
Abstract
PR-104 is a clinical stage bioreductive prodrug that is converted in vivo to its cognate alcohol, PR-104A. This dinitrobenzamide mustard is reduced to activated DNA cross-linking metabolites (hydroxylamine PR-104H and amine PR-104M) under hypoxia by one-electron reductases and independently of hypoxia by the 2-electron reductase aldo-keto reductase 1C3 (AKR1C3). High expression of AKR1C3, along with extensive hypoxia, suggested the potential of PR-104 for treatment of hepatocellular carcinoma (HCC). However, a phase IB trial with sorafenib demonstrated significant toxicity that was ascribed in part to reduced PR-104A clearance, likely reflecting compromised glucuronidation in patients with advanced HCC. Here, we evaluate the activity of PR-104 in HCC xenografts (HepG2, PLC/PRF/5, SNU-398, Hep3B) in mice, which do not significantly glucuronidate PR-104A. Cell line differences in sensitivity to PR-104A in vitro under aerobic conditions could be accounted for by differences in both expression of AKR1C3 (high in HepG2 and PLC/PRF/5) and sensitivity to the major active metabolite PR-104H, to which PLC/PRF/5 was relatively resistant, while hypoxic selectivity of PR-104A cytotoxicity and reductive metabolism was greatest in the low-AKR1C3 SNU-398 and Hep3B lines. Expression of AKR1C3 in HepG2 and PLC/PRF/5 xenografts was in the range seen in 21 human HCC specimens. PR-104 monotherapy elicited significant reductions in growth of Hep3B and HepG2 xenografts, and the combination with sorafenib was significantly active in all 4 xenograft models. The results suggest that better-tolerated analogs of PR-104, without a glucuronidation liability, may have the potential to exploit AKR1C3 and/or hypoxia in HCC in humans.
Abbreviations
| AKR1C3 | = | aldo-keto reductase 1C3 |
| HCC | = | hepatocellular carcinoma |
| POR | = | NADPH:cytochrome P450 oxidoreductase |
| UGT-2B7 | = | UDP-glucuronosyltransferase-2B7 |
| HCR | = | hypoxic cytotoxicity ratio |
| Nrf2 | = | NF-E2-related factor-2 |
Introduction
Tumor hypoxia has been shown to play an important role in cancer progression and resistance to treatment.Citation1-5 However, the severe hypoxia observed in solid tumors also represents an attractive opportunity for the development of selective anti-cancer therapies. A number of hypoxia-activated prodrugs have been developed in an attempt to exploit tumor hypoxia.Citation5 One such compound is PR-104, a water soluble 3,5-dinitrobenzamide mustard phosphate ester pre-prodrug that undergoes systemic hydrolysis in vivo to the corresponding alcohol prodrug PR-104A.Citation6-8 PR-104A is a bioreductive prodrug reduced under conditions of pathological hypoxia (pO2 < 1 mmHg)Citation9 to the cytotoxic hydroxylamine (PR-104H) and amine (PR-104M) derivatives, which are reactive nitrogen mustards that cause cell death through the formation of DNA inter-strand crosslinks.Citation6,10,11
The reduction of PR-104A under hypoxic conditions is catalyzed by members of the diflavin reductase family of proteins, especially NADPH:cytochrome P450 oxidoreductase (POR).Citation12,13 However, PR-104A is also activated in an oxygen-insensitive manner by human aldo-keto reductase 1C3 (AKR1C3).Citation14 This represents an off-target mechanism of activation in relation to the original drug design concept but may be exploitable in cancers with high AKR1C3 activity. We have previously analyzed AKR1C3 expression in 2700 tumors of 19 different types and a wide range of normal tissues, demonstrating that strong AKR1C3 expression was most prevalent in hepatocellular carcinoma (HCC) tumors.Citation14 The increased sensitivity of AKR1C3 staining in HCC relative to most normal tissues, coupled with the high expression of POR in many HCC biopsiesCitation12 and evidence for hypoxiaCitation15 suggested that PR-104 may have therapeutic utility in this clinical setting.
The multi-kinase inhibitor sorafenib has proven activity against advanced HCC,Citation16,17 although improvements in overall survival are modest which has generated much interest in combination of sorafenib with other agents.Citation18 Notably, sorafenib treatment has been shown to increase tumor hypoxia in renal cell carcinoma xenograft models.Citation19,20 In addition, Liang et al. have provided evidence that sustained sorafenib treatment leads to increased intratumoural hypoxia in HCC in patients and mice, and that this treatment-induced hypoxia contributes to sorafenib resistance,Citation21 suggestive of potential for synergy between PR-104 and sorafenib.
These considerations led to the clinical evaluation of PR-104 in combination with sorafenib in a phase IB trial in advanced HCC.Citation22 However, the trial was discontinued because of severe haematological toxicity, likely reflecting the observed low clearance of PR-104A in these patients (either with or without sorafenib) relative to previous studies in non-HCC patients.Citation22 This, in turn, may reflect compromised hepatic metabolism of PR-104A in patients with advanced HCC given that glucuronidation of its alcohol side-chain by UDP-glucuronosyltransferase-2B7 (UGT-2B7) is a major mechanism of PR-104A clearance in humans.Citation23 In contrast, O-glucuronidation of PR-104A is not a significant pathway in mice.Citation23,24 Here, we evaluate activity of PR-104, with and without sorafenib, against human HCC xenografts in mice, in effect modeling a PR-104 analog that is not a glucuronidation substrate. We also explore the contribution of aerobic (AKR1C3-dependent) and hypoxia-selective activation of PR-104A in the same HCC cell lines.
Results
HCC cell lines show varying levels of aerobic sensitivity to PR-104A and its cytotoxic metabolites in vitro
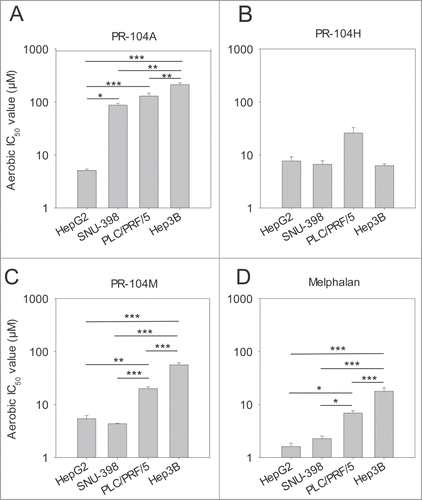
The sensitivity of 4 HCC cell lines to PR-104A or its cytotoxic metabolites, PR-104H and PR-104M, was examined under aerobic conditions using an antiproliferative assay. HepG2 cells were significantly more sensitive to PR-104A with IC50 values that were 17, 25 or 42-fold lower than that for SNU-398, PLC/PRF/5 or Hep3B cells, respectively (). When the same cell lines were exposed to the active metabolites PR-104H or PR-104M some differences in sensitivity were also observed; there was a trend toward lower sensitivity of PLC/PRF/5 to PR-104H () and both PLC/PRF/5 and Hep3B cells were significantly more resistant (∼fold4- and ∼fold10- respectively) to PR-104M (). To determine whether differences in sensitivity to PR-104H and PR-104M between the HCC cell lines reflect differences in intrinsic sensitivity to DNA crosslinking nitrogen mustards, we also exposed cells to the nitrogen mustard melphalan. Sensitivity to melphalan showed a similar pattern to PR-104M, with significantly higher IC50 values for PLC/PRF/5 and Hep3B than HepG2 and SNU-398 ().
Figure 1. Antiproliferative activity of the prodrug PR-104A (A), its DNA crosslinking metabolites PR-104H (B) and PR-104M (C), and the reference DNA crosslinking agent melphalan (D) against HCC cell lines. Cells were exposed to the drugs for 4 hr under aerobic conditions and grown for a further 5 d before staining with sulphorhodamine B. IC50 values are the mean of 3 or 4 independent experiments and error bars are SEM. Significance of pairwise differences are shown as * for P < 0.05, ** for P < 0.01 and *** for P < 0.001. One way ANOVA demonstrated significant differences between cell lines in panel B (P = 0.043) but none of the pairwise differences reached significance (P > 0.05).
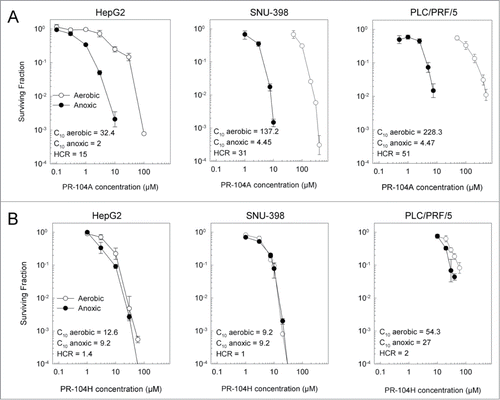
The hypoxic selectivity of PR-104A differs between HCC cell lines in vitro
The sensitivity of the cell lines to PR-104A and PR-104H under both aerobic and hypoxic conditions was also examined using a clonogenic end point. Hep3B cells did not form distinct colonies using this assay so were excluded from this study. Consistent with the antiproliferative assays, PR-104A aerobic sensitivity was greatest for HepG2 cells, with a C10 (concentration for 10% survival) which was 4.1 and 7.1-fold lower than for SNU-398 or PLC/PRF/5 cells respectively (). All three cell lines showed increased sensitivity to PR-104A under anoxic conditions, with the highest hypoxic cytotoxicity ratio (HCR, determined as aerobic C10/hypoxic C10) for PLC-PRF-5 (51-fold) and lowest for HepG2 (15-fold; ). In contrast, anoxia had little or no effect on sensitivity to PR-104H (HCR values 1–2; ). Consistent with the antiproliferative results, PLC/PRF/5 cells were the most resistant to PR-104H in the clonogenic assays, with an aerobic C10 value of 54 µM as compared to 12.6 µM and 9.2 µM for HepG2 and SNU-398 cells respectively ().
Figure 2. Clonogenic survival of HCC cell lines following a 2 hr exposure to PR-104A (A) or PR-104H (B) under aerobic or anoxic conditions. Values show the mean ± range of duplicate experiments.
Aerobic and anoxic PR-104A metabolism in HCC cell lines is consistent with expression levels of known PR-104A reductases
Levels of PR-104A metabolites were evaluated by LC-MS/MS following 1 h exposure of cell monolayers to PR-104A under anoxic or aerobic conditions. All cell lines showed higher levels of the reduced metabolites PR-104H and PR-104M under anoxic conditions () than under aerobic conditions (). The sum of these 2 active metabolites was 9-fold higher under anoxia for HepG2 cells, 27-fold for PLC/PRF/5, 34-fold for SNU-398 and 250-fold for Hep3B; the difference between aerobic and anoxic PR-104A metabolism were highly statistically significant (P < 0.001) in all cases by t-test. The O-glucuronide of PR-104A (PR-104G) was also detected in HepG2 and Hep3B cells and was present at similar levels under both anoxic and aerobic conditions, but not in the other 2 cell lines (). We did not detect the O-glucuronide of PR-104H, previously reported to give an MS/MS transition of 661 > 485Citation23 in any of the HCC cell lines (data not shown).
Figure 3. Metabolism of PR-104A under anoxic or aerobic conditions and expression of known PR-104A metabolizing enzymes in HCC cell lines. (A) Metabolism of PR-104A (100 μM, 1 hr) under anoxia, determined by LC-MS/MS quantitation of metabolites. Values are the mean ± SEM for 6 biological replicates in 2 independent experiments. All pairwise differences in the sum of the active metabolites (PR-104H & PR-104M) between cell lines were statistically significant (P < 0.05; ANOVA with Holm-Sidak). (B) Aerobic metabolism of PR-104A, as for A. All pairwise differences of the sum of PR-104H & PR-104M between cell lines were statistically significant except SNU-398 vs Hep3B. (C) Lysates from the same pretreatment cell populations were analyzed by Western blot to detect POR and AKR1C3. The numerical values shown are the mean ± range for the POR/ β-actin or AKR1C3/β-actin ratio for the 2 experiments. (D) Analysis of AKR1C3 enzymatic activity in aerobic HCC cells using the fluorogenic AKR1C probe coumberone (10 μM). Formation of coumberol in the presence of a specific AKR1C3 inhibitor, SN34037, was used to identify the activity attributable to AKR1C3. Values are mean and errors are SEM for 3 biological replicates. AKR1C3 activity in HepG2 was significantly higher than PLC/PRF/5 (2-tailed P = 0.0004, t-test) and was not detectable in the other lines.
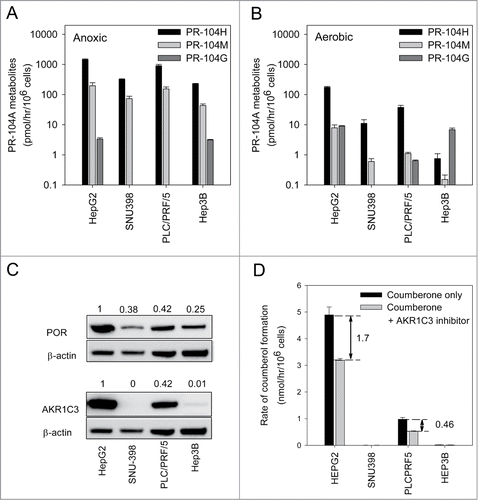
Under either anoxic or aerobic conditions, the sum of PR-104H and PR-104M showed the same trend between cell lines (HepG2 > PLC/PRF/5/ > SNU-398 > Hep3B), although cell line differences were more marked for aerobic metabolism. The rank order of anoxic metabolism was consistent with that of POR expression in the 4 cell lines by Western blotting (). Similarly, the much higher aerobic metabolism of PR-104A in HepG2 and PLC/PRF/5 was consistent with much higher levels of AKR1C3 in these 2 cell lines (). AKR1C3 enzyme activity was also examined using a coumberone-based assay; coumberone is reduced to the fluorescent product coumberol by all 4 human AKR1C enzymes.Citation14 The proportion of coumberol formation attributable to AKR1C3 was elucidated using the specific AKR1C3 inhibitor SN3403728 (). AKR1C3 activity was 3.7-fold higher in HepG2 cells (1.7 ± 0.1 nmol/106 cells/hr) than in PLC/PRF/5 cells (0.46 ± 0.02 nmol/106 cells/hr) and was not detectable in the other lines, consistent with the Western blots.
Hypoxia and AKR1C3 expression in HCC xenografts
Activation of PR-104 in vivo is expected to be dependent on both the extent of tumor hypoxia and the presence of intracellular reductases capable of reducing PR-104A. The extent of tumor hypoxia in each xenograft model was assessed using immunostaining of covalently bound metabolites of the hypoxia marker pimonidazole (). The hypoxic fraction for each tumor was calculated from the ratio of pimonidazole stained area to the total viable area. SNU-398 xenografts showed the largest hypoxic fraction with (20.4 ± 6.8)% of the tumor staining positive for pimonidazole; this was ∼10-fold higher than PLC/PRF/5, the least hypoxic xenograft with a HF of just (1.7 ± 0.9)% ().
Figure 4. Levels of tumor hypoxia and AKR1C3 expression in HCC xenografts. (A) Representative images of pimonidazole binding in HCC xenograft sections. (B) Hypoxic fractions of HCC xenograft sections (mean ± SEM) calculated from imaging of pimonidazole binding (green). Only the difference between SNU-398 and PLC/PRF/5 reached statistical significance (P < 0.05). (C) Determination of AKR1C3 expression in HCC xenografts by immunohistochemistry of xenograft sections and bright-field microscopy (40x). (D) Western blot of AKR1C3, NQO1 and β-actin expression in lysates derived from in vitro cell culture or in vivo tumor xenografts. Murine liver lysate was used as a negative control. Values are the AKR1C3/actin and NQO1/actin ratios by densitometry, and errors are the range for duplicate samples.
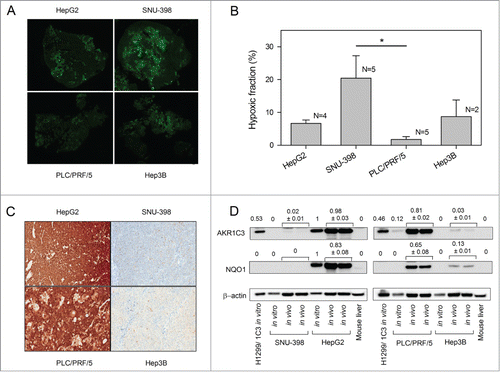
Expression of AKR1C3 was also assessed in HCC xenograft sections by immunohistochemistry. The intensity of AKR1C3 expression in the HCC xenografts was consistent with analysis of in vitro samples, with strong AKR1C3 expression only observed in HepG2 and PLC/PRF/5 xenografts (). The intensity of AKR1C3 expression was quantified using a 4 point scoring system, confirming stronger expression in HepG2 and PLC/PRF/5 than SNU-398 or Hep3B xenografts ().
Table 1. Pathology review of AKR1C3 immunohistochemical staining in HCC xenografts and HCC surgical specimens
We also compared AKR1C3 expression levels in cell lines and tumor lysates by Western blot (). Lysates from H1299 cells stably transfected to over-express AKR1C3, and mouse liver, were used as positive and negative controls respectively. Strong AKR1C3 expression was observed in HepG2 and PLC/PRF/5 tumor lysates. Band densitometry and normalization to actin loading control showed AKR1C3 expression in HepG2 tumor lysates was similar to in vitro cell lines. However, expression of AKR1C3 in lysates of PLC/PRF/5 xenografts was approximately 6-fold higher than the AKR1C3 levels observed in the tissue culture cell line. Weak AKR1C3 bands were also observed in lysates of SNU-398 and Hep3B xenografts, but not in the corresponding cell lines in vitro. To test whether increases in AKR1C3 expression in tumors might reflect upregulation of the NF-E2-related factor-2 (Nrf2) transcription factor, which is known to transcribe AKR1C3 in some lines,Citation14,25 we looked at levels of NQO1 which is well-characterized as a transcriptional target of Nrf2Citation26,27 (). Consistent with AKR1C3 levels, NQO1 expression was not dramatically altered in lysates of HepG2 xenografts but was upregulated in PLC/PRF/5 and Hep3B xenografts relative to the same lines in tissue culture.
AKR1C3 is highly expressed in HCC surgical samples
Analysis of AKR1C3 expression in 21 human HCC specimens by immunohistochemistry demonstrated a high frequency of strong AKR1C3 expression (). AKR1C3 was expressed in ≥ 90% of tumor cells for 19 out of the 21 samples, with 8 of these 19 specimens showing strong (3+) AKR1C3 intensity in ≥ 80% of cancer cells. These data indicate that the intensity of AKR1C3 expression in the HepG2 and PLC/PRF/5 xenografts ( and ) lies within the range of most HCC surgical samples (). Only 2 out of 21 specimens showed low levels of AKR1C3 expression with a similar intensity to SNU-398 and Hep3B xenografts, with ≤ 25% of cells scoring positive for AKR1C3 and the majority of those exhibiting weak (1+) staining intensity ().
Activity of PR-104 and sorafenib against HCC xenografts
We hypothesized that high AKR1C3 expression and aerobic sensitivity of HepG2 to PR-104A in vitro would translate into monotherapy activity of PR-104 in xenografts, and that sorafenib-induced increases in hypoxia might enhance the effectiveness of PR-104 even in HCC xenografts lacking AKR1C3. Mice were stratified to treatment () when tumors reached a mean diameter of 6 mm. Each HCC tumor model was associated with cachexia as evidenced by the mean body weight loss at nadir ranging from 6.7 ± 1.9 % relative to pretreatment weights (SNU-398) to 10.8 ± 0.5% loss (HepG2) in mice treated with vehicle only (). Unsurprisingly, body weight loss and mortality was exacerbated by drug treatment, resulting in 4/29 deaths (including culls because of body weight loss) with sorafenib only, 3/28 deaths with PR-104 only across the 4 tumor xenograft models, but this toxicity was no more severe (4/33 deaths) for the combination
Figure 5. Effect of PR-104, sorafenib and the combination of both drugs on HCC tumor growth. (A) Dosing schema. (B–E) Survival curves of the time taken for tumors to reach RTV4 after treatment with vehicle, sorafenib (80 mg/kg p.o., qdx5), PR-104 (250 mg/kg i.p., qd × 6) or both drugs in combination. Significance of difference from vehicle-only controls was determined by log-rank test with Holm-Sidak multiple comparison post-test analysis.
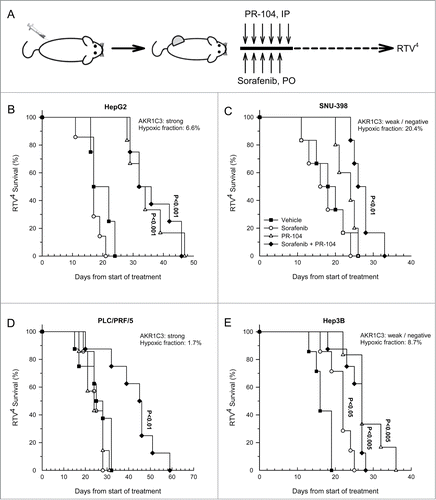
Table 2. Toxicity profile of HCC xenograft data. Body weight loss at nadir is the maximum body weight loss as a percentage of the pretreatment value (mean ± SEM). Exclusions indicate the number of animals excluded from analysis as a result of death from drug toxicity or culling due to excessive body weight loss
Tumor growth inhibition in drug-treated mice was evaluated using the time for a 4-fold increase in tumor volume (RTV4) as endpoint. Considering monotherapy responses to PR-104, the strongly AKR1C3-expressing HepG2 xenografts showed highly significant tumor growth inhibition (P < 0.001), consistent with widespread activation of PR-104 by AKR1C3 in both aerobic and hypoxic regions of the tumor (). The low AKR1C3 tumors showed a non-significant trend to growth delay by PR-104 in the case of SNU-398 () and highly significant activity in Hep3B (). However, PR-104 had no activity in the high AKR1C3-expressing tumor with low hypoxic fraction, PLC/PRF/5 (). Sorafenib alone showed significant activity only against Hep3B (). The combination of PR-104 with sorafenib provided highly significant activity in all 4 tumor models, most strikingly in PLC/PRF/5 in which the combination was clearly synergistic given that neither agent alone showed activity ().
The synergy between sorafenib and PR-104 in the PLC/PRF/5 xenograft model is not due to increases in tumor hypoxia or pERK inhibition
Given that synergy between PR-104 and sorafenib was observed in PLC/PRF/5 xenografts, we asked whether sorafenib increases tumor hypoxia in this model. Mice with PLC/PRF/5 xenografts were administered daily sorafenib (80 mg/kg) for 5 d and treated with the hypoxia marker EF5 on day 6. The extent of tumor hypoxia in each model was assessed using immunostaining of EF5 (). The EF5 positive viable fraction was not significantly different in sorafenib treated mice, and H&E staining did not show evidence of increased necrosis (). Consistent with the unchanged EF5 binding, sorafenib also did not modify the levels of PR-104, PR-104A or its reduced metabolites in PLC/PRF/5 tumors ().
Figure 6. Effects of sorafenib on hypoxia, PR-104 metabolites and ERK phosphorylation in PLC/PRF/5 xenografts, and ERK phosphorylation and PR-104A sensitivity in PLC/PRF/5 cells in culture. (A) EF5 and H&E staining of vehicle or sorafenib-treated (80 mg/kg p.o., qdx5) PLC/PRF/5 tumors. Hypoxia probe EF5 (60 mg/kg, i.p.) was administered 24 hr after the final sorafenib dose, and tumors recovered 4.5 hr later. Representative EF5 and H&E images are shown. (B) Quantitation of area of hypoxia and necrosis from tumors treated with vehicle or sorafenib as in A. (C) Concentrations of PR-104 and its metabolites in tumors 30 min after dosing with PR-104 (250 mg/kg, IV), following pre-treatment with vehicle or sorafenib for 5 d as in A. (D) Western blot of pERK, ERK and β-actin from lysates of tumors from vehicle, PR-104 or PR-104+sorafenib-treated mice, using the same doses and schedules as in . Tumors were excised 30 min after the final PR-104 dose. Each lane is for a single tumor. Ratios of pERK/ERK (N = 4) are shown below the Westerns. (E) Western blot of pERK, ERK and β-actin from lysates of PLC/PRF/5 cells following a 2 h treatment with a range of sorafenib concentrations in vitro. (D) Clonogenic survival of PLC/PRF/5 cells under aerobic or anoxic conditions following a 2 hr exposure to PR-104A either in media only (circles) or following 3 day incubation with 8 μM sorafenib (squares).
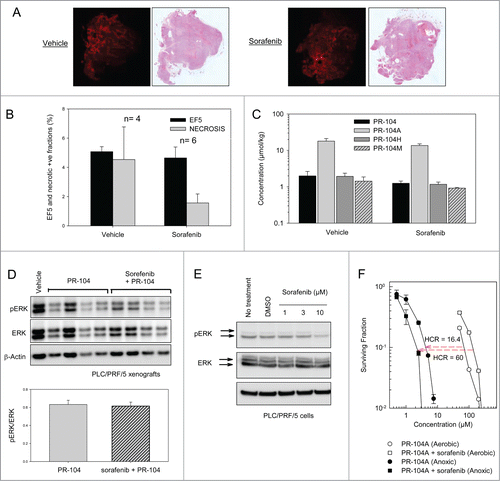
We also examined whether sorafenib at this dose and schedule inhibits phosphorylation of ERK in the PLC/PRF/5 xenograft model. Lysates of tumor samples were analyzed by Western blot to detect phosphorylated ERK (pERK), total ERK and β-actin. Band densitometry of pERK relative to total ERK or β-actin did not show any evidence of pERK inhibition in sorafenib treated tumors versus vehicle or PR-104-treated tumors (). We also examined whether sorafenib could inhibit ERK phosphorylation in PLC/PRF/5 cells in vitro. Consistent with previous reports,Citation28,29 we found a ∼50% inhibition of pERK/ERK ratios by band densitometry after a 2 h incubation with 10 μM sorafenib (). We next examined whether sorafenib could synergize with PR-104A in PLC/PRF/5 cells in vitro using a clonogenic assay. To mimic the prolonged exposure in vivo, cells were grown for 3 d without or with sorafenib (8 μM) which caused 53% reduction in cell number relative to controls by day 3. When cells were then exposed to PR-104A ± sorafenib under anoxic or aerobic conditions, sorafenib appeared to increase sensitivity to PR-104A under anoxic conditions, but it reduced sensitivity under aerobic conditions ().
Discussion
The original rationale in evaluating PR-104 in HCC patientsCitation22 was that this prodrug has the potential to exploit high expression of AKR1C3 as well as hypoxia in these tumors. The present study confirms high and uniform expression of AKR1C3 in most HCC surgical biopsies () and in 2 of the 4 HCC lines we investigated (HepG2 and PLC/PRF/5). The latter lines show correspondingly high levels of the reduced (activated) metabolites of PR-104A, hydroxylamine PR-104H and amine PR-104M (). In addition, we show that PR-104A metabolism and cytotoxicity is enhanced by hypoxia, even in the high AKR1C3 cell lines with extensive metabolic activation under aerobic conditions. It is also notable that the combination of PR-104 with sorafenib is well tolerated in mice, with no increase in toxicity relative to the single agents, and that the combination provides significant activity in all 4 xenograft models (). In these respects the present results validate the assumptions behind the previous clinical study. However, our study also points to a number of challenges that would need to be addressed for PR-104 or a related dinitrobenzamide mustard to find utility in the treatment of HCC.
The first of these challenges is that high AKR1C3 expression in HCC is not universal, with 2 surgical biopsies () and 2 of 4 cell lines (SNU-398, Hep3B) showing very low expression. Thus it would be essential to evaluate AKR1C3 expression in any such clinical study. Secondly, high AKR1C3 expression does not guarantee PR-104A sensitivity; PLC/PRF/5 cells in culture show AKR1C3 protein expression and enzymatic activity approaching that of HepG2 (), but are intrinsically resistant to the activated metabolites PR-104H and PR-104M and to the related DNA crosslinking agent melphalan (). We have not investigated the mechanism of this resistance, beyond demonstrating that it is not due to glucuronidation of PR-104A or its reduced metabolites (). However, the implication is that AKR1C3 expression alone is unlikely to be a reliable predictive biomarker, and that a more complete understanding of the molecular correlates of sensitivity would be needed. The present finding that expression of AKR1C3 (and the classic Nrf2 target gene NQO1) are upregulated in some HCC xenografts (PLC/PRF/5 and Hep3B) relative to monolayer cell cultures (), which likely reflects hypoxic generation of ROS and activation of Nrf2 signaling through multiple mechanisms,Citation30,31 emphasizes the importance of evaluating predictive biomarkers in the autochthonous human tumors rather than in patient-derived cell lines, even though the latter can inform selection of drug combinations in some settings.Citation32
An additional challenge is that the mechanism of synergy between PR-104 and sorafenib (as seen with PLC/PRF/5 xenografts, but not in the other xenograft lines) is not yet understood. The hypoxic fraction in PLC/PRF/5 xenografts following sorafenib treatment was not significantly different to untreated tumors () and we did not detect a reduction in pERK in PLC/PRF/5 xenografts following sorafenib treatment (), even though that finding has been reported in other studies.Citation28,29 Sorafenib did inhibit ERK phosphorylation in PLC/PRF/5 cells in vitro (), as previously reported.Citation28,29 Sorafenib has been shown to inhibit a wide range of receptor tyrosine kinases and activate aspects of the unfolded protein response in other cell line models,Citation33 any of these phenomena might contribute to the observed synergy between PR-104 and sorafenib. Indeed, sorafenib treatment resulted in increased sensitivity of PLC/PRF/5 cells in vitro to PR-104A under anoxic conditions and reduced sensitivity under aerobic conditions (); we have not investigated the underlying mechanism given that it is not clear what impact this in vitro phenomenon would have in PLC/PRF/5 xenografts given their low hypoxic fraction.
Finally, although the present study demonstrates activity of the PR-104/sorafenib combination in all 4 HCC xenograft lines, the doses of PR-104 (250 mg/day for 6 days) provide PR-104A exposure in mice well above that achievable in HCC patients. The 6 day schedule used here () has not been evaluated in humans, but the cumulative PR-104 dose of 1.5 g/kg body weight would represent a dose of 6.6 g/m2 in humans if allometrically scaled based on plasma pharmacokinetics of PR-104A.Citation34 While such cumulative doses are achievable over 2 cycles in haematological oncology patients,Citation35 they are not achievable in the HCC population for which a cumulative dose of 1.54 g/m2 over 2 cycles elicted severe dose-limiting thrombocytopaenia and neutropenia.Citation22 The development of dinitrobenzamide mustards that are insensitive to glucuronidation might help minimize changes in PR-104A clearance in HCC patients with compromised liver function, and might also avoid the dose-limiting gastrointestinal seen at high dose (4 g/m2) in leukemia patients.Citation35 However, it remains to be determined whether activation by AKR1C3 contributes to the apparent difference in PR-104 toxicity between mice (which lack AKR1C3) and humans, and whether prodrug activation by AKR1C3 is a valid therapeutic strategy in humans. In part to address this question, we are currently exploring PR-104 analogs that are activated by either AKR1C3 or hypoxia rather than by both.
Materials and Methods
Compounds
PR-104A, PR-104H, PR-104M, the O-glucuronide of PR-104A (PR-104G) and tetradeuterated derivatives of PR-104A and PR-104H were synthesized and purified as described previously.Citation8,23,36,37 Stocks of PR-104A were prepared in DMSO and stored at −20°C or −80°C. Stocks of PR-104H, PR-104M and PR-104G were prepared in acetonitrile and stored at −80°C. Coumberone and coumberolCitation38 and the AKR1C3 inhibitor SN34037Citation39,40 were synthesized in this laboratory and stored as DMSO stocks at −20°C prior to use. Clinical formulations of PR-104 were supplied by Proacta, Inc. (Lot No 309–01–001); each vial contained 200 mg PR-104 ((2-bromoethyl)-2-{[(2-hydroxyethyl)amino]carbonyl}-4,6-dinitroanilino)ethyl methanesulfonate phosphate ester, 8.5% (w/w) sodium carbonate and 0.8% (w/w) D-mannitol. The added solvent was 2 ml of water for injection and PBS diluent. Sorafenib was purchased from American Custom Chemicals Corporation (San Diego, CA) and formulated as a suspension in Cremophor EL/ethanol (50:50) and diluted 4x with sterile endotoxin-free water, as previously described.Citation28,33
Cell lines
PLC/PRF/5, HepG2, Hep3B and SNU-398 human HCC tumor cells and H1299 human non-small cell lung cancer cells were obtained from American Type Culture Collection (Rockville, MD) and cultured in αMEM (Hep3B in DMEM) containing 10% fetal bovine serum (FBS) in a 5% CO2 incubator at 37°C. All cell lines were grown in the absence of antibiotics for < 3 months from frozen stocks confirmed to be mycoplasma free by PCR-ELISA (Roche). H1299 cells were genetically engineered to over-express AKR1C3 as described previously.Citation14
Antiproliferative (IC50) assays
Cells were harvested, counted and seeded at a density of 2000 (HepG2, SNU-398 and PLC/PRF/5) or 3000 (Hep3B) cells/well in 96 well plates (Nunc). Cells were incubated for 2 h to allow attachment, then exposed to drugs using 3-fold serial dilutions in duplicate, and incubated for a further 4 hr. For PR-104H, which has a half-life of ∼ 10 min in medium at 37°C,Citation41 the dilutions from acetonitrile stock solutions were made immediately before use. Subsequently cells were washed 2x with medium and allowed to regrow for 5 d Cultures were then stained with sulphorhodamine B to measure total cells.Citation42 The absorbance-concentration plots were fitted using 4-parameter logistic regression, and IC50 was determined by interpolation as the drug concentration that reduced staining to 50% of controls on the same plate.
Clonogenic assays
Cell killing was assessed by clonogenic assay (loss of colony forming potential) using a 96-well format for drug exposure as described previously.Citation13 Briefly, cells were harvested, counted and 5 × 106 cells pelleted (220 g, 5 min). Pellets were overlayed with 100 μl media, transferred into a standard culture hood or a 5% H2/palladium catalyst Bactron anaerobic chamber (Sheldon Manufacturing, Cornelius, OR) and resuspended in 2.4 mL of culture medium. Cells (3 × 105 /well in 150 μL) were plated into 96-well plates and incubated at 37°C for 2 hr. PR-104A or PR-104H stock solutions were diluted in media to 2x final concentration. 150 μL of these drug dilutions were transferred to the cultures and the plate incubated at 37°C for 2 hr. Anoxic plates were removed from the anaerobic chamber, cells were trypsinised, transferred to microtubes and centrifuged (220 g, 5 min). Media was aspirated and cells resuspended in 500 μl fresh medium (nominally 6 × 105 cells/ml), and dilutions were prepared to transfer 105, 104, 103 and 102 cells in 450 μl into triplicate 60mm cell culture dishes containing 4.5 ml culture medium (αMEM with 5% FBS). For HepG2, 6-well plates were used for the clonogenic assay, with addition of 104 lethally irradiated (40 Gy, cobalt-60 gamma irradiation) HepG2 cells as feeders. The plates were incubated at 37°C for 13 days, stained with methylene blue and colonies with >50 cells were counted. Surviving fraction was calculated as the ratio of colonies from treated/control wells.
Determination of PR-104A metabolism by LC/MS/MS
Cells (5 × 105 cells per well in 24-well plate) were pre-incubated at 37°C for 2 hr under oxic or anoxic conditions as above, followed by addition of PR-104A to 100 μM. Plates were incubated for a further 1 hr, samples extracted with 2 volumes of ice-cold methanol containing tetra-deuterated stable isotope internal standards, and stored at -80°C prior to analysis by LC–MS/MS as previously described.Citation8 Briefly, samples were analyzed with an Agilent 1200 HPLC coupled with a triple quadrupole mass spectrometer (Model 6410, Agilent Technologies). Separation was achieved using an acetonitrile/water gradient on an Agilent Eclipse XDB-C18 Rapid Resolution HT (50 mm × 2.1 mm, 1.8 µm) column with 6 min run time. The m/z transitions monitored were 499 > 403 (PR-104A), 505 > 409 (PR-104A-d4), 485 > 389 (PR-104H), 491 > 395 (PR-104H-d4), 469 > 373 (PR-104M), 475 > 379 (PR-104M-d4) and 675 > 499 (PR-104G).
Measurement of AKR1C3 activity in cells
AKR1C3 enzyme activity was quantified using a recently validated assayCitation43 in which the AKR1C3 inhibitor SN34037 is used to identify AKR1C3-specific reduction of the pan AKR1C fluorogenic substrate coumberone to fluorescent coumberol.Citation44 Briefly, white 96 well plates (Falcon 35–3296, Becton Dickinson) containing 2 × 104 cells/well in 100 μl phenol red-free media were equilibrated at 37°C in a 5% CO2 incubator for 24 hr. After addition of 20 μl media only or media containing SN34037 (final concentration 1 μM), the plate was incubated for 1 hr. An additional 20 μl of media containing coumberone (final concentration 10 μM) was added to each well and the plates incubated for a further 3 h (HepG2) or 24 hr (other lines). Fluorescence intensity was measured at an excitation of 385 nm and an emission of 510 nm (cutoff 495 nm) on a SpectraMax M2 plate reader (Molecular Devices). The signal was quantitated against a coumberol calibration curve (0.1–10 µM) on the same plate. AKR1C3 activity was calculated as SN34037-sensitive coumberol formation.
Western immunoblotting
Cell lysates were prepared in radioimmunoprecipitation assay buffer and 20 μg of protein was loaded on SDS-PAGE gels (NuPAGE 4–12% gradient gels; Life Technologies, Carlsbad, CA) before transfer to nitrocellulose. Blots were probed with the following primary antibodies: β-actin (MAB1501R; Chemicon, Temecula, CA), AKR1C3 (A6229, Sigma-Aldrich, Australia) or POR (sc25263, Santa Cruz Biotech, Dallas, TX) followed by a HRP-conjugated secondary antibody (sc2055, Santa Cruz Biotech) and detected using chemiluminescent ECL (Supersignal, Thermoscientific) and visualization using an ImageReader LAS-3000 (Fujifilm, Tokyo, Japan). Band densitometry was carried out using ImageJ software (v1.37; National Institutes of Health, Bethseda, MD).
Xenograft models and treatment
All animal studies were approved by the University of Auckland Animal Ethics Committee. Tumors for the growth delay experiments were grown subcutaneously after inoculation of 5 × 106 (HepG2, Hep3B, PLC/PRF/5) or 7.5 × 106 (SNU-398) cells in the flanks of female mice; NOD/SCID mice were used for HepG2 and Hep3B cells, NIH-III nude mice (NIH-Lystbg-JFoxn1nuBtkxid) were used for PLC/PRF/5 cells and Rag-1−/- Balb/c mice were used for SNU-398 cells. Animals were stratified to experimental groups after tumors had achieved a mean diameter of 6 mm. Sorafenib (80 mg/kg) was given daily (days 1–5) by oral gavage. PR-104 (250 mg/kg) in PBS was administered i.p. daily (days 1–6). Control animals received either Cremophor EL/ethanol (50:50) diluted 4x with sterile endotoxin-free water or PBS alone. The median time for tumors to increase in volume 4-fold relative to start of treatment (RTV4) was used to evaluate antitumour efficacy. Animal body weight was measured daily during the treatment period and 3 times weekly thereafter with any animals losing >20% of their initial body weight being culled and recorded as unscheduled deaths for the purpose of Kaplan Meier analysis.
Tumors for the modulation of hypoxia study were grown subcutaneously in both flanks by inoculating 5 × 106 PLC/PRF/5 cells. Treatment started when xenografts reached 150 mm3 and sorafenib (80 mg/kg) was administered daily, p.o., for 5 d On day 6 animals were treated with EF5 at 60 mg/kg, i.p., and 4 h later mice were dosed with PR-104 (250 mg/kg), i.v., and culled after 30 minutes. Half of each tumor was immediately frozen in liquid nitrogen and stored at −80°C for western blotting and for determination of PR-104 and its metabolites by LC/MS/MS as previously.Citation8 The second half was formalin-fixed and paraffin-embedded (FFPE) for H&E and EF5 immunofluorescence staining.
Immunohistochemistry
Single sections 5 μm thick were cut from each tumor and mounted on glass slides. Hypoxic regions were identified by staining of EF5 or pimonidazole protein adducts. Briefly, after de-paraffinization, antigen retrieval (10 mM sodium citrate buffer pH 6.0, 25 min, >100°C) and blocking (10% goat serum, 1 hr, RT), sections were stained with a Cy5-conjugated mouse anti-EF5 (100 μg/ml) antibody (kindly provided by Dr C.J. Koch, Philadelphia, PA) or a fluorescein-conjugated mouse IgG1 monoclonal anti-pimonidazole antibody (dil 1:25, HypoxyprobeTM-1 Plus kit, Hypoxyprobe, Burlington, MA). Whole-section montage images were acquired using a Zeiss LSM710 inverted laser scanning confocal microscope with a 10x objective and a motorized X-Y stage. Tissue sections were subsequently stained with haematoxylin to delineate regions of necrosis.
Immunohistochemistry for AKR1C3 was performed in accordance with protocols validated at Mosaic Laboratories (Lake Forest, CA) as described previously.Citation14 Briefly, following mounting onto slides, deparaffinization and antigen retrieval steps, slides were incubated with anti-AKR1C3 antibody (Sigma) for 30 minutes. Detection was by the Envision+ Mouse HRP Detection Kit (DakoCytomation) and counterstaining with haematoxylin (DakoCytomation). Photomicrographs were acquired with a Spot Insight QE Model 4.2 cooled CCD camera (Diagnostic Instruments) attached to a Nikon Eclipse 50i microscope using a 40x objective magnification. Staining was evaluated by a certified pathologist using a 4 point score: 0 indicates no staining, 1+ indicates weak staining, 2+ indicates moderate staining, and 3+ indicates strong staining.
Statistical analysis
Unless otherwise indicated, values are means and errors are SEM for multiple independent experiments, and significance of differences between groups was tested by 2-tailed Student's t-test (two groups) or one way ANOVA (multiple groups) with evaluation of pairwise differences using the Holm-Sidak method. Significance of activity against xenografts were tested with RTV4 as endpoint, using the Log-Rank test with Holm-Sidak multiple comparison post-test analysis.
Disclosure of Potential Conflicts of Interest
AVP and WRW are inventors on patents related to PR-104.
Acknowledgments
We thank Chenbo Wu, Jie Li and Dr Kevin Sun for assistance with the xenograft model assays.
Funding
This study was funded by the Health Research Council of New Zealand (grant 111103).
References
- Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nature Reviews Cancer 2008; 8:967-75; PMID:18987634; http://dx.doi.org/10.1038/nrc2540
- Bristow RG, Hill RP. Hypoxia, DNA repair and genetic instability. Nat Rev Cancer 2008; 8:180-92; PMID:18273037; http://dx.doi.org/10.1038/nrc2344
- Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer 2008; 8:851-64; PMID:18846101; http://dx.doi.org/10.1038/nrc2501
- Lunt SJ, Chaudary N, Hill RP. The tumor microenvironment and metastatic disease. Clin Exp Metastasis 2009; 26:19-34; PMID:18543068; http://dx.doi.org/10.1007/s10585-008-9182-2
- Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nature Reviews Cancer 2011; 11:393-410; PMID:21606941; http://dx.doi.org/10.1038/nrc3064
- Patterson AV, Ferry DM, Edmunds SJ, Gu Y, Singleton RS, Patel K, Pullen SM, Hicks KO, Syddall SP, Atwell GJ, et al. Mechanism of action and preclinical antitumor activity of the novel hypoxia-activated DNA crosslinking agent PR-104. Clin Cancer Res 2007; 13:3922-32; PMID:17606726; http://dx.doi.org/10.1158/1078-0432.CCR-07-0478
- Patel K, Lewiston D, Gu Y, Hicks KO, Wilson WR. Analysis of the hypoxia-activated dinitrobenzamide mustard phosphate prodrug PR-104 and its alcohol metabolite PR-104A in plasma and tissues by liquid chromatography-mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 2007; 856:302-11; PMID:17644498; http://dx.doi.org/10.1016/j.jchromb.2007.06.035
- Gu Y, Wilson WR. Rapid and sensitive ultra-high-pressure liquid chromatography-tandem mass spectrometry analysis of the novel anticancer agent PR-104 and its major metabolites in human plasma: Application to a pharmacokinetic study. J Chromatogr B Analyt Technol Biomed Life Sci 2009; 877:3181-6; PMID:19709934; http://dx.doi.org/10.1016/j.jchromb.2009.08.009
- Hicks KO, Myint H, Patterson AV, Pruijn FB, Siim BG, Patel K, Wilson WR. Oxygen dependence and extravascular transport of hypoxia-activated prodrugs: comparison of the dinitrobenzamide mustard PR-104A and tirapazamine. Int J Radiat Oncol Biol Phys 2007; 69:560-71; PMID:17869669; http://dx.doi.org/10.1016/j.ijrobp.2007.05.049
- Singleton RS, Guise CP, Ferry DM, Pullen SM, Dorie MJ, Brown JM, Patterson AV, Wilson WR. DNA crosslinks in human tumor cells exposed to the prodrug PR-104A: relationships to hypoxia, bioreductive metabolism and cytotoxicity. Cancer Res 2009; 69:3884-91; PMID:19366798; http://dx.doi.org/10.1158/0008-5472.CAN-08-4023
- Gu Y, Patterson AV, Atwell GJ, Chernikova SB, Brown JM, Thompson LH, Wilson WR. Roles of DNA repair and reductase activity in the cytotoxicity of the hypoxia-activated dinitrobenzamide mustard PR-104A. Molecular Cancer Therapeutics 2009; 8:1714-23; PMID:19509245; http://dx.doi.org/10.1158/1535-7163.MCT-08-1209
- Guise CP, Abbattista MR, Tipparaju SR, Lambie NK, Su J, Li D, Wilson WR, Dachs GU, Patterson AV. Diflavin oxidoreductases activate the bioreductive prodrug PR-104A under hypoxia. Mol Pharmacol 2012; 81:31-40; PMID:21984255; http://dx.doi.org/10.1124/mol.111.073759
- Guise CP, Wang A, Thiel A, Bridewell D, Wilson WR, Patterson AV. Identification of human reductases that activate the dinitrobenzamide mustard prodrug PR-104A: a role for NADPH:cytochrome P450 oxidoreductase under hypoxia. Biochem Pharmacol 2007; 74:810-20; PMID:17645874; http://dx.doi.org/10.1016/j.bcp.2007.06.014
- Guise CP, Abbattista M, Singleton RS, Holford SD, Connolly J, Dachs GU, Fox SB, Pollock R, Harvey J, Guilford P, et al. The bioreductive prodrug PR-104A is activated under aerobic conditions by human aldo-keto reductase 1C3. Cancer Res 2010; 70:1573-84; PMID:20145130; http://dx.doi.org/10.1158/0008-5472.CAN-09-3237
- Wu XZ, Xie GR, Chen D. Hypoxia and hepatocellular carcinoma: The therapeutic target for hepatocellular carcinoma. J Gastroenterol Hepatol 2007; 22:1178-82; PMID:17559361; http://dx.doi.org/10.1111/j.1440-1746.2007.04997.x
- Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008; 359:378-90; PMID:18650514; http://dx.doi.org/10.1056/NEJMoa0708857
- Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS, Luo R, Feng J, Ye S, Yang TS, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol 2009; 10:25-34; PMID:19095497; http://dx.doi.org/10.1016/S1470-2045(08)70285-7
- Abdel-Rahman O, Fouad M. Sorafenib-based combination as a first line treatment for advanced hepatocellular carcinoma: a systematic review of the literature. Crit Rev Oncol Hematol 2014; 91:1-8; PMID:24457121; http://dx.doi.org/10.1016/j.critrevonc.2013.12.013
- Murakami M, Zhao S, Zhao Y, Chowdhury NF, Yu W, Nishijima KI, Takiguchi M, Tamaki N, Kuge Y. Evaluation of changes in the tumor microenvironment after sorafenib therapy by sequential histology and 18F-fluoromisonidazole hypoxia imaging in renal cell carcinoma. Int J Oncol 2012; 41(5):1593-600; PMID:22965141; http://dx.doi.org/10.3892/ijo.2012.1624
- Chang YS, Adnane J, Trail PA, Levy J, Henderson A, Xue D, Bortolon E, Ichetovkin M, Chen C, McNabola A, et al. Sorafenib (BAY 43-9006) inhibits tumor growth and vascularization and induces tumor apoptosis and hypoxia in RCC xenograft models. Cancer Chemother Pharmacol 2007; 59:561-74; PMID:17160391; http://dx.doi.org/10.1007/s00280-006-0393-4
- Liang Y, Zheng T, Song R, Wang J, Yin D, Wang L, Liu H, Tian L, Fang X, Meng X, et al. Hypoxia-mediated sorafenib resistance can be overcome by EF24 through Von Hippel-Lindau tumor suppressor-dependent HIF-1alpha inhibition in hepatocellular carcinoma. Hepatology 2013; 57:1847-57; PMID:23299930; http://dx.doi.org/10.1002/hep.26224
- Abou-Alfa GK, Chan SL, Lin CC, Chiorean EG, Holcombe RF, Mulcahy MF, Carter WD, Patel K, Wilson WR, Melink TJ, et al. PR-104 plus sorafenib in patients with advanced hepatocellular carcinoma. Cancer Chemother Pharmacol 2011; 68:539-45; PMID:21594722; http://dx.doi.org/10.1007/s00280-011-1671-3
- Gu Y, Tingle MD, Wilson WR. Glucuronidation of anticancer prodrug PR-104A: Species differences, identification of human UDP-glucuronosyltransferases and implications for therapy. J Pharmacol Exp Ther 2011; 337:692-702; PMID:21427202; http://dx.doi.org/10.1124/jpet.111.180703
- Gu Y, Atwell GJ, Wilson WR. Metabolism and excretion of the novel bioreductive prodrug PR-104 in mice, rats, dogs and humans. Drug Metab Dispos 2010; 38:498-508; PMID:20019245; http://dx.doi.org/10.1124/dmd.109.030973
- MacLeod AK, McMahon M, Plummer SM, Higgins LG, Penning TM, Igarashi K, Hayes JD. Characterization of the cancer chemopreventive NRF2-dependent gene battery in human keratinocytes: demonstration that the KEAP1-NRF2 pathway, and not the BACH1-NRF2 pathway, controls cytoprotection against electrophiles as well as redox-cycling compounds. Carcinogenesis 2009; 30:1571-80; PMID:19608619; http://dx.doi.org/10.1093/carcin/bgp176
- Venugopal R, Jaiswal AK. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc Natl Acad Sci U S A 1996; 93:14960-5; PMID:8962164; http://dx.doi.org/10.1073/pnas.93.25.14960
- Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 1997; 236:313-22; PMID:9240432; http://dx.doi.org/10.1006/bbrc.1997.6943
- Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D, Wilhelm S, Lynch M, Carter C. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res 2006; 66:11851-8; PMID:17178882; http://dx.doi.org/10.1158/0008-5472.CAN-06-1377
- Liu LP, Ho RL, Chen GG, Lai PB. Sorafenib inhibits hypoxia-inducible factor-1alpha synthesis: implications for antiangiogenic activity in hepatocellular carcinoma. Clin Cancer Res 2012; 18:5662-71; PMID:22929805; http://dx.doi.org/10.1158/1078-0432.CCR-12-0552
- Bryan HK, Olayanju A, Goldring CE, Park BK. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem Pharmacol 2013; 85:705-17; PMID:23219527; http://dx.doi.org/10.1016/j.bcp.2012.11.016
- Cullinan SB, Diehl JA. Coordination of ER and oxidative stress signaling: the PERK/Nrf2 signaling pathway. Int J Biochem Cell Bio 2006; 38:317-32; PMID:16290097; http://dx.doi.org/10.1016/j.biocel.2005.09.018
- Crystal AS, Shaw AT, Sequist LV, Friboulet L, Niederst MJ, Lockerman EL, Frias RL, Gainor JF, Amzallag A, Greninger P, et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 2014; 346:1480-6; PMID:25394791; http://dx.doi.org/10.1126/science.1254721
- Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res 2004; 64:7099-109; PMID:15466206; http://dx.doi.org/10.1158/0008-5472.CAN-04-1443
- Patel K, Choy SF, Hicks KO, Melink TJ, Holford NHG, Wilson WR. A combined pharmacokinetic model for the hypoxia-targeted prodrug PR-104A in humans, dogs, rats and mice predicts species differences in clearance and toxicity. Cancer Chemother Pharmacol 2011; 67:1145-55; PMID:20683596; http://dx.doi.org/10.1007/s00280-010-1412-z
- Konopleva M, Thall PF, Arana Yi C, Borthakur G, Coveler A, Bueso-Ramos C, Benito J, Konoplev S, Gu Y, Ravandi F, et al. Phase I/II study of the hypoxia-activated prodrug PR104 in refractory/relapsed acute myeloid and acute lymphoblastic leukemia. Haematologica 2015; PMID:25682597; http://dx.doi.org/10.3324/haematol.2014.118455
- Yang S, Atwell GJ, Denny WA. Synthesis of asymmetric halomesylate mustards with aziridineethanol/alkali metal halides: application to an improved synthesis of the hypoxia prodrug PR-104. Tetrahedron 2007; 63:5470-6; http://dx.doi.org/10.1016/j.tet.2007.04.044
- Atwell GJ, Denny WA. Synthesis of 3H- and 2H4-labelled versions of the hypoxia-activated pre-prodrug 2-[(2-bromoethyl)-2,4-dinitro-6-[[[2-(phosphonooxy)ethyl]amino]carbonyl]anilino]ethyl methanesulfonate (PR-104). J Labelled Comp Radiopharm 2007; 50:7-12; http://dx.doi.org/10.1002/jlcr.1147
- Yee DJ, Balsanek V, Bauman DR, Penning TM, Sames D. Fluorogenic metabolic probes for direct activity readout of redox enzymes: Selective measurement of human AKR1C2 in living cells. Proc Natl Acad Sci U S A 2006; 103:13304-9; PMID:16938874; http://dx.doi.org/10.1073/pnas.0604672103
- Flanagan JU, Atwell GJ, Heinrich DM, Brooke DG, Silva S, Rigoreau LJ, Trivier E, Turnbull AP, Raynham T, Jamieson SM, et al. Morpholylureas are a new class of potent and selective inhibitors of the type 5 17-b-hydroxysteroid dehydrogenase (AKR1C3). Bioorg Med Chem 2014; 22:967-77; PMID:24411201; http://dx.doi.org/10.1016/j.bmc.2013.12.050
- Jamieson SM, Gu Y, Manesh DM, El-Hoss J, Jing D, MacKenzie KL, Guise CP, Foehrenbacher A, Pullen SM, Benito J, et al. A novel fluorometric assay for aldo-keto reductase 1C3 predicts metabolic activation of the nitrogen mustard prodrug PR-104A in human leukaemia cells. Biochem Pharmacol 2014; 88:36-45; PMID:24434189; http://dx.doi.org/10.1016/j.bcp.2013.12.019
- Foehrenbacher A, Patel K, Abbattista M, Guise CP, Secomb TW, Wilson WR, Hicks KO. The role of bystander effects in the antitumor activity of the hypoxia-activated prodrug PR-104. Front Oncol 2013; 3:263; PMID:24109591
- Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc 2006; 1:1112-6; PMID:17406391; http://dx.doi.org/10.1038/nprot.2006.179
- Jamieson SM, Gu Y, Manesh DM, El-Hoss J, Jing D, MacKenzie KL, Guise CP, Foehrenbacher A, Pullen SM, Benito J, et al. A novel fluorometric assay for aldo-keto reductase 1C3 predicts metabolic activation of the nitrogen mustard prodrug PR-104A in human leukaemia cells. Biochem Pharmacol 2014; 88:36-45; PMID:24434189; http://dx.doi.org/10.1016/j.bcp.2013.12.019
- Halim M, Yee DJ, Sames D. Imaging induction of cytoprotective enzymes in intact human cells: coumberone, a metabolic reporter for human AKR1C enzymes reveals activation by panaxytriol, an active component of red ginseng. J Am Chem Soc 2008; 130:14123-8; PMID:18826220; http://dx.doi.org/10.1021/ja801245y