
Abstract
Constitutive activation of AKT is a frequent occurrence in the development of human T-cell acute lymphocytic leukemia/lymphomas (T-ALLs), due largely to inactivation of PTEN. Up regulation of MYC is also commonly observed in human T-ALLs. We previously demonstrated that expression of a constitutively active form of Lck-Akt2 alone is sufficient to initiate T-cell lymphoma in mice, and that tumor formation typically requires up regulation of Myc or Dlx5 caused by specific chromosomal rearrangements. Furthermore, Lck-Dlx5 mice develop T-ALLs that consistently acquire overexpression of Myc and activation of Akt, the latter due to loss of Pten expression. Proliferation of T-ALL cells from Lck-Dlx5 mice was found to be highly sensitive to the Akt pathway inhibitors BEZ235 and RAD001, as well as to JQ1, an inhibitor of bromodomain proteins, one of which (BRD4) regulates Myc transcription. Additionally, low concentrations of BEZ235 were found to cooperate with JQ1 to enhance cell cycle arrest. Higher concentrations of BEZ235 (≥0.5 µM) promoted cell death, although the addition of JQ1 did not result in a further increase in apoptosis. In contrast, the specific Myc inhibitor 10058-F4 caused apoptosis, and when combined with BEZ235 (≥0.5 µM), an enhanced effect on apoptosis was consistently observed. In addition, BEZ235 and RAD001 potentiated vincristine-induced apoptosis when the cells were treated with both drugs simultaneously, whereas pretreatment with BEZ235 antagonized the cell-killing effect of vincristine. Collectively, these experimental findings provide rationale for the design of novel combination therapies for T-ALL that includes targeting of AKT and MYC.
Introduction
Constitutive activation of AKT, activating mutations of NOTCH1, and up regulation of MYC are major oncogenic drivers in human acute T-cell lymphoblastic leukemia/lymphoma (T-ALL).Citation1 In a mouse model of human T-ALL, expression of a constitutively active form of Akt2 specifically in immature T cells is sufficient to drive T-cell lymphoma formation.Citation2 Tumors from these transgenic mice consistently harbor one or the other of 2 recurrent chromosomal rearrangements that each involve somatic juxtaposition of a T cell receptor (TCR) enhancer and a transcription factor gene, either Dlx5 or Myc – in each case resulting in unregulated expression of Myc protein.Citation3 Myc is thought to be essential to T cell development, and Myc-null T cells fail to proliferate at the CD4/CD8 double-negative stage in mice.Citation4 While overexpression of Myc drives T-ALL in zebrafish, transgenic mice specifically overexpressing Myc in immature T cells, under the control of an Lck promoter, did not develop T-ALL, indicating that Myc is necessary, but not sufficient, to induce T-cell malignancy in mice.Citation5,6 In contrast, Lck-Dlx5 transgenic mice overexpressing Dlx5 in T cells exhibit a high incidence of thymic lymphomas with unregulated Akt activity and Myc overexpression (manuscript in preparation).
ALL accounts for ˜70% of childhood leukemia cases, approximately 15% of which are of T-cell lineage. The frontline treatment regimen in ALL is combination chemotherapy that includes vincristine, prednisone, dexamethasone, asparaginase and daunorubicin, leading to survival rates of 80% in children and 20-40% in adults.Citation7 However, more effective and less toxic therapies are still needed. Consequently, inhibitors of oncogenic pathways are being explored. For example, the dual PI3K/mTOR inhibitor PI-103 was shown to induce cell cycle arrest and apoptosis in T-ALL cell lines.Citation8 Similarly, various γ-secretase inhibitors have been used to treat T-ALL cells. However, only a small fraction of T cell lymphoma cell lines responded to such Notch inhibitors due to the presence of activating mutations of NOTCH1 in the majority of lines tested.Citation9 Targeting MYC has been a challenge, although the bromodomain inhibitor JQ1 has recently shown promise,Citation10 including a recent study demonstrating that JQ1 reduces MYC expression and inhibits the proliferation of relapsed T-ALL cells in vitro.Citation11 The direct MYC inhibitor 10058-F4 suppresses MYC function by interfering with MYC-MAX dimerization and has been shown to induce apoptosis in human acute myeloid leukemia cells.Citation12 However, its clinical applicability is limited by its low potency and short half-life in vivo.Citation13
Here, we report that the Akt pathway inhibitor BEZ235 cooperates with JQ1 in inhibiting the proliferation of T-cell lymphoma cells from Lck-Dlx5 transgenic mice. Furthermore, BEZ235 cooperates with 10058-F4 to promote increased apoptosis in Lck-Dlx5 lymphoma cells. In addition, we show that BEZ235 or RAD001 potentiate vincristine-induced cell death in these lymphoma cells.
Results
T-cell lymphoma cells from Lck-Dlx5 mice acquire up regulation of Myc and loss of Pten expression
We generated 3 transgenic founder lines with Lck promoter-driven Dlx5 expression that exhibit a high incidence of thymic lymphoma. Compared to thymocytes from wild-type mice, lymphoma cells from Lck-Dlx5 mice exhibited up regulation of Myc and constitutive activation of Akt, the latter due to the loss of Pten expression as shown by immunoblot analysis (). Sequence analysis of Pten cDNA revealed variable nonsense and missense mutations in each of the 5 T cell lymphoma lines from Lck-Dlx5 mice that were tested (). The second Pten allele was retained, suggesting that loss of Pten protein expression occurred through a combination of mutation and epigenetic silencing. Array-CGH revealed trisomy 15 in each cell line tested. Since the Myc locus resides on murine chromosome 15, trisomy of this chromosome may have contributed to the up regulation on Myc protein observed in these tumor cells ( and Suppl. Fig. 1). Additionally, we have previously shown that overexpression of Dlx5 transcriptionally regulates the expression of Myc.Citation14
Figure 1. Thymic T-cell lymphoma cells from Lck-Dlx5 mice have elevated Akt activity and elevated Myc expression. Proteins from 3 lymphoma cell lines (F86-786, F86-793 and F86-801) derived from Lck-Dlx5 mice and normal thymocytes from 2 wild type (WT) littermates were analyzed by immunoblotting. Expression of Myc-tagged Dlx5, Myc, phospho-Akt (ser473), total Akt and Pten were evaluated. (A) Note that activation of Akt in T-cell lymphoma cells was associated with loss of Pten expression. Array-CGH profiles of 3 Lxk-Dlx5 lymphoma cell lines (including 2 matching those shown in panel A, F47-0 showed the similar pattern on a separate gel) with recurrent trisomy of chromosome (Chr) 15, location of Myc, in tumor cells (B).
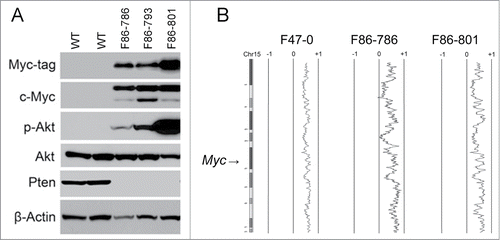
Table 1. Pten mutations in T-cell lymphoma lines from Lck-Dlx5 mice
Lck-Dlx5 tumor cells are highly sensitive to the Akt pathway inhibitor BEZ235
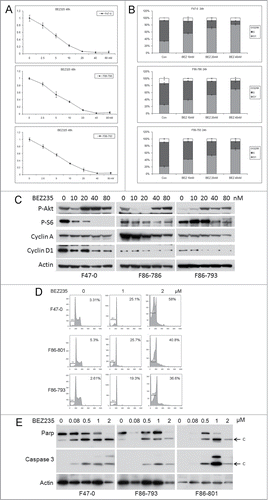
To test whether activation of Akt is essential to the survival and proliferation of T-ALL cells from Lck-Dlx5 mice, the dual Akt pathway inhibitor BEZ235 was used. BEZ235 treatment at low concentrations (0, 2.5, 5, 10, 20, 40, 80 nM) reduced cell viability/proliferation in a dose-dependent manner, as shown by MTS assay (). Additionally, FACS analysis demonstrated that the cell cycle arrest occurred at the G1 phase without induction of a sub-G1 apoptotic population (, Suppl. Fig. 2). Mechanistically, these cells were found to have reduced levels of cyclin A and cyclin D1, but not induction of caspase 3 cleavage, indicating that the low doses of BEZ235 used in these experiments triggered cell cycle arrest but not cell death (). Western blotting also showed that these low concentrations of BEZ235 inhibited mTORC1 but had little or no effect on PI3K, given that phosphorylation of S6, but not Akt, was diminished. The mTOR inhibitor RAD001 had a similar effect on cell proliferation (Suppl. Fig. 3). Moreover, high doses of BEZ235 (≥0.5 µM) causes apoptosis in these cells as shown by an increased proportion of sub-G1 cells and activation of Caspase 3 ().
Figure 2. Low concentrations of BEZ235 induce dose-dependent cell cycle inhibition and decreased cell viability. Three different Lck-Dlx5 lymphoma cell lines were treated with BEZ235 at the indicated concentrations. Cell viability/proliferation were monitored by MTS assay (A). Cell cycle analysis was performed by flow cytometry. Histograms summarizing results from 3 independent flow cytometry experiments are shown in panels (B). Western blot analysis of phospho-Akt, phospho-S6, cyclin A, cyclin D1 and β-actin (C). Higher concentrations of BEZ235 (≥0.5 µM) treatment for 24 h cause apoptosis in these lymphoma cells as shown by an increase in sub-G1 cells (D), and activation of caspase 3 (E).
JQ1 suppresses the proliferation of lymphoma cells from Lck-Dlx5 mice
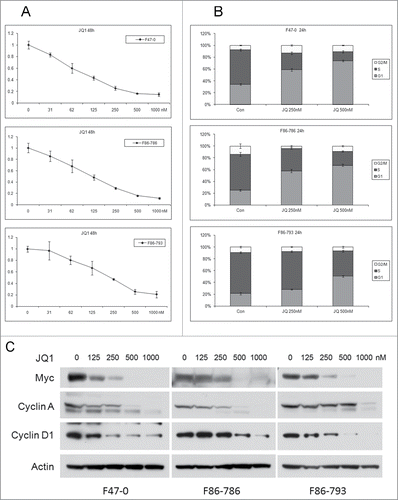
The transcription factor Myc has broad effects on tumor growth and cell proliferation by regulating the expression of various target genes. Myc transcription is up regulated by bromodomain proteins that bind to Myc enhancers. To evaluate the influence of Myc up regulation in lymphoma cells from Lck-Dlx5 mice, we treated cells with the BET bromodomain inhibitor JQ1. MTS assays demonstrated a potent effect of JQ1 on cell proliferation in a dose-dependent manner (). FACS analysis showed that the reduced cell proliferation was due to cell cycle arrest at G1 phase (, Suppl. Fig. 4). Immunoblotting revealed that treatment of cells with JQ1 resulted in reduced expression of Myc protein, as well as down regulation of cyclin A and cyclin D1 ().
Figure 3. Bromodomain inhibitor JQ1 induces dose-dependent cell cycle inhibition and reduced cell viability in lymphoma cells from Lck-Dlx5 mice. Cell lines were treated with JQ1 at the indicated concentrations. Cell viability/proliferation was monitored by MTS assay (A). Cell cycle analysis was performed by flow cytometry (B). Western blot analysis depicting expression of Myc, cyclin A, cyclin D1 and β-actin (C).
BEZ235 cooperates with JQ1 in inducing G1 arrest of Lck-Dlx5 lymphoma cells
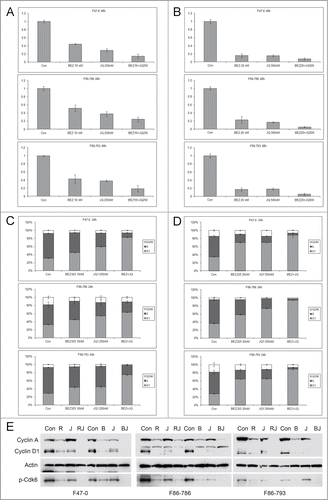
To investigate if BEZ235 can further enhance the G1 arrest induced by JQ1 in Lck-Dlx5 tumor cells, combinational treatments were performed. Low concentrations of BEZ235 (10 and 20 nM) showed a cooperative effect with JQ1 (at 250 and 500 nM) in suppressing cell proliferation () as well as in inducing cell cycle arrest ( and Suppl. Fig. 5A,B). RAD001 at 1 nM showed similar cooperativity with JQ1 in suppressing cell proliferation and inducing cell cycle arrest (Suppl. Fig. 6). Immunoblotting demonstrated that BEZ235 or RAD001, when combined with JQ1, strongly suppressed the expression of cyclin A and cyclin D1 as well as the activity of Cdk6 (). Collectively, these results suggest that Akt/mTor inhibition and Myc inhibition converge to suppress cell cycle progression. However, in the context of apoptosis triggered by higher doses of BEZ235 (≥0.5 µM), the addition of JQ1 slightly antagonized the killing effect of BEZ235 (Suppl. Fig. 7A,B,C).
Figure 4. Low concentrations of BEZ235 and JQ1 cooperate to induce enhanced cell cycle arrest. Lck-Dlx5 lymphoma lines were treated with BEZ235 and JQ1 separately or in combination at the indicated concentrations. Cell viability/proliferation was monitored by MTS assay (A, B), and cell cycle analysis was performed by flow cytometry (C, D). BEZ235 (B, 40 nM) or RAD001 (R, 1 nM), when combined with JQ1 (J, 250 nM) for 24 h, cooperatively suppress expression of cyclin A, cyclin D1 and phospho-Cdk6 (E).
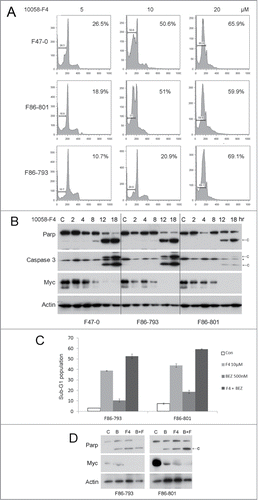
BEZ235 and 10058-F4 cooperatively trigger apoptosis in Lck-Dlx5 tumor cells
Since JQ1 broadly inhibits various targets of bromodomain proteins, we next tested the effects of the selective Myc inhibitor 10058-F4 on Lck-Dlx5 lymphoma cells. We found that this compound indeed induced apoptosis in these cells as shown by sub-G1 DNA peak and caspase3 activation (). Furthermore, 10058-F4 (10 µM) potentiated the apoptotic effect of BEZ235 (0.5 µM) in these cells ().
Figure 5. The specific Myc inhibitor 10058-F4 induces apoptosis in lymphoma cells from Lck-Dlx5 mice and cooperates with BEZ235 to enhance apoptosis in these cells). 10058-F4 along triggers apoptotic sub-G1 DNA condensation (A). 10058-F4 at 20 µM potently promotes apoptosis in Lck-Dlx5 lymphoma cells as early as 12 h after initiating treatment, and 10058-F4 treatment results in cleavage of Parp and caspase 3 as well as decreased expression of Myc in a dose-dependent manner (B). 10 µM 10058-F4 (F4) cooperates with BEZ235 (0.5 µM) in triggering apoptotic sub-G1 DNA condensation (C) as well as Parp cleavage and decreased Myc protein levels (D).
Inhibition of the Akt pathway augments apoptosis induced by vincristine in lymphoma cells from Lck-Dlx5 mice
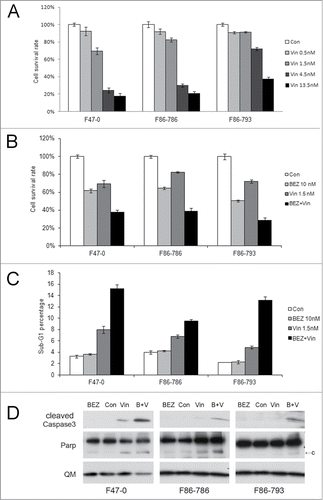
The chemotherapeutic agent vincristine (Oncovin®) is one of the drugs used in combination induction therapy such as the CHOP regimen used in non-Hodgkin's lymphoma and peripheral T-cell lymphoma. We found that vincristine can reduce viability of T-ALL cells from Lck-Dlx5 mice, as shown by MTS assay (). Notably, low doses of vincristine, when combined with BEZ235 or RAD001, more effectively suppresses viability than vincristine alone (, Suppl. Fig. 8,9). This effect was shown to be due to enhanced apoptosis as demonstrated by an increase in sub-G1 cells and cleavage of caspase 3 (). JQ1 could also slightly increase the effects of vincristine; however, combined use of JQ1, RAD001 and vincristine did not render greater efficacy (Suppl. Fig. 10). Moreover, when the cells were pre-treated with BEZ235 or JQ1 for 16 h before adding vincristine, the arrested cells were resistant to vincristine-induced cell death (Suppl. Fig. 11).
Figure 6. Apoptosis induction in Lck-Dlx5 lymphoma cells by vincristine and BEZ235. Vincristine-induced apoptosis is dose-dependent (A). Cells were treated with vincristine (Vin) at the indicated concentrations for 24 h, and cell viability/proliferation was monitored by MTS assay. mTORC1 inhibition potentiates vincristine-induced apoptosis (B). Cells were treated with 10 nM BEZ235 (BEZ) separately or in combination with 1.5 nM vincristine for 24 h, and MTS assay was performed. Apoptotic sub-G1 cell populations were analyzed by using flow cytometry (C). Western blot analysis depicting activation of caspase 3 (D).
Discussion
Previously, we demonstrated that a constitutively active form of Akt2, i.e. Lck-Myr (myristoylated)-Akt2, drives thymic T-cell lymphoma within 12–24 weeks in an Akt dosage-dependent manner.Citation2 In some Lck-Myr-Akt2 founder lines, a recurrent chromosomal inversion implicated the homeobox gene Dlx5 as an oncogene.Citation3 In subsequent work (manuscript in preparation), we found that transgenic Lck-Dlx5 mice develop spontaneous thymic lymphomas that acquire constitutive activation of Akt, due to loss of Pten expression, as well as up regulation of Myc. Such genetic and epigenetic changes are characteristics of other acute T-cell lymphoma animal models as well as human T-ALLs.Citation15-17 We found that the Lck-Dlx5 tumor cells are extremely sensitive to Akt pathway inhibitors, implying that Akt activation plays a critical role in mediating Dlx5-initiated lymphomagenesis. Previously, we reported that Dlx5 augments Akt signaling in human ovarian cancer cells,Citation18 suggesting that overexpression of Dlx5 in Lck-Dlx5 mice may also contribute to the early stages of T-cell lymphomagenesis prior to loss of Pten expression. Also, Myc is a transcriptional target of Dlx5, which may serve as another hit in Dlx5-mediated T-cell oncogenesis.Citation14 These findings suggest that the design of therapies targeting the AKT pathway and DLX5/MYC simultaneously could provide improved therapeutic efficacy in tumors with activation of these cooperating oncogenes.
mTOR/Raptor is a major target of AKT signaling that controls protein synthesis and lipid metabolism.Citation19 The p70 S6 kinase (p70 S6K) is a substrate of mTORC1,Citation20 and inhibition of p70 S6K results in G1 cell cycle arrest.Citation21,22 Our data demonstrate that low concentrations of BEZ235 behave in a manner similar to RAD001, which inhibits phosphorylation of p70 S6K, but not of AKT. At higher concentrations, however, BEZ235 is known to be a potent dual inhibitor of mTORC1/2 and PI3K.Citation23
The protein product of the Myc oncogene forms an active dimer with Max that binds to the E-box motif of its target genes. This process recruits RNA polymerase to the target promoter and initiates transcription.Citation24,25 Many pro-proliferation and growth-related genes are known to be Myc targets, including cyclin D2, CDK4, ODC, eIF2A and LDHA.Citation26 We found that the bromodomain inhibitor JQ1 inhibits cell cycle progression of Lck-Dlx5 lymphoma cells with concomitant reduction of Myc expression. When combined with low doses of BEZ235 or RAD001, JQ1 induction of G1 arrest was enhanced, suggesting that combinatorial therapies employing Akt and bromodomain inhibitors might have enhanced efficacy while permitting the use of less toxic doses than with single agent therapies. However, the specific Myc inhibitor 10058-F4 was found to be a potent inhibitor of cell survival and potentiated BEZ235-triggered apoptosis in Lck-Dlx5 lymphoma cells. Moreover, inhibitors of oncogenic addiction may sensitize tumor cells to standard chemotherapeutic agents such as vincristine. Notably, however, dose scheduling must be considered, as we found that BEZ235 sensitizes Lck-Dlx5 lymphoma cells to vincristine-induced apoptosis only when treatment was simultaneous. When BEZ235 was given 16 h prior to treatment with vincristine, an antagonistic effect was observed. These findings indicate that inhibition of AKT signaling is required for the initiation of apoptosis by vincristine. Collectively, our findings provide rationale for the design of novel combination therapies for T-ALL that would include targeting of AKT and MYC together with conventional therapeutic agents.
Materials and Methods
Reagents
Antibodies against cyclins A (#sc-596), D1(#sc-246), D2(#sc-181) and D3(#sc-9566) and β-actin (#sc-1615) were purchased from Santa Cruz Biotech. Antibodies against c-Myc (#5605), Pten (#9552), phospho-AKT (#9271), and phospho-S6 (#4858) were from Cell Signaling (#4858). BEZ235 (#S1009), RAD001(#S1120), 10058-F4 (#S7153) and JQ1 (#S7110) were from Selleckchem.
Cell culture
Tumor cells were released from the thymus by gently forcing tissue through a 100-gauge mesh. Cells were maintained in ISCOVE'S MEM supplemented with 15% FBS plus penicillin and streptomycin, 2 mM L-glutamine, MEM sodium pyruvate, non-essential amino acids, and β-mercaptoethanol (Life Technologies).
Cell viability assay
Cell viability/proliferation was measured by MTS assay (Promega, Madison WI). Briefly, cells were seeded at 2 × 104 cells/well in 96-well plates overnight, followed by treatment with various inhibitors for 24 or 48 h. MTS reagent (20 μl) was added to each well and then incubated for 1 to 4 h for color development. OD value at 490 nm was measured using a 96-well micro-plate reader (BioRad).
Cell cycle analysis
Cells were fixed with 70% ice-cold ethanol for 2 h at 4°C, washed twice with ice-cold PBS, and then stained with propidium iodine solution for 20 min. Samples were subjected to FACS (Beckman) analysis using FlowJo software.
Western blot analysis
Total cellular protein was extracted by using cell lysis buffer supplemented with 2 mM PMSF (Cell Signaling, #9803). Cellular debris was removed by centrifuging at 15,000 x g for 15 min at 4°C. Protein concentration was determined using Bradford reagent. Protein lysates (50 μg/well) were loaded into Tris-Glycine buffered SDS-PAGE gels (Invitrogen, #EC6038BOX), and separated protein was then transferred onto PVDF membranes (Millipore, #IPVH00010). The membranes were incubated with primary antibodies diluted in 5% non-fat milk or BSA overnight at 4°C on a shaker. After washing with TBST buffer 4 times, membranes were incubated with HRP-coupled secondary antibody for 1 h at room temperature. Membranes were then treated with ECL to excite chemiluminescence for film exposure.
Array-CGH analysis
Agilent mouse 244k chips were used to assess genomic imbalances in lymphoma cells from Lck-Dlx5 mice, according to the manufacturer's protocol.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary Material
Download Zip (1.6 MB)Acknowledgments
We thank the Genomics, Flow Cytometry, Laboratory Animal, and DNA Sequencing Facilities at Fox Chase Cancer Center for assistance.
Funding
This work was supported by the National Cancer Institute grants CA077429, CA083638 and CA06927, and an appropriation from the Commonwealth of Pennsylvania.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Palomero T, Ferrando A. Oncogenic NOTCH1 control of MYC and PI3K: challenges and opportunities for anti-NOTCH1 therapy in T-cell acute lymphoblastic leukemias and lymphomas. Clin Cancer Res 2008; 14:5314-7; PMID:18765521; http://dx.doi.org/10.1158/1078-0432.CCR-07-4864
- Timakhov RA, Tan Y, Rao M, Liu Z, Altomare DA, Pei J, Wiest DL, Favorova OO, Knepper JE, Testa JR. Recurrent chromosomal rearrangements implicate oncogenes contributing to T-cell lymphomagenesis in Lck-MyrAkt2 transgenic mice. Genes Chromosomes Cancer 2009; 48:786-94; PMID:19530243; http://dx.doi.org/10.1002/gcc.20683
- Tan Y, Timakhov RA, Rao M, Altomare DA, Xu J, Liu Z, Gao Q, Jhanwar SC, Di Cristofano A, Wiest DL, et al. A novel recurrent chromosomal inversion implicates the homeobox gene Dlx5 in T-cell lymphomas from Lck-Akt2 transgenic mice. Cancer Res 2008; 68:1296-302; PMID:18316591; http://dx.doi.org/10.1158/0008-5472.CAN-07-3218
- Douglas NC, Jacobs H, Bothwell AL, Hayday AC. Defining the specific physiological requirements for c-Myc in T cell development. Nat Immunol 2001; 2:307-15; PMID:11276201; http://dx.doi.org/10.1038/86308
- Langenau DM, Traver D, Ferrando AA, Kutok JL, Aster JC, Kanki JP, Lin S, Prochownik E, Trede NS, Zon LI, et al. Myc-induced T cell leukemia in transgenic zebrafish. Science 2003; 299:887-90; PMID:12574629;http://dx.doi.org/10.1126/science.1080280
- Rudolph B, Hueber AO, Evan GI. Reversible activation of c-Myc in thymocytes enhances positive selection and induces proliferation and apoptosis in vitro. Oncogene 2000; 19:1891-900; PMID:10773879; http://dx.doi.org/10.1038/sj.onc.1203508
- Inaba H, Greaves M, Mullighan CG. Acute lymphoblastic leukaemia. Lancet 2013; 381:1943-55; PMID:23523389; http://dx.doi.org/10.1016/S0140-6736(12)62187-4
- Chiarini F, Fala F, Tazzari PL, Ricci F, Astolfi A, Pession A, Pagliaro P, McCubrey JA, Martelli AM. Dual inhibition of class IA phosphatidylinositol 3-kinase and mammalian target of rapamycin as a new therapeutic option for T-cell acute lymphoblastic leukemia. Cancer Res 2009; 69:3520-8; PMID:19351820; http://dx.doi.org/10.1158/0008-5472.CAN-08-4884
- O'Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, Hardwick J, Welcker M, Meijerink JP, Pieters R, et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J Exp Med 2007; 204:1813-24; PMID:17646409; http://dx.doi.org/10.1084/jem.20070876
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. Selective inhibition of BET bromodomains. Nature 2010; 468:1067-73; PMID:20871596; http://dx.doi.org/10.1038/nature09504
- Roderick JE, Tesell J, Shultz LD, Brehm MA, Greiner DL, Harris MH, Silverman LB, Sallan SE, Gutierrez A, Look AT, et al. c-Myc inhibition prevents leukemia initiation in mice and impairs the growth of relapsed and induction failure pediatric T-ALL cells. Blood 2014; 123:1040-50; PMID:24394663; http://dx.doi.org/10.1182/blood-2013-08-522698
- Huang MJ, Cheng YC, Liu CR, Lin S, Liu HE. A small-molecule c-Myc inhibitor, 10058-F4, induces cell-cycle arrest, apoptosis, and myeloid differentiation of human acute myeloid leukemia. Exp Hematol 2006; 34:1480-9; PMID:17046567; http://dx.doi.org/10.1016/j.exphem.2006.06.019
- Guo J, Parise RA, Joseph E, Egorin MJ, Lazo JS, Prochownik EV, Eiseman JL. Efficacy, pharmacokinetics, tisssue distribution, and metabolism of the Myc-Max disruptor, 10058-F4 [Z,E]-5-[4-ethylbenzylidine]-2-thioxothiazolidin-4-one, in mice. Cancer Chemother Pharmacol 2009; 63:615-25; PMID:18509642; http://dx.doi.org/10.1007/s00280-008-0774-y
- Xu J, Testa JR. DLX5 (distal-less homeobox 5) promotes tumor cell proliferation by transcriptionally regulating MYC. J Biol Chem 2009; 284:20593-601; PMID:19497851; http://dx.doi.org/10.1074/jbc.M109.021477
- Weng AP, Millholland JM, Yashiro-Ohtani Y, Arcangeli ML, Lau A, Wai C, Del Bianco C, Rodriguez CG, Sai H, Tobias J, et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev 2006; 20:2096-109; PMID:16847353; http://dx.doi.org/10.1101/gad.1450406
- Kindler T, Cornejo MG, Scholl C, Liu J, Leeman DS, Haydu JE, Fröhling S, Lee BH, Gilliland DG. K-RasG12D-induced T-cell lymphoblastic lymphoma/leukemias harbor Notch1 mutations and are sensitive to gamma-secretase inhibitors. Blood 2008; 112:3373-82; PMID:18663146; http://dx.doi.org/10.1182/blood-2008-03-147587
- Sanda T, Li X, Gutierrez A, Ahn Y, Neuberg DS, O'Neil J, Strack PR, Winter CG, Winter SS, Larson RS, et al. Interconnecting molecular pathways in the pathogenesis and drug sensitivity of T-cell acute lymphoblastic leukemia. Blood 2010; 115:1735-45; PMID:20007543; http://dx.doi.org/10.1182/blood-2009-07-235143
- Tan Y, Cheung M, Pei J, Menges CW, Godwin AK, Testa JR. Upregulation of DLX5 promotes ovarian cancer cell proliferation by enhancing IRS-2-AKT signaling. Cancer Res 2010; 70:9197-206; PMID:21045156; http://dx.doi.org/10.1158/0008-5472.CAN-10-1568
- Huang K, Fingar DC. Growing knowledge of the mTOR signaling network. Semin Cell Dev Biol 2014; 36:79-90; PMID:25242279; http://dx.doi.org/10.1016/j.semcdb.2014.09.011
- Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, Schreiber SL. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 1994; 369:756-8; PMID:8008069; http://dx.doi.org/10.1038/369756a0
- Chung J, Kuo CJ, Crabtree GR, Blenis J. Rapamycin-FKBP specifically blocks growth-dependent activation of and signaling by the 70 kd S6 protein kinases. Cell 1992; 69:1227-36; PMID:1377606; http://dx.doi.org/10.1016/0092-8674(92)90643-Q
- Kawamata S, Sakaida H, Hori T, Maeda M, Uchiyama T. The upregulation of p27Kip1 by rapamycin results in G1 arrest in exponentially growing T-cell lines. Blood 1998; 91:561-9; PMID:9427710
- Serra V, Markman B, Scaltriti M, Eichhorn PJ, Valero V, Guzman M, Botero ML, Llonch E, Atzori F, Di Cosimo S, et al. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res 2008; 68:8022-30; PMID:18829560; http://dx.doi.org/10.1158/0008-5472.CAN-08-1385
- Blackwood EM, Eisenman RN. Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 1991; 251:1211-7; PMID:2006410; http://dx.doi.org/10.1126/science.2006410
- Prendergast GC, Ziff EB. Methylation-sensitive sequence-specific DNA binding by the c-Myc basic region. Science 1991; 251:186-9; PMID:1987636; http://dx.doi.org/10.1126/science.1987636
- Xiong J, Du Q, Liang Z. Tumor-suppressive microRNA-22 inhibits the transcription of E-box-containing c-Myc target genes by silencing c-Myc binding protein. Oncogene 2010; 29:4980-8; PMID:20562918; http://dx.doi.org/10.1038/onc.2010.241