
Abstract
In the present study, we examined the mechanisms of oxaliplatin-induced drug resistance in human colorectal cancer cell lines HT29 and HCT116. Our results demonstrate a significant autophagy expression in CRC cells after an oxaliplatin treatment. Administration of oxaliplatin to human CRC cells significantly enhanced the expression of HMGB1, which regulated the autophagy response and negatively regulate the cell apoptosis. Moreover, a decreased oxaliplatin -induced autophagy response and an increased apoptosis level were detected in stable CRC cells harboring HMGB1 shRNA. Then we noted that HMGB1 significantly induced extracellular signal-regulated kinase (ERK)/Extracellular signal-regulated kinase kinase (MEK) phosphorylation. Taken together, these data suggest that HMGB1-mediated autophagy modulates sensitivity of colorectal cancer cells to oxaliplatin via MEK/ERK signaling pathway.
Abbreviation
| CRC | = | Colorectal cancer |
| HMGB1 | = | High–mobility group box 1 protein |
| 5-FU | = | 5-fluorouracil |
| 3-MA | = | 3-methyladenosine |
| shRNA | = | short hairpin RNA |
| CCK8 | = | Cell Counting Kit-8 |
| ERK | = | extracellular signal-regulated kinase |
| MEK | = | extracellular signal-regulated kinase kinase |
Introduction
Colorectal cancer (CRC) is now a major health issue in the industrialized countries and the third most commonly diagnosed cancer worldwide.Citation1 The number of new cases of colorectal cancer keeps increasing and 50% of newly diagnosed patients develop metastatic disease.Citation2 At present, major drugs for chemotherapy of colon cancer are oxaliplatin and 5-fluorouracil (5-FU). 5-FU, for a long time, was the only drug used in advanced colorectal cancer therapy until 2 novel chemotherapeutic agents, oxaliplatin and irinotecan in combination with 5-FU, were used and shown the clinical benefit.Citation3,4 Oxaliplatin, a third generation of platinum-based compound with a wide use to the metastatic colorectal cancer, disrupts DNA replication and transcription by covalently binding to DNA, generating platinum-DNA adducts.Citation5
Despite the advanced cancer therapy, the prognosis of CRC remains poor with a median overall survival of 12 to 18 months and the response to the chemotherapy using 5-FU plus is only 50%.Citation6,7 Eventually, all metastatic CRC will have resistance effect to the oxaliplatin when it comes to the progression of 8 months.Citation8 Resistance to oxaliplatin becomes a big concern in the chemotherapy of the metastatic CRC. A lot of efforts have been put on the oxaliplatin resistance study and a diverse array of hypotheses on the mechanism are reported like reduced apoptosis,Citation2 DNA repair,Citation9 drug detoxification,Citation10 etc. Lately, some studies have shown that autophagy is a response to certain forms of therapies including some chemotherapies and radiation therapy.Citation11,12 Autophagy, a catabolic process in which subcellular membranes undergo dynamic morphological changes that result in the degradation of cellular proteins and cytoplasmic organelles, is implicated in many physiological and pathophysiological conditions.Citation13 However, the exact roles of autophagy in affecting the efficacy of oxaliplatin in colon cancer remain unknown. In the chemotherapy with oxaliplatin, how autophagy determines and regulates the sensitivity of oxaliplatin in CRC still needs to be clearly defined.
High–mobility group box 1 protein (HMGB1), a well-known regulator of autophagy, is a highly conserved nuclear protein which functions as a chromatin-binding factor and promotes assembly of many transcriptional protein complexes. Besides the nuclear roles, HMGB1 also acts as an extracellular signaling molecule biding to RAGE and TLRs which leads to cell differentiation, cell migration, tumor progression and inflammation.Citation14,15
So far, the role of autophagy regulated by HMGB1 in the resistance of chemotherapy with oxaliplatin in CRC was still unknown. In this study, we want to investigate that; first, to assess whether oxaliplatin can cause autophagy in 2 CRC cell lines; second, whether this autophagy induced by oxaliplatin is regulated by HMGB1; third, to determine the signaling pathway involved in this process. Our data reveals that oxaliplatin-induced autophagy protects the CRC cells from the toxicity of this drug and this process is regulated by HMGB1 via the ERK/MEK pathway. This finding also support HMGB1 as a potential therapeutic target for enhancing the efficacy of oxaliplatin in CRCs.
Results
Autophagy induced by oxaliplatin protect the colorectal cancer cells from the cytotoxicity of oxaliplatin
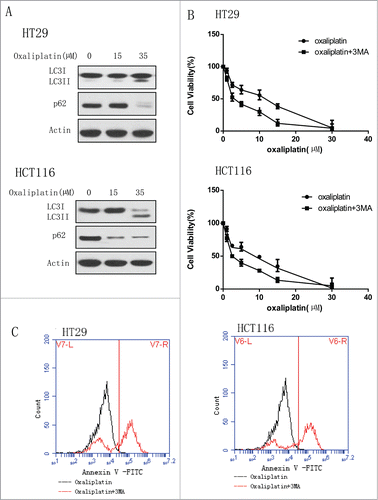
We used the human CRC cell lines HT29 and HCT116 to determine if oxaliplatin induces autophagy and this autophagy will affect the sensitivity of cancer cells to this drug. We first tested the activity of autophagy in colorectal cancer cells treated with oxaliplatin. Treatment of the human colorectal cancer cell lines HT29 and HCT116 with oxaliplatin caused a dose-dependent activation of autophagy. Western blotting analyses showed that the conversion of LC3 I to LC3II was increased and p62 level was decreased (). Then, we wanted to determine whether autophagy induced by oxaliplatin will protect the cancer cells from the cytotoxicity of oxaliplatin. We treated the human CRC cell lines HT29 and HCT116 with oxaliplatin alone or did the co-treatment with oxaliplatin and inhibitors of autophagy, 3-MA (). We examined the cell viability by CCK8 and found that the group treated with only oxaliplatin are more resistant to the drug compared to the group of co- treatment, indicating the protective role of autophagy in cancer cells exposed to the oxaliplatin. Moreover, consistent with the results above, the co-treatment with 3-MA enhanced the CRC cells apoptosis ().
Figure 1 (See previous page). Oxaliplatin induces the autophagy in colorectal cancer cells and inhibition of autophagy enhances the chemotherapy sensitivity. (A) HT29 and HCT116 were treated with the indicated concentration of oxaliplatin for 24 hours. At the end of treatment, cell lysates were prepared, resolved by SDS-PAGE, and subjected to Western blot analysis using anti-LC3, anti-p62 and anti-actin antibodies, respectively. Actin was used as a loading control. (B) HT29 and HCT116 cells were treated with the indicated concentrations of oxaliplatin for 24 h in the presence or absence of 3-MA (2 mM).At the end of treatment, cell viability was measured by CCK8. (C) HT29 and HCT116 were treated with oxaliplatin (35 µM) for 24 hours in the presence or absence of 3-MA (2 mM) and then the level of apoptosis was determined by flow cytometric analysis of Annexin V staining.
The expression and release of HMGB1 in colorectal cancer cells are increased upon the oxaliplatin treatment
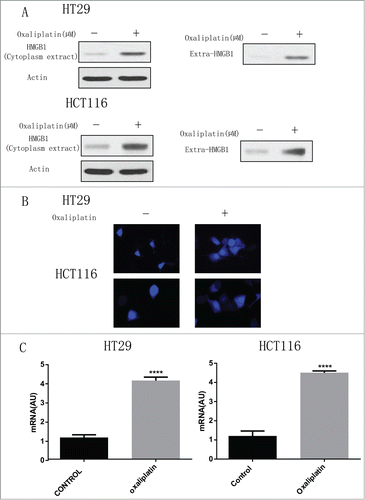
Since HMGB1 is well known as an important regulator of autophagy,Citation14,16 we wanted to determine the level of HMGB1 in CRC cells treated by oxaliplatin. First, we examined the effects of the anticancer agent oxaliplatin on the expression of HMGB1. We treated these 2 human CRC cell lines with oxaliplatin (35 µM) for 24 h, followed by the western blotting assay (). Oxaliplatin increased the level of HMGB1 in the cytoplasm and significantly enhanced expression of HMGB1 in supernatant, (). Consistent with the protein gel blotting results, we also noted that the release of HMGB1 was significantly elevated in both HT29 and HCT116 cells after treated with oxaliplatin at 35 µM for 30 min from microscopic analysis (). Moreover, real-time PCR showed that HMGB1 level was largely increased after the treatment of oxaliplatin for 24 hours (). These results suggest that oxaliplatin promote the HMGB1 expression in CRC cells.
Figure 2. Oxaliplatin induces HMGB1 expression in colorectal cancer cells. (A) HT29 and HCT116 cells were treated with 35 µM of oxaliplatin for 24h. At the end of treatment, Cytoplasmic fractions, and concentrated conditioned media, were prepared, resolved by SDS-PAGE, and subjected to Western blot analysis of HMGB1. Actin was used as a loading control. Oxaliplatin at 35 µM induce HMGB1 release from nuclear. (B) HT29 and HCT116 cells were treated with or without oxaliplatin at 35 µM for 30 min and then HMGB1 level was assayed microscopic analysis (n = 3; P < 0.05). (C) HT29 and HCT116 were treated with or without oxaliplatin (35 µM) for 24 hours and then HMGB1 mRNA level was measured by real-time PCR (n = 3, P < 0.05, untreated group was set as 1).
HMGB1 negatively regulates the sensitivity of colorectal cancer cells to oxaliplatin through autophagy pathway
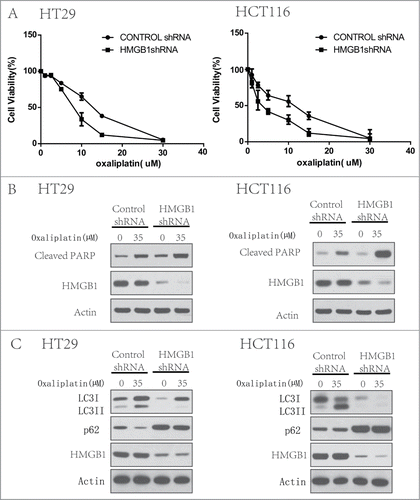
To determine the effect of HMGB1 in the regulation of sensitivity to oxaliplatin, in CRC cells, we transfected a shRNA from Sigma targeting HMGB1 into HT29 and HCT116 cells. HMGB1 shRNA transfection caused a significant decrease of both HMGB1 protein in these 2 cell lines (). At the end of 48 h treatment with oxaliplatin (35 µM), we tested the cell viability by CCK8. demonstrated that knockdown of HMGB1 expression in these cells significantly enhanced the cell sensitive to oxaliplatin compared to that in the cells transfected with the control siRNA. And this was largely relevant with an increased level of apoptosis () and partially block of autophagy (). These data indicates that HMGB1 increases the resistance of CRC cells to oxaliplatin and play an important role in regulating both autophagy and apoptosis.
Figure 3. Inhibition of HMGB1 increases chemotherapy sensitivity with oxaliplatin by regulating autophagy and apoptosis of colorectal cancer cells. HT29 and HCT116 cells were transfected with a control RNA or a shRNA targeting HMGB1, followed by treatment with 35 µM of oxaliplatin or vehicle for 24 h. At the end of treatment, cell viability was measured by CCK8 (A), cell lysates were prepared, resolved by SDS-PAGE, and subjected to Western blot analysis of HMGB1, cleaved-PARP (B) and LC3, p62 (C). Actin was used as a loading control. Results shown are the representative of 3 independent experiments.
HMGB1 regulates autophagy in colorectal cancer cells through MEK/ERK pathway
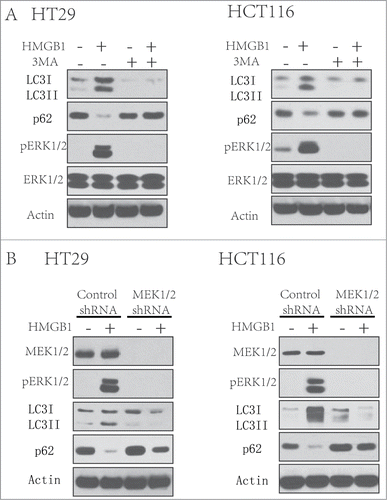
The extracellular signal-regulated kinase (ERK) cascade, which was the first MAPK to be defined, is activated by various stimuli, and functions in the regulation of cell proliferation, differentiation, survival, apoptosis and so on.Citation17 Extracellular signal-regulated kinase (MEK) acts as an upstream key protein of ERK. Nevertheless, whether this signaling pathway functions in autophagy induced by HMGB1 in chemotherapy of CRC cells was previously unknown. In this study, we then try to determine the role of MEK/ERK pathway in HMGB1-induced autophagy in CRC cell lines. We treated the CRC cells using HMGB1 (1 µg/ml) for 24 hours in the presence or absence of 3-MA(2 mM).We found that the autophagy and ERK1/2 pathway was activated by HMGB1 protein in both CRC cell lines (). And the autophagy inhibitor, 3-MA, inhibited HMGB1-induced ERK1/2 phosphorylation (). In next experiments, we knocked down the expression of MEK1/2 in HT29 and HCT116 cells with MEK1/2 shRNA and then treated these cells with HMGB1 (1 µg/ml) for 24 hours. Knockdown of MEK1/2 either in HT29/HCT116 cells can lead to decreased level of HMGB1-induced autophagy and ERK pathway activation, comparing to that in the cells transfected control shRNA (). These results indicate a key role of MEK/ERK pathway in regulating the HMGB1 induced autophagy.
Figure 4. MEK/ERK pathway is involved in the HMGB1-induced autophagy. (A) HT29 and HCT116 cells were treated with 3-MA (2 mM) after HMGB1 (1 µg/ml) treatment for 24 hours. At the end of treatment, LCI/II and p62 were analyzed by Western blot. (B) HT29 and HCT116 cells were transfected with a control RNA or a shRNA targeting MEK1/2 for 48 hours, followed by the treatment of HMGB1(1 µg/ml) for 24 hours. At the end of treatment, whole cell lysates were prepared, resolved by SDS-PAGE, and subjected to Western blot analysis. Actin was used as a loading control.
Discussion
Since last decade, notable progress has been made in the field of diagnosis, treatment and molecular mechanisms of CRC. The median survival for patients after advanced therapies has largely increased and is now crossing the 2 year level.Citation18 However, most of the CRC patients develop resistance to chemotherapy drugs like oxaliplatin and the cure rate remains low.Citation19 Drug resistance now becomes a big issue in the treatment of colon cancer.Citation16,20,21 Ten years ago, people already found 2 mechanisms which may be related to the oxaliplatin resistance: increased level of GSH induced by γ-glutamyl transpeptidaseCitation22 and a reduction in platinum accumulation and DNA-platinum adduct levels.Citation23 To date, many genes have already been discovered to be related to the oxaliplatin resistance. The expression level of glutathione and ERCC-1 seem to affect both of the inherent and acquired resistance to oxaliplatin.Citation23 In addition, DNA polymerase βCitation24 and phosphoserine aminotransferase PSAT1Citation25 are also shown the effects on the oxaliplatin resistance. Nevertheless, the molecular mechanism of the oxaliplatin resistance in colorectal cancer still needs to be clarified.
Autophagy is a process associated with the regulation of cancer development and progression. HMGB1, an important regulator of autophagy, is still not well characterized in its molecular mechanisms on how to regulate the sensitivity of CRC cells to anticancer drugs. So, first we examined the level of autophagy in CRC cells with the treatment of different dose of oxaliplatin. We found that the autophagy was induced by oxaliplatin in a dose-dependent manner and it will protect the cancer cells from the cytotoxicity of oxaliplatin. Then, we examined the effects of the oxaliplatin on the expression of HMGB1. We treated these 2 human CRC cell lines with oxaliplatin and detected the level of HMGB1 by western blotting (), microscopic analysis () and real-time PCR (). Our results suggest that oxaliplatin promote the HMGB1 expression in CRC cells and HMGB1 negatively regulates the sensitivity of colorectal cancer cells to oxaliplatin through autophagy pathway (). Furthermore, we found that the MEK/ERK pathway is involved in the regulation of HMGB1 induced autophagy response (). However, the response of these 2 CRC cell lines are sometimes different perhaps caused by the specific character of some genes. Additional experiments need to clarify what downstream gene will play an important role inside it.
In summary, our study shows that HMGB1 is released in the CRC cell lines after the chemotherapy with oxaliplatin and negatively regulate the sensitivity to this anticancer drug by activating the MEK/ERK pathway. As an important regulator of autophagy, HMGB1 also affects the apoptosis response at the same time. These findings give us promising evidence that the inhibition of autophagy and this pathway could have a therapeutic effect to the conventional oxaliplatin chemotherapy. Further studies are needed to characterize the HMGB1 function of downstream genes in CRC cells and to identify our hypothesis in vivo mice models.
Materials and Methods
Cell lines and culture
We used the human CRC cell lines, HT29 and HCT116 from American Type Culture collection (Manassas, VA, USA) for our experiments. Both cell lines were cultured in McCoy's 5A (Gibco, USA) or Iscove's modified Dul-becco's medium supplemented with 10% heat-inactivated fetal bovine serum (FBS), 2 mM glutamine and an antibiotic-antifungal mixture in a humidified incubator with 5% CO2 and 95% air. Both cell lines were confirmed according to American Type Culture collection instruction.
Antibodies and reagents
Oxaliplatin and 3-methyladenosine (3-MA) were purchased from Sigma (St. Louis, MO, USA); the recombinant HMGB1 protein was from Eli Lilly Company (Indianapolis, IN, USA). Nuclear and Cytoplasmic Extraction Reagents and Western blot reagents were obtained from Pierce Biotechnology (Rockford, IL, USA). The antibodies to cleaved-PARP, HMGB1, Phospho-extracellular signal-regulated kinase 1 and 2 (ERK1/2), ERK1/2, MEK1/2 and actin were obtained from Cell Signaling Technology (Danvers, MA, USA); anti-LC3I/II was purchased from Novus (Littleton, CO, USA); anti-p62 was from Santa Cruz biotechnology (Santa Cruz, CA, USA).
Small interfering RNA (siRNA) transfection
Short hairpin RNA (shRNA) against HMGB, MEK1/2 (Sigma) and control shRNA were transfected into cells by the Lipofectamine 2000 Transfection Reagent (Life Technologies, Carlsbad, CA, USA) according to the manufacturer's instructions. The concentration of siRNA was chosen by the dose-response studies.
Western blotting
The methods have been described previously.Citation26,27 The whole cell lysates were prepared with ice- cold cell lysis buffer and cleared by centrifugation. The total protein concentration was measured with the bicinchoninic acid assay Kit (Bio-Rad Laboratories). Proteins were resolved on a denaturing 8–12% SDS-PAGE gel and followed by transferring to polyvinylidene fluoride membranes by electroblotting. The membrane was then blocked in PBST containing 5% dried milk at room temperature for 1 hour, incubated with primary antibodies at 4°C overnight. Blots were incubated with appropriate secondary antibodies at room temperature for 1 hour the next day. The signals were detected by ECL reagent. Actin protein was used as an equivalent loading.
Cell viability assay
Cells were plated at a density of 5×104 cells/well in 96-well plates in 100 ml medium. After treatment, cell viability was assessed by the Cell Counting Kit-8 (Dojindo, Japan) test according to the manufacturer's instructions.Citation28 The trypan blue assay was also performed for the parallel analysis (data not included).
Apoptosis assays
The degree of apoptosis in CRC cells was measured by 1) an Annexin V –propidium iodide Apoptosis Detection Kit (BD PharMingen) by flow cytometric assay. This assay includes staining the cells with annexin V-FITC and propidium iodide to detect early apoptosis or late apoptosis/necrosis; 2) Western blot analysis of the cleaved PARP.
Measurement of HMGB1 release
HMGB1 released into cell culture supernatant was measured by protein gel blotting and immunofluorescence confocal microscopy. Cells were plated in 12-well plates containing sterile coverslips, allowed for 24 hours growth, and starved in serum free medium for 3 hours. After stimulation with 35 µM Oxaliplatin for 24 h at 37°C, the cells were fixed with 4% paraformaldehyde, permeabilized in 0.1% Triton X-100 in PBS and blocked in 3% BSA. Cells were stained with anti-HMGB1 antibodies, and probed with an Alexa Fluor 488-conjugated or 546-conjugated secondary antibody. Coverslips were mounted and visualized with confocal laser scanning microscopy (Olympus FV1000). Twenty-five images were analyzed.
Quantitative real-time PCR
mRNA level was determined by quantitative real-time polymerase chain reaction. RNA was extracted from 1×107 cells with TRIzol (Invitrogen) and cDNA from different samples were amplified with specific primers for HMGB1 (upper strand: 5′-TATGGCAAAAGCGGACAAGG-3′, lower strand: 5′- CTTCGCAACATCACCAATGGA-3′; GAPDH, upper strand: 5′-ATCAGCAATGCCTCCTGCAC-3′, lower strand: 5′-CGTCAAAGGTGGAGGAGTGG-3′)Citation29 Data were normalized to GAPDH expression and the untreated group was set as 1.
Statistical analysis
Data showed in this study are the mean values±SEM of at least 3 different independent experiments. Results were analyzed by student's t-test for the comparisons between the 2 groups. A significant difference was considered to be present at P < 0.05.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This research was supported by grants from the National Natural Science Foundation of China (30860257, 81101188 and 810701297), and Yunnan Provincial Department of Education (Number: ZD2011007).
References
- Bardelli A, Siena S. Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J Clin Oncol 2010; 28:1254-61; PMID:20100961; http://dx.doi.org/10.1200/JCO.2009.24.6116
- Gourdier I, Del Rio M, Crabbe L, Candeil L, Copois V, Ychou M, Auffray C, Martineau P, Mechti N, Pommier Y, et al. Drug specific resistance to oxaliplatin is associated with apoptosis defect in a cellular model of colon carcinoma. FEBS Lett 2002; 529:232-6; PMID:12372606; http://dx.doi.org/10.1016/S0014-5793(02)03347-1
- Napier MP, Ledermann JA. Novel chemotherapeutic agents in colorectal cancer. Eur J Surg Oncol 2000; 26:605-10; PMID:11034814; http://dx.doi.org/10.1053/ejso.2000.0955
- Waters J, Cunningham D. The changing face of chemotherapy in colorectal cancer. Br J Cancer 2001; 84:1-7; PMID:11139304; http://dx.doi.org/10.1054/bjoc.2000.1552
- Saif MW, Choma A, Salamone SJ, Chu E. Pharmacokinetically guided dose adjustment of 5-fluorouracil: a rational approach to improving therapeutic outcomes. J Natl Cancer Inst 2009; 101:1543-52; PMID:19841331; http://dx.doi.org/10.1093/jnci/djp328
- Adlard JW, Richman SD, Seymour MT, Quirke P. Prediction of the response of colorectal cancer to systemic therapy. Lancet Oncol 2002; 3:75-82; PMID:11902527; http://dx.doi.org/10.1016/S1470-2045(02)00648-4
- Poston GJ, Figueras J, Giuliante F, Nuzzo G, Sobrero AF, Gigot JF, Nordlinger B, Adam R, Gruenberger T, Choti MA, et al. Urgent need for a new staging system in advanced colorectal cancer. J Clin Oncol 2008; 26:4828-33; PMID:18711170; http://dx.doi.org/10.1200/JCO.2008.17.6453
- Goldberg RM, Sargent DJ, Morton RF, Fuchs CS, Ramanathan RK, Williamson SK, Findlay BP, Pitot HC, Alberts SR. A randomized controlled trial of fluorouracil plus leucovorin, irinotecan, and oxaliplatin combinations in patients with previously untreated metastatic colorectal cancer. J Clin Oncol 2004; 22:23-30; PMID:14665611; http://dx.doi.org/10.1200/JCO.2004.09.046
- Kweekel DM, Gelderblom H, Guchelaar HJ. Pharmacology of oxaliplatin and the use of pharmacogenomics to individualize therapy. Cancer Treat Rev 2005; 31:90-105; PMID:15847979; http://dx.doi.org/10.1016/j.ctrv.2004.12.006
- Wakasugi T, Izumi H, Uchiumi T, Suzuki H, Arao T, Nishio K, Kohno K. ZNF143 interacts with p73 and is involved in cisplatin resistance through the transcriptional regulation of DNA repair genes. Oncogene 2007; 26:5194-03; PMID:17297437; http://dx.doi.org/10.1038/sj.onc.1210326
- Livesey KM, Tang D, Zeh HJ, Lotze MT. Autophagy inhibition in combination cancer treatment. Curr Opin Investig Drugs 2009; 10:1269-79; PMID:19943199
- White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res 2009; 15:5308-16; PMID:19706824; http://dx.doi.org/10.1158/1078-0432.CCR-07-5023
- Liu L, Yang M, Kang R, Wang Z, Zhao Y, Yu Y, Xie M, Yin X, Livesey KM, Lotze MT, et al. HMGB1-induced autophagy promotes chemotherapy resistance in leukemia cells. Leukemia 2011; 25:23-31; PMID:20927132; http://dx.doi.org/10.1038/leu.2010.225
- Tang D, Kang R, Cheh CW, Livesey KM, Liang X, Schapiro NE, Benschop R, Sparvero LJ, Amoscato AA, Tracey KJ, et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 2010; 29:5299-310; PMID:20622903; http://dx.doi.org/10.1038/onc.2010.261
- Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol 2005; 5:331-42; PMID:15803152; http://dx.doi.org/10.1038/nri1594
- Livesey KM, Kang R, Vernon P, Buchser W, Loughran P, Watkins SC, Zhang L, Manfredi JJ, Zeh HJ 3rd, Li L, et al. p53/HMGB1 complexes regulate autophagy and apoptosis. Cancer Res 2012; 72:1996-2005; PMID:22345153; http://dx.doi.org/10.1158/0008-5472.CAN-11-2291
- Shaul YD, Seger R. The MEK/ERK cascade: from signaling specificity to diverse functions. Biochim Biophys Acta 2007; 1773:1213-26; PMID:17112607; http://dx.doi.org/10.1016/j.bbamcr.2006.10.005
- Cunningham D, Atkin W, Lenz HJ, Lynch HT, Minsky B, Nordlinger B, Starling N. Colorectal cancer. Lancet 2010; 375:1030-47; PMID:20304247; http://dx.doi.org/10.1016/S0140-6736(10)60353-4
- Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin 2014; 64:9-29; PMID:24399786; http://dx.doi.org/10.3322/caac.21208
- Xiu P, Dong X, Xu Z, Zhu H, Liu F, Wei Z, Zhai B, Kanwar JR, Jiang H, Li J, et al. Secretory clusterin contributes to oxaliplatin resistance by activating Akt pathway in hepatocellular carcinoma. Cancer Sci 2013; 104:375-82; PMID:23279642; http://dx.doi.org/10.1111/cas.12088
- Ekblad L, Johnsson A. Cetuximab sensitivity associated with oxaliplatin resistance in colorectal cancer. Anticancer Res 2012; 32:783-6; PMID:22399593
- el-akawi Z, Abu-hadid M, Perez R, Glavy J, Zdanowicz J, Creaven PJ, Pendyala L. Altered glutathione metabolism in oxaliplatin resistant ovarian carcinoma cells. Cancer Lett 1996; 105:5-14; PMID:8689632; http://dx.doi.org/10.1016/0304-3835(96)04245-0
- Hector S, Bolanowska-Higdon W, Zdanowicz J, Hitt S, Pendyala L. In vitro studies on the mechanisms of oxaliplatin resistance. Cancer Chemother Pharmacol 2001; 48:398-406; PMID:11761458; http://dx.doi.org/10.1007/s002800100363
- Iwatsuki M, Mimori K, Yokobori T, Tanaka F, Tahara K, Inoue H, Baba H, Mori M. A platinum agent resistance gene, POLB, is a prognostic indicator in colorectal cancer. J Surg Oncol 2009; 100:261-6; PMID:19330779; http://dx.doi.org/10.1002/jso.21275
- Vie N, Copois V, Bascoul-Mollevi C, Denis V, Bec N, Robert B, Fraslon C, Conseiller E, Molina F, Larroque C, et al. Overexpression of phosphoserine aminotransferase PSAT1 stimulates cell growth and increases chemoresistance of colon cancer cells. Mol Cancer 2008; 7:14; PMID:18221502; http://dx.doi.org/10.1186/1476-4598-7-14
- Ji C, Yang YL, Yang Z, Tu Y, Cheng L, Chen B, Xia JP, Sun WL, Su ZL, He L, et al. Perifosine sensitizes UVB-induced apoptosis in skin cells: new implication of skin cancer prevention? Cell Signal 2012; 24:1781-9; PMID:22584119; http://dx.doi.org/10.1016/j.cellsig.2012.05.003
- Ji C, Yang B, Yang Z, Tu Y, Yang YL, He L, Bi ZG. Ultra-violet B (UVB)-induced skin cell death occurs through a cyclophilin D intrinsic signaling pathway. Biochem Biophys Res Commun 2012; 425:825-9; PMID:22892127; http://dx.doi.org/10.1016/j.bbrc.2012.07.160
- Hamamoto R, Furukawa Y, Morita M, Iimura Y, Silva FP, Li M, Yagyu R, Nakamura Y. SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat Cell Biol 2004; 6:731-40; PMID:15235609; http://dx.doi.org/10.1038/ncb1151
- Liu F, Zhang Y, Peng Z, Gao H, Xu L, Chen M. High expression of high mobility group box 1 (hmgb1) predicts poor prognosis for hepatocellular carcinoma after curative hepatectomy. J Transl Med 2012; 10:135; PMID:22747650; http://dx.doi.org/10.1186/1479-5876-10-135