
Abstract
PTEN has been studied in several tumor models as a tumor suppressor. In this study, we explored the role of PTEN in the inhibition state of polarized M2 subtype of macrophage in tumor microenvironment (TME) and the underlying mechanisms. To elucidate the potential effect in TME, RAW 264.7 macrophages and 4T1 mouse breast cancer cells were co-cultured to reconstruct tumor microenvironment. After PTEN was down-regulated with shRNA, the expression of CCL2 and VEGF-A, which are definited to promote the formation of M2 macrophages, have a dramatically increase on the level of both gene and protein in co-cultured RAW 264.7 macrophages. And at the same time, NHERF-1 (Na+/H+ exchanger regulating factor-1), another tumor suppressor has a similar tendency to PTEN. Q-PCR and WB results suggested that PTEN and NHERF-1 were consistent with one another no matter at mRNA or protein level when exposed to the same stimulus. Coimmunoprecipitation and immunofluorescence techniques confirmed that PTEN and NHERF-1 were coprecipitated, and NHERF-1 protein expression was properly reduced with rCCL2 effect. In addition, cell immunofluorescence images revealed a profound transferance, in co-cultured RAW 264.7 macrophages, an up-regulation of NHERF-1 could promote the PTEN marked expression on the cell membrane, and this form for the interaction was not negligible. These observations illustrate PTEN with a certain synergy of NHERF-1, as well as down-regulation of CCL2 suppressing M2 macrophage transformation pathway. The results suggest that the activation of PTEN and NHERF-1 may impede the evolution of macrophages beyond the M1 into M2 phenotype in tumor microenvironment.
Abbreviations
| CAFs | = | cancer associated fibroblasts |
| CM | = | complete medium |
| CXCL | = | the chemokine (C-X-C motif) ligand |
| CXCR3 | = | Chemokine (C-X-C motif) receptor 3 |
| FAK | = | focal adhesion kinase |
| NHERF-1 | = | Na+/H+ exchanger regulating factor1 |
| PTEN | = | phosphatase and tensin homolog deletedon chromosome 10 |
| SCC | = | squamous cell carcinoma |
| TAMs | = | tumor-associated macrophages |
| TSN | = | tumor culture supernatant |
Introduction
It is now generally accepted that tumor-associated macrophages (TAMs) have an M2 phenotype which shows mostly pro-tumoral functions and promotes tumor cells to grow.Citation1,2 Our previous studies were spent to hammer away at researching the specific molecular mechanisms about M1 to M2 macrophage phenotype transformation and reported the transcription factor Fos family member Fra-1 induced the generation of M2 subtype macrophages.Citation3 PTEN (phosphatase and tensin homolog deleted on chromosome 10) is regarded as the new tumor suppressor.Citation4,5 The function of PTEN is displayed through suppressing PI3K, thus the process of PIP2 transform into PIP3 would be blocked, leading the downstream Akt not to be phosphorylated, namely inhibit PI3K/Akt/mTOR (mammalian target of rapamycin, mTOR)Citation6,7 and other kinds of signal pathways participating in tumor cell proliferation, evasion of apoptosis and resistance to chemotherapy.Citation8 We learned through literatures that the mRNA expression of chemokines including CCL2, CCL4, CCL3, CXCL, CXCL5, and chemokine receptor CXCR3, was increased in carcinoma tissues of Pten knock out (KO) mice or in cancer cells of siRNA-mediated inhibition of PTEN gene expression,Citation9-11 which have been linked to play precisely domesticated role of the M2 phenotype and thus will likely facilitate development of microenvironment to enhance tumor infiltrating, metastasis and deterioration.Citation12,13 A considerable amount of information displayed, the decline of PTEN protein in cancer was known to heighten the expression of the angiogenic factor VEGF-A, and there was a negative correlation between PTEN and VEGF-A.Citation14 The exact mechanisms about PTEN regulates VEGF-A suggested that the defect of PTEN could not inhibit angiogenesis by regulating the expression of hypoxia-inducible factor (HIF-1α) and VEGF-A through PI3K/AKT activation in cancer cells.Citation14 In addition, VEGF-A could skew macrophage differentiation toward the M2 phenotype, and subsequently promote macrophage production of VEGF-A and angiogenesis in the tumor microenvironment.Citation15
The purpose of this study was to determine the potential regulation between PTEN and M2 macrophage polarization in tumor microenvironment we reconstructed in vitro. We found that PTEN could inhibit the polarization from innate macrophage to an M2 phenotype via decreasing the expression of CCL2 and VEGF-A. Moreover, we identified another tumor suppressor-NHERF-1-as a key regulator of suppressing M2 macrophage polarization via associated with PTEN, playing an important role in this process.
Materials and Methods
Cell lines and preparation of tumor culture supernatant (TSN)
Primary Mφs were obtained from female BALB/c mice at 6–8 weeks of age. RAW 264.7 macrophage cell line was purchased from School of Basic Medicine, Peking Union Medical College (Beijing, China); Murine 4T1 breast carcinoma cells were kindly provided by Dr Ostrand-Rosenberg S (University of Maryland, College Park, Maryland, USA); MDA-MB-231 cells and U937 cells were purchased from ATCC. Both cell lines were grown in IMEM (GIBCO, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (MDgenics, St Louis, MO, USA), 100 units/ml penicillin and 100 mg/ml streptomycin. For co-culture experiments, RAW264.7 cells were co-cultured with 4T1 cells at a 1:4 ratio in complete medium (CM) for 24 h, 48 h, 72 h or 96 h. In inducing experiments, RAW264.7 cells were incubated with supernatant of 4T1 cells (TSN, tumor culture supernatant, diluted 1:2.5) for 36 h (added at hours 0, 12 and 24) and rCCL2 (250 ng/ml) for 0, 12, and 24 h. In neutralization experiment, co-cultured cells were maintained in CM with rabbit anti-mouse CCL2 (500 ng/ml) or isotype IgG antibody (500 ng/ml) (added at hours 0, 12 and 24). TSN was harvested and centrifuged at 2500 rpm/min for 25 min to remove cells and debris, and then frozen at -80 degrees Celsius as long as 5 to 8 days, though it should ideally be used within 2 or 3 days. RAW264.7 cells co-cultured with supernatants of 4T1 (diluted 1:2.5) for 36 h accomplish transformation (TSN-exposed RAWs). As above, we also used the human macrophage cell line U937 and the human breast cancer MDA-MB-231 culture supernatants (TSN-exposed U937s) to mimic the human tumor microenvironment in vitro according to the same condition.
Immunohistochemical and immunofluorescence analyses
Samples of gastric carcinoma and breast cancers (6 of each) were obtained from Tianjin Cancer Hospital of Tianjin Medical University and coded anonymously in accordance with local ethical guidelines. Paraffin-embedded and formalin-fixed samples were cut into 5μm sections, and then processed for immunohistochemistry as previously described.Citation16 Following incubation with anti-CD68 antibody (Ab, Boster, Wuhan, China) and anti-PTEN Ab (Bioworld, Shanghai, China), immunoactivity was detected with diaminobenzidine (DAB). For immunofluorescence analysis, the treated cells were fixed and then perforated with 0.5% Triton X-100, and then were blocked for 1 h prior to staining with rabbit anti-PTEN Ab (1:200), anti-p-PTEN Ab (1:200), rabbit anti-NHERF1 Ab (1:300) (Cell Signaling Technology, Beijing, China) for overnight at 4 degrees Celsius. Immunoreactive proteins were detected by incubating with TRITC-conjugated goat anti-rabbit IgG (H+L) (1:100, ZSGB-BIO, Beijing, China), the nuclear stained with a DAPI in dark. Images were assessed with a confocal microscope (Olympus, FV1000).
Transfection experiments
For stable PTEN gene transfection, the DNA fragment encoding membrane PTEN was amplified from PTEN cDNA by PCR using following primers:5′ EcoR I- PTEN primer (5′-GCCGAATTCGACTTTTGTAATTTGTGTA-3′), 3′ PTEN-Xho I primer (5′-CCGCTCGAGCAGTCGCTGCAACCATCCA-3′). PTEN cDNA was cloned into mammalian expression vector pMD18-T (Takara, Dalian, China) using the EcoR I / Xho I restriction sites and into pEGFP-N1 vector (Clontech, Shanghai, China).After amplification in E.coli strain DH5α (Invitrogen, Shanghai, China), plasmid DNA was purified using Qiagen Plasmid Midi Kit (Qiagen,Beijing, China), then transfected pEGFP-N1 empty vector or pEGFP-N1 coding for PTEN(pEGFP-PTEN) into TSN-exposed RAWs using Lipofectamine 2000 according to the manufacturer's instructions. To establish stable PTEN-knockdown TSN-exposed RAWs by vector based RNAi system, RNAi-mediated PTEN knockdown was accomplished by shRNA produced by the DNA-based shRNA-expressing retroviral vector (pSuper-Retro). The vectors were from Clontech, Shanghai, China. The knockdown experiment was conducted with Lipofectamine 2000. The PTEN shRNA target sequence is GTTGGCGAGT GT TTTGTGAA G. Additionally, transient transfection of TSN-exposed RAWs with NHERF-1-siRNA and PTEN-siRNA was performed, the cells were transfected with siRNA specific for mice NHERF-1 and PTEN (RiboBio Co, Guangzhou, China) or the negative control siRNA using HiperFect transfection reagent (Qiagen, Beijing, China) according to the manufacturer's specifications. NHERF-1 and PTEN siRNA targeted the following mRNA sequence 5′-CAG AAG GAG AAC AGT CGT GAA-3′and 5′-GGCGCUAUGUGUAUUAUUAdTdT-3′, respectively. The expression of PTEN and NHERF-1 in transfected cells was observed by fluorescence microscope or western blotting. Transfections were carried out in duplicate and repeated at least 3 times in independent experiments.
PTEN and NHERF-1 enrichment using inhibitor or cytokine treatment
TSN-exposed RAWs were treated with LY294002 (30 μM, Promega, Madison, WI, USA) for 12 h to accomplish PTEN enrichment, while were stimulated with murine LPS and IFNγ (200 ng/ml and 200 U/ml, Sigma, Shanghai, China) for 16 h to carry out the enrichment of NHERF-1.Citation17
RT-PCR and quantitative PCR (Q-PCR) analysis
Total RNA and cDNA were generated as described in the RT-PCR analysis, and quantitative PCR was performed with the QuantiFast SYBR Green PCR kit (Qiagen, Valencia, CA, USA) and detected by the DA7600 Real-time Nucleic Acid Amplification Fluorescence Detection System (Da An Gene, Guangdong, China). For each sample, theΔΔCt values were calculated according to the derived equation
2−ΔΔCt (the Ct value of the target gene minus the Ct value of the housekeeping gene β-actin). All Q-PCR results were expressed as fold changes in the mRNA expression compared with the control cells. The represented data were from 3 independent experiments performed in triplicate. The specific primers to amplify the genes were as follows: PTEN (forward: 5′-TTGAAGACCA TAACCCACC-3′; reverse: 5′- AGTTCCGCCA CTGAACAT-3′), CCL2 (forward: 5′- GAGGAAGGCC AGCCCAGCAC-3′; reverse: 5′- TGGATGCTCC AGCCGGCAAC-3′), HIF-1α (forward: 5′-GCATCTCCAT CTTCTACCCC-3′; reverse:5′- TTTTGCTCCG TTCCATTCT-3′), VEGF-A (forward: 5′- GCTACTGCCG TCCGATTGAG-3′; reverse: 5′- GTGCTG-GCTT TGGTGAGGTT-3′), NHERF-1 (forward: 5′- AGGTCAATGG TGTCTGCA-3′; reverse: 5′- CTTTAGCCAC AGCCAAGGA-3′), β-actin (forward: 5′-CGTTGACATC CGTAAA-GACC-3′; reverse: 5′- AACAGTCCGC CTAGAAGCAC-3′).
Measurement of CCL2 concentration by ELISA
A total of 5 × 105 transfected cells were seeded in 24-well plates and the supernatants were harvested at hours 0, 12, 24, 36 and 48, centrifuged at 2500 rpm for 25 min, and then transferred to a new micro-centrifuge tube. The concentrations of CCL2 were measured by ELISA kits (Dakewe, Beijing, China).
Immunoblotting
For protein gel blot analysis, total cell lysates were separated by SDS-PAGE (10% acrylamide). Immunoblotting was carried out with rabbit anti mouse specific antibodies or rabbit anti-mouse housekeeping genes (anti-PTEN and anti-p-PTEN, Bioworld, Shanghai, China; anti-NHERF-1, Cell Signaling Technology, Beijing, China; anti-VEGF, anti-HIF-1α, anti-GAPDH and anti-β-actin, Santa Cruz, CA, USA). After being washed, HRP-conjugated anti-rabbit IgG secondary antibodies (Santa Cruz, CA, USA) were incubated with the membranes, and then were washed and detected with Immobilon Western Chemiluminescent HRP substrate (Millipore, Bedford, MA, USA). For coimmunoprecipitation and immunoblotting analyses, TSN-exposed RAWs without/with transfected PTEN gene were stimulated with LPS and IFNγ (200 ng/ml and 200 U/ml) for 16 h to carry out the enrichment of NHERF-1 alone and the enrichment of both NHERF-1 and PTEN, respectively. The cell proteins were extracted, NHERF-1 was immunoprecipitated and detected by western blot analysis with rabbit anti- NHERF-1 (1:400). The immuno-complex was captured by gently adding anti-PTEN-Protein A/G-Agarose plus beads (Gendepot, Beijing, China) and subsequently subjected to protein gel blotting (10% SDS-PAGE).
Migration assay
All cell migration assays were performed with transwell inserts (8-μm pore size; Corning, Shanghai, China) as described previously.Citation18 Under different external stimuli, the migrated cells were quantized through 12 to 15 random fields under a light microscope (Nikon, C-HGFi). The remaining cell-attached-dye was dissolved in 33.3% acetic acid solution in ddH2O. The dissolved dye solution was transferred into a 96-well plate and read OD value in 450 nm.
Flow cytometry analysis of M2 phenotype polarization
The treated cells were trypsinized and stained with FITC conjugated anti-mouse CD206 (Biolegend, San Diego, CA, USA) or with mouse IgG-FITC (BD Biosciences, San Jose, CA, USA) and analyzed on BD FACS Canto II. Data were analyzed with BD FACSDiva Software v 6.1.2.
Determination of cell growth rate
4T1 cells were cultured in different conditional medium and counted with a hemocytometer under light microscope at hours 12, 24, 36, 48 and 60 at an initial concentration of 104 cells/ml. Triple 24-well plates were counted for each time point to determine the growth rate.
Statistical analysis
Results are presented in the form of “mean ± SE” and were analyzed with SigmaStat10.0 software (SPSS). The differences between 2 groups were assessed by the Student t test. The differences among 3 or more groups were evaluated by a one-way ANOVA followed by the Dunnett test. P < 0.05 was considered statistically significant.
Results
Immunohistochemical characteristics of the tumorspecimen proofed distinct expression patterns of PTEN in actual macrophages in cancer tissues
To investigate the potential role of PTEN in macrophages of tumor microenvironment, we first examine its expression and distribution in serial sections of human cancerous tissues stained for PTEN and CD68 (marker for macrophages, Mφs). By immunohistochemical staining with a polyclonal rabbit anti-mouse PTEN, we showed that PTEN was expressed by CD68+ Mφs with only one thin continual layer of paraffin section in gastric and breast cancerous tissues (, PTEN, arrowheads). The cancer tissue origin of these CD68-positive cells was confirmed by positive staining for Mφs (, Mφs, and arrowheads), that is, most macrophages in gastric and breast cancer were also positively stained with PTEN. Thus, we concluded that PTEN was expressed in both the cancer cells and actual macrophages in cancer tissues. We next tried to knockdown PTEN in macrophages and analyzed the effect of cancer culture supernatants in reconstruct cancer microenvironment. Is this really a model of actual cancer microenvironment? The distinct expression patterns of PTEN and CD68 (marker for Mφs) in serial sections of human cancerous tissues we have showed in can answer this.
Figure 1. Tumor-associated macrophages (Mφ) differentially expressed PTEN in vivo and in vitro. (A) Distinct expression patterns of tumor-infiltrating CD68-positive and PTEN-positive cells in human tumor samples. Consecutive sections of gastric or breast carcinoma were used for immunohistochemical identification of CD68- positive and PTEN-positive cells. Positive cells were stained brown, nonspecific immunoglobulin was used as a negative control. (B, C) The level of PTEN in primary macrophages and RAW 264.7 cells transfected with shRNA and pEGFP specific for mouse PTEN or the negative control were analyzed by Q-PCR and WB, GAPDH was used as an internal control.
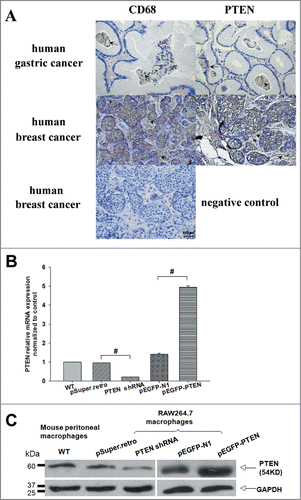
To evaluate the potential effect of PTEN on macrophages phenotype in tumor microenvironment, we established different cell models accompanied by decreased and increased level of PTEN, and defined its expression in primary macrophages and RAW 264.7 cells of our reconstructing tumor microenvironment in vitro by RT-PCR (Data not shown), Q-PCR () and western blotting(). As shown in transfected pEGFP-PTEN protein was stably expressed, while shPTEN cells showed significantly decreased expression compare with control cells.
Knockdown PTEN in RAW 264.7 macrophages induced CCL2 to up-regulate significantly and the culture supernatants promoted the migration of 4T1 cells
shPTEN induced CCL2 to up-regulate significantly in RAW264.7 cells that was the point-cut across the whole study. There was evidence that in squamous cell carcinoma (SCC) tissues of Pten knockout mice, the expression of CCL2 mRNA elevated significantly.Citation19 However, in TME whether PTEN impacts the expression and secretion of CCL2 in macrophages is not clear. By ELISA experiments we found that CCL2 secretion reached the highest level after cultured for 36 h, and CCL2 concentration in PTEN-loss co-RAWs increased significantly from 128 ± 4.4 pg/ml to 272 ± 6.1 pg/ml at 36 h (P < 0.05, n = 6, ). Once again, we confirmed this result using stable over-express PTEN in high expression model().
Figure 2. Interference of PTEN boosted CCL2 production and then promoted 4T1 tumor cells invasion and VEGF-A expression in vitro. (A) Production of CCL2 in TSN-exposed RAWs with shPTEN and pEGFP-PTEN was determined by ELISA. Supernatants were obtained for the indicated time periods, #P < 0.05 vs. control. (B) The 4T1 cells were added into the upper chamber and incubated for 24 h with different stimuli in culture medium. The migrated cells were quantified in 10 random fields at × 100 magnification. #P < 0.05. All data were representatives of at least 3 independent experiments. (C, D) mRNA and protein were extracted from TSN-exposed RAWs and U937 treated with siPTEN and LY294002, then validated the expression of PTEN, VEGF-A, and HIF-1α by Q-PCR and WB. Q-PCR and immunoblotting data were representatives of 3 separate experiments, #P < 0.05.
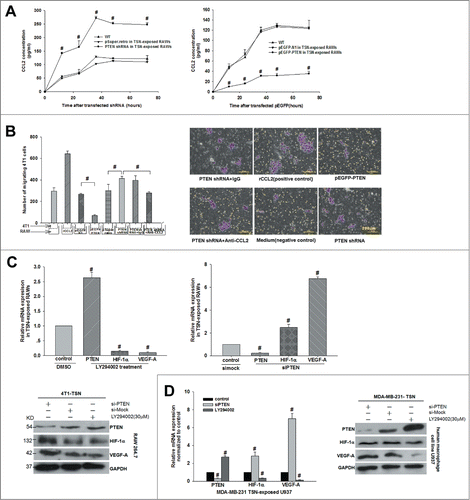
To verify whether the growing secretion of CCL2 in the culture supernatants induced by shPTEN further urged the polarization of M2 macrophages, and then provide 4T1 tumor cells more suitable soil for tumor to migrate, we implemented transwell migration about shPTEN, pEGFP-PTEN, shPTEN rescue functional experiments. After being cultured for 36 hours, 4T1 cells in the shPTEN group had migrated much more than control, increased 2.17-fold relative to negative control group (P < 0.05), and the number of migrated 4T1 cells in the group which additional adding rCCL2(250 ng/ml) was regarded as a positive control ().
siPTEN in both RAW 264.7 macrophages and U937 human macrophages increased VEGF-A expression
In light of these results above, we further explored the expression of a series of coefficient molecules such as HIF-1α, CCL2, VEGF-A, and PDGF in siPTEN macrophages via Q-PCR and protein gel blot. And we impressively observed that siPTEN in both RAW 264.7 macrophages and U937 human macrophages dramatically increased VEGF-A expression. As shown in , after PTEN was silenced, the expression level of PTEN mRNA decreased from 1 to 0.22 ± 0.07 (P < 0.05, n = 6), while the expression of VEGF-A increased to 6.80 ± 0.15 fold (P < 0.05, n = 8). Here, LY294002 acts in vivo as a highly selective inhibitor of PI3K and specifically abolishes PI3K activity but does not inhibit other lipid or protein kinase. So, we also validated the effect of LY294002 on the expression of HIF-1α/VEGF through up-regulating PTEN. The data revealed that LY294002 (30 μM) treatment enhanced the level of PTEN mRNA from 1 to 2.63 ± 0.19 fold, but the level of VEGF-A mRNA dropped obviously (P < 0.05). In the protein level, we found the same variation tendency between PTEN and VEGF-A ().
In addition, the stable expression tendency in the gene and protein level was also observed in the human macrophage cell line TSN-exposed U937 in which U937 co-cultured with human breast cancer MDA-MB-231 cells under the same corresponding condition ().
NHERF-1 could promote PTEN to express markedly on the cell membrane, which is where PTEN does its work
In this study, we found that HIF-1α might affect PTEN expression, at the meanwhile, from the literatures we learned that NHERF-1 was relative to hydrogen ion exchange, which could recruit PTEN to the plasma membrane where PTEN does its work.Citation20 Speaking of which,we could not help asking whether other physical and chemical properties of tumor microenvironment also influenced PTEN expression, and, if they did, whether NHERF-1 was proper to join in them. To clarify this question, we changed pH value and O2 concentration, and then detected the mRNA and protein expression of PTEN and NHERF-1. Surprisingly, we found that PTEN and NHERF-1 showed consistent trend either at the mRNA () or protein level ().
Figure 3. Association of PTEN with NHERF-1. (A, B) mRNA expression of NHERF-1 and PTEN in TSN-exposed RAWs with different treatment was detected by Q-PCR and WB was performed to detect the protein expression of NHERF-1 and PTEN. (C) TSN-exposed RAWs were stimulated with IFNγ and LPS or left in media for 16 h, lysed, and analyzed by western blot and then quantificated the increase expression of NHERF-1. (D) IP: IB was performed to determine the possible intracellular association between PTEN and NHERF-1. Lane1: NHERF-1 lysate; Lane2: NHERF-1 and PTEN lysate; Lane3: IP PTEN, IB NHERF-1 in NHERF-1 lysate; Lane4: IP PTEN, IB NHERF-1 in NHERF-1 and PTEN lysate. Q-PCR and immunoblotting data was representative of 3 separate experiments, #P < 0.05.
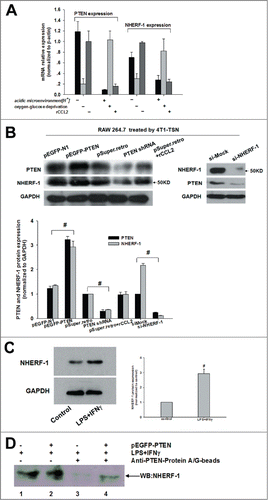
To define the possible synergies of these 2 proteins, furthermore, we did the coimmunoprecipitation (IP) and immunoblotting (IB) analyses. Prior to the IP:IB experiments, we estimated that NHERF-1 expression was increased upon IFNγ and LPS stimulation.Citation17 () And then our IP: IB results revealed only both PTEN and NHERF-1 immuo-complex could be captured by adding anti-PTEN-Protein A/G-Agarose beads (). It was clear that PTEN robustly associated with NHERF-1 and it was supposed that this association was mediated via the binding of PTEN carboxyl-terminal PDZ motif with the PDZ1 of NHERF-1.Citation21,22 (DNAssist version 2.2 software).
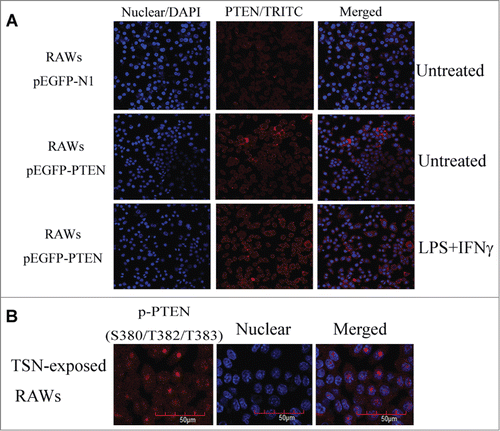
To further elucidate how NHERF-1 affected PTEN, we performed immunofluorescence labeling experiments, and the confocal images exhibited how NHERF-1 could affect the location of PTEN. The microscopy revealed that in macrophages, the upregulation of NHERF-1 helped PTEN translocate to the plasma membrane, not cytoplasm or nuclear, and this expression transference was very crucial. () Many molecules participated in a series of molecular events in the cell in different forms, to be complete, we should add a sub-rule to this, PTEN phosphorylation protein (p-PTEN) was less active form of PTEN for its mainly expressing in nucleus of cells.Citation23 And it only was dephosphorylated to become PTEN, and shifted from nucleus to cell membrane, could it play a synergistic antitumor role independently or with other molecules.Citation24 We tested whether p-PTEN protein expressed in nucleus of TSN-exposed RAWs, as showed in , confocal microscopy images specified the localization of p-PTEN through fluorescence intensity.
Figure 4. Up-regulating NHERF-1 increased PTEN binding to plasma membrane. (A) TSN-exposed RAWs with different treatment were fixed and stained on glass cover slips. The distribution of PTEN (red) in cells was analyzed by confocal microscopy. DAPI, blue. (B) The indicated p-PTEN was determined in nucleus of TSN-exposed RAWs by immunofluorescence analyses.
NHERF-1 might be synergistically involved in the regulation of PTEN in M2 macrophages polarization and CCL2 counteracted this regulation
To go on to investigate the possible influence of PTEN and NHERF-1 on M2 macrophages phenotype polarization, we used FACS to undertake the experiment of M1 or M2 phenotype identification. The experimental results showed when PTEN and NHERF-1 were enriched synergistically in RAW 264.7 cells, the number of CD206+ macrophages (M2 phenotype) went down from (13 ± 1.10 ) × 105 to (5.4 ± 0.58) × 105 (P < 0.05, n = 8) (). After rCCL2 treatment, the number of CD206+ macrophages (M2 phenotype) number increased again from 5.02% ± 0.75% to 17.87% ± 1.08% (P < 0.05, n = 6) (). However, when adding LPS and rM-CSF, we couldn't obtain the same recovery of the number of CD206+ macrophages (Data not shown). These results suggested CCL2 might affect the cooperating function of PTEN together with NHERF-1 on polarization of M2 macrophages.
Figure 5. PTEN cooperated with NHERF-1 impeded M2 phenotype polarization and CCL2 impaired the inhibition. (A, B) CD206+ cells were determined by flow cytometry in co-RAWs with different treatments (pEGFP-PTEN+LPS+IFNγ or rCCL2) and compared with negative control, respectively. The flow cytometry data were representatives of at least 4 separate experiments, results were presented as means ± SEM. #P < 0.05; ##P < 0.01. (C, D, E) Transfection of PTEN gene with/without enriched NHERF-1 in TSN-exposed RAWs and their phenotype identification of surface markers (M1 phenotype genes [iNOS] and M2 phenotype genes [Arg1]) was primitively determined, then subsequently detected TSN-exposed RAWs stimulated with rCCL2 (250 ng/ml) for 24 h compared with control group by Western blot assay. Further their supernatant effects on cell growth in 4T1 cells were figured up. Immunoblotting data were representatives of 3 separate experiments, *P < 0.05; **P < 0.01. Cells were counted every day. Data for the total number of cells (mean for triplicate cultures).
![Figure 5. PTEN cooperated with NHERF-1 impeded M2 phenotype polarization and CCL2 impaired the inhibition. (A, B) CD206+ cells were determined by flow cytometry in co-RAWs with different treatments (pEGFP-PTEN+LPS+IFNγ or rCCL2) and compared with negative control, respectively. The flow cytometry data were representatives of at least 4 separate experiments, results were presented as means ± SEM. #P < 0.05; ##P < 0.01. (C, D, E) Transfection of PTEN gene with/without enriched NHERF-1 in TSN-exposed RAWs and their phenotype identification of surface markers (M1 phenotype genes [iNOS] and M2 phenotype genes [Arg1]) was primitively determined, then subsequently detected TSN-exposed RAWs stimulated with rCCL2 (250 ng/ml) for 24 h compared with control group by Western blot assay. Further their supernatant effects on cell growth in 4T1 cells were figured up. Immunoblotting data were representatives of 3 separate experiments, *P < 0.05; **P < 0.01. Cells were counted every day. Data for the total number of cells (mean for triplicate cultures).](/cms/asset/f46ef0b3-2530-483d-873d-661d00cbab5c/kcbt_a_1002353_f0005_c.gif)
Consistent with these results, the expression of PTEN appeared to be associated with NHERF-1 to affect M2 macrophage polarization. To lastly confirm the role of PTEN associated with NHERF-1 in regulating M2 macrophage polarization and to confirm the role of CCL2 influence the regulating function. We transfected the PTEN gene into RAW 264.7 cells without/ with the over-expression of NHERF-1 represent the function of PTEN alone () and both PTEN and NHERF-1 (). Additionally, we enriched NHERF-1 using LPS and IFNγ in RAW 264.7 cells representing the function of NHERF-1 alone (). As shown in , both transfected PTEN or/and NFERF-1 RAW 264.7 cells stably downregulated M2 phenotype proteins Arg-1, up-regulated M1 phenotype proteins iNOS, instead. We compared the growth rate of 4T1 cells incubated with supernatant from RAWs expressing PTEN or NHERF-1 with control. 4T1 cells showed significantly decreased growth rate (P < 0.05), while, supernatant from RAWs expressing PTEN and NHERF-1, 4T1 cells showed only half of that for control after 36 hours of culture(P < 0.01), moreover, the eliminating function of CCL2 seemed to be much more significant during this process (P < 0.01).
Discussion
The PTEN/PI3K/Akt signaling pathway may play a very critical role in the carcinogenesis of many cancers. Herein, we have shown that PTEN deletion in the macrophages of tumor microenvironment initiates tumor cell migration by increasing M2 macrophage polarization.Citation25 In the current study, we found that upon deletion of PTEN in macrophages, cytokines that were known to promote M2 macrophage to polarize, such as CCL2 was over-expressed. There is enhanced recruitment of M2 macrophage, which results in increased angiogenesis through up-regulation of VEGF-A as well as immune suppression in the tumor stroma. Similar results were observed in our cell models, when PTEN was disrupted, VEGF-A expression increased significantly. We also supposed that PTEN deletion increased VEGF-A expression by activating the PI3K/Akt/HIF-1α signaling pathway, but not through the protein tyrosine phosphatase-initiated focal adhesion kinase (FAK) dephosphorylation.Citation19,26,27 These findings suggest that PTEN defect also contributes significantly to the deterioration of tumor microenvironment. Therefore, on one hand upregulation of CCL2 by PTEN loss would certainly exert its tumor suppressive effects on tumor initiating cells and enhance the recruitment of M2 macrophage in the tumor. On the other hand the deletion of PTEN in macrophages of the tumor microenvironment would induce VEGF-A and promote VEGF-dependent angiogenesis.
It should be noted that the cytokines network regulating the polarization of M2 macrophages is much more complex than we have already explored. Even so, our current study provided important evidence that similar regulatory loop might also apply to microenvironment in vitro. It has been reported by other groups that IL-6, M-CSF, CCL2 and PDGF-B were significantly upregulated in M2 macrophages in vitro or in vivo.Citation3,11,16 These data were in part accord with our current conclusions.
Consistent with a very recent study by other group showing that PTEN interacted directly with NHERF-1,Citation20,21 we demonstrated that PTEN bound NHERF-1in vitro. In our study, that PTEN and NHERF-1 protein had interaction under different stimuli was revealed. NHERF-1 is regarded as another tumor suppressor,Citation28 and our finding that NHERF-1 modification of PTEN increases PTEN binding to the plasma membrane may explain why a small fraction of PTEN acts through the dynamic interaction with the inner face of plasma membrane. The structure of PTEN clues about the protein's function, some data indicates that the interaction between NHERF-1 and PTEN takes place between PDZ1 domain of NHERF-1 and the PDZ motif in C2 domain of PTEN.Citation20,29,30 Then PDZ1 domain of NHERF-1 can recruit PTEN to the membrane and then form the complex formation. NHERF-1 modification of PTEN occurs within seconds, and PTEN binds to the plasma membrane for a few hundred milliseconds, which is sufficient to dephosphorylate PIP3. Other research teams also have shown that PDGF regulates the PI3K signaling pathway through the PTEN signal transduction pathway mediated by NHERF-1.Citation21,31 These information seemed to support PTEN and NHERF-1 fragments might interact, but the interaction of PTEN and NHERF-1 complete molecules in the cells were lack of direct experimental data.Citation20,22,32 In our current experiment, first, we directly demonstrated the interaction of PTEN and NHERF-1 molecules in the cells through co-immunoprecipitation. The finding above provided a theoretical basis for the elucidation of PTEN in macrophage, which was involved in the regulation of M2 macrophage phenotype polarization through associating with other molecules in tumor microenvironment.
More importantly, our research team found that increased PTEN expression actually was based on up-regulating NHERF-1 expression. With confocal microscope, we examined PTEN protein mainly expressed in cytoplasm and the level of expression was relatively small in background level. After the cells were stably transected PTEN, the expression of PTEN protein were relatively high in cytoplasm. Impressively, after processing with LPS+IFNγ to up-regulate NHERF-1 expression, we could clearly see the expression of PTEN significantly increased, and mainly distributed onto membrane. This phenomenon at least illustrates this point visually, while, only by raising expression quantity of NHERF-1 and PTEN protein synchronously, can the purpose of PTEN enrichment massively on the membrane be reached. Or, more specifically, it is only by raising expression quantity of NHERF-1, can increase the expression quantity of PTEN on membrane. Here, combining with NHERF1 becomes a necessary condition that PTEN may behave as a tumor suppressor.Citation21 To our knowledge, this is the first report to describe the significance of NHERF-1 expression in relation to the effect of PTEN on M2 macrophage polarization.
In the present study, we focused on the absence of PTEN promoted tumor metastasis and PTEN played a critical role in a vicious spiral of the tumor microenvironment. Moreover, currently studies have promulgated NHERF-1 plays a vital role in breast cancer, involved in cell adhesion, cell migration, signal transduction and membrane function of protein orientation process.Citation28,33 Our study revealed that NHERF-1 was a critical role for PTEN to inhibit M2 macrophage polarization in microenvironment. Specifically emphasized here, NHERF-1 and PTEN coordination might impede CCL2 regulating M2 macrophage polarization pathway. According to our research data from FACS and 4T1 growth rate tests, it avoided the tumor microenvironment in vicious circle. Correspondingly, it has been confirmed that CCL2 is critical for cancer associated fibroblasts. (CAFs)-enhanced tumor microenvironment of oral SCC.Citation34 This implied that CCL2 was essential enough to determine tumor progression, but further studies were required to address this issue.
In summary, we have demonstrated that PTEN deletion initiates tumor microenvironment M2 macrophages phenotype polarization by increasing CCL2 and VEGF-A. Our results also indicate that PTEN deletion in macrophages could promote M2 macrophage to polarize, and this function of PTEN needs the regulation of NHERF-1, In other words, PTEN should be interacted with NHERF-1 and then could complete to translocate to membrane. These innovative findings in our study have significant implications regarding the effective therapeutic strategies targeting tumor microenvironment both PTEN and NHERF-1 for the treatment of cancers.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Qing-Shan Wang and Yujiao Yang for the preliminary studies, Profs Zhiping Xie and Yongzhe Che for helpful discussion, and Yanan Chen for immunofluorescence technical assistance.
Funding
This work was supported by grants from the National Natural Science Foundation of China (NSFC) (Nos. 81171975).
Reference
- Sica A, Saccani A, Mantovani A. Tumor-associated macrophages: a molecular perspective. Int Immunopharmacol 2002; 2:1045-54; PMID:12349942; http://dx.doi.org/10.1016/S1567-5769(02)00064-4
- Talmadge JE, Donkor M, Scholar E. Inflammatory cell infiltration of tumors: Jekyll or Hyde. Cancer Metast Rev 2007; 26:373-400; PMID:17717638; http://dx.doi.org/10.1007/s10555-007-9072-0
- Wang Q, Ni H, Lan L, Wei X, Xiang R, Wang Y. Fra-1 protooncogene regulates IL-6 expression in macrophages and promotes the generation of M2d macrophages. Cell Res 2010; 20:701-12; PMID:20386569; http://dx.doi.org/10.1038/cr.2010.52
- Li J. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997; 275:1943-7; PMID:9072974; http://dx.doi.org/10.1126/science.275.5308.1943
- Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet 1997; 15:356-62; PMID:9090379; http://dx.doi.org/10.1038/ng0497-356
- Georgescu MM. PTEN tumor suppressor network in PI3K-Akt pathway control. Genes Cancer 2010; 1:1170-7; PMID:21779440; http://dx.doi.org/10.1177/1947601911407325
- Vignot S, Faivre S, Aguirre D, Raymond E. mTOR-targeted therapy of cancer with rapamycin derivatives. Ann Oncol 2005; 16:525-37; PMID:15728109; http://dx.doi.org/10.1093/annonc/mdi113
- Tamura M. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science 1998; 280:1614-7; PMID:9616126; http://dx.doi.org/10.1126/science.280.5369.1614
- Hsu CJ, Wu MH, Chen CY, Tsai CH, Hsu HC, Tang CH. AMP-activated protein kinase activation mediates CCL3-induced cell migration and matrix metalloproteinase-2 expression in human chondrosarcoma. Cell Commun Signal 2013; 11:68-83; PMID:24047437; http://dx.doi.org/10.1186/1478-811X-11-68
- Tsuyada A, Chow A, Wu J, Somlo G, Chu P, Loera S, Luu T, Li AX, Wu X, Ye W, et al. CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res 2012; 72:2768-79; PMID:22472119; http://dx.doi.org/10.1158/0008-5472.CAN-11-3567
- Zhang J, Patel L, Pienta KJ. CC chemokine ligand 2 (CCL2) promotes prostate cancer tumorigenesis and metastasis. Cytokine Growth Factor Rev 2010; 21:41-8; PMID:20005149; http://dx.doi.org/10.1016/j.cytogfr.2009.11.009
- Tarassishin L, Lim J, Weatherly DB, Angeletti RH, Lee SC. Interleukin-1-induced changes in the glioblastoma secretome suggest its role in tumor progression. J Proteomics 2014; 99:152-68; PMID:24503185; http://dx.doi.org/10.1016/j.jprot.2014.01.024
- Ohnuki H, Jiang K, Wang D, Salvucci O, Kwak H, Sanchez-Martin D, Maric D, Tosato G. Tumor-infiltrating myeloid cells activate Dll4/Notch/TGF-beta signaling to drive malignant progression. Cancer Res 2014; 74:2038-49; PMID:24520074; http://dx.doi.org/10.1158/0008-5472.CAN-13-3118
- Bao B, Ali S, Ahmad A, Azmi AS, Li Y, Banerjee S, Kong D, Sethi S, Aboukameel A, Padhye SB, et al. Hypoxia-induced aggressiveness of pancreatic cancer cells is due to increased expression of VEGF, IL-6 and miR-21, which can be attenuated by CDF treatment. PloS One 2012; 7:e50165; PMID:23272057; http://dx.doi.org/10.1371/journal.pone.0050165
- Perrot-Applanat M, Di Benedetto M. Autocrine functions of VEGF in breast tumor cells: adhesion, survival, migration and invasion. Cell Adhesion Migr 2012; 6:547-53; PMID:23257828; http://dx.doi.org/10.4161/cam.23332
- Yang Y, Qin J, Lan L, Li N, Wang C, He P, Liu F, Ni H, Wang Y. M-CSF cooperating with NFkappaB induces macrophage transformation from M1 to M2 by upregulating c-Jun. Cancer Biol Ther 2014; 15:99-107; PMID:24100343; http://dx.doi.org/10.4161/cbt.26718
- Davis AS, Vergne I, Master SS, Kyei GB, Chua J, Deretic V. Mechanism of inducible nitric oxide synthase exclusion from mycobacterial phagosomes. PLoS Pathog 2007; 3: e186; PMID:18069890; http://dx.doi.org/10.1371/journal.ppat.0030186
- Cheng J, Huo DH, Kuang DM, Yang J, Zheng L, Zhuang SM. Human macrophages promote the motility and invasiveness of osteopontin-knockdown tumor cells. Cancer Res 2007; 67:5141-7; PMID:17545592; http://dx.doi.org/10.1158/0008-5472.CAN-06-4763
- Bian Y, Hall B, Sun ZJ, Molinolo A, Chen W, Gutkind JS, Waes CV, Kulkarni AB. Loss of TGF-beta signaling and PTEN promotes head and neck squamous cell carcinoma through cellular senescence evasion and cancer-related inflammation. Oncogene 2012; 31:3322-32; PMID:22037217; http://dx.doi.org/10.1038/onc.2011.494
- Molina JR, Agarwal NK, Morales FC, Hayashi Y, Aldape KD, Cote G, Georgescu MM. PTEN, NHERF1 and PHLPP form a tumor suppressor network that is disabled in glioblastoma. Oncogene 2012; 31:1264-74; PMID:21804599; http://dx.doi.org/10.1038/onc.2011.324
- Takahashi Y, Morales FC, Kreimann EL, Georgescu MM. PTEN tumor suppressor associates with NHERF proteins to attenuate PDGF receptor signaling. EMBO J 2006; 25:910-20; PMID:16456542; http://dx.doi.org/10.1038/sj.emboj.7600979
- Molina JR, Morales FC, Hayashi Y, Aldape KD, Georgescu MM. Loss of PTEN binding adapter protein NHERF1 from plasma membrane in glioblastoma contributes to PTEN inactivation. Cancer Res 2010; 70:6697-703; PMID:20736378; http://dx.doi.org/10.1158/0008-5472.CAN-10-1271
- Vazquez F, Grossman SR, Takahashi Y, Rokas MV, Nakamura N, Sellers WR. Phosphorylation of the PTEN tail acts as an inhibitory switch by preventing its recruitment into a protein complex. J Biol Chem 2001; 276:48627-30; PMID:11707428; http://dx.doi.org/10.1074/jbc.C100556200
- Gomes AM, Soares MV, Ribeiro P, Caldas J, Povoa V, Martins LR, Melão A, Serra-Caetano A, de Sousa AB, Lacerda JF, et al. Adult B-cell acute lymphoblastic leukemia cells display decreased PTEN activity and constitutive hyperactivation of PI3K/Akt pathway despite high PTEN protein levels. Haematologica 2014; 99:1062-8; PMID:24561792; http://dx.doi.org/10.3324/haematol.2013.096438
- Cambien B. Signal transduction involved in MCP-1-mediated monocytic transendothelial migration. Blood 2001; 97:359-66; PMID:11154209; http://dx.doi.org/10.1182/blood.V97.2.359
- Pedrero JM, Carracedo DG, Pinto CM, Zapatero AH, Rodrigo JP, Nieto CS, Gonzalez MV. Frequent genetic and biochemical alterations of the PI 3-K/AKT/PTEN pathway in head and neck squamous cell carcinoma. Int J Cancer J 2005; 114:242-8; PMID:15543611; http://dx.doi.org/10.1002/ijc.20711
- Schoenleber SJ, Kurtz DM, Talwalkar JA, Roberts LR, Gores GJ. Prognostic role of vascular endothelial growth factor in hepatocellular carcinoma: systematic review and meta-analysis. Br J Cancer 2009; 100:1385-92; PMID:19401698; http://dx.doi.org/10.1038/sj.bjc.6605017
- Georgescu MM. NHERF1: molecular brake on the PI3K pathway in breast cancer. Breast Cancer Res 2008; 10:106-7; PMID:18430260; http://dx.doi.org/10.1186/bcr1992
- Morales FC, Takahashi Y, Momin S, Adams H, Chen X, Georgescu MM. NHERF1/EBP50 head-to-tail intramolecular interaction masks association with PDZ domain ligands. Mol Cell Biol 2007; 27:2527-37; PMID:17242191; http://dx.doi.org/10.1128/MCB.01372-06
- Yang L, Wang Y, Chen P, Hu J, Xiong Y, Feng D, Liu H, Zhang H, Yang H, He J. Na(+)/H(+) exchanger regulatory factor 1 (NHERF1) is required for the estradiol-dependent increase of phosphatase and tensin homolog (PTEN) protein expression. Endocrinology 2011; 152:4537-49; PMID:21990315; http://dx.doi.org/10.1210/en.2011-1207
- Pan Y, Weinman EJ, Dai JL. Na+/H+ exchanger regulatory factor 1 inhibits platelet-derived growth factor signaling in breast cancer cells. Breast Cancer Res 2008; 10:R5; PMID:18190691; http://dx.doi.org/10.1186/bcr1846
- Georgescu MM, Morales FC, Molina JR, Hayashi Y. Roles of NHERF1/EBP50 in cancer. Curr Mol Med 2008; 8:459-68; PMID:18781953; http://dx.doi.org/10.2174/156652408785748031
- Cardone RA, Greco MR, Capulli M, Weinman EJ, Busco G, Bellizzi A, Casavola V, Antelmi E, Ambruosi B, Dell'Aquila ME, et al. NHERF1 acts as a molecular switch to program metastatic behavior and organotropism via its PDZ domains. Mol Biol Cell 2012; 23:2028-40; PMID:22496422; http://dx.doi.org/10.1091/mbc.E11-11-0911
- Li X, Xu Q, Wu Y, Qinqiao F. A CCL2/ROS autoregulation loop is critical for cancer associated fibroblasts-enhanced tumor growth of oral. Carcinogenesis 2014; 35:1362-70; PMID:24531940; http://dx.doi.org/10.1093/carcin/bgu046