
Abstract
Mutations in epidermal growth factor receptor (EGFR) rendering it constitutively active is one of the major causes for metastatic non-small-cell lung cancer (NSCLC), and EGFR-targeted therapies utilizing tyrosine kinase inhibitors (TKIs) are often used clinically as the first-line treatment. But approximately half of NSCLC patients develop resistance to these therapies, where the MET proto-oncogene is amplified by EGFR through the hypoxia-inducible factor (HIF)-1α. Here we report that endothelial PAS domain-containing protein 1 (EPAS1), with 48% sequence identity to HIF-1α, specifically binds to TKI-resistant T790M EGFR, but not to wild-type EGFR, in NSCLC cell lines. Expression of EPAS1 enhances amplification of MET when simultaneously expressed with T790M EGFR but not with wild-type EGFR, and this enhancement is independent of ligand binding domain of EGFR. MET amplification requires EPAS1, since EPAS1 knock-down reduced MET levels. When NSCLC cells expressing T790M EGFR were treated with TKIs, reduced EPAS1 levels significantly enhanced the drug effect, whereas over-expression of EPAS1 increased the drug resistant effect. This EPAS1-dependent TKI-resistance was abolished by knocking-down MET, suggesting that EPAS1 does not cause TKI-resistance itself but functions to bridge EGFR and MET interactions. Our findings suggest that EPAS1 is a key factor in the EGFR-MET crosstalk in conferring TKI-resistance in NSCLC cases, and could be used as a potential therapeutic target in TKI-resistant NSCLC patients.
Abbreviations
| EGFR | = | epidermal growth factor receptor |
| NSCLC | = | non-small-cell lung cancer |
| TKIs | = | tyrosine kinase inhibitors |
| EPAS1 | = | endothelial PAS domain-containing protein 1 |
| HIF)-1α. | = | hypoxia-inducible factor |
Introduction
Lung cancer is the leading cause of cancer death in the world, and non-small-cell lung cancer (NSCLC) accounts for the majority of lung tumor diagnoses and often presents as advanced or metastatic diseaseCitation1,2 Mutations in epidermal growth factor receptor (EGFR) that renders it constitutively active have been identified in a considerable percentage of NSCLC patients.Citation3-6.EGFR, also known as HER/ERBB, is a subclass of the receptor tyrosine kinase family and has 4 members: EGFR or HER1/ERBB1, HER2/ERBB2, HER3/ERBB3 and HER4/ERBB4. They are all comprised of an extracellular ligand binding domain, a transmembrane motif and a cytoplasmic tyrosine kinase domain.Citation7 Upon ligand binding, EGFR is activated and in turn stimulate many intracellular signaling pathways, including mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K)/AKT (protein kinase B) pathways, that significantly influence cell survival and proliferation.Citation8 Therefore instead of cytotoxic chemotherapy, EGFR tyrosine kinase inhibitors (TKIs) such as erlotinib and gefitinib, have been chosen clinically as the first-line treatment for NSCLC patients with activating EGFR mutations.Citation9-11 However, in approximately half of NSCLC cases, tumors developed EGFR-TKI resistance, and the T790M mutation in EGFR has been shown to be responsible,Citation12-14 but the exact reason for conferring the drug-resistance is not yet clear.
EGFR has been shown to frequently interact with the MET proto-oncogene, which is also a receptor tyrosine kinase and activates the same MAPK and PI3K/AKT downstream pathways as EGFR.Citation15,16 In NSCLC cells, MET activation promotes metastasis and invasive growth.Citation17,18.Recent reports have suggested an association between EGFR signaling pathway and MET activation.Citation19-21 Abnormal EGFR activation results in increased MET phosphorylation in thyroid carcinoma cells.Citation22 EGFR function is essential for cell invasiveness in MET-mediated colon cancer.Citation23 MET amplification also caused acquired resistance to EGFR TKIs.Citation24 The mechanisms for crosstalk between EGFR and MET remain largely elusive, except that EGFR regulates MET levels in NSCLC cells through the hypoxia-inducible factor (HIF)-1α pathway independently of hypoxia.Citation25
Here we have identified endothelial PAS domain-containing protein 1 (EPAS1), which has 48% sequence identity to HIF-1α, as a novel player mediating EGFR and MET interactions. We found that EPAS1 directly interacts with EGFR containing TKI-resistant T790M mutation, but not with wild-type EGFR, in NSCLC cell lines. EPAS1 and T790M EGFR interaction is sufficient to trigger amplification of MET signaling independent of EGF ligand binding. Moreover, TKI-resistance of NSCLC cells expressing T790M EGFR requires both EPAS1 and MET signaling pathway. We hypothesize EPAS1 plays a key role in regulating EGFR and MET crosstalk, and poses as a potential therapeutic target in TKI-resistant NSCLC cases.
Results
EPAS1 is a HIF-1α homolog and directly interacts with T790M EGFR in NSCLC cells
Previous report has demonstrated that HIF-1α is involved in the regulation of MET levels by EGFR.Citation25 To explore if there are other factors mediating EGFR-MET interaction, we carried out a genome-wide search for HIF-1α homologs and EPAS1 (NCBI Reference Sequence: NP_001421.2) was returned as the top hit with 48% amino acid sequence identity (). EPAS1 was first discovered to be a transcription factor of the basic helix-loop-helix domain family, and was highly expressed in vascularized organs including the lung.Citation26 When expressed in NSCLC cell line HCC827, EPAS1 localized mainly inside the nucleus, with lesser distribution throughout the cytoplasm (), suggesting besides the role of transcription factors, it performs other cellular functions.
Figure 1. EPAS1 is a HIF-1α homolog and interacts with T790M EGFR in HCC827 cells. (A) Sequence alignment of HIF-1α (top row) and EPAS1 (bottom row), identical amino acid highlighted in yellow. (B) Localization of EPAS1 in HCC827 cells. Top panel: HA-tagged EPAS1 (HA-EPAS1) expressed in HCC827 cells labeled with HA-probe (green) is concentrated in the nucleus marked with DAPI (blue) as well as in the cytoplasm. Bottom panel: HCC827 cells expressing control plasmid (HA) labeled the same as top panel. (C) EPAS1 interacts with T790M EGFR but not wild-type EGFR. HCC827 cells expressing HA-EPAS1, Myc-tagged wild-type EGFR (Myc-EGFR) and/or Myc-tagged T790M EGFR (Myc-T790M) in combination as indicated were subject to immunoprecipitation followed by western blot analysis using antibodies against either HA or Myc. Top panel: 1/10 of input. Middle panel: IP with anti-HA antibody. Bottom panel: IP with anti-Myc antibody.
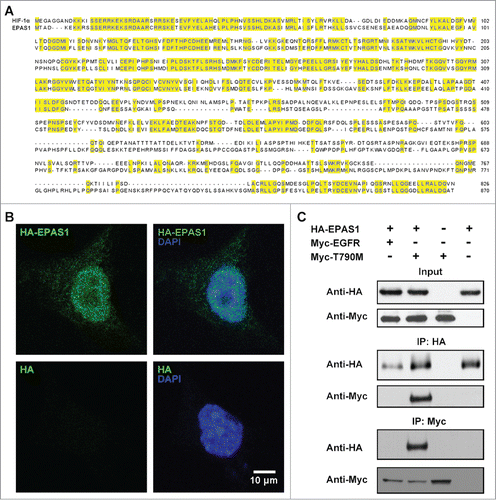
Due to its high resemblance to HIF-1α in sequence as well as tissue expression pattern,Citation26 we suspected EPAS1 may also function to regulate EGFR and MET interactions. To assess whether EPAS1 is directly involved in EGFR signaling pathway, we co-expressed HA-tagged EPAS1 (HA-EPAS1) and wild-type EGFR (Myc-EGFR) in HCC827 cells and performed co-immunoprecipitation assay using anti-HA as bait. No wild-type EGFR was detected in the precipitate (, middle panel, lane 1 from left). To our surprise, the EGFR variant (Myc-T790M), which contains the threonine 790 to methionine mutation responsible for adapted resistance in TKI therapies treating NSCLC, was pulled down together with EPAS1 (, middle panel, lane 2). This specific interaction between EPAS1 and T790M EGFR was validated to be specific, since Myc-T790M was only detected in the pull-down in the presence of HA-EPAS1 (, middle and bottom panel, lane 3), and vice versa (, middle and bottom panel, lane 4). To rule out the possibility whether this selective interaction between EPAS1 and T790M EGFR was a cell line specific effect, we did the same expression and co-immunoprecipitation assay in another NSCLC cell line A549 (Fig. S1). As expected, EPAS1 only interacted with T790M but not wild-type EGFR in A549 cells, meaning the binding between these 2 proteins is a bona fide interaction across different cell lines.
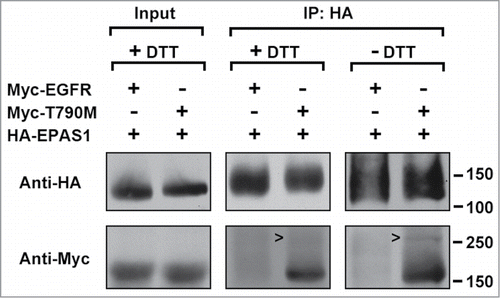
Next we investigated whether the interaction between EPAS1 and T790M EGFR was a direct binding or not, through protein crosslinking assay using dithio-bismaleimidoethane (DTME) as the crosslinker. HCC827 cells expressing HA-EPAS1 were also transfected with either wild-type or T790M EGFR and protein lysates were subjected to immunoprecipitation with HA antibody after the crosslinking. Same as the previous experiment, T790M but not wild-type EGFR was pull-down together with HA-EPAS1 (, middle panel, + DTT). Because DTME is a thiol-cleavable crosslinker, removing DTT from the sample loading buffer could preserve the covalent bond between crosslinked protein pairs, causing them to migrate slower in SDS-PAGE. Indeed at non-reducing conditions (- DTT), a band could be seen migrating around 250 kDa in the protein precipitate of T790M EGFR and HA-EPAS1 (, right panel, open arrow heads), but was absent from the equivalent lane at reducing condition (, middle panel, + DTT). Judged by its mobility this single band came from the direct crosslinking of HA-EPAS1 and T790M EGFR.
Figure 2. EPAS1 directly binds T790M EGFR in protein crosslinking assay. HCC827 cells co-expressing HA-EPAS1 with either wild-type (Myc-EGFR) or T790M (Myc-T790M) Myc-tagged EGFR were incubated with crosslinker DTME (see Materials and Methods) and subjected to immunoprecipitation using antibody against HA, followed by protein gel blot using antibodies against either HA (top row) or Myc (bottom row). Left panel shows input at reducing condition (+ DTT). Middle panel: immunoprecipitation with anti-HA, and proteins were eluted using SDS-PAGE sample buffer with DTT to cleave the DTME crosslinker. Right panel: immunoprecipitation with anti-HA, and proteins were eluted in the absence of DTT to maintain direct protein-protein crosslinking. Notice the open arrow heads pointing to a Myc-positive band migrating above the 250 kDa marker in the most right lane but missing from the middle panel.
EPAS1 and T790M EGFR interaction up-regulates MET pathway independent of EGF ligand binding
In NSCLC cases, aberrant activation of MET is the major cause for resistance to EGFR TKIs,Citation27 because MET shares the same downstream pathway as EGFR.Citation15,16 To test whether MET reacts to the interaction of EPAS1 and T790M EGFR, we expressed T790M EGFR and EPAS1 in HCC827 cells simultaneously and examined MET protein levels using anti-c-Met antibody. As previously reported, expression of wild-type and T790M EGFR alone was sufficient to trigger MET amplification,Citation25 even in the absence of EPAS1 (, lanes 1 and 2 from left). EPAS1 expression combined with wild-type EGFR had no further effects on the level of MET (, lane 3), however when EPAS1 was co-expressed with T790M EGFR, MET amplification was greatly enhanced (, lane 4, comparing with lane 2 and 3). In cells expressing just EPAS1, MET was only mildly activated (, lane 5, comparing with lane 2 and 4). These results have shown that EPAS1 and T790M EGFR interaction synergizes to up-regulate MET signaling pathway. The same results were also acquired from A549 cells (Fig. S2), again indicating this EPAS1 and T790M EGFR synergistic up-regulation of MET indeed reflected a general mechanism in NSCLC cells.
Figure 3. EPAS1 interaction with T790M EGFR up-regulates MET independent of ligand binding. (A) Co-expression of T790M but not wild-type EGFR with EPAS1 increased MET levels. Whole cell lysates from HCC827 cells expressing HA-EPAS1, Myc-tagged wild-type EGFR (Myc-EGFR) and/or Myc-tagged T790M EGFR (Myc-T790M) in combination as indicated were subject to western blot analysis, using antibodies against Myc, HA and MET as labeled on the left side of each panel. Anti-actin was used as loading control in the bottom panel. (B) MET amplification through EPAS1 and T790M EGFR interaction is independent of EGF ligand binding. T790M EGFR full length (Myc-T790M) and a truncated version lacking the ligand binding domain (ΔN-T790M) were expressed in HCC827 cells at the absence and presence of HA-EPAS1. Whole cell lysates were subject to protein gel blot analysis using antibodies Myc, HA and MET as labeled on left side of each panel. Full length (FL) and the truncated version of T790M (ΔN) EGFR migrate differently as indicated by open arrow heads on the right side of top panel. Anti-actin antibody was used as a loading control in the bottom panel.
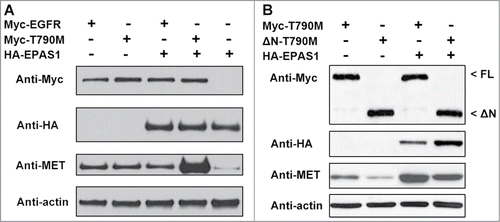
Although MET and EGFR both regulate MAPK and PI3K/AKT downstream pathways, the 2 tyrosine receptor kinases are activated by distinct ligands: hepatocyte growth factor (HGF) for MET and epidermal growth factor (EGF) for EGFR. Given the observation that EPAS1 is mainly expressed inside the cell and shows no localization to compartments of the secretory pathway ( and data not shown), as well as the fact that threonine 790 lies within the cytoplasmic domain,Citation28 the mechanism for EPAS1 binding to T790M EGFR to up-regulate MET is more likely to operate independently of the extracellular ligand binding domain. To test this hypothesis, we constructed a truncated version of T790M EGFR, where amino acids 2 to 620 coding for the ligand binding domain was deleted from its open reading frame, namely ΔN-T790M. When expressed in cells, ΔN-T790M migrated as a smaller protein band recognized by anti-Myc antibody in western blot, compared with the full length version Myc-T790M (, top panel, open arrow heads on the right side, comparing FL and ΔN). Both full length and truncated EGFR were still able to activate MET in the absence of EPAS1 (, lanes 1 and 2 from left). When EPAS1 is introduced into the cells, the levels of MET were again greatly up-regulated for both EGFR proteins (, lanes 3 and 4), with slightly less extent for ΔN-T790M compared with the full length version. Combined, the above results strongly suggest that EPAS1 and T790M EGFR interaction up-regulates MET pathway, through mechanism independent of EGF ligand binding.
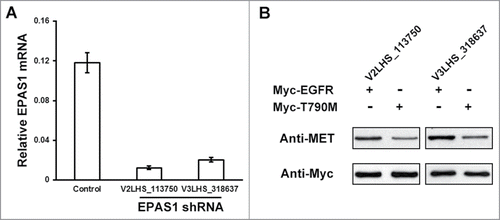
Next we asked the question whether EPAS1 expression at physiological condition is sufficient to function with T790M EGFR in amplifying MET, using small interfering RNA (siRNA) to knock down endogenous EPAS1 in HCC827 cells. Two individual siRNA targeting EPAS1, named V2LHS_113750 and V3LHS_318637, were introduced into the cells respectively, as well as a non-silencing control. The EPAS1 mRNA levels were measured by quantitative real-time PCR and normalized to GAPDH mRNA levels. As a result, both shRNAs were able to degrade EPAS1 expression significantly compared with non-silencing control (). In the absence of extra EGFR, we didn't observe any change in MET levels by knocking down of endogenous EPAS1 (data not shown), which is consistent with our result that EPAS1 alone has little effect on MET (, lane 5). However, MET levels was noticeably reduced when T790M EGFR was indeed introduced in HCC827 cells stably down-regulating endogenous EPAS1, compared that of wild-type EGFR expression (, comparing lanes 1 with 2 and 3 with 4, respectively). These results further indicate that interacting with TKI-resistant EGFR to amplify MET signaling is an intrinsic function of EPAS1 protein.
Figure 4. Endogenous EPAS1 is required for MET activation through T790M EGFR. (A) RNAi knockdown of endogenous EPAS1 expression. Lentiviral-encoded non-silencing control shRNA, and 2 independent shRNAs targeting EPAS1 (V2LHS_113750 and V3LHS_318637) were packaged and transduced into HCC827 cells, and then selected for puromycin resistance. Total RNA levels were extracted and analyzed by quantitative real-time PCR and mRNA levels of EPAS1 were normalized to GAPDH. Data shown were calculated from average of 3 individual experiments, and error bar represented standard error of the mean. (B) Endogenous EPAS1 regulates MET amplification through T790M EGFR. Wild-type (Myc-EGFR) and T790M (Myc-T790M) EGFR were expressed respectively in EPAS1 down-regulated HCC827 cells (V2LHS_113750 for left panel, V3LHS_318637 for right panel). MET protein levels were analyzed by western blot, and anti-Myc antibody was used as a loading control in the bottom panel.
EPAS1 mediates T790M EGFR and MET pathways to confer TKI-resistance in NSCLC cells
To assess whether interaction of EPAS1 to T790M EGFR has physiological functions in NSCLC, we performed MTS proliferation and growth assay in HCC827 cells co-expressing EPAS1 and T790M EGFR, by treating them with increasing concentrations of either gefitinib or erlotinib. Cells that express both EPAS1 and T790M exhibited EC50 (concentration of drug at which cell growth is inhibited by 50% of control) of gefitinib at over 10 μM (, blue line at top panel), and erlotinib at 5 μM (, blue line at bottom panel). As comparison, cells expressing T790M EGFR alone were evidently more sensitive to both TKIs, with EC50 of gefitinib at over 0.1 μM (, red line at top panel), and erlotinib at about 0.6 μM (, red line at bottom panel). This indeed indicated EPAS1 expression increased TKI-resistance in NSCLC cells expressing T790M EGFR. To validate this result we performed colony formation assay to assess the survival of NSCLC cells co-expressing T790M EGFR and EPAS1 with TKI treatment. We treated HCC827 cells with 0.1 μM gefitinib and 0.6 μM erlotinib respectively, and compared the colony numbers (Fig. S3A). As expected HCC827 cells co-expressing T790M EGFR and EPAS1 exhibited strong resistance to both TKIs, whereas cells expressing T790M EGFR alone were much more sensitive (Fig. S3B). Colony formation assay was also performed on A549 cells and generated the similar results (Fig. S4).
Figure 5. EPAS1 is necessary and sufficient to confer TKI resistance in HCC827 cells expressing T790M EGFR. (A) EPAS1 expression increased resistance to TKIs in HCC827 cells expressing T790M EGFR. Plasmid expressing EPAS1 (EPAS1 O/E, blue) or empty plasmid (red) were co-transfected with T790M EGFR into HCC827 cells. Twenty-four hours after transfection, the cells were subjected to MTS proliferation and growth assays as described in Materials and Methods in increasing concentrations of gefitinib (top panel) or erlotinib (bottom panel). (B) Knockdown of endogenous EPAS1 increased sensitivity of HCC827 cells expressing T790M EGFR to TKIs. HCC827 cells stably expressing shRNA V2LHS_113750 targeting EPAS1 (EPAS1 RNAi, blue) and non-silencing control (red) were transfected with T790M EGFR. Twenty-four hours after transfection the cells were subjected to MTS proliferation and growth assays as described in Materials and Methods in increasing concentrations of gefitinib (top panel) or erlotinib (bottom panel).
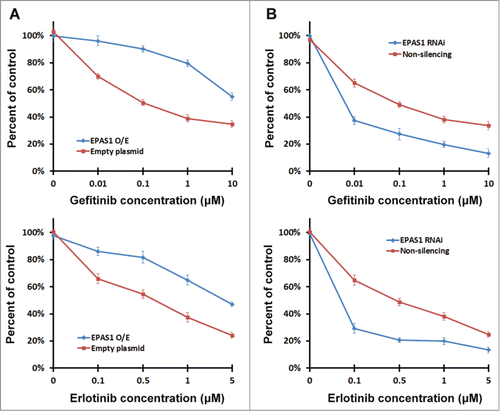
We also found that in cells with reduced levels of EPAS1, the results were similar. Cells transfected with T790M EGFR with physiological levels of EPAS1 (, non-silencing, red line) have EC50 of gefitinib at 0.1 μM (, red line at top panel) and EC50 of erlotinib at 0.5 μM (, red line at bottom panel). Whereas in cells lacking endogenous EPAS1, the EC50 for gefitinib was approximately 0.007 μM and for erlotinib was about 0.08 μM (, EPAS1 RNAi, blue line), indicating enhanced sensitivity to TKI treatments. Together these results show that EPAS1 functions to confer TKI-resistance through T790M EGFR in NSCLC cells.
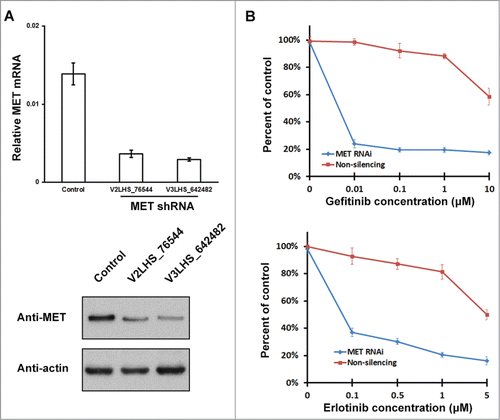
Although binding of EPAS1 to T790M activated MET pathway (), the enhanced TKI-resistance we observed above could still be explained as a direct result of EPAS1 expression independent of MET activation. In order to rule out this possibility, we knocked down endogenous MET expression by RNAi and repeated the MTS proliferation and growth assay. Two shRNAs targeting MET were tested, both considerably reduced MET messenger and protein levels (), and V3LHS_642482 was used in the following experiment. As expected, growth of cells stably down-regulating MET were severely inhibited by both gefitinib and erlotinib, with their EC50 at 0.005 μM and 0.09 μM respectively (, blue lines in both panels). While in non-silencing controls, EPAS1 and T790M EGFR interaction has given cells very strong TKI-resistance, with estimated EC50 for both TKIs beyond the highest concentration used in the assay (, red lines in both panels). Therefore, the function of EPAS1 interacting with T790M to confer resistance to TKIs is through the activation of MET pathway rather than expression of EPAS1 itself.
Figure 6. EPAS1 mediated TKI-resistance requires MET function. (A) RNAi knockdown of endogenous MET expression. Lentiviral-encoded non-silencing control shRNA, and 2 independent shRNAs targeting MET (V2LHS_76544 and V3LHS_642482) were packaged and transduced into HCC827 cells, and then selected for puromycin resistance. Levels of MET mRNA and protein were analyzed by quantitative real-time PCR (top panel) and protein gel blot (bottom panel). mRNA levels were normalized to GAPDH, and calculated from average of 3 individual experiments with standard error of the mean shown. Anti-actin antibody was used as a loading control in the bottom panel. The cell line transduced with non-silencing control and V3LHS_642482 was used for subsequent MTS proliferation and growth assay. (B) Knockdown of MET reduced TKI-resistance in HCC827 cells expressing EPAS1 and T790M EGFR. HCC827 cells stably knocking-down MET (MET RNAi, blue) and non-silencing control (red) were co-transfected with EPAS1 and T790M EGFR, and subject to MTS proliferation and growth assay in increasing concentrations of gefitinib (top panel) or erlotinib (bottom panel).
Discussion
EPAS1 was first discovered as a basic helix-loop-helix/PAS domain transcription factor and is almost ubiquitously expressed in tissues including the lung.Citation26 Loss of EPAS1 resulted in disturbed catecholamine homeostasis and heart failure during embryonic development.Citation29 It was also essential for spermatogenesis in postnatal testis in mice.Citation30 We have identified EPAS1 as novel factor mediating the crosstalk between EGFR and MET pathways, both of which are involved in NSCLC.Citation3-6,17,18 Our result is the first demonstration of EPAS1 involvement in NSCLC. Nevertheless, the interplay between EGFR and MET pathways to confer drug resistance is not limited in NSCLC. Overexpression of the EGFR ligand transforming growth factor-α (TGF-α) leads to MET activation and cetuximab resistance.Citation31 MET amplification was also the cause of resistance for colorectal cancer patients treated with anti-EGFR antibody therapy.Citation32 These observations underlie the key role of EGFR and MET in promoting resilient tumor growth and it will be interesting to check whether EPAS1 also plays similar role in other types of cancers.
EPAS1 directly binds to EGFR harboring the T790M mutation (), an acquired secondary mutation that resulted in resistance to EGFR TKIs in about 50% of NSCLC patients, who initially responded to the therapy.Citation12-14 T790M mutation was also found to increase the activity of EGFR itself,Citation33 and some NSCLC patients who never received TKI treatments had spontaneous T790M mutation.Citation34 This could be explained by our finding that the direct binding of EPAS1 to T790M EGFR could significantly elevate MET levels (), which could intensify the same downstream pathways shared with EGFR.Citation15,16 But the exact molecular mechanism for which EPAS1 directs T790M EGFR and MET pathway is unclear, which could probably be revealed by mapping the interaction domain on EPAS1 to T790M EGFR. In a recent report, Engelman et al. induced T790M into H3255 and H1975 cells, which didn't harbor the mutation initially.Citation35 As a result, these cell lines with acquired T790M exhibited greatly elevated resistance to not only gefitinib but other EGFR inhibitors. Our similar findings in both HCC827 and A549 suggested that EPAS1 interacts with T790M EGFR to confer TKI resistance could reflect a more generalized mechanism in NSCLC.
One observation worth pointing out is the interaction between EPAS1 and T790M EGFR persisted even in the absence of EGF ligand binding domain (). EGFR cytoplasmic domain consists of the juxtamembrane domain, tyrosine kinase domain (TKD) spanning from glutamate 685 to isoleucine 953, followed by the regulatory region.Citation28 The EGFR-TKD is likely to be the major site for TKI-resistance related mechanism to operate, because not only T790M mutation lies within this region, but in fact gefitinib and erlotinib, both EGFR specific TKIs used in this study, bind to the TKD on EGFR.Citation36,37 Gefitinib and erlotinib both exhibit much higher affinity to the active conformation of the EGFR-TKD over the inactive form. We propose that T790M mutation causes the EGFR-TKD to assume an intermediate conformation that favors the kinase activity but reduces affinity to the TKIs, giving the NSCLC cells resistance to the treatment. This conformational change could also explain the adapted binding of EPAS1 to EGFR. Crystallographic studies on T790M EGFR with TKIs would provide definitive answers to these questions.
Several studies were performed on the synergizing effect of EGFR and c-Met inhibitors on NSCLC cell lines. Using MTT assays, Puri et al. found in NSCLC cell lines that IC50 of EGFR inhibitor Tyrphostin to be 1 μM and IC50 of c-Met inhibitor SU11274 to be 4 μM respectively, and a combination of 0.5 μM Tyrphostin and 2 μM SU11274 inhibited cell growth by 65.2%, clearly indicating a synergistic effect.Citation38 Combination of another EGFR inhibitor gefitinib and SU11274 also exhibited synergistic effect in the same study. In a similar study, Nakagawa et al. also measured the growth of several NSCLC cell lines when subjected to EGFR-T790M inhibitor WZ4002 and c-Met inhibitor E7050 using MTT assays.Citation39 Separately, E7050 exhibited little inhibitory effect on cell proliferation, and WZ4002 was only able to sensitize NSCLC cells in the absence of c-Met overexpression. Whereas WZ4002 and E7050 combined exhibited strong synergistic effect to inhibit cell growth, giving IC50 far less than 1 μM in all NSCLC cell lines examined. Almost identical synergistic effect of EGFR and c-Met inhibitors were reported in mouse model of lung cancer.Citation40 EGFR-T790M inhibitor WZ4002 and c-Met inhibitor crizotinib were administered separately and combined to EGFR/c-Met-expressing or EGFR-only mice. WZ4002 was only able to suppress tumor growth in mice without c-Met transgene, and crizotinib had no effect in either transgenic mice. Whereas combination therapy of WZ4002 and crizotinib showed strong tumor regression in both EGFR/c-Met and EGFR-only transgenic mice. Together these studies all clearly demonstrated that EGFR and c-Met inhibitors are able to synergize in inhibiting cancer cell proliferation and tumor growth. The reason behind this synergy of 2 kinds of TKIs is that EGFR and c-Met pathways crosstalk in promoting lung cancer, therefore both of these 2 pathways need to be repressed simultaneously to achieve the maximum therapeutic outcome.
In summary the significance of our study is that we have identified EPAS1 as a direct interactor of EGFR-T790M, and the binding of EPAS1 to EGFR-T790M activates c-Met. Our finding provides the mechanistic link between EGFR-T790M TKI-resistant mutant and c-Met activation, therefore could serve as potential therapeutic target especially in TKI-resistant NSCLC cases. By further understanding the molecular machinery responsible for EPAS1 affinity to EGFR-T790M, drugs could be designed to inhibit the specific process where EPAS1 mediates EGFR and MET pathways, providing new therapeutic target in NSCLC cases.
Materials and Methods
Cell culture and antibodies
NSCLC HCC827 and A549 cells were obtained from the American Type Culture Collection (Manassas) and passaged for less than 6 months after receipt for completion of the studies. Cells were maintained in DMEM containing 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM L-glutamine at 37°C in 5% CO2, supplemented with assay specific reagents. Mouse monoclonal HA-probe (F-7), rabbit anti-c-Myc (C-19) and rabbit anti-c-Met (C-28) antibodies were purchased from Santa Cruz Biotechnology Inc.. Rabbit anti-actin antibody (ab1801) was purchased from Abcam. Alexa Fluor 488 goat anti-mouse, HRP-linked anti-rabbit and anti-mouse secondary antibodies were purchased from Life Technologies.
Plasmids
EPAS1 cDNA coding region (NCBI Reference Sequence: NM_001430.4) was cloned into the pCMV-HA vector with an HA-tag in the N-terminal. The human EGFR cDNA coding region (GenBank accession no. X00588.1) and the ligand binding domain (amino acid 2–620) deletion mutant EGFR were cloned into the pcDNA3.1 vector with a Myc-tag in the N-terminal. The EGFR T790M missense mutation was constructed with Quick Change Site-Directed Mutagenesis kit (Stratagene) using primers 5′- CTCACCTCCACCGTGCAACTCATCATGCAGCTCATGCCCTTCGGCTGCCTC-3′ according to manufacturer's instructions.
Immunofluorescence and microscopy
HCC827 cells transfected with HA-tagged EPAS1 were grown on glass coverslips and fixed in 3% formaldehyde for 10 min, followed by permeabilization using 0.2% Triton X-100 for 5 min. After fixation and permeabilization, cells were labeled with blocking buffer (0.2% fish gelatin) with mouse monoclonal HA-probe as primary antibody and Alexa Fluor 488 goat anti-mouse as secondary antibody for 20 min at room temperature. DAPI was used to visualize DNA. Cells were washed 3 times with PBS after each antibody labeling step. Coverslips were mounted using ProLong Gold Antifade reagent (Life Technologies) and visualized using Zeiss LSM 510 META inverted microscope (Carl Zeiss MicroImaging) with a Plan-Apochromat 100ċ Ph3 objective and standard PMT. Images were archived using LSM 5 Image Examiner (Carl Zeiss MicroImaging) and Adobe Photoshop 7.0 (Adobe Systems).
Immunoprecipitation and immunoblotting
Protein lysates were extracted using RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, and protease inhibitors). Lysates were centrifuged at 16,000 g for 10 min at 4°C. The supernatants were rotated for 2 hours at 4°C with Protein A Sepharose beads (Sigma-Aldrich) coupled to anti-HA or anti-Myc antibodies. After incubation samples were thoroughly washed 5 times with lysis buffers and then twice with a final wash buffer (50 mM Tris, pH 7.5, and 50 mM NaCl). Proteins were eluted by adding SDS-PAGE sample buffer followed by boiling at 98°C for 5 min and were analyzed by SDS-PAGE and transferred to nitrocellulose membrane using semi-dry transfer unit (Bio-Rad Laboratories). The membrane was then immersed in blocking buffer (PBS, 0.1% Tween-20) with 5% nonfat milk for 20 min and then incubated with appropriate primary antibody overnight at 4°C. After washes with blocking buffer, blots were incubated with HRP-conjugated secondary antibody (Life Technologies) for 20 min, washed again with blocking buffer, and visualized using Luminata Forte Western HRP Substrate (EMD Millipore).
Protein crosslinking assay
HCC827 cells co-transfected with EPAS1 with either wild-type or T790M EGFR were collected by centrifugation at 800 g for 10 min at 4°C and resuspended in PBS buffer. Crosslinker dithio-bismaleimidoethane (Thermo Scientific) was dissolved in DMSO immediately prior to use, and added to the cell suspension at a final concentration of 0.1 mM, followed by incubation for 2 hours at 4°C. Protein lysates were extracted using RIPA buffer, and subjected to immunoprecipitation. Proteins were eluted by adding SDS-PAGE sample buffer with or without DTT and analyzed by SDS-PAGE, followed by protein gel blot analysis.
RNA interference (RNAi) methods and Quantitative real-time PCR
For RNAi knock-down experiments, lentiviral shRNA constructs V2LHS_113750 and V3LHS_318637 targeting EPAS1, and V2LHS_76544 and V3LHS_642482 targeting MET, as well as GIPZ Non-silencing Lentiviral shRNA Control were purchased from GE Dharmacon. Cells were transduced with the packaged virus and subsequently maintained with 1 µg/ml puromycin for stable knockdown. To assess the efficiency of RNAi assay, total RNA was isolated from cells using the RNeasy MiniPrep Kit (Qiagen). One μg of total RNA was reverse-transcribed with Superscript II First-Strand Synthesis kit (Life Technologies) as recommended by the manufacturer. GAPDH mRNA levels were measured for normalization and all data were presented as relative expression.
Cell proliferation, growth and survival assays
Growth and inhibition of growth were assessed by 3-(4, 5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay according to previously described methods.Citation41 The cells were treated with TKIs for 72 hours, and the number of cells was scored as percentage relative to mock treatment. Colony formation assay was performed according to previously described methods.Citation42 Each concentration of drug was performed 5 times. All data were presented as mean ± SEM.
Statistical analysis
All data were presented as mean ± SEM. Two-tailed student's t-test was used to establish significant differences between groups. Data were determined to be statistically different when P < 0.05.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary Material
Download Zip (586.3 KB)Acknowledgments
The authors thank professor Jia-bao Liu for his professional suggestions and the center lab of Shijiazhuang No.1 Hospital and Research Center of Hebei Medical University for their help in the experiments.
Funding
This work was supported by the Priority Academic Program Development of Hebei General Higher Education Institutions (No. 2005–52).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clini 2013; 63:11–30; PMID:23335087; http://dx.doi.org/10.3322/caac.21166
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clini 2011; 61:69–90; PMID:21296855.
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004; 350:2129–39; PMID:15118073; http://dx.doi.org/10.1056/NEJMoa040938.
- Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004; 304:1497–500; PMID:15118125; http://dx.doi.org/10.1126/science.1099314.
- Janne PA, Engelman JA, Johnson BE. Epidermal growth factor receptor mutations in non-small-cell lung cancer: implications for treatment and tumor biology. J Clini Oncol 2005; 23:3227–34; PMID:15886310; http://dx.doi.org/10.1200/JCO.2005.09.985.
- Soria JC, Mok TS, Cappuzzo F, Janne PA. EGFR-mutated oncogene-addicted non-small cell lung cancer: current trends and future prospects. Cancer Treat Rev 2012; 38:416–30; PMID:22119437; http://dx.doi.org/10.1016/j.ctrv.2011.10.003.
- Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer 2005; 5:341–54; PMID:15864276; http://dx.doi.org/10.1038/nrc1609.
- Pao W, Miller VA. Epidermal growth factor receptor mutations, small-molecule kinase inhibitors, and non-small-cell lung cancer: current knowledge and future directions. J Clini Oncol 2005; 23:2556–68; PMID:15767641; http://dx.doi.org/10.1200/JCO.2005.07.799.
- Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, Campos D, Maoleekoonpiroj S, Smylie M, Martins R, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med 2005; 353:123–32; PMID:16014882; http://dx.doi.org/10.1056/NEJMoa050753.
- Kris MG, Natale RB, Herbst RS, Lynch TJ, Jr., Prager D, Belani CP, Schiller JH, Kelly K, Spiridonidis H, Sandler A, et al. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. Jama 2003; 290:2149–58; PMID:14570950; http://dx.doi.org/10.1001/jama.290.16.2149.
- Thatcher N, Chang A, Parikh P, Rodrigues Pereira J, Ciuleanu T, von Pawel J, Thongprasert S, Tan EH, Pemberton K, Archer V, et al. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer). Lancet 2005; 366:1527–37; PMID:16257339; http://dx.doi.org/10.1016/S0140-6736(05)67625-8.
- Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2005; 2:e73; PMID:15737014; http://dx.doi.org/10.1371/journal.pmed.0020073.
- Kwak EL, Sordella R, Bell DW, Godin-Heymann N, Okimoto RA, Brannigan BW, Harris PL, Driscoll DR, Fidias P, Lynch TJ, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci U S A 2005; 102:7665–70; PMID:15897464; http://dx.doi.org/10.1073/pnas.0502860102.
- Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med 2005; 352:786–92; PMID:15728811; http://dx.doi.org/10.1056/NEJMoa044238.
- Ponzetto C, Bardelli A, Zhen Z, Maina F, dalla Zonca P, Giordano S, Graziani A, Panayotou G, Comoglio PM. A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell 1994; 77:261–71; PMID:7513258; http://dx.doi.org/10.1016/0092-8674(94)90318-2.
- Sipeki S, Bander E, Buday L, Farkas G, Bacsy E, Ways DK, Faragó A. Phosphatidylinositol 3-kinase contributes to Erk1/Erk2 MAP kinase activation associated with hepatocyte growth factor-induced cell scattering. Cell Signal 1999; 11:885–90; PMID:10659996; http://dx.doi.org/10.1016/S0898-6568(99)00060-1.
- Qiao H, Hung W, Tremblay E, Wojcik J, Gui J, Ho J, Klassen J, Campling B, Elliott B. Constitutive activation of met kinase in non-small-cell lung carcinomas correlates with anchorage-independent cell survival. J Cell Biochem 2002; 86:665–77; PMID:12210733; http://dx.doi.org/10.1002/jcb.10239.
- Bredin CG, Liu Z, Klominek J. Growth factor-enhanced expression and activity of matrix metalloproteases in human non-small cell lung cancer cell lines. Anticancer Res 2003; 23:4877–84; PMID:14981939.
- Jo M, Stolz DB, Esplen JE, Dorko K, Michalopoulos GK, Strom SC. Cross-talk between epidermal growth factor receptor and c-Met signal pathways in transformed cells. J Biol Chem 2000; 275:8806–11; PMID:10722725; http://dx.doi.org/10.1074/jbc.275.12.8806.
- Huang PH, Mukasa A, Bonavia R, Flynn RA, Brewer ZE, Cavenee WK, Furnari FB, White FM. Quantitative analysis of EGFRvIII cellular signaling networks reveals a combinatorial therapeutic strategy for glioblastoma. Proc Natl Acad Sci U S A 2007; 104:12867–72; PMID:17646646; http://dx.doi.org/10.1073/pnas.0705158104.
- Guo A, Villen J, Kornhauser J, Lee KA, Stokes MP, Rikova K, Possemato A, Nardone J, Innocenti G, Wetzel R, et al. Signaling networks assembled by oncogenic EGFR and c-Met. Proc Natl Acad Sci U S A 2008; 105:692–7; PMID:18180459; http://dx.doi.org/10.1073/pnas.0707270105.
- Bergstrom JD, Westermark B, Heldin NE. Epidermal growth factor receptor signaling activates met in human anaplastic thyroid carcinoma cells. Experimental cell research 2000; 259:293–9; PMID:10942601; http://dx.doi.org/10.1006/excr.2000.4967.
- Pai R, Nakamura T, Moon WS, Tarnawski AS. Prostaglandins promote colon cancer cell invasion; signaling by cross-talk between two distinct growth factor receptors. FASEB J 2003; 17:1640–7; PMID:12958170; http://dx.doi.org/10.1096/fj.02-1011com.
- Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007; 316:1039–43; PMID:17463250; http://dx.doi.org/10.1126/science.1141478.
- Xu L, Nilsson MB, Saintigny P, Cascone T, Herynk MH, Du Z, Nikolinakos PG, Yang Y, Prudkin L, Liu D, et al. Epidermal growth factor receptor regulates MET levels and invasiveness through hypoxia-inducible factor-1alpha in non-small cell lung cancer cells. Oncogene 2010; 29:2616–27; PMID:20154724; http://dx.doi.org/10.1038/onc.2010.16.
- Tian H, McKnight SL, Russell DW. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev 1997; 11:72–82; PMID:9000051; http://dx.doi.org/10.1101/gad.11.1.72.
- Smyth EC, Sclafani F, Cunningham D. Emerging molecular targets in oncology: clinical potential of MET/hepatocyte growth-factor inhibitors. OncoTargets and therapy 2014; 7:1001–14; PMID:24959087; http://dx.doi.org/10.2147/OTT.S44941.
- Burgess AW, Cho HS, Eigenbrot C, Ferguson KM, Garrett TP, Leahy DJ, Lemmon MA, Sliwkowski MX, Ward CW, Yokoyama S. An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol Cell 2003; 12:541–52; PMID:14527402; http://dx.doi.org/10.1016/S1097-2765(03)00350-2.
- Tian H, Hammer RE, Matsumoto AM, Russell DW, McKnight SL. The hypoxia-responsive transcription factor EPAS1 is essential for catecholamine homeostasis and protection against heart failure during embryonic development. Genes Dev 1998; 12:3320–4; PMID:9808618; http://dx.doi.org/10.1101/gad.12.21.3320.
- Gruber M, Mathew LK, Runge AC, Garcia JA, Simon MC. EPAS1 Is Required for Spermatogenesis in the Postnatal Mouse Testis. Biol Reprod 2010; 82:1227–36; PMID:20181618; http://dx.doi.org/10.1095/biolreprod.109.079202.
- Troiani T, Martinelli E, Napolitano S, Vitagliano D, Ciuffreda LP, Costantino S, Morgillo F, Capasso A, Sforza V, Nappi A, et al. Increased TGF-α as a mechanism of acquired resistance to the anti-EGFR inhibitor cetuximab through EGFR-MET interaction and activation of MET signaling in colon cancer cells. Clini Cancer Res 2013; 19:6751–65; PMID:24122793; http://dx.doi.org/10.1158/1078-0432.CCR-13-0423.
- Bardelli A, Corso S, Bertotti A, Hobor S, Valtorta E, Siravegna G, Sartore-Bianchi A, Scala E, Cassingena A, Zecchin D, et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov 2013; 3:658–73; PMID:23729478; http://dx.doi.org/10.1158/2159-8290.CD-12-0558.
- Bell DW, Gore I, Okimoto RA, Godin-Heymann N, Sordella R, Mulloy R, Sharma SV, Brannigan BW, Mohapatra G, Settleman J, et al. Inherited susceptibility to lung cancer may be associated with the T790M drug resistance mutation in EGFR. Nature genetics 2005; 37:1315–6; PMID:16258541; http://dx.doi.org/10.1038/ng1671.
- Toyooka S, Kiura K, Mitsudomi T. EGFR mutation and response of lung cancer to gefitinib. N Engl J Med 2005; 352:2136; author reply; PMID:15901872; http://dx.doi.org/10.1056/NEJM200505193522019.
- Engelman JA, Mukohara T, Zejnullahu K, Lifshits E, Borras AM, Gale CM, Naumov GN, Yeap BY, Jarrell E, Sun J, et al. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J Clini Invest 2006; 116:2695–706; PMID:16906227; http://dx.doi.org/10.1172/JCI28656.
- Yun CH, Boggon TJ, Li Y, Woo MS, Greulich H, Meyerson M, Eck MJ. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell 2007; 11:217–27; PMID:17349580; http://dx.doi.org/10.1016/j.ccr.2006.12.017.
- Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem 2002; 277:46265–72.; PMID:12196540; http://dx.doi.org/10.1074/jbc.M207135200.
- Puri N, Salgia R. Synergism of EGFR and c-Met pathways, cross-talk and inhibition, in non-small cell lung cancer. J Carcinog 2008; 7:9; PMID:19240370; http://dx.doi.org/10.4103/1477-3163.44372.
- Nakagawa T, Takeuchi S, Yamada T, Nanjo S, Ishikawa D, Sano T, Kita K, Nakamura T, Matsumoto K, Suda K, et al. Combined therapy with mutant-selective EGFR inhibitor and Met kinase inhibitor for overcoming erlotinib resistance in EGFR-mutant lung cancer. Mol Cancer Ther 2012; 11:2149–57; PMID:22844075; http://dx.doi.org/10.1158/1535-7163.MCT-12-0195.
- Xu L, Kikuchi E, Xu C, Ebi H, Ercan D, Cheng KA, Padera R, Engelman JA, Jänne PA, Shapiro GI, et al. Combined EGFR/MET or EGFR/HSP90 inhibition is effective in the treatment of lung cancers codriven by mutant EGFR containing T790M and MET. Cancer Res 2012; 72:3302–11; PMID:22552292; http://dx.doi.org/10.1158/0008-5472.CAN-11-3720.
- Tracy S, Mukohara T, Hansen M, Meyerson M, Johnson BE, Janne PA. Gefitinib induces apoptosis in the EGFRL858R non-small-cell lung cancer cell line H3255. Cancer Res 2004; 64:7241–4; PMID:15492241; http://dx.doi.org/10.1158/0008-5472.CAN-04-1905.
- Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nature Protoc 2006; 1:2315–9; PMID:17406473; http://dx.doi.org/10.1038/nprot.2006.339.