
Abstract
Accumulating evidence suggests that changes in methylation patterns may help mediate the sensitivity or resistance of cancer cells to ionizing radiation. The present study provides evidence for the involvement of radioresistance-induced DNA methylation changes in tumor radioresistance. We established radioresistant laryngeal cancer cells via long-term fractionated irradiation, and examined differences in DNA methylation between control and radioresistant laryngeal cancer cells. Interestingly, we found that the promoter-CpG islands of 5 previously identified radioresistance-related genes (TOPO2A, PLXDC2, ETNK2, GFI1, and IL12B) were significantly altered in the radioresistant laryngeal cancer cells. Furthermore, the demethylation of these gene promoters with a DNA methyltransferase inhibitor (5-aza-2'-deoxycytidine) increased their transcription levels. Treatment with 5-aza-2'-deoxycytidine also sensitized the radioresistant laryngeal cancer cells to irradiation, indicating that changes in DNA methylation contributed to their radioresistance. Of the tested genes, the expression and activity levels of TOPO2A were tightly associated with the radioresistant phenotype in our system, suggesting that the hypermethylation of TOPO2A might be involved in this radioresistance. Collectively, our data suggest that radiation-induced epigenetic changes can modulate the radioresistance of laryngeal cancer cells, and thus may prove useful as prognostic indicators for radiotherapy.
Abbreviations
| DNMT | = | DNA methyltransferases |
| 5-Aza | = | 5-aza-2′-deoxycytidine |
| TOPO2A | = | Topoisomerase II α |
| Hep-2 | = | Human laryngeal squamous cell carcinoma |
| RR-Hep-2 | = | Radioresistant Hep-2 |
| HRP | = | Horseradish peroxidase |
| RT-PCR | = | Reverse Transcription-polymerase Chain Reaction |
| PI | = | Propidium iodide |
| BSA | = | Bovine Serum Albumin |
Introduction
Epigenetics is the study of heritable changes in gene expression that take place without alterations in the DNA sequence.Citation1 One of the best-studied epigenetic phenomena is the methylation of cytosine residues located within CpG dinucleotides.Citation2-5 Three DNA methyltransferases (DNMTs; DNMT1, DNMT3a and DNMT3b) can add methyl groups to the 5' cytosine of a CpG dinucleotide,Citation6 aberrant DNA methylation is found in 2 distinct forms: hypermethylation and hypomethylation. Previous studies have unequivocally demonstrated that DNA hypermethylation typically occurs at CpG islands and many cytosine-guanine-rich regions, and is associated with gene silencing. For example, genes with high levels of methylcytosine in their promoter regions are usually transcriptionally silent,Citation7 and DNA hypermethylation of CpG islands in the promoter and first exon is critical to the functional suppression of genes associated with cancer development.Citation5,8-11 Conversely, aberrant promoter hypermethylation of tumor-suppressor genes is a hallmark of cancer and is thought to contribute to carcinogenesis.Citation12,13
The existing data suggest that aberrant DNA methylation is an attractive mechanism for the propagation of genomic instability following ionizing radiation.Citation14 For almost 100 years, radiotherapy with ionizing radiation (either alone or in combination with chemotherapy) has been widely used to treat cancer; however, the curative effect of clinical radiotherapy is limited by injury to normal tissues and the radioresistance of some human tumors.Citation15,16 Multiple studies have identified aberrant DNA methylation patterns following the exposure of plants, rodents, and rodent or human cell lines to ionizing radiation. Furthermore, post-irradiation changes in DNA methylation have been suggested to correlate with the initiation of radioresistance.Citation1
Laryngeal cancer is generally treated by radiotherapy in conjunction with surgery and/or chemotherapy,Citation4 but tumor cells can acquire radioresistance during clinical radiotherapy based on fractionated radiation treatment.Citation13,14 However, while we have a partial understanding of the mechanisms underlying cellular radioresistance, the molecular events and markers underlying the acquired radioresistance of laryngeal cancer remain to be clarified.Citation17-20
We previously used fractionated irradiation to establish radioresistant laryngeal cancer cells (RR-Hep-2 cells) as a model system for studying the radioresistant phenotype. Using these radioresistant cells, we systematically screened a laryngeal cancer expressed sequence tag (EST) database to identify gene profiles associated with tumor radioresistance,Citation17,21-23 and then used an in silico analysis to confirm several radioresistance-related genes. Here, we used a high-throughput technique for methylation analysis (pyrosequencing)Citation24 to further investigate the methylation status of the promoter regions of these genes in control and RR-Hep-2 cells. Our results revealed that RR-Hep-2 cells had higher levels of CpG methylation in the promoters of the genes encoding ethanolamine kinase 2 (ETNK2), growth factor independent 1 transcription repressor (GFI1), interleukin 12B (IL12B), plexin domain containing 2 (PLXDC2), and topoisomerase II α (TOPO2A). For TOPO2A, the average extent of methylation was ∼5.9 times higher in RR-Hep-2 than control Hep-2 cells, and methylation was positively associated with cellular radioresistance. Moreover, treatment with the DNA methyltransferase inhibitor, 5-aza-2′-deoxycytidine (5-Aza), which triggered demethylation of the 5 gene promoters in question, was associated with increased mRNA expression of the encoded genes, as well as enhanced radiosensitivity among treated RR-Hep-2 cells. Together, these findings strongly suggest that radiation-induced DNA promoter hypermethylation can play a role in the radioresistance of laryngeal cancer cells, and thus may be useful as a prognostic and/or therapeutic indicator for radiotherapy in this and other cancers.
Results
Radioresistant laryngeal cancer cells show altered expression of DNA methyltransferases
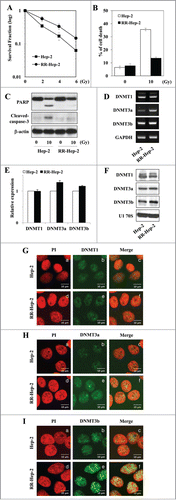
We used long-term fractionated irradiation to establish radioresistant laryngeal cancer Hep-2 cells (RR-Hep-2 cells; for details, see the Materials and Methods section). We observed increased survival () and decreased radiation-induced cell death () among RR-Hep-2 cells compared with parental Hep-2 cells, confirming the radioresistant phenotype. To analyze whether this acquired radioresistance was associated with epigenetic alterations, we examined the expression levels of the cellular maintenance (DNMT1) and de novo (DNMT3a and DNMT3b) DNA methyltransferases. These enzymes are responsible for maintaining DNA methylation patterns in the mammalian genome, and dysregulation of their expression and/or activity has been associated with altered DNA methylation.Citation25,26 As shown in , RT-PCR analysis showed that the DNMT3a and DNMT3b mRNA were more highly expressed in RR-Hep-2 cells than in Hep-2 cells. In addition, western blot () and immunofluorescence analyses indicated that DNMT3a () and DNMT3b () were increased in RR-Hep-2 cells compared to Hep-2 cells. Taken together, our data suggest that DNA methylation may be altered in our radioresistant laryngeal cancer cell model.
Figure 1. Analysis of survival and DNMT expression in our radioresistant Hep-2 human laryngeal cancer cell line (RR-Hep-2). (A) Clonogenic survival fractions of parental Hep-2 and RR-Hep-2 cells were determined following exposure to the indicated doses of radiation. Parental Hep-2 and RR-Hep-2 cells were treated with or without 10 Gy radiation and then cultured for 24 h. (B) Cell viability was determined with the FACScan flow cytometer and data are presented as the percentage of PtdIns-positive cells. (C) The protein levels of cleaved PARP and cleaved caspase-3 were determined by Western blotting; β-actin was employed as the loading control. (D) The transcript levels of DNMT1, DNMT3a and DNMT3b were determined by conventional RT-PCR and (E) quantified by real time PCR. GAPDH was used as the loading or internal control. (F) The protein levels of DNMT1, DNMT3a and DNMT3b were determined by Western blotting; U1 70S was employed as the loading control. (G-I) The localizations of (G) DNMT1, (H) DNMT3a and (I) DNMT3b were examined by immunofluorescence staining using an FITC-conjugated secondary antibody and propidium iodide staining (PI, for nuclei) followed by confocal microscopy. Panels: (a and d) PtdIns (red) staining; (b and e) localization of DNMTs (green); and (c and f) merged images of DNMTs and PI. Data are presented as results of a typical experiment from 4 independent experiments. The error bars represent standard deviations.
Methylation patterns at the promoter-region CpG islands of radioresistance-related genes in Hep-2 and RR-Hep-2 cells
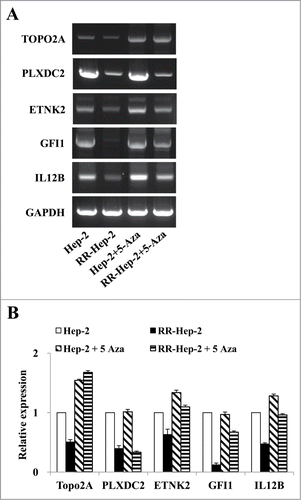
We previously screened a laryngeal cancer EST database to identify gene profiles associated with tumor radioresistance.Citation17 Here, we used bisulfate pyrosequencing to analyze the methylation patterns at the promoter regions of 5 of the identified radioresistance-related genes. The PCR primers amplified fragments containing 3∼6 CpG sites in each target promoter. Bisulfite-modified gDNA was prepared, PCR amplification was performed, and the methylation percentage at each primer was calculated by averaging the degree of methylation at the CpG sites formulated during pyrosequencing. As shown in , higher methylation was detected at all tested CpG sites for RR-Hep-2 cells compared to Hep-2 cells. Next, we used RT-PCR to examine the expression levels of the target genes in Hep-2 and RR-Hep-2 cells. As shown in , TOPO2A, PLXDC2, ETNK2, GFI1, and IL12B were highly expressed in Hep-2 cells, but not in RR-Hep-2 cells. Taken together, our results indicate that the 5 tested radioresistance-related genes showed promoter hypermethylation and decreased mRNA expression in our radioresistant laryngeal cancer cells.
Figure 2. Pyrosequencing of promoter CpG islands in the promoters of 5 radioresistance-related genes in Hep-2 and RR-Hep-2 cells. The fractions of methylated CpG positions were compared between cell lines for (A) TOPO2A, (B) PLXDC2, (C) ETNK2, (D) GFI1 and (E) IL12B. The bars on the far right represent the means of all the methylated positions examined herein. The data presented represent a typical result or average values with standard deviations from 3 independent experiments.
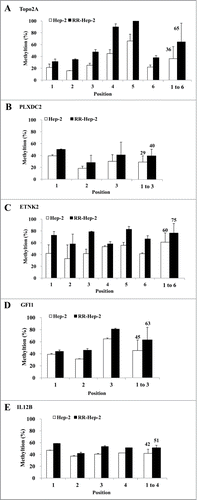
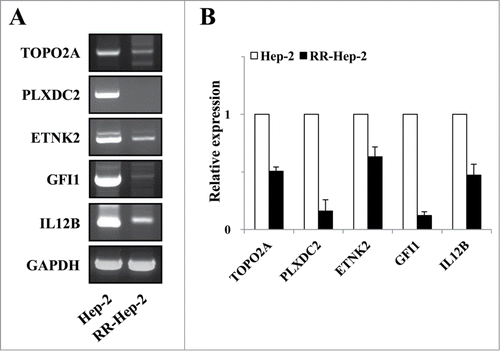
Figure 3. The mRNA expression levels of the 5 target radioresistance-related genes in Hep-2 and RR-Hep-2 cells. (A) RT-PCR analysis of the TOPO2A, PLXDC2, ETNK2, GFI1 and IL12B genes in Hep-2 and RR-Hep-2 cells. (B) The mRNA levels of TOPO2A, PLXDC2, ETNK2, GFI1, and IL12B were measured by quantitative real-time PCR.
Demethylation of Hep-2 and RR-Hep-2 cells increases transcription of the target radioresistance-related genes
Since the downregulation of TOPO2A, PLXDC2, ETNK2, GFI1, and IL12B mRNA levels in RR-Hep-2 cells appears to be related to altered promoter methylation Hep-2 and RR-Hep-2 cells were treated with 5 μM of 5-Aza (a DNA methyltransferase inhibitor) for 72 h. Bisulfate pyrosequencing showed that the methylation levels of all examined CpG sites were decreased in inhibitor-treated RR-Hep-2 and Hep-2 cells (). Interestingly, TOPO2A showed particularly significantly decreases in the methylation percentages following inhibitor treatment, from 36% to 18% in Hep-2 cells and from 65% to 16% in RR-Hep-2 cells (). Furthermore, RT-PCR analysis revealed that the mRNA expression levels of TOPO2A, ETNK2, GFI1, and IL12B were upregulated following 5-Aza treatment. PLXDC2 does not follow the same trend as the other 4: Its gene expression is not affected by 5-Aza treatment (). Collectively, our data suggest that 5-Aza treatment of Hep-2 and RR-Hep-2 cells led to demethylation of thetarget-gene promoters, increasing the transcription of the target radioresistance-related genes.
Figure 4. The methylation levels of the 5 target gene promoters following 5-Aza treatment of Hep-2 and RR-Hep-2 cells. Cells were treated with or without 5-Aza (5 μM) for 72 h, and the fractions of methylated CpG sites were compared for (A) TOPO2A, (B) PLXDC2, (C) ETNK2, (D) GFI1 and (E) IL12B. The bars on the far right indicate the means of all methylated positions. The data presented represent a typical result or average values with standard deviations from 3 independent experiments.
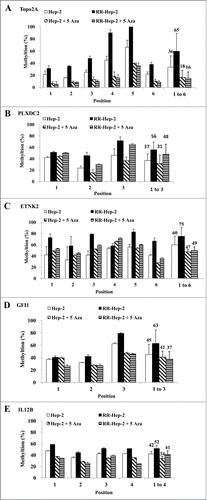
Figure 5. The mRNA expression levels of the 5 radioresistance-related genes in Hep-2 and RR-Hep-2 cells were positively associated with 5-Aza treatment. Hep-2 and RR-Hep-2 cells were treated with or without 5-Aza (5 μM) for 72 h, and (A) RT-PCR analysis was used to assess the mRNA expression levels of TOPO2A, PLXDC2, ETNK2, GFI1, and IL12B. (B) The mRNA levels of TOPO2A, PLXDC2, ETNK2, GFI1, and IL12B were measured by quantitative real-time PCR.
Five-Aza treatment increases the expression and activity of TOPO2A and radiosensitizes the radioresistant laryngeal cancer cells
TOPO2A, which is a nuclear enzyme that regulates many cellular processes, including replication, transcription and sister chromatid decatenation, has been proposed to confer at least some of the inter-individual variation in chromatid radiosensitivity.Citation27 To examine the cellular effects of 5-Aza (5 μM)-induced TOPO2A expression in Hep-2 and RR-Hep-2 cells, we conducted clonogenic survival assays. Our results revealed that 5-Aza treatment significantly reduced the survival of Hep-2 and RR-Hep2 cells exposed to various doses of radiation (), compared with their non-irradiated counterparts. Furthermore, the colony formation in response to 4-Gy irradiation was dramatically lower in 5-Aza-treated RR-Hep-2 cells compared to untreated controls (). These results indicate that DNA methylation may modulate radiosensitivity in these laryngeal cancer cells.
Figure 6. The 5-Aza-mediated induction of TOPO2A reduces the survival of Hep-2 and RR-Hep-2 cells and increases TOPO2A activity. (A) Hep-2 and RR-Hep-2 cells were treated with or without 5-Aza (5 μM), and clonogenic survival fractions were determined following exposure to the indicated doses of radiation. (B) RR-Hep-2 cells were treated with 5-Aza (5 μM) and then further treated with or without 4-Gy irradiation. Colony formation was visualized by trypan blue staining. (C) The protein levels of TOPO2A were determined by Western blotting; β-actin was employed as the loading control. (D) TOPO2A decatenation activity was measured in nuclear extracts from Hep-2 and RR-Hep-2 cells treated with or without 5 μM of 5-Aza for 72 h. (E) Hep-2 and RR-Hep-2 cells were treated with or without 5-Aza (5 μM) for 72 h and fixed with 4% (v/v) paraformaldehyde. Cells were stained for TOPO2A using an FITC-conjugated secondary antibody, and propidium iodide (PtdIns) was used to visualize nuclei. Panels: (a, d, g, and j) PI (red) staining; (b, e, h, and k) localization of TOPO2A (green); and (c, f, i and l) merged images of TOPO2A and PtdIns. The data presented represent a typical result or average values with standard deviations from 3 independent experiments. *P < 0.05.
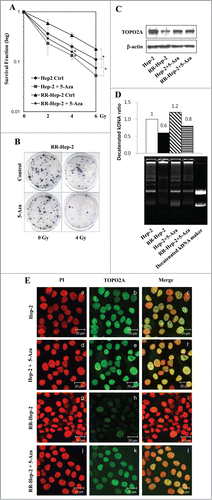
To further examine the association between TOPO2A expression and increased resistance to irradiation, we next treated parental Hep-2 and RR-Hep-2 cells with 5-Aza for 72, and then used Western blotting and immunofluorescence to assess TOPO2A protein levels. Our results revealed that TOPO2A protein levels were generally lower in RR-Hep-2 cells than in parental Hep-2 cells (, top panel, lane 2; and , panel h), and that these levels increased in both cell lines following 5-Aza treatment (, top panel, lane 4; and , panel k). We also assessed the decatenation activity of TOPO2A in these cells, which was assessed in nuclear extracts following ATP-independent relaxation of supercoiled DNA. The enzyme activity of TOPO2A increased following 5-Aza treatment of Hep-2 and RR-Hep-2 cells compared to untreated controls from 1 to 1.2, and from 0.6 to 0.8, respectively (, top), and showed the formation of decatenated kDNA (, bottom). Taken together, these data suggest that changes in DNA methylation can modulate the radiosensitivity of laryngeal cancer cells via alterations in TOPO2A expression and activity.
Discussion
Exposure to ionizing radiation may lead to epigenetic effects, and radiation-induced DNA methylation changes may be critical to some aspects of radioresistance in cancer. The precise underlying pathways and mechanisms, however, remain enigmatic. Here, we analyzed DNA methylation at certain target genes in radioresistant laryngeal cancer cells, and examined the possible association between radiation-induced DNA methylation changes and tumor radioresistance. We report that radiation-induced DNA methylation changes in the promoters of 5 previously identified radioresistance-related genes (including TOPO2A) are associated with the radioresistance of laryngeal cancer cells.
Most of the existing research in the field of cancer epigenetics has focused on the promoter hypermethylation of tumor suppressor genes in radiosensitive and radioresistant cancer cell lines.Citation26,28-30 Variations in DNA methylation patterns have been observed between cell lines differing in gene status,Citation31 but no previous study has examined the DNA methylation patterns of Hep-2 and RR-Hep-2 cells. Here, we used pyrosequencing (a sensitive real-time sequencing-by-synthesis technique) to assess changes in the regional genomic methylation patterns of these cells, and discovered differences in the DNA methylation levels between Hep-2 and RR-Hep-2 cells.
The DNA methylation of CpG dinucleotides is believed to be carried out by methyltransferase.Citation31 Consistent with this, we observed significant DNA hypermethylation in RR-Hep-2 cells (), and significant increases in the expression levels of the de novo (DNMT3a and 3b) DNA methyltransferases (). We speculate that this might account for the observed increases in the methylation levels at the promoters of 5 tested target genes and the corresponding decreases in their mRNA expression levels in RR-Hep-2 cells (). Furthermore, the application of a demethylating agent was found to rescue the expression levels of these target genes in our system. Quantitative analyses using pyrosequencing and RT-PCR identified correlations between decreased promoter demethylation and increased mRNA levels for all 5 target genes.
Among the studied genes, the methylation percentage of TOPO2A significantly decreased (from 65% to 16%) in RR-Hep-2 cells treated with 5-Aza (). DNA topoisomerases, such as TOPO2A, play essential roles in RNA transcription and DNA replication by relieving DNA torsional tension (via swiveling) and resolving intertwined DNA molecules (via unknotting and decatenation).Citation32-35 A previous study showed that TOPO2A contributes to forming chromatid breaks in radiation-exposed cells.Citation27 Here, we report that TOPO2A protein levels were increased by 5-Aza treatment of RR-Hep-2 cells (), and that the promoter demethylation of TOPO2A was closely mirrored by an increase of its decatenation activity in Hep-2 and RR-Hep-2 cells (). This indicates that the demethylation-agent-induced reactivation of TOPO2A expression in resistant tumor cells could potentially facilitate radiotherapy in certain tumor types (i.e., those in which TOPO2A expression has been lost).
The DNA methylation profiles of certain genetic loci have been found to be good biomarkers for cancer radioresistance, offering a number of advantages over other biomarkers. For example: DNA is much more stable than RNA and can survive routine processing for histopathology; DNA methylation changes have been observed very early during cancer development;Citation24 methylation always occurs in the same DNA location and is therefore easier to detect than a gene mutation;Citation36 changes in DNA methylation patterns can be correlated with RNA expression levels;Citation24,37 and since methylation appears to be a general process affecting the whole genome, it is only necessary to analyze a small number of CpG islands to get a sense of a cell's methylation status.Citation36 Our present results show that methylation is an important molecular mechanism in RR-Hep-2 cells, and thus could prove useful as a prognostic and/or diagnostic marker.
In conclusion, we herein report new evidence supporting the involvement of DNA methylation changes in tumor radioresistance, and show that this involves the promoter-methylation-mediated inactivation of TOPO2A in our system. Additional studies are needed to further examine the involvement of DNA methylation and other epigenetic mechanisms in radiation-induced DNA hypermethylation and cancer radioresistance. Such epigenetic changes could be exploited to improve the efficacy of radiotherapy and/or as a novel approach for early diagnosis, prognosis and risk assessment. Notably, the high frequency of methylation in RR-Hep-2 cells suggests that this tumor could be a good target for the development of novel demethylation-based therapies.
Materials and Methods
Cell culture and the radioresistant Hep-2 cell line
Human laryngeal squamous cell carcinoma (Hep-2) cells were purchased from the American Type Culture Collection (Manassas, VA, USA) and grown in DMEM (Gibco-BRL, Rockville, MD, USA) supplemented with 10% FBS. Radioresistant Hep-2 (RR-Hep-2) cells were established as previously described.Citation17 All cells were cultured in a humidified incubator at 37°C and 5% CO2. Cells were irradiated using a 137Cs-ray source (Atomic Energy of Canada, Mississauga, Canada) at a dose rate of 3.81 Gy/min.
Clonogenic assay
After irradiation, cell survival was determined by a clonogenic assay, as previously described.Citation17 Briefly, cells were treated with a single dose of radiation ranging from 0 to 6 Gy. Immediately following irradiation, cells were trypsinized, diluted, and seeded to triplicate 60-mm tissue culture dishes at various cell densities (200 cells for control, 400 cells for 2 Gy, 1500 cells for 4 Gy, and 3000 cells for 6 Gy). After 10–14 d, the colonies were fixed with methanol and stained with trypan blue solution. Colonies containing > 50 cells were counted as survivors using a colony counter (Imaging Products, Chantilly, VA, USA).
Cell death analysis
Cells were seeded at a density of 2 × 105 cells per 60-mm dish, treated with 10 Gy of radiation and then cultured under the indicated experimental conditions. Apoptotic cell death was determined by protein gel blot analysis of cleaved poly ADP-ribose polymerase (PARP) and Cleaved caspase-3 (Santa Cruz Biotechnology). To quantify cell death, cells were analyzed using a FACScan flow cytometer (Becton Dickson), as previously described in reference 17.
Western blot analysis
Total cell lysates were prepared in lysis buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 5 mM EDTA, 1% [v/v] Triton X-100, 1% [w/v] sodium deoxycholic acid, 30 mM Na2HPO4, 50 mM NaF, and 1 mM Na3VO4) containing a freshly added protease inhibitor cocktail (GenDEPOT, Katy, TX, USA). Cytoplasmic and nuclear extracts were prepared using a NE-PER Nuclear and Cytoplasmic Extraction Kit (Pierce). Extracts (30 μg) were mixed with SDS sample buffer, boiled for 5 min, separated by 8% or 10% (w/v) SDS-PAGE and transferred to nitrocellulose membranes (Millipore). Blots were incubated with antibodies against DNMT1, DNMT3a, DNMT3b (Abcam), Topo2A and α-actinin (Santa Cruz Biotechnology).
Reverse transcription-polymerase chain reaction (RT-PCR) and Real-time PCR
Total RNA was isolated using the QIAzol reagent (Qiagen, Hilden, Germany) and quantified by formaldehyde-agarose gel electrophoresis. Single-strand cDNA was synthesized from RNA (2 μg) using 0.27 μg of oligo dT and the amfiRivert reverse transcriptase (GenDEPOT). The desired cDNA fragments were PCR amplified using the following specific primer pairs: DNMT1, 5′-ATG AGC AGC CCA TCT TCC TG -3′ (sense) and 5′-CCT CAT CGT CT CTG CCT CC-3′ (antisense); DNMT3a, 5′-CCC AGG CAG CCA TTA AGG AA -3′ (sense) and 5′-GCG ATC ATC TCC CTC CTT GG-3′ (antisense); DNMT3b, 5′-GGC TGT TTG TCT TGT GGC AG -3′ (sense) and 5′-GGA ATG GCA GGG TAC AGC TT-3′ (antisense); and GAPDH (glyceraldehyde-3-phosphate dehydrogenase), 5′-TGC TGA GTA TGT CGT GGA GTC TA-3′ (sense) and 5′-AGT GGG AGT TGC TGT TGA AGT CG-3′ (antisense). The Opticon system for real-time PCR (Bio-Rad Laboratories, CA, USA) was used to measure the amount of gene expression. Each reaction mixture contained 300 nM of forward and reverse primer, 200 nM of HOT FIREPol EvaGreen DNA ploymerase (Solis BioDyne, Tartu, Estonia) in a total volume of 20 μl.
Nested PCR
The presence of GFI1 and IL12B was assessed by nested PCR using the primers listed in . For the first-round PCR, 1 μl of the reverse-transcribed product was amplified with HOT FIREPol DNA polymerase (Solis BioDyne, Tartu, Estonia) in a total volume of 20 μl containing the provided master mix buffer. To increase the sensitivity and specificity, a second-round PCR was performed using the first-round PCR product (1 μl) as the template. The amplified products were resolved by 2% agarose gel electrophoresis and visualized with ethidium bromide (0.1 μg/ml) staining.
Table 1. Gene pyrosequencing conditions and primers
Table 2. PCR conditions and primers
DNA extraction and sodium-bisulfite modification
Genomic DNA (gDNA) was extracted from the cultured cells using a QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). The bisulfite reaction was carried out on 2 μg gDNA, the reaction volume was adjusted to 50 μl with sterile water, and 130 μl of the provided CT conversion reagent was added. The sample tubes were placed in a thermal cycler (MJ Research, Waltham, MA, USA), incubated for 15 min at 37˚C and for 16 h at 50˚C, and then stored at 4˚C. The resulting bisulfite-modified DNA was purified using an EZ DNA Methylation Kit (Zymo Research, Orange, CA, USA) and eluted with 40 μl of the provided M-Elution buffer. Finally, 1-μl aliquots were used as templates for PCR.
Pyrosequencing analysis
Pyrosequencing primers (one biotinylated and one non-biotinylated) were designed to amplify 3 to 6 CpG dinucleotides in each target-gene promoter, and PCR was carried out in a volume of 50 μl with ≤ 50 ng converted gDNA, 2 U Hot/Start Taq polymerase (Solis BioDyne, Estonia), 2 μl of 10 pmol/μl non-biotinylated primer and 2 μl of 10 pmol/μl biotinylated primer. The utilized primers were designed using the PSQ Assay Design Program (Biotage, Uppsala, Sweden), and are listed in . Amplification was carried out according to the general guidelines suggested by the pyrosequencing protocol, as follows: denaturing at 95˚C for 5 min; followed by 45 cycles of 95˚C for 30 sec, 53∼59˚C for 30 sec and 72˚C for 30 sec; and a final extension at 72˚C for 10 min. The quality and purity of the PCR (10 μl) products were confirmed by 2% agarose gel electrophoresis and ethidium bromide staining. Pyrosequencing was performed on a PyroMark ID system using streptavidin Sepharose HP beads (Amersham Biosciences, Uppsala, Sweden) and a Pyro Gold Kit (Biotage, Charlottesville, VA, USA); experiments were performed according to the manufacturer's instructions without further optimization. The methylation percentage was calculated as the average degree of CpG site methylation. All pyrosequencing experiments were performed twice.
Demethylation
Cells (1×105) were seeded to 60-mm dishes in DMEM containing 10% FBS. After the cells had attached to the dishes, they were treated with 5 μM of 5-Aza (Sigma–Aldrich) for 72 h.
DNA decatenation assay
Unless otherwise noted, the reagents utilized for this assay were purchased from TopoGEN (Port Orange, FL, USA). Topoisomerase II was extracted from the nuclei of Hep-2 or RR-Hep-2 cells. The specific topoisomerase II decatenation activity was calculated based on the ability of a given nuclear extract (containing the α and β isoforms) or a defined amount of purified enzyme to completely decatenate a given amount of catenated input DNA (100 ng kDNA) in a particular amount of time. Reactions were started by adding ATP (final concentration, 450 μM); samples were incubated 37˚C for 20 minutes; and reactions were terminated by placing the samples on ice and adding 4 μl stop/gel loading buffer. Samples (20 μl) were resolved by 1% agarose gel electrophoresis; the gels were analyzed under a UV transilluminator; and decatenated kDNA products were quantified using the ImageJ software.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by Basic Science research Program (Grant no. 2014R1A1A2053524) and the Nuclear Research and Development Program through a National Research Foundation of Korea (NRF; 2012M2B2B1055642 and 2012M2A2A7012494) grant funded by the Korean government.
References
- Aypar U, Morgan WF, Baulch JE. Radiation-induced genomic instability: are epigenetic mechanisms the missing link? Int J Radiat Biol; 87:179-91; PMID:21039330; http://dx.doi.org/10.3109/09553002.2010.522686.
- Jones PA, Gonzalgo ML. Altered DNA methylation and genome instability: a new pathway to cancer? Proc Natl Acad Sci U S A 1997; 94:2103-5; PMID:9122155; http://dx.doi.org/10.1073/pnas.94.6.2103.
- Rizwana R, Hahn PJ. CpG methylation reduces genomic instability. J Cell Sci 1999; 112 (Pt 24):4513-9; PMID:10574701.
- Ehrlich M. DNA methylation in cancer: too much, but also too little. Oncogene 2002; 21:5400-13; PMID:12154403; http://dx.doi.org/10.1038/sj.onc.1205651.
- Pogribny I, Raiche J, Slovack M, Kovalchuk O. Dose-dependence, sex-and tissue-specificity, and persistence of radiation-induced genomic DNA methylation changes. Biochem Biophys Res Commun 2004; 320:1253-61; PMID:15249225; http://dx.doi.org/10.1016/j.bbrc.2004.06.081.
- Ha PK, Califano JA. Promoter methylation and inactivation of tumour-suppressor genes in oral squamous-cell carcinoma. Lancet Oncol 2006; 7:77-82; PMID:16389187; http://dx.doi.org/10.1016/S1470-2045(05)70540-4.
- Herceg Z. Epigenetics and cancer: towards an evaluation of the impact of environmental and dietary factors. Mutagenesis 2007; 22:91-103; PMID:17284773; http://dx.doi.org/10.1093/mutage/gel068.
- Jones PL, Wolffe AP. Relationships between chromatin organization and DNA methylation in determining gene expression. Semin Cancer Biol 1999; 9:339-47; PMID:10547342; http://dx.doi.org/10.1006/scbi.1999.0134.
- Baylin SB, Esteller M, Rountree MR, Bachman KE, Schuebel K, Herman JG. Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Hum Mol Genet 2001; 10:687-92; PMID:11257100; http://dx.doi.org/10.1093/hmg/10.7.687.
- Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science 2001; 293:1089-93; PMID:11498579; http://dx.doi.org/10.1126/science.1063443.
- Lee ES, Issa JP, Roberts DB, Williams MD, Weber RS, Kies MS, El-Naggar AK. Quantitative promoter hypermethylation analysis of cancer-related genes in salivary gland carcinomas: comparison with methylation-specific PCR technique and clinical significance. Clin Cancer Res 2008; 14:2664-72; PMID:18451230; http://dx.doi.org/10.1158/1078-0432.CCR-07-1232.
- Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet 2002; 3:415-28; PMID:12042769; http://dx.doi.org/10.1038/nrg962.
- Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, Lam WL, Schubeler D. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet 2005; 37:853-62; PMID:16007088; http://dx.doi.org/10.1038/ng1598.
- Kaup S, Grandjean V, Mukherjee R, Kapoor A, Keyes E, Seymour CB, Mothersill CE, Schofield PN. Radiation-induced genomic instability is associated with DNA methylation changes in cultured human keratinocytes. Mutat Res 2006; 597:87-97; PMID:16417911; http://dx.doi.org/10.1016/j.mrfmmm.2005.06.032.
- Jung IL, Kang HJ, Kim KC, Kim IG. PTEN/pAkt/p53 signaling pathway correlates with the radioresponse of non-small cell lung cancer. Int J Mol Med; 25:517-23; PMID:20198299.
- Fu XY, Zhang SW, Ran RQ, Shen ZH, Gu JX, Cao SL. Restoration of the p16 gene is related to increased radiosensitivity of p16-deficient lung adenocarcinoma cell lines. J Cancer Res Clin Oncol 1998; 124:621-6; PMID:9860291; http://dx.doi.org/10.1007/s004320050224.
- Kim JS, Chang JW, Yun HS, Yang KM, Hong EH, Kim DH, Um HD, Lee KH, Lee SJ, Hwang SG. Chloride intracellular channel 1 identified using proteomic analysis plays an important role in the radiosensitivity of HEp-2 cells via reactive oxygen species production. Proteomics; 10:2589-604; PMID:20461716; http://dx.doi.org/10.1002/pmic.200900523.
- Weichselbaum RR, Beckett MA, Schwartz JL, Dritschilo A. Radioresistant tumor cells are present in head and neck carcinomas that recur after radiotherapy. Int J Radiat Oncol Biol Phys 1988; 15:575-9; PMID:3417487; http://dx.doi.org/10.1016/0360-3016(88)90297-0.
- Joiner MC, Lambin P, Malaise EP, Robson T, Arrand JE, Skov KA, Marples B. Hypersensitivity to very-low single radiation doses: its relationship to the adaptive response and induced radioresistance. Mutat Res 1996; 358:171-83; PMID:8946022; http://dx.doi.org/10.1016/S0027-5107(96)00118-2.
- Kim JJ, Tannock IF. Repopulation of cancer cells during therapy: an important cause of treatment failure. Nat Rev Cancer 2005; 5:516-25; PMID:15965493; http://dx.doi.org/10.1038/nrc1650.
- Seve M, Chimienti F, Devergnas S, Favier A. In silico identification and expression of SLC30 family genes: an expressed sequence tag data mining strategy for the characterization of zinc transporters' tissue expression. BMC Genomics 2004; 5:32; PMID:15154973; http://dx.doi.org/10.1186/1471-2164-5-32.
- Oh J, Lee J, Woo JM, Choi E, Park I, Han C, Baek N, Lee H, Kim do H, Cho C. Systematic identification and integrative analysis of novel genes expressed specifically or predominantly in mouse epididymis. BMC Genomics 2006; 7:314; PMID:17166261; http://dx.doi.org/10.1186/1471-2164-7-314.
- Kim JS, Ryoo ZY, Chun JS. Cytokine-like 1 (Cytl1) regulates the chondrogenesis of mesenchymal cells. J Biol Chem 2007; 282:29359-67; PMID:17644814; http://dx.doi.org/10.1074/jbc.M700965200.
- Gokul G, Gautami B, Malathi S, Sowjanya AP, Poli UR, Jain M, Ramakrishna G, Khosla S. DNA methylation profile at the DNMT3L promoter: a potential biomarker for cervical cancer. Epigenetics 2007; 2:80-5; PMID:17965599; http://dx.doi.org/10.4161/epi.2.2.3692.
- Jeltsch A. Molecular enzymology of mammalian DNA methyltransferases. Curr Top Microbiol Immunol 2006; 301:203-25; PMID:16570849.
- Loree J, Koturbash I, Kutanzi K, Baker M, Pogribny I, Kovalchuk O. Radiation-induced molecular changes in rat mammary tissue: possible implications for radiation-induced carcinogenesis. Int J Radiat Biol 2006; 82:805-15; PMID:17148264; http://dx.doi.org/10.1080/09553000600960027.
- Terry SY, Riches AC, Bryant PE. A role for topoisomerase II α in the formation of radiation-induced chromatid breaks. Br J Cancer 2008; 99:670-4; PMID:18665175; http://dx.doi.org/10.1038/sj.bjc.6604514.
- Fackler MJ, McVeigh M, Mehrotra J, Blum MA, Lange J, Lapides A, Garrett E, Argani P, Sukumar S. Quantitative multiplex methylation-specific PCR assay for the detection of promoter hypermethylation in multiple genes in breast cancer. Cancer Res 2004; 64:4442-52; PMID:15231653; http://dx.doi.org/10.1158/0008-5472.CAN-03-3341.
- Widschwendter M, Siegmund KD, Muller HM, Fiegl H, Marth C, Muller-Holzner E, Jones PA, Laird PW. Association of breast cancer DNA methylation profiles with hormone receptor status and response to tamoxifen. Cancer Res 2004; 64:3807-13; PMID:15172987; http://dx.doi.org/10.1158/0008-5472.CAN-03-3852.
- Jones PA. Overview of cancer epigenetics. Semin Hematol 2005; 42:S3-8; PMID:16015502; http://dx.doi.org/10.1053/j.seminhematol.2005.05.001.
- Chaudhry MA, Omaruddin RA. Differential DNA methylation alterations in radiation-sensitive and resistant cells. DNA Cell Biol; 31:908-16; PMID:22185261; http://dx.doi.org/10.1089/dna.2011.1509.
- Wang JC. DNA topoisomerases. Annu Rev Biochem 1985; 54:665-97; PMID:2992360; http://dx.doi.org/10.1146/annurev.bi.54.070185.003313.
- Osheroff N, Zechiedrich EL, Gale KC. Catalytic function of DNA topoisomerase II. Bioessays 1991; 13:269-73; PMID:1654050; http://dx.doi.org/10.1002/bies.950130603.
- Hsieh TS. DNA topoisomerases. Curr Opin Cell Biol 1992; 4:396-400; PMID:1323315; http://dx.doi.org/10.1016/0955-0674(92)90004-V.
- Leteurtre F, Kohlhagen G, Fesen MR, Tanizawa A, Kohn KW, Pommier Y. Effects of DNA methylation on topoisomerase I and II cleavage activities. J Biol Chem 1994; 269:7893-900; PMID:8132507.
- Catto JW, Azzouzi AR, Rehman I, Feeley KM, Cross SS, Amira N, Fromont G, Sibony M, Cussenot O, Meuth M, et al. Promoter hypermethylation is associated with tumor location, stage, and subsequent progression in transitional cell carcinoma. J Clin Oncol 2005; 23:2903-10; PMID:15753461; http://dx.doi.org/10.1200/JCO.2005.03.163.
- Laird PW. The power and the promise of DNA methylation markers. Nat Rev Cancer 2003; 3:253-66; PMID:12671664; http://dx.doi.org/10.1038/nrc1045.