one proteomics method and in more than one scientific report.
Validation and qualification of biomarkers require concerted action and involvement of multiple stakeholders from clinicians, researchers, to pharma companies, and patient organizations.
Declaration of interest
The author has no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Anderson NL. The clinical plasma proteome: a survey of clinical assays for proteins in plasma and serum. Clin Chem. 2010;56(2):177–185.
- Landegren U, Vänelid J, Hammond M, et al. Opportunities for sensitive plasma proteome analysis. Anal Chem. 2012;84(4): 1824–1830.
- Borrebaeck CAK. Precision diagnostics: moving towards protein biomarker signatures of clinical utility in cancer. Nat Rev Cancer. 2017;17(3):199–204.
- Patil H, Saxena SG, Barrow CJ, et al. Chasing the personalized medicine dream through biomarker validation in colorectal cancer. Drug Discov Today. 2017;22(1):111–119.
- Pritzker K. Biomarker imprecision in precision medicine. Expert Rev Mol Diagn. 2018;18(8):685–687.
- Califf RM. Biomarker definitions and their applications. Exp Biol Med (Maywood). 2018;243(3):213–221.
- FDA-NIH Biomarker Working Group. BEST (Biomarkers, endpoints, and other tools) resource. 2016.
- Romitti PA, Zhu Y, Puzhankara S, et al. Prevalence of Duchenne and Becker muscular dystrophies in the United States. Pediatrics. 2015;135(3):513–521.
- Giegerich E, Stuntz M. Duchenne Muscular Dystrophy prevalence in the US: a novel incidence-based modeling approach using system dynamics. Value Health. 2019;22:S244–S244.
- Bladen CL, Salgado D, Monges S, et al. The TREAT-NMD DMD global database: analysis of more than 7,000 Duchenne Muscular Dystrophy mutations. Hum Mutat. 2015;36(4):395–402.
- Falzarano MS, Scotton C, Passarelli C, et al. Duchenne Muscular Dystrophy: from diagnosis to therapy. Molecules. 2015;20(10):18168–18184.
- Emery AEH. Muscular dystrophy into the new millennium. Neuromuscul Disord. 2002;12(4):343–349.
- Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne Muscular Dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9(1):77–93.
- Van Ruiten HJA, Marini Bettolo C, Cheetham T, et al. Why are some patients with Duchenne Muscular Dystrophy dying young: an analysis of causes of death in North East England. Eur J Paediatr Neurol. 2016;20(6):904–909.
- Landfeldt E, Thompson R, Sejersen T, et al. Life expectancy at birth in Duchenne Muscular Dystrophy: a systematic review and meta-analysis. Eur J Epidemiol. 2020;71(3):304.
- Koenig M, Monaco AP, Kunkel LM. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell. 1988;53(2):219–228.
- Campbell KP. Three muscular dystrophies: loss of cytoskeleton-extracellular matrix linkage. Cell. 1995;80(5):675–679.
- Le Rumeur E, Winder SJ, Hubert J-F. Dystrophin: more than just the sum of its parts. Biochim Biophys Acta. 2010;1804(9):1713–1722.
- Gumerson JD, Research DMB. The dystrophin-glycoprotein complex in the prevention of muscle damage. J Biomed Biotechnol. 2011;83:ID210797.
- Feige P, Rudnicki MA. Muscle stem cells. Curr Biol. 2018;28(10):R589–R590.
- Serrano AL, Mann CJ, Vidal B, et al. Cellular and molecular mechanisms regulating fibrosis in skeletal muscle repair and disease. Curr Top Dev Biol. 2011;96:167–201.
- Chang NC, Chevalier FP, Rudnicki MA. Satellite cells in muscular dystrophy - lost in polarity. Trends Mol Med. 2016;22(6):479–496.
- Feige P, Brun CE, Ritso M, et al. Orienting muscle stem cells for regeneration in homeostasis, aging, and disease. Cell Stem Cell. 2018;23(5):653–664.
- Duguez S, Duddy W, Johnston H, et al. Dystrophin deficiency leads to disturbance of LAMP1-vesicle-associated protein secretion. Cell. Mol. Life Sci. 2013;70(12):2159–2174.
- Straub V, Rafael JA, Chamberlain JS, et al. Animal models for muscular dystrophy show different patterns of sarcolemmal disruption. J Cell Biol. 1997;139(2):375–385.
- McArdle A, Edwards R, Jackson MJ. Time-course of changes in plasma-membrane permeability in the dystrophin-deficient Mdx mouse. Muscle Nerve. 1994;17(12):1378–1384.
- Hodgetts S, Radley H, Davies M, et al. Reduced necrosis of dystrophic muscle by depletion of host neutrophils, or blocking TNFalpha function with etanercept in mdx mice. Neuromuscul Disord. 2006;16(9–10):591–602.
- Panovský R, Pešl M, Holeček T, et al. Cardiac profile of the Czech population of Duchenne Muscular Dystrophy patients: a cardiovascular magnetic resonance study with T1 mapping. Orphanet J Rare Dis. 2019;14(1):10.
- Barp A, Bello L, Caumo L, et al. Muscle MRI and functional outcome measures in Becker muscular dystrophy. Sci Rep. 2017;7(1):16060.
- Barnard AM, Willcocks RJ, Triplett WT, et al. MR biomarkers predict clinical function in Duchenne Muscular Dystrophy. Neurology. 2020;94(9):e897-e909.
- Barnard AM, Willcocks RJ, Finanger EL, et al. Skeletal muscle magnetic resonance biomarkers correlate with function and sentinel events in Duchenne Muscular Dystrophy. PLoS ONE. 2018;13(3):e0194283.
- Willcocks RJ, Rooney WD, Triplett WT, et al. Multicenter prospective longitudinal study of magnetic resonance biomarkers in a large Duchenne Muscular Dystrophy cohort. Ann Neurol. 2016;79(4):535–547.
- Warman Chardon J, Diaz-Manera J, Tasca G, et al. MYO-MRI diagnostic protocols in genetic myopathies. Neuromuscul Disord. 2019;29(11):827–841.
- Hathout Y, Marathi RL, Rayavarapu S, et al. Discovery of serum protein biomarkers in the mdx mouse model and cross-species comparison to Duchenne Muscular Dystrophy patients. Hum Mol Genet. 2014;23(24):6458–6469.
- Nadarajah VD, Mertens B, Dalebout H. Serum peptide profiles of Duchenne Muscular Dystrophy (DMD) patients evaluated by data handling strategies for high resolution content. J Proteom Bioinform [ Internet]. 2012;5(4):096.
- Ayoglu B, Chaouch A, Lochmüller H, et al. Affinity proteomics within rare diseases: a BIO-NMD study for blood biomarkers of muscular dystrophies. EMBO Mol Med. 2014;6(7):918–936.
- Cynthia Martin F, Hiller M, Spitali P, et al. Fibronectin is a serum biomarker for Duchenne Muscular Dystrophy. Proteomics Clin Appl. 2014;8(3–4):269–278.
- Catapano F, Domingos J, Perry M, et al. Downregulation of miRNA-29, −23 and −21 in urine of Duchenne Muscular Dystrophy patients. Epigenomics. 2018;10(7):875–889.
- Roberts TC, Coenen-Stass AML, Wood MJA. Assessment of RT-qPCR normalization strategies for accurate quantification of extracellular microRNAs in murine serum. PLoS ONE. 2014;9(2):e89237.
- Roberts TC, Godfrey C, McClorey G, et al. Extracellular microRNAs are dynamic non-vesicular biomarkers of muscle turnover. Nucleic Acids Res. 2013;41(20):9500–9513.
- Hrach HC, Mangone M. miRNA profiling for early detection and treatment of Duchenne Muscular Dystrophy. Int J Mol Sci. 2019;20(18):4638.
- Coenen-Stass AML, Wood MJA, Roberts TC. Biomarker Potential of Extracellular miRNAs in Duchenne Muscular Dystrophy. Trends Mol Med. 2017;23(11):989–1001.
- Tsonaka R, Signorelli M, Sabir E, et al. Longitudinal metabolomic analysis of plasma enables modeling disease progression in Duchenne Muscular Dystrophy mouse models. Hum Mol Genet. 2020;29(5):745–755.
- Boca SM, Nishida M, Harris M, et al. Discovery of metabolic biomarkers for Duchenne Muscular Dystrophy within a natural history study. PLoS ONE. 2016;11(4):e0153461.
- Spitali P, Hettne K, Tsonaka R, et al. Cross-sectional serum metabolomic study of multiple forms of muscular dystrophy. J Cell Mol Med. 2018;22(4):2442–2448.
- Szigyarto CA-K, Spitali P. Biomarkers of Duchenne Muscular Dystrophy: current findings. DNND. 2018;8:1–13.
- Yasmineh WG, Ibrahim GA, Abbasnezhad M, et al. Isoenzyme distribution of creatine kinase and lactate dehydrogenase in serum and skeletal muscle in Duchenne Muscular Dystrophy, collagen disease, and other muscular disorders. Clin Chem. 1978;24(11):1985–1989.
- Kiessling WR, Beckmann R. Serum levels of myoglobin and creatine kinase in Duchenne Muscular Dystrophy. Klin Wochenschr. 1981;59(7):347–348.
- Pernice W, Guggolz MA, Guggolz M, et al. A mathematical analysis of creatine kinase activity in the course of Duchenne Muscular Dystrophy. Muscle Nerve. 1986;9(4):333–340.
- Percy ME, Chang LS, Murphy EG, et al. Serum creatine kinase and pyruvate kinase in Duchenne Muscular Dystrophy carrier detection. Muscle Nerve. 1979;2(5):329–339.
- Cardon MW. 50 years ago in the journal of pediatrics: an assessment of the creatine kinase test in the detection of carriers of Duchenne Muscular Dystrophy. J Pediatr. 2017;186:63.
- Nakamura A, Kobayashi M, Kuraoka M, et al. Initial pulmonary respiration causes massive diaphragm damage and hyper-CKemia in Duchenne Muscular Dystrophy dog. Sci Rep. 2013;3(1):359.
- Burch PM, Pogoryelova O, Palandra J, et al. Reduced serum myostatin concentrations associated with genetic muscle disease progression. J Neurol. 2017;264(3):541–553.
- Oonk S, Spitali P, Hiller M, et al. Comparative mass spectrometric and immunoassay-based proteome analysis in serum of Duchenne Muscular Dystrophy patients. Proteomics Clin Appl. 2015;10(3):290–299.
- Sun G, Haginoya K, Chiba Y, et al. Elevated plasma levels of tissue inhibitors of metalloproteinase-1 and their overexpression in muscle in human and mouse muscular dystrophy. J Neurol Sci. 2010;297(1–2):19–28.
- Zelikovich AS, Quattrocelli M, Salamone IM, et al. Moderate exercise improves function and increases adiponectin in the mdx mouse model of muscular dystrophy. Sci Rep. 2019;9(1):281.
- Crowe KE, Shao G, Flanigan KM, et al. N-terminal α dystroglycan (αDG-N): a potential serum biomarker for Duchenne Muscular Dystrophy. J Neuromuscul Dis. 2016;3(2):247–260.
- Colussi CC, Banfi CC, Brioschi MM, et al. Proteomic profile of differentially expressed plasma proteins from dystrophic mice and following suberoylanilide hydroxamic acid treatment. Proteomics Clin Appl. 2009;4(1):71–83.
- Guiraud S, Edwards B, Squire SE, et al. Identification of serum protein biomarkers for utrophin based DMD therapy. Sci Rep. 2017;7:43697.
- Saito K, Koboyashi D, Komatsu M et al. A sensitive assay of tumor necrosis factor a in sera from Duchenne muscular dystrophy patients. Clin Chem 2000;46(10):1703e4
- Füzéry AK, Levin J, Chan MM, et al. Translation of proteomic biomarkers into FDA approved cancer diagnostics: issues and challenges. Clin Proteomics. 2013;10(1):13.
- Dochez V, Caillon H, Vaucel E, et al. Biomarkers and algorithms for diagnosis of ovarian cancer: CA125, HE4, RMI and ROMA, a review. J Ovarian Res. 2019;12(1):28–29.
- McDonagh TA, Cunningham AD, Morrison CE, et al. Left ventricular dysfunction, natriuretic peptides, and mortality in an urban population. Heart. 2001;86(1):21–26.
- Dowling P, Murphy S, Zweyer M, et al. Emerging proteomic biomarkers of X-linked muscular dystrophy. Expert Rev Mol Diagn. 2019;19(8): 739–755.
- Richterich R, Rosin S, Aebi U, et al. Progressive muscular dystrophy. V. The identification of the carrier state in the Duchenne type by serum creatine kinase determination. Am J Hum Genet. 1963;15(2):133–154.
- Pearce MJ, Walton NJ, Pennington R. Serum enzyme studies in muscle disease. III. serum creatine kinase activity in relatives of patients with the Duchenne type of muscular dystrophy. J Neurol Neurosurg Psychiatry. 1964;27(3):181–185.
- Mokuno K, Riku S, Matsuoka Y, et al. Serum muscle-specific enolase in progressive muscular dystrophy and other neuromuscular diseases. J Neurol Sci. 1984;63(3):345–352.
- Kato K, Okagawa Y, Suzuki F, et al. Immunoassay of human-muscle enolase subunit in serum - a novel marker antigen for muscle diseases. Clin Chim Acta. 1983;131(1–2):75–85.
- Carter ND, Heath R, Jeffery S, et al. Carbonic anhydrase III in Duchenne Muscular Dystrophy. Clin Chim Acta. 1983;133(2):201–208.
- Mokuno K, Riku S, Matsuoka Y, et al. Serum carbonic anhydrase III in progressive muscular dystrophy. J Neurol Sci. 1985;67(2):223–228.
- Ozawa E, Hagiwara Y, Yoshida M. Creatine kinase, cell membrane and Duchenne Muscular Dystrophy. Mol Cell Biochem. 1999;190(1–2):143–151.
- Carberry S, Zweyer M, Swandulla D, et al. Profiling of age-related changes in the tibialis anterior muscle proteome of the mdx mouse model of dystrophinopathy. J Biomed Biotechnol. 2012;691641–691641:2011.
- Guevel L, Lavoie JR, Perez-Iratxeta C, et al. Quantitative proteomic analysis of dystrophic dog muscle. J Proteome Res. 2011;10(5):2465–2478.
- Rayavarapu S, Coley W, Cakir E, et al. Identification of disease specific pathways using in vivo SILAC proteomics in dystrophin deficient mdx mouse. Mol Cell Proteomics. 2013;12(5):1061–1073.
- Lewis C, Jockusch H, Ohlendieck K. Proteomic profiling of the dystrophin-deficient MDX heart reveals drastically altered levels of key metabolic and contractile proteins. J Biomed Biotechnol. 2010;2010(3):648501–648520.
- Alagaratnam S, Mertens BJA, Dalebout JC, et al. Serum protein profiling in mice: identification of Factor XIIIa as a potential biomarker for muscular dystrophy. Proteomics. 2008;8(8):1552–1563.
- Rouillon J, Poupiot J, Zocevic A, et al. molecular FAH, 2015. Serum proteomic profiling reveals fragments of MYOM3 as potential biomarkers for monitoring the outcome of therapeutic interventions in muscular dystrophies. Hum Mol Genet. 2015;24(17):4916–4932.
- Pietrowska M, Wlosowicz A, Gawin M, et al. MS-based proteomic analysis of serum and plasma: problem of high abundant components and lights and shadows of albumin removal. Adv Exp Med Biol. 2019;1073(5):57–76.
- Kuhn E, Addona T, Keshishian H, et al. Developing multiplexed assays for troponin I and interleukin-33 in plasma by peptide immunoaffinity enrichment and targeted mass spectrometry. Clinl Chem. 2009;55(6):1108–1117.
- Murphy S, Dowling P, Zweyer M, et al. Proteomic profiling of mdx-4cv serum reveals highly elevated levels of the inflammation-induced plasma marker haptoglobin in muscular dystrophy. Int J Mol Med. 2017;39(6):1357–1370.
- Granger J, Siddiqui J, Copeland S, et al. Albumin depletion of human plasma also removes low abundance proteins including the cytokines. Proteomics. 2005;5(18):4713–4718.
- Seam N, Gonzales DA, Kern SJ, et al. Quality control of serum albumin depletion for proteomic analysis. Clin Chem. 2007;53(11):1915–1920.
- Uhlén M, Fagerberg L, Hallström BM, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347(6220):1260419.
- Signorelli M, Ayoglu B, Johansson C, et al. Longitudinal serum biomarker screening identifies malate dehydrogenase 2 as candidate prognostic biomarker for Duchenne Muscular Dystrophy. J Cachexia, Sarcopenia Muscle. 2019;87:149.
- Schwenk JM, Nilsson P. Antibody suspension bead arrays. Methods Mol Biol. 2011;723:29–36.
- Breen EJ, Tan W, Khan A. The statistical value of raw fluorescence signal in luminex xMAP based multiplex immunoassays. Sci Rep. 2016;6(1):26996.
- Algenäs C, Agaton C, Fagerberg L, et al. Antibody performance in western blot applications is context-dependent. Biotechnol J. 2014;9(3):435–445.
- Bourbeillon J, Orchard S, Benhar I, et al. Minimum information about a protein affinity reagent (MIAPAR). Nat Biotech. 2010;28(7):650–653.
- Uhlén M, Bandrowski A, Carr S, et al. A proposal for validation of antibodies. Nat Methods. 2016;13(10): 823.
- Edfors F, Hober A, Linderbäck K, et al. Enhanced validation of antibodies for research applications. Nat Commun. 2018;9(1):4130.
- Doerr A. Enhanced antibody validation. Nat Methods. 2018;15(12):1001.
- Hathout Y, Brody E, Clemens PR, et al. Large-scale serum protein biomarker discovery in Duchenne Muscular Dystrophy. Proc Natl Acad Sci USA. 2015;112(23):7153–7158.
- Voleti S, Olivieri L, Hamann K, et al. Troponin I Levels Correlate With Cardiac MR LGE and Native T1 Values in Duchenne Muscular Dystrophy Cardiomyopathy and Identify Early Disease Progression. Pediatr Cardiol. 2020;10.1007/s00246-020-02372-5.
- Hathout Y, Liang C, Ogundele M, et al. Disease-specific and glucocorticoid-responsive serum biomarkers for Duchenne Muscular Dystrophy. Sci Rep. 2019;9(1):12167.
- Anderson J, Seol H, Gordish-Dressman H, et al. Interleukin 1 receptor-like 1 protein (ST2) is a potential biomarker for cardiomyopathy in Duchenne Muscular Dystrophy. Pediatr Cardiol. 2017;38(8):1606–1612.
- Parolo S, Marchetti L, Lauria M, et al. Combined use of protein biomarkers and network analysis unveils deregulated regulatory circuits in Duchenne Muscular Dystrophy. PLoS ONE. 2018;13(3):e0194225.
- Zatz M, Rapaport D, Vainzof M, et al. Serum creatine-kinase (CK) and pyruvate-kinase (PK) activities in Duchenne (DMD) as compared with Becker (BMD) muscular dystrophy. J Neurol Sci. 1991;102(2):190–196.
- Mokuno K, Riku S, Sugimura K, et al. Serum creatine kinase isoenzymes in Duchenne Muscular Dystrophy determined by sensitive enzyme immunoassay methods. Muscle Nerve. 1987;10(5):459–463.
- Baird MF, Graham SM, Baker JS, et al. Creatine-kinase- and exercise-related muscle damage implications for muscle performance and recovery. J Nutr Metab. 2012;960363:2012.
- Magal M, Dumke CL, Urbiztondo ZG, et al. Relationship between serum creatine kinase activity following exercise-induced muscle damage and muscle fibre composition. J Sports Sci. 2010;28(3):257–266.
- Kobayashi YM, Rader EP, Crawford RW, et al. Endpoint measures in the mdx mouse relevant for muscular dystrophy pre-clinical studies. Neuromuscul Disord. 2012;22(1):34–42.
- Aartsma-Rus A, Ferlini A, Vroom E. Biomarkers and surrogate endpoints in Duchenne: meeting report. Neuromuscul Disord. 2014;24(8):743–745.
- Timonen A, Lloyd-Puryear M, Neonatal DHJO. Duchenne Muscular Dystrophy newborn screening: evaluation of a new GSP® neonatal creatine kinase-MM Kit in a US and Danish population. Int J Neonatal Screen. 2019;5(27). DOI:10.3390/ijns5030027
- Moat SJ, Bradley DM, Salmon R, et al. Newborn bloodspot screening for Duchenne Muscular Dystrophy: 21 years experience in Wales (UK). Eur J Hum Genet. 2013;21(10):1049–1053.
- Mendell JR, Shilling C, Leslie ND, et al. Evidence-based path to newborn screening for Duchenne Muscular Dystrophy. Ann Neurol. 2012;71(3):304–313.
- Murphy S, Zweyer M, Mundegar RR, et al. Proteomic serum biomarkers for neuromuscular diseases. Expert Rev Proteomic. 2018;15(3):277–291.
- Spitali P, Hettne K, Tsonaka R, et al. Tracking disease progression non‐invasively in Duchenne and Becker muscular dystrophies. J Cachexia, Sarcopenia Muscle. 2018;80(4):706–726.
- Ohta M, Itagaki Y, Itoh N, et al. Carbonic anhydrase III in serum in muscular dystrophy and other neurological disorders: relationship with creatine kinase. Clin Chem. 1991;37(1):36–39.
- Cheeran D, Khan S, Khera R, et al. Predictors of death in adults with Duchenne Muscular Dystrophy–associated cardiomyopathy. J Am Heart Assoc. 2017;6(10):121.
- Matsumura T, Saito T, Fujimura H, et al. Cardiac troponin I for accurate evaluation of cardiac status in myopathic patients. Brain Dev. 2007;29(8):496–501.
- Townsend DW, Turner I, Yasuda S, et al. Chronic administration of membrane sealant prevents severe cardiac injury and ventricular dilatation in dystrophic dogs. J Clin Invest. 2010;120(4):1140–1150.
- Nadarajah VD, van Putten M, Chaouch A, et al. Serum matrix metalloproteinase-9 (MMP-9) as a biomarker for monitoring disease progression in Duchenne Muscular Dystrophy (DMD). Neuromuscul Disord. 2011;21(8):569–578.
- Lourbakos A, Yau N, de Bruijn P, et al. Evaluation of serum MMP-9 as predictive biomarker for antisense therapy in Duchenne. Sci Rep. 2017;7(1):17888.
- Soslow JH, Xu M, Slaughter JC, et al. The role of matrix metalloproteinases and tissue inhibitors of metalloproteinases in Duchenne Muscular Dystrophy cardiomyopathy. J Card Fail. 2019;25(4):259–267.
- Zocevic A, Rouillon J, Wong B, et al. Evaluation of the serum matrix metalloproteinase-9 as a biomarker for monitoring disease progression in Duchenne Muscular Dystrophy. Neuromuscul Disord. 2015;25(5):444–446.
- Spitali P, Aartsma-Rus A, T’ Hoen PAC. Response to: evaluation of the serum matrix metalloproteinase-9 as a biomarker for monitoring disease progression in Duchenne Muscular Dystrophy. Neuromuscul Disord. 2015;25(5):5.
- Rodríguez-Cruz M, Cruz-Guzmán OR, Escobar RE, et al. Leptin and metabolic syndrome in patients with Duchenne/Becker muscular dystrophy. Acta Neurol Scand. 2016;133(4):253–260.
- Cruz-Guzmán ODR, Rodríguez-Cruz M, Escobar-Cedillo RE. Systemic inflammation in Duchenne Muscular Dystrophy: association with muscle function and nutritional status. Biomed Res Int. 2015;2015(3):891972–891977.
- John HA, Purdom IF. Elevated plasma levels of haptoglobin in Duchenne Muscular Dystrophy: electrophoretic variants in patients with a severe form of the disease. Electrophoresis. 1989;10(7):489–493.
- Flanigan KM, Voit T, Rosales XQ, et al. Pharmacokinetics and safety of single doses of drisapersen in non-ambulant subjects with Duchenne Muscular Dystrophy: results of a double-blind randomized clinical trial. Neuromuscul Disord. 2014;24(1):16–24.
- de Carvalho SC, Matsumura CY, Santo Neto H, et al. Identification of plasma interleukins as biomarkers for deflazacort and omega-3 based Duchenne Muscular Dystrophy therapy. Cytokine. 2018;102:55–61.
- Dharnidharka VR, Kwon C, Stevens G. Serum cystatin C is superior to serum creatinine as a marker of kidney function: a meta-analysis. Am J Kidney Dis. 2002;40(2):221–226.
- Viollet L, Gailey S, Thornton DJ, et al. Utility of cystatin C to monitor renal function in Duchenne Muscular Dystrophy. Muscle Nerve. 2009;40(3):438–442.
- Strandberg K, Ayoglu B, Roos A, et al. Blood-derived biomarkers correlate with clinical progression in Duchenne Muscular Dystrophy. J Neuromuscul Dis. 2020;7(3).
- van der Burgt YEM. Protein biomarker discovery is still relevant and has entered a new phase. EBioMedicine. 2019;43:15.
- Drucker E, Krapfenbauer K. Pitfalls and limitations in translation from biomarker discovery to clinical utility in predictive and personalised medicine. Epma J. 2013;4(1):7.
- Wilson JL, Altman RB. Biomarkers: delivering on the expectation of molecularly driven, quantitative health. Exp Biol Med (Maywood). 2018;243(3):313–322.
- Rodrigues M, Yokota T. An overview of recent advances and clinical applications of exon skipping and splice modulation for muscular dystrophy and various genetic diseases. Methods Mol Biol. 1828;31–55:2018.
- Bushby K, Finkel R, Wong B, et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve. 2014;50(4):477–487.
- Mora M, Angelini C, Bignami F, et al. The EuroBioBank network: 10 years of hands-on experience of collaborative, transnational biobanking for rare diseases. Eur J Hum Genet. 2015;23(9):1116–1123.
- Graham C, Dawkins H, Terry S, et al. Current trends in biobanking for rare diseases: a review. BSAM. 2014;2:49–61.
- Gainotti S, Torreri P, Wang CM, et al. The RD-connect registry & biobank finder: a tool for sharing aggregated data and metadata among rare disease researchers. Eur J Hum Genet. 2018;26(5): 631–643.
- Greco V, Piras C, Pieroni L, et al. Direct assessment of plasma/Serum sample quality for proteomics biomarker investigation. Methods Mol Biol. 2017;1619(3):3–21.
- Menezes MP, North KN. Inherited neuromuscular disorders: pathway to diagnosis. J Paediatr Child Health. 2012;48(6):458–465.
- Deenen JCW, Arnts H, van der Maarel SM, et al. Population-based incidence and prevalence of facioscapulohumeral dystrophy. Neurology. 2014;83(12):1056–1059.
- Gudde AEEG, González-Barriga A, van den Broek WJAA, et al. A low absolute number of expanded transcripts is involved in myotonic dystrophy type 1 manifestation in muscle. Hum Mol Genet. 2016;25(8):1648–1662.
- Mahmood OA, Jiang XM. Limb-girdle muscular dystrophies: where next after six decades from the first proposal (Review). Mol Med Rep. 2014;9(5):1515–1532.
- Bushby K. Report on the 12th ENMC sponsored international workshop–the “limb-girdle” muscular dystrophies. Neuromuscul Disord. 1992;2:3–5.
- van der Kooi AJ, Barth PG, Busch HF, et al. The clinical spectrum of limb girdle muscular dystrophy. A survey in The Netherlands. Brain. 1996;119(Pt 5):1471–1480.
- Dunnen Den JT, Grootscholten PM, Bakker E, et al. Topography of the Duchenne Muscular Dystrophy (DMD) gene: FIGE and cDNA analysis of 194 cases reveals 115 deletions and 13 duplications. Am J Hum Genet. 1989;45(6):835–847.
- Becker PE, Kiener F. A new x-chromosomal muscular dystrophy. Arch Psychiatr Nervenkr Z Gesamte Neurol Psychiatr. 1955;193(4):427–448.
- Burch PM, Pogoryelova O, Goldstein R, et al. Muscle-derived proteins as serum biomarkers for monitoring disease progression in three forms of muscular dystrophy. J Neuromuscul Dis. 2015;2(3):241–255.
- Ibrahim GA, Zweber BA, Awad EA. Muscle and serum enzymes and isoenzymes in muscular-dystrophies. Arch Phys Med Rehab. 1981;62(6):265–269.
- Coenen-Stass AML, McClorey G, Manzano R, et al. Identification of novel, therapy-responsive protein biomarkers in a mouse model of Duchenne Muscular Dystrophy by aptamer-based serum proteomics. Sci Rep. 2015;5(1):e38–11.
- Aartsma-Rus A, Morgan J, Lonkar P, et al. Report of a TREAT-NMD/world duchenne organisation meeting on dystrophin quantification methodology. J Neuromuscul Dis. 2019;6(1):147–159.
- Gaasterland CMW, van der Weide MCJ, Du Prie-Olthof MJ, et al. The patient’s view on rare disease trial design - a qualitative study. Orphanet J Rare Dis. 2019;14(1):31–39.
- Verhaart IEC, T Hoen PAC, Roos M, et al. Meeting on data sharing for Duchenne 21–22 March 2019 Amsterdam, the Netherlands. Neuromuscul Disord. 2019;29(10):800–810.
- Aartsma-Rus A, Ferlini A, McNally EM, et al. Workshop participants. 226th ENMC international workshop:: towards validated and qualified biomarkers for therapy development for Duchenne Muscular Dystrophy 20–22 January 2017, Heemskerk, The Netherlands. Neuromuscul Disord. 2018;28(1):77–86.
- Zurriaga Ó, Martínez J, Corrochano V, et al. Rare disease registries and biobanks: A way forward. Arbor. 2018;194(789):469.