
Abstract
The pediatric solid tumor neuroblastoma (NB) often depends on the anti-apoptotic protein, Mcl-1, for survival through Mcl-1 sequestration of pro-apoptotic Bim. High affinity Mcl-1 inhibitors currently do not exist such that novel methods to inhibit Mcl-1 clinically are in high demand. Receptor tyrosine kinases (RTK) regulate Mcl-1 in many cancers and play a role in NB survival, yet how they regulate Bcl-2 family interactions in NB is unknown. We found that NB cell lines derived to resist the Bcl-2/-xl/-w antagonist, ABT-737, acquire a dependence on Mcl-1 and show increased expression and activation of the RTK, EGFR. Mcl-1 dependent NB cell lines derived at diagnosis and from the same tumor following relapse also have increased EGFR expression compared to those dependent on Bcl-2. Inhibition of EGFR by shRNA or erlotinib in Mcl-1 dependent NBs disrupts Bim binding to Mcl-1 and enhances its affinity for Bcl-2, restoring sensitivity to ABT-737 as well as cytotoxics in vitro. Mechanistically treatment of NBs with small molecule inhibitors of EGFR (erlotinib, cetuximab) and ERK (U0126) increases Noxa expression and dephosphorylates Bim to promote Bim binding to Bcl-2. Thus, EGFR regulates Mcl-1 dependence in high-risk NB via ERK-mediated phosphorylation of Bim such that EGFR/ERK inhibition renders Mcl-1 dependent tumors now reliant on Bcl-2. Clinically, EGFR inhibitors are ineffective as single agent compounds in patients with recurrent NB, likely due to this transferred survival dependence to Bcl-2. Likewise, EGFR or ERK inhibitors warrant further testing in combination with Bcl-2 antagonists in vivo as a novel future combination to overcome therapy resistance in the clinic.
Abbreviations
| AKT | = | protein kinase B |
| Bcl-2 | = | B-cell lymphoma-2 |
| BH3 | = | Bcl-2 homology domain 3 |
| co-IP | = | co-immunoprecipitation |
| EGFR | = | epidermal growth factor receptor |
| ERK | = | extracellular signal related kinase |
| HR NB | = | high-risk neuroblastoma |
| LPP | = | lambda protein phosphatase |
| Mcl-1 | = | Myeloid cell leukemia-1 |
| NB | = | neuroblastoma |
| RTK | = | receptor tyrosine kinase |
| TK | = | tyrosine kinase |
| WCL | = | whole cell lysate |
Introduction
Patients with high-risk neuroblastoma (HR NB) are treated with chemotherapy, autologous stem cell transplant, radiation, surgery and biotherapy and despite the increasing intensity of treatment, survival is still <50%.Citation1 All cancers, including HR NB, depend on apoptosis suppression to survive the myriad of stressors they encounter, including chemotherapy.Citation2 Therefore, therapies targeting aberrant apoptosis should prove highly effective in HR NBs to subvert chemotherapy resistance, the main cause of progressive disease. Chemotherapy activates mitochondrial apoptosis to induce tumor cell death and the Bcl-2 family proteins regulate this process.Citation3 BH3 proteins, a pro-death subset of the Bcl-2 family (Bim, Bid, Puma) are activated by chemotherapy and initiate apoptosis by binding to and activating pro-apoptotic Bax and Bak on the mitochondria to induce mitochondrial pore formation and the release of apoptosis effector molecules like cytochrome C. Anti-apoptotic members (Bcl-2, Mcl-1, Bcl-xl, Bcl-w, A1/Bfl1) prevent apoptosis by neutralizing these BH3 proteins, preventing them from activating Bak and/or Bax.
We have shown that the pro-apoptotic BH3 protein, Bim, is activated and sequestered by anti-apoptotic Bcl-2 members in HR NB cell lines and primary NB tumors both at diagnosis and at relapse, defining their principal survival dependency.Citation4 Co-immunoprecipitation (co-IP) of pro-apoptotic Bcl-2 members and BH3 profiling of isolated NB mitochondria applied to HR NBs at diagnosis demonstrate a selective dependence on either Bcl-2 or Mcl-1 exclusively to sequester Bim Citation4 and accurately predicted NB responses to the Bcl-2/Bcl-xl/Bcl-w antagonist, ABT-737 (Abbvie), in vitro and in vivo.Citation4,5 While Bcl-2 dependent NB xenografts in mice could be cured with a short combination of cyclophosphamide and ABT-737,Citation4 Mcl-1 dependent NB xenografts were resistant to the combination as ABT-737 does not target Mcl-1. Furthermore, HR NBs assayed at relapse following therapy revealed that they maintained Bim priming to Bcl-2 or Mcl-1 as at diagnosis. Thus survival dependence in HR NB appears to be fixed and a prevalence of NBs depend on Mcl-1 to survive.Citation5
HR NBs reliant on Mcl-1 for survival are both chemotherapy as well as ABT-737 resistant in vitro and in vivo.Citation5,6 We have also shown that HR NB cell lines that acquire resistance to ABT-737 under selective pressure do so by becoming Mcl-1 dependent, suggesting Mcl-1 dependence is a biomarker for an extreme therapy resistant phenotype.Citation4 Given Mcl-1 imparts a less favorable outcome by driving resistance to chemotherapy upfront and emergent Bcl-2 antagonist resistance in HR NB and other tumors, novel therapeutic approaches to inhibiting Mcl-1 are needed.Citation7 Drugs that selectively inhibit Mcl-1 with high affinity currently do not exist. Even “pan-Bcl-2 family inhibitors” fail to target Mcl-1 with great affinity and often show off target effects.Citation8 Mcl-1 is a protein with a very short half-life as it is highly regulated both transcriptionally and post-translationally. We hypothesized that we could restore chemotherapy sensitivity and ABT-737 sensitivity by identifying and targeting proteins positively regulating Mcl-1 activity in HR NB.
Tyrosine kinases (e.g., MEK/ERK, PI3/AKT/MTOR, MAPK) and their cognate receptors (EGFR, IGFR, TRKB) regulate the transcription and post-translational stability of Bcl-2 family members in many tumor modelsCitation9,10 and have also been implicated in NB pathogenesis,Citation11-14 yet their role in Bcl-2 family protein regulation in HR NB is not well defined. We thus investigated whether receptor tyrosine kinases (RTKs) regulate Mcl-1 to promote Mcl-1 dependence in HR NB cell lines derived from both diagnostic and relapsed tumors and in NBs that acquire resistance to Bcl-2 antagonists. To that end, we now demonstrate that EGFR regulates Bim:Mcl-1 interactions to promote Mcl-1 dependence in HR NBs and that successful small molecule inhibition of EGFR or its effector molecule, ERK, can restore responses to chemotherapy and ABT-737.
Results
EGFR regulates Mcl-1 dependence in HR NB cell lines with acquired ABT-737 resistance
To characterize adaptive changes causing resistance to ABT-737, we created a model of acquired ABT-737 resistance by incubating the Bcl-2 dependent NB cell line, CHLA-15, in increasing concentrations of ABT-737 and selecting for clones capable of growth in > 200 nM (CHLA-15-ABTR; CHLA-15 ABT-737 Resistant, where CHLA-15 ABT-737 IC50 is normally 20 nM).Citation4 In CHLA-15-ABTR compared to CHLA-15, Bcl-2 and Mcl-1 protein expression are unchanged and expression of the pro-apoptotic BH3 proteins, Bim, Noxa and Puma are slightly decreased (). Interestingly, Bim, which is originally sequestered by Bcl-2 in CHLA-15, converts to binding exclusively to Mcl-1 in CHLA-15-ABTR, as demonstrated by co-immunoprecipitation (co-IP) (), suggesting a post-translational modification may mediate Bcl-2 family interactions creating the shift toward Mcl-1 dependence.
Figure 1. (A) Western blot analysis of Bcl-2 family protein expression between an acquired model of ABT-737 Resistance (CHLA-15-ABTR) and the parent cell line, CHLA-15. (B) Co-IP showing Bim bound to Bcl-2 in CHLA-15 and to Mcl-1 in CHLA-15-ABTR. (C) Receptor tyrosine kinase human phosphoprotein microarray identifies differences in RTK expression in CHLA-15-ABTR vs. CHLA-15; RTK's blotted for are represented in duplicate side by side dots on the membrane for each RTK; ***; white stars designate positive controls. (D) Western blots of cells grown at steady-state confirm increased EGFR, pEGFR and downstream EGFR effectors in CHLA-15-ABTR.
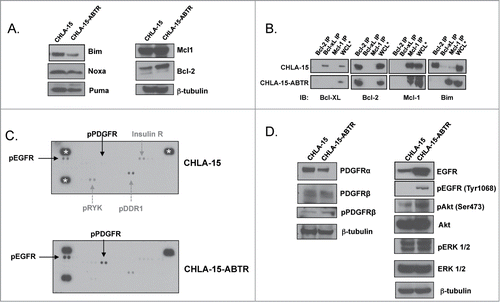
We then evaluated for differences in the activation of 49 different RTKs between the ABT-737 sensitive and ABT-737 resistant CHLA-15 and CHLA-15-ABTR cell lines, respectively, using a human RTK phospho-protein microarray. Results show that phosphorylated RTK proteins such as DDR1, RYK, and Insulin receptor are decreased and other RTK's such as EGFR and PDGFR-β are increased in CHLA-15-ABTR compared to CHLA-15 (). Given that increased RTK expression and phosphorylation in many tumor models promote proliferation and therapy resistance, we focused on the RTK's that are increased in CHLA-15-ABTR on the microarray, EGFR and PDGFR. Western blot analysis shows that PDGFR-α, PDGFR-β and phosphorylated (p)-PDGFR-β expression are relatively unc-hanged in CHLA15-ABTR compared to CHLA-15, whereas EGFR, pEGFR and downstream EGFR targets, pAKT and pERK, are increased in keeping with the array findings ().
To confirm that increased EGFR is not a stochastic event that occurs randomly in the process of selecting for acquired ABT-737 resistance, we evaluated other ABT-737 resistant clones derived from a second Bcl-2 dependent HR NB cell line, SMS-SAN. Selection of the ABT-737 resistant clones of SMS-SAN (deemed SAN-ABTR) was performed in the same manner as CHLA-15-ABTR.Citation4 We previously showed that SAN-ABTR converts to Mcl-1 dependence through Mcl-1 sequestration of Bim Citation4 and now confirm that separate subclones, SAN-ABTR #1, SAN-ABTR #3, growing in 200 nM ABT-737 and selected from different incubation wells, have increased EGFR and pEGFR compared to the parent cell line SMS-SAN ().
Figure 2. (A) Different subpopulations of SMS-SAN selected for ABT-737 resistance (SAN-ABTR) at different times from the Bcl-2 dependent SMS-SAN growing in > 150 nM of ABT-737 (parent cell line IC50 5 nM). (B) CHLA-15-ABTR cells were transfected with EGFR siRNA for 24 hours, then treated with ABT-737 for 24 additional hours at which time cell viability was assessed by WST-1. Mcl-1 dependent CHLA-15-ABTR (C) and Bcl-2 dependent CHLA-15 (D) were treated with erlotinib and ABT-737 simultaneously and assessed for changes in viability at 48 hours by WST-1 assay. *; statistically significant difference, p< 0.05, 95% Ci.
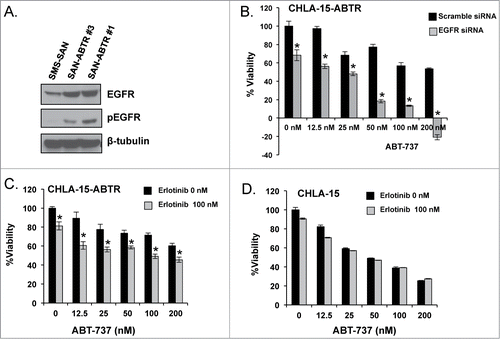
Inhibition of EGFR by small interfering (si)-RNA in CHLA-15-ABTR restores ABT-737 induced cell death, further alluding to a functional role for EGFR in acquired Mcl-1 dependence and Bcl-2 antagonist resistance (). When CHLA-15-ABTR is challenged with the small molecule EGFR inhibitor, erlotinib, there is significantly enhanced cytotoxicity in combination with ABT-737 compared to ABT-737 treatment alone, although not to the extent genetic knockdown produced (). Treatment of CHLA-15 with the same combination confirms that EGFR inhibition alone or in combination with ABT-737 has no effect on this Bcl-2 dependent cell line and does not affect its exquisite sensitivity to ABT-737 ().
Mcl-1 dependent NBs derived at diagnosis and following relapse show increased EGFR expression
We have previously shown that NB cell lines derived from HR NB tumors at diagnosis have heterogeneous Bcl-2 dependence patterns, with Bim bound to Bcl-2 in some subsets and Bim bound exclusively to Mcl-1 in other subsets to prevent apoptosis.Citation5 Previously characterized Mcl-1 dependent NB cell lines (represented by SK-N-BE(2)) are not just resistant to ABT-737 but are more resistant to chemotherapy agents as well when compared to Bcl-2 dependent HR NBs (represented by SMS-SAN; and.Citation5,6)
Figure 3. (A) WST-1 evaluation for survival following a 48-hour treatment with chemotherapy. Western blot analysis of EGFR signaling (B) and Bcl-2 family (C) proteins of untreated whole cell lysate from Bcl-2 dependent SMS-SAN and Mcl-1 dependent SK-N-BE(2). (D) Western analysis for EGFR and pEGFR performed in a panel of HR NB cell lines previously characterized for Bim binding patterns (Bcl-2 or Mcl-1 dependenceCitation4). Paired NB cell lines derived from the same patient's tumor at diagnosis and following cytotoxic therapy and relapse, with relapsed cell line designated with a *; SK-N-BE(1)/SK-N-BE(2)*, CHLA-15/CHLA-20*. NLF was incubated for 48 hours in the presence (+ FBS) or absence (- FBS) of serum and evaluated by western for EGFR and pEGFR.
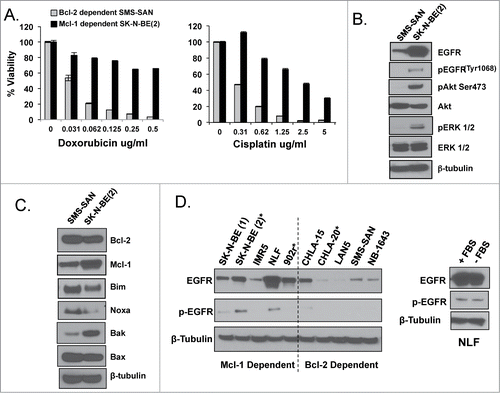
HR NB cell lines as well as primary tumors have increased EGFR expression at diagnosis and following cytotoxic therapy and relapse.Citation11,13 We thus assessed whether EGFR is expressed differently in Mcl-1 dependent NB cell lines compared to those dependent on Bcl-2. Indeed, Mcl-1 dependent SK-N-BE(2) has increased EGFR, pEGFR, pAKT and pERK protein expression compared to Bcl-2 dependent SMS-SAN (). Interestingly, similar to CHLA-15-ABTR, SK-N-BE(2) has diminished Bim and Noxa expression compared to SMS-SAN (). The low level of Noxa in SK-N-BE(2) is likely due to the acquired p53 mutation in this relapsed tumor from which the cell line was derived, that prevents p53 downstream effectors like Noxa from being transcribed.Citation15 The diminished Bak expression in SMS-SAN is likely due to decreased Mcl-1 expression in that cell line, as Mcl-1 protein is known to sequester and stabilize Bak.Citation16 Although both are MYCN amplified cell lines, SK-N-BE(2) and SMS-SAN are very heterogeneous biologically and genetically, as SK-N-BE(2) was derived from a heavily pretreated relapsed tumor and SMS-SAN was derived from a tumor at diagnosis, such that Bcl-2 dependence patterns may be unrelated to their EGFR expression. We thus evaluated a larger panel of previously characterized Mcl-1 and Bcl-2 dependent HR NB cell lines Citation4 and confirmed higher EGFR protein expression in Mcl-1 dependent NBs compared to the Bcl-2 dependent panel, with 2 of 5 Mcl-1 dependent NBs showing EGFR activation by increased pEGFR (). Persistent p-EGFR expression in the absence of serum (the source of EGF) supports that p-EGFR is constitutively active in these Mcl-1 dependent NBs ().
We have shown that NB cell line pairs derived from the same patient's tumor at diagnosis and following cytotoxic therapy and relapse maintain identical Bim binding patterns to Mcl-1 or Bcl-2, despite the acquisition of a more therapy resistant phenotype.Citation4 Interestingly, EGFR protein expression is increased in Mcl-1 dependent SK-N-BE(2) (post-relapse) compared to SK-N-BE(1) (at diagnosis). In contrast the Bcl-2 dependent recurrent tumor cell line CHLA-20 expresses much less EGFR than its pre-relapse homolog, CHLA-15 (). Our results show that EGFR expression while high in Mcl-1 dependent tumors at diagnosis, is increased even more in the same tumor following relapse and therefore may be necessary to maintain survival and promote progression of Mcl-1 dependent NBs compared to those dependent on Bcl-2. We thus sought to determine whether there is a functional relationship between EGFR expression and Mcl-1 dependence in HR NBs.
EGFR regulates de novo Bim binding to Mcl-1 in HR NBs
To assess for a functional role for EGFR on Bcl-2 family protein interactions, we stably inhibited EGFR expression by short hairpin (sh)-RNA in SK-N-BE(2), a HR NB cell line with Bim:Mcl-1 interactions at steady state (). Co-IP results show that Bim moves from Mcl-1 over to the binding pocket of Bcl-2 following EGFR knockdown in SK-N-BE(2) (). Western blot analysis confirms inhibition of pEGFR and its downstream targets pAKT and pERK in SK-N-BE(2)-shEGFR, as well as relatively unchanged levels of Bcl-2, Mcl-1, and Bim protein (). Thus changes in Bim binding are not due to alterations in pro-survival Bcl-2 protein expression following EGFR inhibition. We did not evaluate Noxa given the p53 mutation prevents Noxa expression in wild type SK-N-BE(2) irrespective of changes in EGFR. Following EGFR knockdown, SK-N-BE(2)-shEGFR cells are re-sensitized to ABT-737 with an IC50 decreased significantly compared to the parental cell line transfected with scramble shRNA (47 nM vs. 628 nM, respectively; ).
Figure 4. (A) Western analysis of EGFR protein expression with densitometry analyzed EGFR normalized to β-tubulin expression for SK-N-BE(2) wild type verses shEGFR-transfected cell lines. (B) Co-IP of pro-survival Bcl-2 proteins followed by immunoblot for pro-survival proteins and Bim, showing Bim moves from Mcl-1 over to the binding pocket of Bcl-2 following EGFR inhibition. (C) Western blot analysis for changes in Bcl-2 family and EGFR signaling proteins following EGFR knockdown in SK-N-BE(2)-shEGFR.
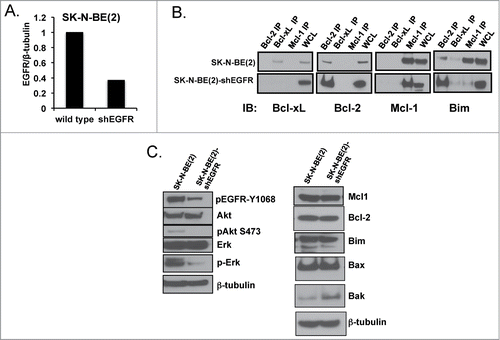
Figure 5. SK-N-BE(2) cells become exquisitely sensitive to both ABT-737 (A) and cytotoxic drugs (B & C) compared to scramble shRNA-transfected SK-N-BE(2) cells. Viability was assessed using WST-1 assay after 48 hours of incubation. All experiments were performed in technical triplicate and standard deviations represent the average of 2 separate biologic experiments. Standard deviations not shown have a less than 5% difference between biologic replicates.
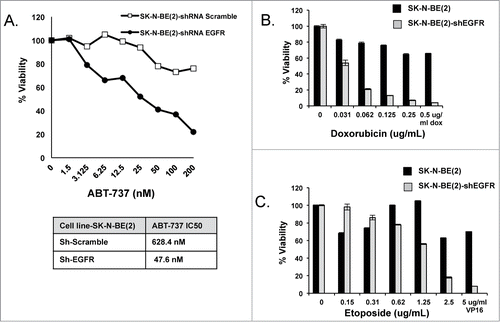
Mcl-1 imparts cytotoxic resistance and poor survival in HR NB models and primary tumors, which led us to investigate whether EGFR also regulates cytotoxic induced cell death via Mcl-1 effects. Indeed, upon EGFR stable inhibition, cell death in response to cytotoxic agents commonly used to treat HR NB is significantly enhanced in the SK-N-BE(2)-shEGFR cells compared to SK-N-BE(2) (). SK-N-BE(2) was derived from a recurrent tumor following cytotoxic therapy and has altered apoptotic machinery via both p53 mutation and Mcl-1 dependence, thus overcoming cytotoxic resistance by targeting EGFR alone in this cell line stresses the importance of EGFR and Mcl-1 in apoptosis suppression in relapsed NB.
Bim is post-translationally modified by EGFR via ERK in Mcl-1 dependent NBs
Mcl-1 dependent cell lines were treated with the EGFR small molecule or antibody inhibitors, erlotinib and cetuximab, respectively, in combination with ABT-737. Molecular inhibition of EGFR enhances ABT-737 induced cell death in the Mcl-1 dependent cell line, NLF (). In comparison to genetic inhibition of EGFR, molecular inhibition of EGFR does not consistently restore ABT-737 sensitivity in all Mcl-1 dependent cell lines tested. For example, NLF and CHLA-15-ABTR are sensitive to erlotinib and ABT-737 combinations ( and ) whereas SK-N-BE(2) is completely resistant (). This may be due to superior EGFR inhibition imparted by erlotinib in tumors containing somatic mutations in the tyrosine kinase (TK) domain of EGFR.Citation17 We sequenced the TK domain of EGFR in SK-N-BE(2) and found that it does not carry the TK domain mutations in Exons 18–21 that are associated with enhanced sensitivity to erlotinib, potentially explaining the dichotomous response of SK-N-BE(2) to EGFR inhibition by shRNA versus molecular inhibition of the kinase.
Figure 6. Mcl-1 dependent NB cells NLF (A) and SK-N-BE(2) (B) were exposed to EGFR inhibitors, erlotinib or cetuximab, and ABT-737 simultaneously and evaluated after 48 hours for changes in survival by WST-1. (C) NLF cells were exposed to given concentrations of U0126, erlotinib, or LY294002 for 24 hours, harvested for protein, and evaluated for changes in EGFR signaling and Bcl-2 family protein expression. (D) Protein from an Mcl-1 co-IP was treated with Lambda Protein Phosphatase (LPP) and then evaluated for changes in Bim:Mcl-1 interactions by immunoblot.
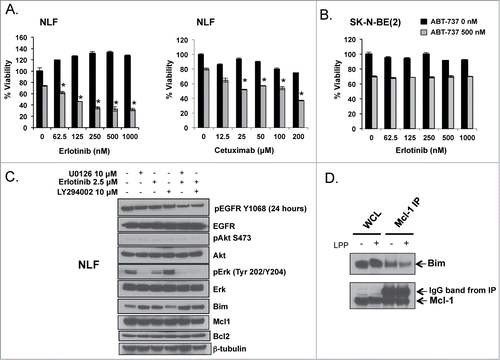
Thus to further investigate one of the mechanisms by which EGFR regulates Bim:Mcl-1 interactions in Mcl-1 dependent NBs, we evaluated the downstream activity of EGFR following erlotinib treatment of the erlotinib-sensitive, Mcl-1 dependent cell line NLF. We treated NLF for 24 hours with EGFR, PI3K and MEK/ERK inhibitors, erlotinib, LY294002 and U0126, respectively, and evaluated for changes in Bcl-2 family protein expression. Following inhibition of ERK or EGFR, Bim protein expression increases and migrates faster/further in the gel, suggesting that Bim has lost a post-translational modification, leading to protein stability and increased expression. Bim expression in the LY294002 treated arm of the experiment (lane 4) shows no change in expression level or size from untreated cells in (lane 1)(), suggesting that EGFR is altering Bim:Mcl-1 interactions via ERK mediated post-translational modification of Bim. In contrast, Mcl-1 and Bcl-2 protein expression do not change following EGFR, ERK, or PI3K inhibition (), supporting that Bim is one of the key proteins regulated by EGFR to promote Mcl-1 binding and dependence.
To investigate whether changes in Bim binding following EGFR inhibition were due to changes in Bim phosphorylation, we employed the serine/tyrosine/threonine dephosphorylating enzyme, lambda phosphatase (LPP). Heating of whole cell lysate to activate LPP inhibited our ability to accurately pull down Bcl-2 or Mcl-1 after the dephosphorylation event to assess for Bim moving dynamically from Mcl-1 over to Bcl-2. We alternatively immunoprecipitated Mcl-1 and treated the Mcl-1 IP with LPP to show that the amount of Bim bound to Mcl-1 decreases following LPP induced protein dephosphorylation (). Mcl-1 expression and migration are not affected by EGFR, ERK or PI3K small molecule inhibition as seen in , supporting that LPP is dephosphorylating Bim residue(s) to impede Bim binding to Mcl-1.
ERK mediated phosphorylation of Bim promotes Bim binding to Mcl-1 in HR NBs
We treated NLF with UO126, LY294002, and erlotinib and performed co-IPs of pro-survival Bcl-2 proteins to evaluate for changes in Bim binding. Following PI3K inhibition, some Bim moves from Mcl-1 over to Bcl-2, however, Bim largely remains bound to Mcl-1 (). In contrast, following both EGFR and ERK inhibition, Bim moves almost entirely from binding Mcl-1 to become exclusively bound to Bcl-2 (). Treatment of NLF with U0126 restores ABT-737 sensitivity similarly to erlotinib, where LY294002 treatment does not ( and data not shown). Therefore, ERK-mediated phosphorylation of Bim enhances its affinity to Mcl-1 in HR NB cell lines that can be disrupted with ERK and EGFR targeting small molecule antagonists.
Figure 7. A. NLF was treated with UO126 (10 μM), LY294002 (2.5 μM), or erlotinib (10 μM) for 24 hours followed by co-IP of anti-apoptotic members to evaluate for changes in Bim binding. B. U0126 synergizes with ABT-737, supporting ERK as the downstream EGFR effector regulating Mcl-1 dependence in HR NB. C. Noxa expression increases following EGFR inhibition with erlotinib but is unchanged by treatment with the MEK/ERK inhibitor U0126.
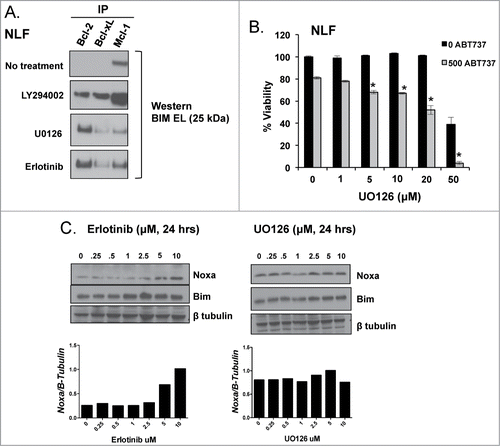
EGFR-mediated downregulation of Noxa may enhance Bim binding to Mcl-1
Noxa is a BH3 protein that binds exclusively to Mcl-1.Citation18 Noxa cannot bind to or activate Bax/Bak directly, but facilitates apoptosis by binding to Mcl-1 and preventing Mcl-1 sequestration of activator BH3 proteins like Bim, Bid, and Puma. Noxa expression is increased in other Bcl-2 dependent tumors and has been shown to saturate Mcl-1 binding sites to promote Bim binding to Bcl-2.Citation19 Given Noxa expression is increased in Bcl-2 dependent SMS-SAN and decreased in Mcl-1 dependent CHLA15-ABTR and SK-N-BE(2), we sought to determine whether Noxa expression changes following small molecule inhibition of EGFR and ERK in NLF. Following a 24-hour exposure to erlotinib, Noxa protein expression increases in NLF (). U0126 treatment does not upregulate Noxa protein expression (). Given the lack of an effective Noxa antibody for immunoprecipitation, the interaction and changes in Noxa binding to Mcl-1 following EGFR inhibition could not be confirmed. We have shown that ERK inhibition alone is sufficient to induce Bim's displacement from Mcl-1, thus EGFR-mediated inhibition of Noxa via non-ERK related pathways may play a less significant role in promoting Mcl-1 dependence in HR NBs.
Discussion
Our past investigations have shown that Bcl-2 family proteins play a crucial part in NB tumor response to chemotherapy and that a critical determinant of chemotherapy resistance and Bcl-2 antagonist resistance is a functional dependence on the anti-apoptotic protein, Mcl-1. We now show that EGFR regulates Mcl-1 dependence not just in NBs that acquire ABT-737 resistance in vitro, but in Mcl-1 dependent NB cell lines derived from tumors at diagnosis and from tumors following cytotoxic therapy and relapse. We confirmed this by showing that multiple Mcl-1 dependent NB cell lines as well as different Mcl-1 dependent clones of acquired ABT-737 resistance have increased EGFR and altered Bim:Mcl-1 interactions following genetic inhibition of EGFR. Primary NB tumors also have increased EGFRCitation11 and Mcl-1 dependence patterns,Citation4 which supports that this phenomenon is not an isolated event nor artificially derived from cell culture selection or environment.
Our results demonstrate that EGFR regulates de novo Mcl-1 dependence via ERK-mediated phosphorylation of Bim that increases the affinity of Bim for the hydrophobic pocket of Mcl-1 (). While Ho, et al., showed that the proliferative effects of EGFR in HR NBs depends on downstream PI3K/AKT signaling,Citation11 we now show that EGFR attenuates apoptosis in HR NBs through downstream MEK/ERK signaling. In other cellular models, ERK-mediated phosphorylation of Bim inhibits Bim from binding to Bax and promotes its proteosomal degradation, without affecting Bim:Mcl-1 or Bim:Bcl-2 interactions.Citation20 In contrast, we show that while Bim protein expression is slightly decreased in Mcl-1 dependent NBs, EGFR and/or ERK prevent apoptosis by phosphorylating Bim to enhance its binding to Mcl-1, despite near equivalent Mcl-1 and Bcl-2. While differences in cell types and the multiple upstream causes of ERK activation may affect its differential regulation of Bim in these cell models, kinase mediated phosphorylation of different amino acids of the Bim protein may also affect its stability and Bcl-2 interactions.Citation21,22 We are currently evaluating the effect on phosphorylation of specific Bim residues following ERK and EGFR activation in HR NBs to enlighten us as to how Bim is regulated differently by EGFR and ERK in our tumor model.
Figure 8. Schema showing EGFR regulation of Bim:Mcl-1 interactions in Mcl-1 dependent HR NB at steady state (left) and how Mcl-1 dependent NB cell survival is affected by molecular/genetic inhibition of EGFR or ERK alone (middle) or in combination with ABT-737 (right). The role of Noxa has not been confirmed by our results and so is left in white.
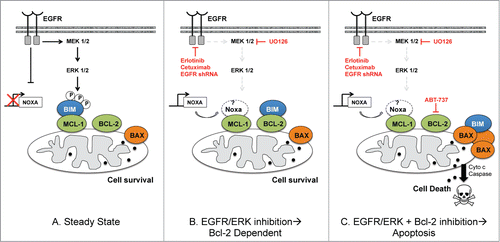
While Noxa expression increases in response to EGFR inhibition in some Mcl-1 dependent NB cell lines, its expression is not affected by inhibition of MEK/ERK, yet ERK inhibition alone is sufficient to induce Bim's displacement from Mcl-1. Furthermore, other NB cell lines that are still sensitive to EGFR genetic inhibition have low to no Noxa expression, such as SK-N-BE(2). This suggests that other factors, such as p53 and MDM2 aberrations may dictate whether Noxa expression levels play a role in the Mcl-1 survival bias regulated by EGFR.
The mechanism by which EGFR is upregulated in Mcl-1 dependent NBs is not well understood. While EGFR mRNA is present in most primary neuroblastoma's, HER family receptor expression does not correlate with MYCN status, stage or risk of the disease.Citation11 Furthermore, next generation sequencing of a large subset of primary high-risk NB tumors failed to show any genomic aberrations in EGFR.Citation23 Thus, EGFR expression is most likely regulated post-translationally and understanding EGFR post-translational modification and stability will be more informative as to choosing optimal compounds to target EGFR clinically in conjunction with Bcl-2 antagonists.
Previous preclinical studies showed that the EGFR small molecule inhibitor gefitinib decreases proliferation of tumor cells in a panel of HR NB cell lines, which led to the use of gefitinib as well as erlotinib in Phase I pediatric clinical trials for relapsed pediatric solid tumors, including neuroblastoma.Citation24,25 These clinical studies failed to show clear responses in patients with recurrent or refractory NB, and conclusions were made that gefitinib and erlotinib may have little utility in HR NB treatment.Citation24,25 Our data suggests that the lack of clinical efficacy of EGFR inhibitors when given as monotherapy to NB patients may be due to the increased role of Bcl-2 that was not taken into account, as Bcl-2 sequesters Bim following EGFR inhibitor-mediated displacement of Bim from Mcl-1 (See ). Ultimately our results imply that EGFR or ERK small molecule inhibitors will be less effective against HR NBs unless one subverts the Bcl-2 survival bias by combining these agents with a Bcl-2 antagonist ().
Combining a Bcl-2 antagonist with an EGFR or ERK inhibitory compound is a novel treatment combination that should not affect normal cells as they are not dependent on such pathways for survival. These results strongly support that the mechanism of EGFR or ERK inhibition and their therapeutic combination with Bcl-2 blockade should be evaluated further in vivo and moved forward clinically as a more effective combination therapy in Mcl-1 dependent tumors like HR NB. Importantly we have evaluated whether the erlotinib and ABT-737 combination antagonizes the potent effects of ABT-737 on Bcl-2 dependent NB cell lines in vitro, and showed that Bcl-2 dependent NBs maintain exquisite sensitivity to ABT-737 (). Thus the combination of an EGFR or ERK inhibitor with a Bcl-2 antagonist could be employed for all patients with HR NB regardless of Bcl-2 addiction pattern and prove equally effective.
Notably certain Mcl-1 dependent NBs were insensitive to erlotinib even when shRNA inhibition of EGFR showed a functional inhibition of the Mcl-1 phenotype, as seen in SK-N-BE(2). While this may be partially due to the lack of a mutation in the EGFR TK domain in SK-N-BE(2), further sequencing of erlotinib-sensitive cell lines, like NLF, revealed that NLF also lacks mutations in EGFR yet it maintains high pEGFR at baseline and is still sensitive to erlotinib and ABT-737 combinations. Furthermore, our results show that many of the Mcl-1 dependent HR NB cell lines have very high EGFR protein levels but yet little to no phosphorylated EGFR at baseline, suggesting that EGFR may also regulate Mcl-1 via a mechanism that is independent of the canonical kinase signaling we identified and chemically inhibited in NLF. Supporting this theory, other research contends that the EGFR protein itself interacts at the mitochondria to regulate program cell death signaling directly, limiting the efficacy of kinase driven inhibitors such as erlotinib and gefitinib.Citation26,27 Investigations as to direct mitochondrial and Bcl-2 family interactions by the EGFR protein itself are underway in our lab as well as investigations of next generation EGFR inhibitors in HR NB models. Accordingly, this data supports that alternative compounds that dephosphorylate Bim and enhance Noxa expression (Bortezimib,Citation28 HDAC inhibitors,Citation29 and CDK5 inhibitorsCitation30) should also prove equally capable of disrupting EGFR controlled Mcl-1:Bim interactions and warrant further investigation in combination with Bcl-2 antagonists to bring out their full potential as novel therapeutics for patients with HR NB.
Materials and Methods
Cell lines
Neuroblastoma cell lines with MYCN amplification [IMR5 Citation31: NLF, SK-N-BE(2),Citation32 SMS-SAN Citation33], and without [CHLA-15 Citation34] are used. CHLA-15-ABTR, an ABT-737 resistant clone was derived by incubating the CHLA-15 cell line in increasing concentrations of ABT-737 and selecting for resistant clones, method as previously described.Citation4 Neural cells were grown in RPMI-1640 (Life Technologies) supplemented with 10%–20% fetal bovine serum (20% for CHLA-15), 2 mM L-glutamine, 1% OPI, 100 U/ml of penicillin. Tissue culture was at 37° C in a humidified atmosphere of 5% CO2. All cell lines were confirmed as unique using STR-based genotyping (AmpFISTR, Applied Biosciences) and identity-confirmed using the COG cell line genotype database (www.cogcell.org). All putative isogenic cell line pairs, selected drug resistant clones, as well as transfected clones were similarly confirmed.
Transfection of EGFR short hairpin (sh)RNA
EGFR gene expression was stably inhibited in SK-N-BE(2) using shRNA (cat# TG320326 from Origene, containing puromycin resistance gene). The EGFR-shRNA and control vectors were transfected into Phoenix Packaging cells (courtesy of Larry Boise, PhD, Winship Cancer Institute, Emory University) using LipofectAMINE 2000 (Invitrogen, NY), according to the manufacturer's protocol. ShEGFR or empty vector-containing MMV viral particles were collected from the supernatant and then used to transduce SK-N-BE(2). After tranduction, cells were incubated at 37°C for 48 h. Transfectants were selected and maintained in puromycin (2 μg/mL).
Phosphokinase microarray
The presence of phosphorylated receptor tyrosine kinase proteins in CHLA-15 and CHLA-15-ABTR cells were assessed using the Human Phospho-Receptor Tyrosine Kinase (Phospho-RTK) Array kit (# ARY001B, R&D systems) as per the manufacture instructions.
Co-immunoprecipitation (co-IP)
Co-IPs were performed by lysing cells in CHAPS buffer, with 200 ug of protein lysate added to antibody-matrix complex as described previously.Citation4 To detect effects on protein binding through changes in phosphorylation, Mcl-1 protein was immunoprecipitated as described previously.Citation4 The Mcl-1 protein;Mcl-1 antibody matrix was then pelleted with centrifugation, and resuspended in a lambda phosphatase (LPP, Promega, Sweden) containing solution (4 uL LPP:4 uL 10X Buffer: 32 uL co-IP solution + 1 M DTT (Millipore)) and incubated at 37°C for 40 min. An equal portion of Mcl-1 protein;Mcl-1 antibody matrix was resuspended in IP buffer alone (without LPP) and incubated at 37°C for 40 min to control for the effects of heating on protein interactions. The matrix was then centrifuged to pellet the immunoprecipitated proteins and washed 3 times to discard proteins that were liberated from Mcl-1 by dephosphorylation. The proteins bound to matrix were released from antibody matrix complex using 2X RIPA buffer, run on Nu-PAGE 10% BisTris gels (Invitrogen), transferred to PVDF membranes and detected for Mcl-1 and Bim. To control for changes in protein expression caused by LPP treatment alone, 25 μg of whole cell lysate (WCL) were incubated in LPP solution (10 μL protein lysate, 1 μL 10x buffer, 1 μL LPP enzyme) or buffer solution alone and incubated at 37°C for 40 min. Of note, the whole cell lysate controls (WCL) in IP experiments are used to show the absence /presence of the protein in the whole cell prior to IP as well as the kDa of the expected protein in the IP. These WCL lanes are often oversaturated on films in order to detect the small amount of the same protein bound to the protein in the IP. Therefore WCL as well as proteins in the IP cannot be used to compare total protein expression levels between different co-IP's or different treatments IP. Western blots with proper loading controls have been performed in separate experiments to address such questions.
Cytotoxicity assays
2 × 104 NB cells/well were plated in 96 well plates and allowed to adhere for 24 hours. The wells were then treated with ABT-737, cytotoxic agent (doxorubicin or etoposide) or vehicle controls in RPMI-based media in triplicate. After 48 hrs, WST-1 was added and absorbance at 590 nM was recorded. For siRNA experiment, CHLA-15-ABTR cells were transiently transfected with 5 ug of shEGFR DNA (cat# TG320326, Origene, MD) using Lipofectamine (for 24 hours) then treated with ABT-737 (for an additional 24 hrs). Viability was then measured by WST-1 as previously described.Citation4
Statistical considerations
Test for significant differences between treatment arms of WST-1 experiments employed a one-way analysis of variance (Anova) or Student's T-test, using Prism Software (Graphpad) analysis, where statistical significance is defined as p-value < 0.05.
Mutational analysis of EGFR
DNA was extracted from NB cells using the GenElute mammalian genomic DNA purification kit (G1N70, Sigma) according to manufacturer's instructions. Samples were then PCR amplified using the custom designed EGFR primers for Exons 8–21. Each reaction contained 1× PCR buffer, 1.5 mmol/L MgCl2, 0.2 mmol/L of each dNTP, 5 pmol of forward primer, and 5 pmol of reverse primer, 1.0 Unit of FastStart DNA polymerase (Roche), 25 ng of template DNA, and dH2O to 25 μl final volume. Cycling conditions were as follows: 95°C 3 minutes, 10× (95°C 1 minute, 60°C 1 minute [-0.5°C pre cycle], 72°C 1 minute), 10× (95°C 1 minute, 55°C 1 minute, 72°C 1 minute), 72°C 7 minutes, 4°C hold. Following amplification, the PCR product was purified using Edge Biosystems ExcelaPure UF PCR Purification System. Cleaned PCR products were cycle sequenced using Big Dye v3.1 reagents (Applied Biosystems) according to manufacturer's protocol. Sequencing products were purified with Performa DTR Gel Filtration Columns (Edge Biosystems) and sequenced on an Applied Biosystems 3130XL. Sequences were aligned and analyzed using Applied Biosystems SeqScape software. Antibodies. Anti-Mcl-1 (BD PharMingen; BDB559027), anti-Bcl-2 (Dako; M088701–2 and Santa Cruz Biotechnology: sc-492), anti-Bcl-xL (clone 7B2.5; gift of L.Boise,PhD, Emory University), anti-PDGFRα (Santa Cruz biotechnology; sc-338), anti-PDGFRβ (sc-80991), anti-phospho PDGFR(sc-12907), anti-EGFR (#2646, Cell Signaling Technology, CST), anti-phospho tyrosine 1068 EGFR(#3777 CST), anti-phospho AKT(S473)(#9271 CST),anti-AKT(#9272 CST), anti-phospho-ERK ½ (#91026 CST), anti-ERK ½(#9102 CST) anti-Bak (#3814CST), anti-Bax (#2772 CST), anti-Puma (#4976 CST), anti-Bim (Millipore Corporation: AB17003), anti-Noxa (abcam #ab13654), β-tubulin(Sigma #T8328) were used.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank the Emory Integrated Genomics Core for EGFR sequencing and STR verification of NB cell lines, C. Patrick Reynolds, MD, PhD, Texas Tech University Health Sciences Center, for his contribution of the paired diagnostic/relapsed neuroblastoma cell lines, Abbvie for the use of ABT-737, and Larry Boise, PhD., Winship Cancer Center, Emory University, for his insightful review of our work.
Funding
This work was supported by NIH K08-CA128925, CURE Childhood Cancer Foundation, and Hyundai Hope on Wheels (to K.C.G).
References
- Matthay KK, Reynolds CP, Seeger RC, Shimada H, Adkins ES, Haas-Kogan D, Gerbing RB, London WB, Villablanca JG. Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: a children's oncology group study. J Clin Oncol 2009; 27:1007-13; PMID:19171716; http://dx.doi.org/10.1200/JCO.2007.13.8925
- Goldsmith KC, Hogarty MD. Targeting programmed cell death pathways with experimental therapeutics: opportunities in high-risk neuroblastoma. Cancer Lett 2005; PMID:15927359
- Walensky LD. BCL-2 in the crosshairs: tipping the balance of life and death. Cell Death Differ 2006; 13:1339-50; PMID:16763614; http://dx.doi.org/10.1038/sj.cdd.4401992
- Goldsmith KC, Gross M, Peirce S, Luyindula D, Liu X, Vu A, Sliozberg M, Guo R, Zhao H, Reynolds CP, et al. Mitochondrial Bcl-2 family dynamics define therapy response and resistance in neuroblastoma. Cancer Res 2012; 72:2565-77; PMID:22589275; http://dx.doi.org/10.1158/0008-5472.CAN-11-3603
- Goldsmith KC, Lestini BJ, Gross M, Ip L, Bhumbla A, Zhang X, Zhao H, Liu X, Hogarty MD. BH3 response profiles from neuroblastoma mitochondria predict activity of small molecule Bcl-2 family antagonists. Cell Death Differ 2010; 17:872-82; PMID:19893570; http://dx.doi.org/10.1038/cdd.2009.171
- Lestini BJ, Goldsmith KC, Fluchel MN, Liu X, Chen NL, Goyal B, Pawel BR, Hogarty MD. Mcl1 downregulation sensitizes neuroblastoma to cytotoxic chemotherapy and small molecule Bcl2-family antagonists. Cancer Biol Ther 2009; 8; PMID:19556859
- Chen S, Dai Y, Harada H, Dent P, Grant S. Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res 2007; 67:782-91; PMID:17234790; http://dx.doi.org/10.1158/0008-5472.CAN-06-3964
- van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, Willis SN, Scott CL, Day CL, Cory S, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 2006; 10:389-99; PMID:17097561; http://dx.doi.org/10.1016/j.ccr.2006.08.027
- Boisvert-Adamo K, Longmate W, Abel EV, Aplin AE. Mcl-1 is required for melanoma cell resistance to anoikis. Mol Cancer Res 2009; 7:549-56; PMID:19372583; http://dx.doi.org/10.1158/1541-7786.MCR-08-0358
- Jourdan M, De Vos J, Mechti N, Klein B. Regulation of Bcl-2-family proteins in myeloma cells by three myeloma survival factors: interleukin-6, interferon-α and insulin-like growth factor 1. Cell Death Differ 2000; 7:1244-52; PMID:11175262; http://dx.doi.org/10.1038/sj.cdd.4400758
- Ho R, Minturn JE, Hishiki T, Zhao H, Wang Q, Cnaan A, Maris J, Evans AE, Brodeur GM. Proliferation of human neuroblastomas mediated by the epidermal growth factor receptor. Cancer Res 2005; 65:9868-75; PMID:16267010; http://dx.doi.org/10.1158/0008-5472.CAN-04-2426
- Fulda S. The PI3K/Akt/mTOR pathway as therapeutic target in neuroblastoma. Current cancer drug targets 2009; 9:729-37; PMID:19754357; http://dx.doi.org/10.2174/156800909789271521
- Meyers MB, Shen WP, Spengler BA, Ciccarone V, O'Brien JP, Donner DB, Furth ME, Biedler JL. Increased epidermal growth factor receptor in multidrug-resistant human neuroblastoma cells. J Cell Biochem 1988; 38:87-97; PMID:2464605; http://dx.doi.org/10.1002/jcb.240380203
- Opel D, Poremba C, Simon T, Debatin KM, Fulda S. Activation of Akt predicts poor outcome in neuroblastoma. Cancer Res 2007; 67:735-45; PMID:17234785; http://dx.doi.org/10.1158/0008-5472.CAN-06-2201
- Tweddle DA, Malcolm AJ, Bown N, Pearson AD, Lunec J. Evidence for the development of p53 mutations after cytotoxic therapy in a neuroblastoma cell line. Cancer Res 2001; 61:8-13; PMID:11196202
- Zhai D, Jin C, Huang Z, Satterthwait AC, Reed JC. Differential regulation of Bax and Bak by anti-apoptotic Bcl-2 family proteins Bcl-B and Mcl-1. J Biol Chem 2008; 283:9580-6; PMID:18178565; http://dx.doi.org/10.1074/jbc.M708426200
- Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, et al. EGF receptor gene mutations are common in lung cancers from "never smokers" and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A 2004; 101:13306-11; PMID:15329413; http://dx.doi.org/10.1073/pnas.0405220101
- Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2002; 2:183-92; PMID:12242151; http://dx.doi.org/10.1016/S1535-6108(02)00127-7
- Morales AA, Kurtoglu M, Matulis SM, Liu J, Siefker D, Gutman DM, Kaufman JL, Lee KP, Lonial S, Boise LH. Distribution of Bim determines Mcl-1 dependence or codependence with Bcl-xL/Bcl-2 in Mcl-1-expressing myeloma cells. Blood 2011; 118:1329-39; PMID:21659544; http://dx.doi.org/10.1182/blood-2011-01-327197
- Ley R, Balmanno K, Hadfield K, Weston C, Cook SJ. Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, Bim. J Biol Chem 2003; 278:18811-6; PMID:12646560; http://dx.doi.org/10.1074/jbc.M301010200
- Ley R, Ewings KE, Hadfield K, Howes E, Balmanno K, Cook SJ. Extracellular signal-regulated kinases 1/2 are serum-stimulated “Bim(EL) kinases7rdquo; that bind to the BH3-only protein Bim(EL) causing its phosphorylation and turnover. J Biol Chem 2004; 279:8837-47; PMID:14681225; http://dx.doi.org/10.1074/jbc.M311578200
- Mouhamad S, Besnault L, Auffredou MT, Leprince C, Bourgeade MF, Leca G, Vazquez A. B cell receptor-mediated apoptosis of human lymphocytes is associated with a new regulatory pathway of Bim isoform expression. J Immunol 2004; 172:2084-91; PMID:14764673; http://dx.doi.org/10.4049/jimmunol.172.4.2084
- Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, Carter SL, Cibulskis K, Hanna M, Kiezun A, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet 2013; 45(3):279-84; PMID:23334666; http://dx.doi.org/10.1038/ng.2529. Epub 2013 Jan 20
- Jakacki RI, Hamilton M, Gilbertson RJ, Blaney SM, Tersak J, Krailo MD, Ingle AM, Voss SD, Dancey JE, Adamson PC. Pediatric phase I and pharmacokinetic study of erlotinib followed by the combination of erlotinib and temozolomide: a Children's Oncology Group Phase I Consortium Study. J Clin Oncol 2008; 26:4921-7; PMID:18794549; http://dx.doi.org/10.1200/JCO.2007.15.2306
- Daw NC, Furman WL, Stewart CF, Iacono LC, Krailo M, Bernstein ML, Dancey JE, Speights RA, Blaney SM, Croop JM, et al. Phase I and pharmacokinetic study of gefitinib in children with refractory solid tumors: a Children's Oncology Group Study. J Clin Oncol 2005; 23:6172-80; PMID:16135484; http://dx.doi.org/10.1200/JCO.2005.11.429
- Cao X, Zhu H, Ali-Osman F, Lo HW. EGFR and EGFRvIII undergo stress- and EGFR kinase inhibitor-induced mitochondrial translocalization: a potential mechanism of EGFR-driven antagonism of apoptosis. Molecular cancer 2011; 10:26; PMID:21388543; http://dx.doi.org/10.1186/1476-4598-10-26
- Zhu H, Cao X, Ali-Osman F, Keir S, Lo HW. EGFR and EGFRvIII interact with PUMA to inhibit mitochondrial translocalization of PUMA and PUMA-mediated apoptosis independent of EGFR kinase activity. Cancer Lett 2010; 294:101-10; PMID:20153921; http://dx.doi.org/10.1016/j.canlet.2010.01.028
- Rizzatti EG, Mora-Jensen H, Weniger MA, Gibellini F, Lee E, Daibata M, Lai R, Wiestner A. Noxa mediates bortezomib induced apoptosis in both sensitive and intrinsically resistant mantle cell lymphoma cells and this effect is independent of constitutive activity of the AKT and NF-kappaB pathways. Leuk Lymphoma 2008; 49:798-808; PMID:18398749; http://dx.doi.org/10.1080/10428190801910912
- Chen S, Dai Y, Pei XY, Grant S. Bim upregulation by histone deacetylase inhibitors mediates interactions with the Bcl-2 antagonist ABT-737: evidence for distinct roles for Bcl-2, Bcl-xL, and Mcl-1. Mol Cell Biol 2009; 29:6149-69; PMID:19805519; http://dx.doi.org/10.1128/MCB.01481-08
- Akiyama T, Dass CR, Choong PF. Bim-targeted cancer therapy: a link between drug action and underlying molecular changes. Mol Cancer Ther 2009; 8:3173-80; PMID:19934277; http://dx.doi.org/10.1158/1535-7163.MCT-09-0685
- Tumilowicz JJ, Nichols WW, Cholon JJ, Greene AE. Definition of a continuous human cell line derived from neuroblastoma. Cancer Res 1970; 30:2110-8; PMID:5459762
- Reynolds CP, Biedler JL, Spengler BA, Reynolds DA, Ross RA, Frenkel EP, Smith RG. Characterization of human neuroblastoma cell lines established before and after therapy. J Natl Cancer Inst 1986; 76:375-87; PMID:3456456
- Schwab M, Alitalo K, Klempnauer KH, Varmus HE, Bishop JM, Gilbert F, Brodeur G, Goldstein M, Trent JM. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature 1983; 305:245-8; PMID:6888561; http://dx.doi.org/10.1038/305245a0
- Keshelava N, Seeger RC, Groshen S, Reynolds CP. Drug resistance patterns of human neuroblastoma cell lines derived from patients at different phases of therapy. Cancer Res 1998; 58:5396-405; PMID:9850071