
Abstract
Lung cancers express non-neuronal, cholinergic autoparacrine loop, which facilitates tumor growth. Interruption of M3 muscarinic cholinergic signaling has been reported to inhibit small cell lung cancer (SCLC) growth. The purpose of this study is to investigate if blocking autoparacrine muscarinic cholinergic signaling could inhibit non-small cell lung cancer (NSCLC) growth and possible underlying mechanisms. Our results showed that PC9 and A549 cells expressed all 5 subtypes of muscarinic receptor (mAChR) and blocking M2 mAChR (M2R) signaling using selective antagonist methoctramine or short hairpin RNA (shRNA) inhibited tumor cell proliferation in vitro and in vivo. Consistent with AChR agonists stimulating p44/42 MAPK (Erk1/2) and Akt phosphorylation, blocking M2R signaling decreased MAPK and Akt phosphorylation, indicating that non-neuronal ACh functions as an autoparacrine growth factor signaling in part through activation of M2R and downstream MAPK and Akt pathways. Importantly, further studies revealed that blocking M2R signaling also reversed epithelial-mesenchymal transition (EMT) in vitro and in vivo, indicating that non-neuronal ACh promotes EMT partially through activation of M2R. These findings demonstrate that M2R plays a role in the growth and progression of NSCLC and suggest M2R antagonists may be an efficacious adjuvant therapy for NSCLC.
Abbreviations
| NSCLC | = | non-small cell lung cancer |
| SCLC | = | small cell lung cancer |
| ACh | = | acetylcholine |
| AChR | = | acetylcholine receptor |
| mAChR | = | muscarinic receptor |
| nAChR | = | nicotinic receptor |
| M2R | = | M2 mAChR |
| EMT | = | epithelial-mesenchymal transition |
| ChAT | = | choline acetyltransferase |
| MAPK | = | mitogen-activated protein kinase |
| shRNA | = | short hairpin RNA |
Introduction
Lung cancer is the world leading cause of cancer-related mortality and the number of cases and deaths related to lung cancer is rising worldwide. Lung cancer is classified into small cell lung cancer (SCLC) which accounts for approximately 15–20 percent of cases and non-small cell lung cancer (NSCLC) which accounts for the remaining cases.Citation1,2 A significant proportion of NSCLC cases are detected only in the advanced stage after the onset of metastasis, leading to a remarkably low 5-year survival rate in patients. Therefore, further understandings of the biology of lung cancer and novel therapeutic approaches are urgently needed. The recent discovery that lung cancers synthesize and secrete acetylcholine (ACh) that acts as an autoparacrine growth factor provides potential new targets to inhibit lung cancer growth.Citation3,4
It is well established that ACh is an important neurotransmitter in the central and peripheral nervous systems and acts via activation of ACh receptors (AChRs).Recently, it has been found that ACh is also widely synthesized by a variety of non-neuronal cell types including airway epithelial cells,Citation5,6 keratinocytes,Citation7 small and large intestine, gall bladder,Citation8 glial,Citation9 vascular endothelial,Citation10 immune cells and most common cancer cells such as NSCLC, SCLC, colon, glial and ovarian carcinomas.Citation11-16 Although the primary function has not yet been completely elucidated, the expression of ACh in many different cell types suggests that its roles will be fundamental and will present novel therapeutic targets.
The non-neuronal ACh acts as a local autoparacrine hormone and stimulates cell growth through either nicotinic cholinergic or muscarinic cholinergic pathway. Multiple studies have shown that activation of nAChRs by nicotine, which is an exogenous ligand for nAChRs, stimulates tumor cells growth in breast,Citation17 lung,Citation18 colon,Citation19 bladder Citation20 and cervical cancer.Citation21 The muscarinic receptors (mAChRs) are G-protein-coupled receptors, and 5 subtypes of mAChRs (M1-M5) have been identified.Citation22 Muscarinic stimulation of cancer growth has been reported for lung,Citation16,23 colon,Citation24 breastCitation25 and prostate cancers.Citation26 In SCLC, tumor growth has been associated with stimulation of M3 mAChR through mitogen-activated protein kinase (MAPK)-dependent mechanisms.Citation27M3 mAChR antagonists can inhibit SCLC growth in vitro and in vivo. In NSCLC, very little is known about which subtypes mediate the proliferative effects of muscarinic activation. Hua and co-workersCitation28 have shown that, R2HBJJ, as a novel mAChR antagonist, can block the local cholinergic loop and inhibit tumor cell growth by antagonizing predominantly M3 and M1 muscarinic receptors in some NSCLC cell lines but not others.
In this study, we first show that all 5 subtypes of mAChR are expressed in NSCLC cell lines A549 and PC9 and muscarinic activation leads to tumor cell growth by increasing p44/42 MAPK and Akt phosphorylation. Antagonizing M1 or M3 mAChR does not have inhibitory effects on tumor growth, while blocking M2 mAChR (M2R) signaling using M2R selective antagonist methoctramine or specific shRNA can inhibit tumor growth and also decrease MAPK and Akt phosphorylation in vitro and in vivo. Moreover, we demonstrate that blocking M2R signaling reverses epithelial-mesenchymal transition (EMT) in vitro and in vivo. To the best of our knowledge, this is the first study to investigate the roles of M2R signaling in the growth and progression of NSCLC.
Results
NSCLC cell lines expressed functional muscarinic receptors
If a cholinergic autoparacrine loop is functional in NSCLC, then cells must express choline acetyltransferase (ChAT), the enzyme that synthesizes ACh, and AChRs for ACh to provide autoparacrine cholinergic stimulation to NSCLC cell growth. The expression of 5 subtypes of mAChRs (M1-M5) and ChAT were examined by RT-PCR in human NSCLC cell lines A549 and PC9. As shown in , mRNAs of M1-M5 and ChAT were detected in both cell lines.
Figure 1. Expression of functional muscarinic receptors in NSCLC cell lines. (A) mRNA expression of mAChRs and choline acetyltransferase (ChAT) in PC9 and A549 cells. RT-PCR was performed on total RNA prepared from the indicated cell lines. GAPDH was used as loading control. (B) Pilocarpine stimulated cell proliferation in PC9 and A549 cells. Cells were treated with 50 μM pilocarpine for 72 h. Cell proliferation was determined by CCK8 assay. Cells treated with solvent (DMSO) were used as a control. (C) Western blot showed that pilocarpine increased MAPK and Akt phosphorylation in PC9 and A549 cells. Cells were treated with 50 μM pilocarpine for 72 h. β-actin was used as loading control for Western blot. (D) Quantification of Western blots shown in C. Data were shown as mean±s.e.m.**, P < 0.01, ***, P < 0.001, compared with control.
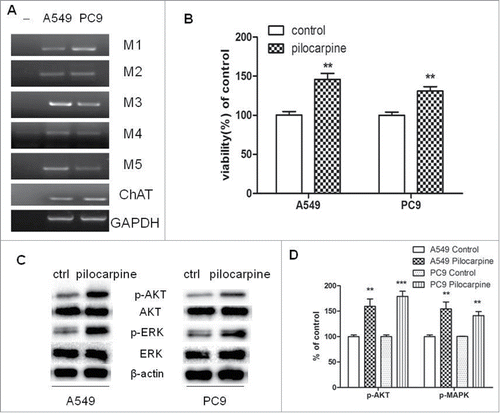
To further demonstrate NSCLC cells express a functional cholinergic system which stimulates tumor cell growth, we examined the effect of muscarinic receptor agonist pilocarpine on NSCLC cell proliferation. As shown in , pilocarpine stimulated the growth of both cell lines. In addition, the potential role of p44/42 MAPK and Akt in mediating effects of cholinergic stimulation on the growth of NSCLC cells was evaluated by measuring pilocarpine-induced phosphorylation of MAPK and Akt. As shown in , phosphorylation of p44/42 MAPK and Akt was induced by pilocarpine in both cell lines. These results imply that both cell lines have functional muscarinic receptors, which can be activated to stimulate cell growth through MAPK and Akt pathways.
Selective M2R antagonist methoctramine inhibited cell proliferation and phosphorylation of MAPK and Akt in NSCLC cell lines
Next, the ability of subtype-selective muscarinic receptor antagonists to inhibit the growth of NSCLC cell lines in vitro was determined. As shown in , selective M2R antagonist methoctramine inhibited A549 and PC9 cell proliferation in a dose-dependent manner, whereas M1 antagonist pirenzepine and M3 antagonist 4-DAMP or darifenacin had no effects on cell proliferation (Fig. S1). The only source of ligand for M2R in these studies was endogenous ACh released by tumor cells, suggesting that non-neuronal ACh functions as an autoparacrine growth factor signaling in part through activation of M2R in NSCLC cell lines.
Figure 2. Effects of M2R antagonist methoctramine on cell proliferation. Cells were treated with the indicated concentrations of methoctramine for 72 h. (A and B) Methoctramine inhibited A549 (A) and PC9 (B) cell proliferation in a concentration-dependent manner. Cell proliferation was determined by CCK8 assay. Cells treated with solvent (DMSO) were used as a control. (C and D) Western blot showed that methoctramine decreased MAPK and Akt phosphorylation in A549 (C) and PC9 (D) cells. β-actin was used as loading control for Western blot. (E and F) Quantification of Western blots shown in C and D, respectively. Data were shown as mean±s.e.m. *, P < 0.05; **, P < 0.01; ***, P < 0.001, compared with control.
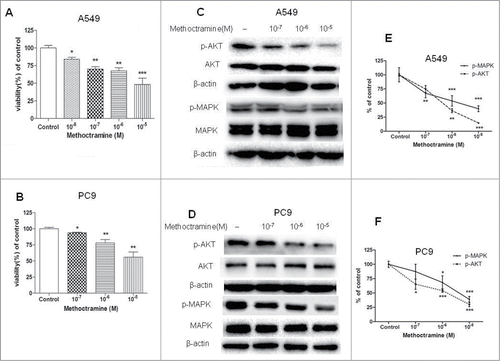
Next, we examined whether basal phosphorylation of MAPK and Akt stimulated by cholinergic autoparacrine loop was partially through activation of M2R. If so, then addition of M2R antagonist alone should decrease MAPK and Akt phosphorylation. As shown in , addition of methoctramine alone to A549 and PC9 cells inhibited MAPK and Akt phosphorylation in a dose-dependent manner, suggesting autoparacrine ACh stimulates cell growth by activating M2R and downstream MAPK and Akt pathways. As expected, methoctramine also inhibited AChR agonist carbachol and mAChR agonist pilocarpine-induced phosphorylation of MAPK and Akt in NSCLC cells in a dose-dependent manner (Fig. S2 and S3).
Methoctramine reversed epithelial-mesenchymal transition (EMT) in NSCLC cells
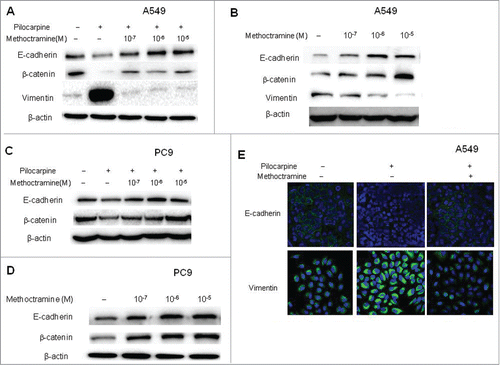
Epithelial-mesenchymal transition (EMT) is known to be one of the vital steps for the acquisition of malignant phenotype. This transition allows cell to acquire migratory properties and metastasize to a new location. EMT involves the repression of epithelial-specific adhesion molecules like E-cadherin and β-catenin with a concomitant expression of mesenchymal proteins like vimentin and fibronectin. It is well-known that growth factors such as TGF-β and EGF can induce EMT. Since endogenous, non-neuronal ACh functions as an autoparacrine growth factor, we wanted to examine if it could induce EMT in NSCLC cells. First, mAChR activation by pilocarpine caused an increase in the levels of vimentin with a concomitant decrease of epithelial markers E-cadherin and β-catenin, and blocking M2R with methoctramine reversed those changes in a dose-dependent manner in A549 cells ( and S4). Similar results were also seen in PC9 cells (), except that vimentin cannot be detected in PC9 cells due to their epithelial phenotype. Thus, EMT was induced by exogenous activation of mAChR partially through M2R in NSCLC cells. These results were further confirmed by immunofluorescence experiments in A549 cells (). As shown in , E-cadherin was located on the surface of cells while vimentin was located in the cytoplasm of cells. Pilocarpine treatment induced EMT by repressing the expression of E-cadherin and simultaneously inducing the expression of vimentin while methoctramine treatment reversed pilocarpine-induced EMT by increasing E-cadherin and decreasing vimentin. Next, whether endogenous ACh could promote EMT through M2R was examined. Addition of methoctramine alone reversed EMT in a dose-dependent manner in A549 ( and S4) and PC9 cells ( and S4), which suggested that autoparacrine ACh promotes EMT partially through M2R in NSCLC cells.
Figure 3. Effects of M2R antagonist methoctramine on EMT. Cells were treated with 50 μM pilocarpine and/or methoctramine of indicated concentrations for 72 h. (A and C), Western blot showed that pilocarpine induced EMT, while methoctramine reversed pilocarpine-induced EMT in a concentration-dependent manner in A549 (A) and PC9 (C) cells. (B and D) Western blot showed that methoctramine alone reversed EMT in a concentration-dependent manner in A549 (B) and PC9 (D) cells. Cells treated with solvent (DMSO) were used as a control. β-actin was used as loading control for Western blot. (E) Confocal microscopy images showed membrane expression of E-cadherin (yellow, upper lane) and cytoplasm expression of vimentin (yellow, lower lane). Nuclei were stained with Hoest 33342 (blue). Pilocarpine (10 μM) induced EMT (middle lane), while methoctramine (1 μM) reversed pilocarpine-induced EMT (right lane) in A549 cells.
M2R-specific shRNA decreased cell proliferation and reversed EMT in NSCLC cells
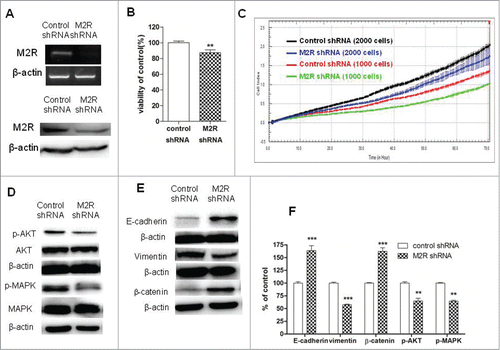
To further demonstrate that non-neuronal ACh and M2R were involved in cell proliferation and EMT in NSCLC, A549 cells were transduced with M2R-specific shRNA lentiviral particles and stable M2R-knockdown cells were selected. Firstly, RT-PCR and Western blot analysis were used to examine the levels of M2R expression. As shown in , mRNA and protein levels of M2R were significantly decreased in M2R-knockdown cells. Secondly, cell proliferation and phosphorylation of MAPK and Akt were examined. Specific knock-down of M2R led to a slower proliferation of A549 cells () and decreased phosphorylation of MAPK and Akt (), thus confirming that non-neuronal ACh functions as an autoparacrine growth factor and stimulates phosphorylation of MAPK and Akt in part through activation of M2R. Thirdly, knockdown of M2R with shRNA increased the levels of E-cadherin and β-catenin and decreased the level of vimentin (), which confirmed the role of M2R in EMT in NSCLC cells.
Figure 4. Effects of M2R knockdown by shRNA on cell proliferation and EMT in A549 cells. A549 cells were transduced with human M2R shRNA lentiviral particles (M2 shRNA) or control shRNA lentiviral particles (control shRNA) and stable cells were selected with puromycin. (A) RT-PCR (upper lane) and Western blot (lower lane) showed that the expression of M2R was downregulated by shRNA lentiviral particles transduction. (B and C) A549 M2 shRNA cells had a slower proliferation rate than A549 control shRNA cells. Cell proliferation was determined by CCK8 assay (B) and RTCA (C). For CCK8 assay, cells were cultured in DMEM supplemented with 1% FBS for 7 d For RTCA, 1000 or 2000 cells were plated in each well in 16-well plates in 100 μL volume. (D) Western blot showed that MAPK and Akt phosphorylation was decreased in A549 M2 shRNA cells compared to A549 control shRNA cells. (E) Western blot showed that EMT was reversed in A549 M2 shRNA cells compared to A549 control shRNA cells. β-actin was used as loading control for Western blot. (F) Quantification of Western blots shown in D and E. Data were shown as mean±s.e.m. **, P < 0.01, ***, P < 0.001, compared with control.
Blocking M2R signaling inhibited A549 xenograft growth and reversed EMT in vivo
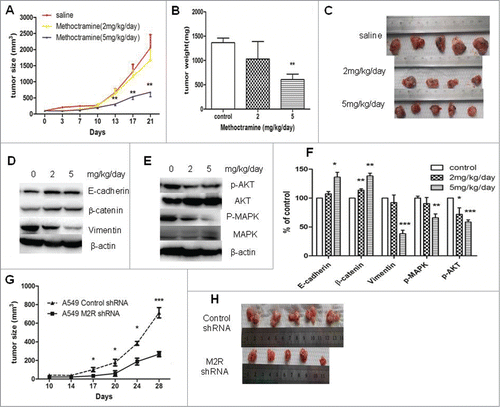
To further investigate the roles of autoparacrine ACh and M2R in NSCLC, the effects of M2R antagonist methoctramine or M2R-knockdown on NSCLC tumor growth were examined in nude mice xenografts. As shown in , consistent with the in vitro effects, methoctramine significantly inhibited A549 tumor growth in a dose-dependent manner. High dose methoctramine treatment (5mg/kg/day) significantly inhibited A549 tumor growth. In the tumors removed from 3-week methoctramine-treated mice, MAPK and Akt phosphorylation was decreased in a dose-dependent manner as shown in . Moreover, 3-week methoctramine treatment also led to EMT reversal in a dose-dependent manner (). Those results are consistent with the in vitro effects of methoctramine. No significant effects of methoctramine treatment on overall animal weight were observed. Moreover, M2R-knockdown A549 cells also showed a lower growth rate than control A549 cells in vivo, as shown in . Those results indicated that blocking M2R signaling could be an efficacious adjuvant therapy for NSCLC.
Figure 5. Effects of blocking M2R signaling on the growth of A549 tumor xenografts in nude mice. (A–C), Methoctramine treatment inhibited tumor growth in a dose-dependent manner. A549 cells were injected s.c. into the left flank of each nude mouse. Methoctramine treatment with indicated dosage started one week after injection. Control group was given the same amount of saline daily. (A) Tumor volume growth curve. Tumor volumes and mice body weight were measured 3 times a week. (B) Tumor weight. Tumors were removed from mice after 3-week treatment and weighed. (C) Photographs of tumors removed from mice after 3-week treatment. (D and E) Western blot showed that 3-week methoctramine treatment decreased MAPK and Akt phosphorylation (D) and reversed EMT (E) in tumor xenografts in a dose-dependent manner. (F) Quantification of Western blots shown in D and E. (G and H) A549 M2 shRNA cells showed a slower growth rate than A549 control shRNA cells in nude mice. Same amount of A549 M2 shRNA cells or A549 control shRNA cells were injected s.c. into the left flank of each nude mice. (G) Tumor volume growth curve. Tumor volumes and mice body weight were measured twice a week. (H) Photographs of tumors removed from mice after 4 weeks. For animal experiment, each group had 4–5 mice and each experiment was repeated twice. Data were shown as mean±s.e.m.*, P < 0.05;**, P < 0.01; ***, P < 0.001, compared with control.
Discussion
Many studies have demonstrated that ACh and other components of cholinergic signaling, including ChAT, cholinesterase and cholinergic receptors, are present in a variety of non-neuronal tissues. Compared to normal lung, cholinergic signaling is markedly upregulated in lung tumor. In NSCLC, studies have shown that ChAT is upregulated while cholinesterases are downregulated which leads to an increased ACh content in tumor tissue.Citation14,29,30 In the local environment of tumors, concentrations of ACh at surface receptors may be even higher due to high cell densities in solid masses and proximity of secretion events to receptor location. Therefore, upregulated autoparacrine cholinergic pathway presents potential new targets to block tumor growth.
A particularly attractive target in the cholinergic pathway is muscarinic receptor, because muscarinic receptor antagonists are in wide clinical use and generally well tolerated while nicotinic receptor antagonists can have significant effects on blood pressure. In SCLC, M3 mAChR appears to be mainly involved in tumor progression. Studies have shown that M3 mAChR antagonists inhibit SCLC cell proliferation by decreasing MAPK phosphorylation in vitro and in vivo,Citation27,31 whereas M1 and M2/M4 antagonists showed no such effects on SCLC cells. In NSCLC, which mAChR subtypes mainly mediate tumor growth responses to endogenous ACh secretion and underlying mechanisms have yet to be elucidated. Hua et al has reported that R2HBJJ, which is a novel mAChR antagonist and has higher affinity to M3 and M1 mAChRs than M2 mAChR, has an inhibitory growth effect on H1299, H460, H157 and H520 cells, but not on A549 or BEP2D cells.Citation28 In this study, we showed that in NSCLC A549 and PC9 cell lines all 5 muscarinic receptor subtypes and ChAT are present (). Exogenous muscarinic agonist pilocarpine stimulated cell growth by increasing MAPK and Akt phosphorylation (), which demonstrated that muscarinic receptor signaling pathways in both cell lines are functional. Next, we determined which mAChR subtypes mainly mediate tumor growth responses to endogenous ACh secretion. Since the only source of ligand for M2R was endogenous ACh released by tumor cells, we examined the inhibitory effects of muscarinic subtype-selective antagonists on cell proliferation by adding them alone to tumor cells. M2R antagonist methoctramine had a significant inhibitory effect on tumor cell growth (). M2R knockdown using specific shRNA also inhibited cell growth (). However, neither M1 antagonist pirenzepine nor M3 antagonists (4-DAMP or darifenacin) had such effects on cell proliferation (Fig. S1). The results from this study and previous work by Hua Citation28 suggested that non-neuronal ACh may act as autoparacrine growth factor through different muscarinic receptors in different lung cancer cell types.
The M2 muscarinic receptors are coupled to Gi and upon activation lead to decreased adenylate cyclase activity and decreased cAMP. Cyclic AMP can stimulate cell growth in many cell types while inhibiting cell growth in others.Citation32 MAPK and Akt are 2 targets of cAMP that are associated with cell proliferation. MAPK and Akt can be activated or inhibited by cAMP, in a cell specific manner, to dictate the growth effects of cAMP. It has been reported that cholinergic stimulation leads to cell proliferation through MAPK and Akt pathways.Citation27,33 In this study, in parallel with stimulation of A549 and PC9 cell proliferation by either carbachol or pilocarpine, phosphorylation of MAPK and Akt was increased (Fig. S2 and S3). Methoctramine can inhibit MAPK and Akt phosphorylation induced by carbachol or pilocarpine in a dose-dependent manner (Fig. S2 and S3). More importantly, methoctramine also decreased basal phosphorylation of MAPK and Akt in a dose-dependent manner as shown in , suggesting that non-neuronal ACh plays a role in maintaining basal phosphorylation of MAPK and Akt in NSCLC. Knockdown of M2R with shRNA confirmed this, causing a similar decrease in basal phosphorylation of MAPK and Akt () as did methoctramine. Of note, studies have shown that interaction of nicotine with nicotinic receptors also leads to increased Akt and MAPK phosphorylation Citation33; thus, ACh secreted by lung tumors may also stimulate Akt and MAPK phosphorylation through interaction with nAChR. Therefore, in smokers with lung cancer, cholinergic-mediated proliferative pathways will be stimulated by exogenous nicotine and endogenous ACh.
Lung cancer is usually diagnosed until advanced stage, and metastasis is the main reason of treatment failure. Epithelial-mesenchymal transition (EMT) is a critical phenotypic alteration of cancer cells that triggers invasion and metastasis. In this study, our data indicated that non-neuronal ACh secreted by NSCLC is involved in EMT in A549 and PC9 cells. As shown in , activation of muscarinic receptors using pilocarpine induced EMT. Blocking M2R with methoctramine reversed pilocarpine-induced EMT. More importantly, methoctramine alone reversed EMT under basal conditions (), that is, the only source of M2R ligand is ACh secreted by tumor cells. These data suggested that non-neuronal ACh promotes EMT in part through activation of M2R. Knockdown of M2R with specific shRNA confirmed this (). Multiple lines of evidence indicate that nicotine can also induce EMT in lung cancer cells Citation34-36; thus non-neuronal ACh secreted by lung tumor may also induce EMT through activation of nAChR.
An important finding of this study is that blocking M2R signaling inhibits the growth of NSCLC xenograft tumors in vivo. As shown in , the growth of A549 xenograft tumors was inhibited by methoctramine in a dose-dependent manner. In addition, M2R-knockdown A549 cells had a slower growth rate than control A549 cells in vivo (). These data demonstrate that ACh secreted by NSCLC stimulates tumor growth in vivo partially through activation of M2R, suggesting M2R antagonist methoctramine may be an efficacious adjuvant therapy for NSCLC. However, methoctramine can block M2R in heart,Citation37 limiting its clinical use. Inhibition of tumor growth by methoctramine in vivo was also associated with decrease of MAPK and Akt phosphorylation in xenografts (), suggesting combinational inhibition of MAPK and Akt may also be a potential chemotherapeutic regimen for NSCLC. Moreover, methoctramine reversed EMT in A549 xenograft tumors (), which is consistent with its in vitro effect and indicates it might also be useful to slow tumor progression and metastasis.
In conclusion, the current study reveals that NSCLC A549 and PC9 cells express an autoparacrine cholinergic system which stimulates the growth of tumor cells. Antagonizing the local cholinergic loop by blocking M2R signaling inhibits tumor cell growth by decreasing MAPK and Akt phosphorylation and reverses EMT in vitro and in vivo. This suggests that selective M2R antagonists may have clinical utility to slow the growth and progression of NSCLC.
Materials and Methods
Reagents
Pilocarpine, carbachol, 4-diphenyl-acetoxy-N-methyl-piperidine (4-DAMP), pirenzepine, methoctramine, and darifenacin were purchased from Sigma-Aldrich (St. Louis, MO, USA). Antibodies used for Western blotting were purchased from Cell Signaling Technology (Danvers, MA, USA) for p44/42 MAPK (extracellular signal-regulated kinase 1/2), phosphor-p44/42 MAPK (Thr202/ Tyr204), Akt, phosphor-Akt (Ser473), E-cadherin, β-catenin, vimentin and β-actin. M2R antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
NSCLC cell culture
Human NSCLC A549 cells were purchased from the Cell Bank of Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China) and PC9 cells were gifts from Dr. Qianggang Dong in Shanghai Cancer Institute, Shanghai Jiao Tong University School of Medicine. Both cell lines were routinely cultured in DMEM (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Invitrogen), 100 μg/ml penicillin and 100 U/ml streptomycin sulfate in a humidified incubator at 37°C with 5% CO2.
Cell proliferation assay
The proliferative capacity of cells was assessed using the cell counting kit-8 (CCK8) (Dojindo, Shanghai, China) and Real Time Cellular Analysis (RTCA) using the xCELLigence system (Roche, Indianapolis, IN, USA) according to the manufacturer's instructions. For CCK8 assay, cells were seeded at a density of 3000 cells/well in 96-well plates. After incubated with serum-free DMEM for 24 h, the cells were treated with agonist carbachol (50 μM) or pilocarpine (50 μM) or antagonists (10−8–10−5M) in DMEM containing 1% FBS for 72 h. For the combination administration, the agonist was added 1 h before antagonist. Each experiment was performed in 4 or 5 replicates and repeated for 3 times. Cells treated with solvent (DMSO) were used as a control, with viability set at 100%.
For RTCA, following background reading, 1000 or 2000 cells were plated in each well in 16-well plates in 100 μL volume. Continuous impedance measurements were taken every 15 to 30 minutes for more than 60 hours. Each assay was done in triplicate or quadruplicate and repeated twice.
RT-PCR
Total RNA was extracted from cells using TRIzol reagent (Invitrogen). RNA was reverse transcribed to synthesize cDNA by using SuperScript II Reverse transcriptase (Invitrogen) according to the manufacturer's instructions. The primers used for PCR were listed in . GAPDH was used as the loading control. The PCR products were resolved on a 1.5% agarose gel stained with ethidium bromide.
Table 1. Primer list for PCR
Western blot
Cancer cells were lysed in RIPA buffer supplemented with protease inhibitor cocktail (Sigma-Aldrich). Equal amounts of lysates were separated on 10% SDS-PAGE and then transferred to PVDF membranes (Millipore, Billerica, MA, USA). Membranes were blocked with PBS buffer containing 5% non-fat milk and 0.1% Tween 20 (PBST) and then incubated with primary antibodies (1:500–1:1000) overnight at 4°C. After being washed 3 times with PBST, membranes were incubated with peroxidase-conjugated secondary antibodies (1:3000) for 1 h and then washed with PBST again and developed with ECL (Pierce, Rockford, IL, USA). β-actin was used as an internal control for loading control. Relative quantities of proteins in Western blotting were analyzed using Image J Software (US National Institutes of Health).
Short hairpin RNA (shRNA) lentiviral particles transduction
Human M2R shRNA lentiviral particles were purchase from Santa Cruz Biotechnology. Lentiviral particles transduction was performed according to manufacturer's instructions. Briefly, lentiviral particles were added to the cells (a 1:40 dilution of lentivirus stock in DMEM) and after overnight the viral particle-containing medium was replaced with fresh complete DMEM medium. After 24 hours the medium was changed again and cells were incubated in complete DMEM medium containing puromycin (5 μg/ml) in order to select transduced cells. In the control group, the cells were transduced with control shRNA lentiviral particles (Santa Cruz Biotechnology) exactly the same way.
Immunofluorescence and confocal microscopy
Cells were fixed in 4% paraformaldehyde, permeabilized in 0.2% Triton X-100, blocked in 1% BSA, and then stained with antibody (1:100) at 4°C overnight. After being washed 3 times with PBS, cells were incubated with FITC-conjugated secondary antibodies (1:100) for 1 h, and then washed with PBS again and incubated with Hoest 33342 for nuclear counterstaining. Cells were visualized with a Zeiss LSM710 (Zeiss, Thornwood, NY, USA) confocal microscope.
Nude mice studies
Mice (nu/nu, 20 ± 2g) were purchased from the Shanghai Laboratory Animal Center (Chinese Academy of Sciences, Shanghai, China) and housed in environmentally controlled, specific pathogen-free conditions for 1 week before the study. All experimental procedures were reviewed and approved in accordance with the guidelines for the care and use of laboratory animals at Shanghai Jiao Tong University. Cells (A549, A549 control shRNA and A549 M2R shRNA) were grown as described above and resuspended in DMEM at a concentration of 2.5 × 10Citation7/ml. A 200 μl aliquot of this cell suspension was injected s.c. into the left flank of each mouse. For methoctramine treatment experiment, tumors were then allowed to grow for 1 week, and then drug administration was initiated and continued for the next 3 weeks. Methoctramine was dissolved in saline and administered by i.p. at doses of 2 and 5 mg/kg/day. Control animals received the same volume of saline daily. The tumor volume and mice body weight were monitored 3 times a week. Tumor volumes (mm3) were calculated as length ×widthCitation2/2. At the end of the study, the mice were sacrificed and the tumors were removed and frozen for protein analyses.
Statistical analysis
All data are presented as the mean±SEM. Statistical analysis was conducted using GraphPad Prism 5.0 software (La Jolla, CA, USA). Differences between groups were examined using Student's t-test. Differences were considered significant if P value was less than 0.05.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental_Figures___Captions.zip
Download Zip (252 KB)Funding
This work was supported by National Natural Science Foundation of China (81173084 and 81372522), Science and Technology Commission of Shanghai Municipality (12ZR1416000 and 12140901400), and International Science & Technology Cooperation Program of China (2011DFA33180).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Savas P, Hughes B, Solomon B. Targeted therapy in lung cancer: IPASS and beyond, keeping abreast of the explosion of targeted therapies for lung cancer. J Thorac Dis 2013; 5:S579-92; PMID:24163750
- Zarogoulidis K, Zarogoulidis P, Darwiche K, Boutsikou E, Machairiotis N, Tsakiridis K, Katsikogiannis N, Kougioumtzi I, Karapantzos I, Huang H, et al. Treatment of non-small cell lung cancer (NSCLC). J Thorac Dis 2013; 5:S389-96; PMID:24102012
- Grando SA, Kawashima K, Kirkpatrick CJ, Meurs H, Wessler I. The non-neuronal cholinergic system: basic science, therapeutic implications and new perspectives. Life Sci 2012; 91:969-72; PMID:23141771; http://dx.doi.org/10.1016/j.lfs.2012.10.004
- Beckmann J, Lips KS. The non-neuronal cholinergic system in health and disease. Pharmacology 2013; 92:286-302; PMID:24296914; http://dx.doi.org/10.1159/000355835
- Proskocil BJ, Sekhon HS, Jia Y, Savchenko V, Blakely RD, Lindstrom J, Spindel ER. Acetylcholine is an autocrine or paracrine hormone synthesized and secreted by airway bronchial epithelial cells. Endocrinology 2004; 145: 2498-506; PMID:14764638; http://dx.doi.org/10.1210/en.2003-1728
- Reinheimer T, Bernedo P, Klapproth H, Oelert H, Zeiske B, Racké K, Wessler I. Acetylcholine in isolated airways of rat, guinea pig, and human: species differences in role of airway mucosa. Am J Physiol 1996; 270:L722-8; PMID:8967505
- Grando SA, Kist DA, Qi M, Dahl MV. Human keratinocytes synthesize, secrete, and degrade acetylcholine. J Invest Dermatol 1993; 101:32-6; PMID:8331294; http://dx.doi.org/10.1111/1523-1747.ep12358588
- Klapproth H, Reinheimer T, Metzen J, Münch M, Bittinger F, Kirkpatrick CJ, Höhle KD, Schemann M, Racké K, Wessler I. Non-neuronal acetylcholine, a signaling molecule synthezised by surface cells of rat and man. Naunyn Schmiedebergs Arch Pharmacol 1997; 355:515-23; PMID:9109369; http://dx.doi.org/10.1007/PL00004977
- Wessler I, Reinheimer T, Klapproth H, Schneider FJ, Racké K, Hammer R. Mammalian glial cells in culture synthesize acetylcholine. Naunyn Schmiedebergs Arch Pharmacol 1997; 356:694-7; PMID:9402051; http://dx.doi.org/10.1007/PL00005107
- Haberberger RV, Bodenbenner M, Kummer W. Expression of the cholinergic gene locus in pulmonary arterial endothelial cells. Histochem Cell Biol 2000; 113:379-87; PMID:10883397
- Shah N, Khurana S, Cheng K, Raufman JP. Muscarinic receptors and ligands in cancer. Am J Cell Physiol 2009; 296:C221-32; PMID:19036940; http://dx.doi.org/10.1152/ajpcell.00514.2008
- Wessler I, Kirkpatrick CJ, Racke K. Non-neuronal acetylcholine, a locally acting molecule, widely distributed in biological systems: expression and function in humans. Pharmacol Ther 1998; 77:59-79; PMID:9500159; http://dx.doi.org/10.1016/S0163-7258(97)00085-5
- Song P, Sekhon HS, Proskocil B, Blusztajn JK, Mark GP, Spindel ER. Synthesis of acetylcholine by lung cancer. Life Sci 2003; 72:2159-68; PMID:12628474; http://dx.doi.org/10.1016/S0024-3205(03)00078-X
- Song P, Sekhon HS, Fu XW, Maier M, Jia Y, Duan J, Proskosil BJ, Gravett C, Lindstrom J, Mark GP, et al. Activated cholinergic signaling provides a target in squamous cell lung carcinoma. Cancer Res 2008; 68:4693-700; PMID:18559515; http://dx.doi.org/10.1158/0008-5472.CAN-08-0183
- Cheng K, Samimi R, Xie G, Shant J, Drachenberg C, Wade M, Davis RJ, Nomikos G, Raufman JP. Acetylcholine release by human colon cancer cells mediates autocrine stimulation of cell proliferation. Am J Physiol Gastrointest Liver Physiol 2008; 295:G591-7; PMID:18653726; http://dx.doi.org/10.1152/ajpgi.00055.2008
- Song P, Sekhon HS, Jia Y, Keller JA, Blusztajn JK, Mark GP, Spindel ER. Acetylcholine is synthesized by and acts as an autocrine growth factor for small cell lung carcinoma. Cancer Res 2003; 63:214-21; PMID:12517800
- Chen CS, Lee CH, Hsieh CD, Ho CT, Pan MH, Huang CS, Tu SH, Wang YJ, Chen LC, Chang YJ, et al. Nicotine-induced human breast cancer cell proliferation attenuated by garcinol through downregulation of the nicotinic receptor and cyclin D3 proteins. Breast Cancer Res Treat 2011,125:73-87; PMID:20229177; http://dx.doi.org/10.1007/s10549-010-0821-3
- Paleari L, Catassi A, Ciarlo M, Cavalieri Z, Bruzzo C, Servent D, Cesario A, Chessa L, Cilli M, Piccardi F, et al. Role of alpha7-nicotinic acetylcholine receptor in human non-small cell lung cancer proliferation. Cell Prolif 2008; 41:936-59; PMID:19040571; http://dx.doi.org/10.1111/j.1365-2184.2008.00566.x
- Wong HP, Yu L, Lam EK, Tai EK, Wu WK, Cho CH. Nicotine promotes cell proliferation via a7-nicotinic acetylcholine receptor and catecholamine-synthesizing enzymes-mediated pathway in human colon adenocarcinoma HT-29 cells. Toxicol Appl Pharma 2007; 221:261-7; http://dx.doi.org/ 10.1016/j.taap.2007.04.002
- Chen RJ, Ho YS, Guo HR, Wang YJ. Rapid activation of stat3 and ERK1/2 by nicotine modulates cell proliferation in human bladder cancer cells. Toxicol Sciences 2008; 104:283-93; PMID:18448488; http://dx.doi.org/10.1093/toxsci/kfn086
- Calleja-Macias IE, Kalantari M, Bernard HU. Cholinergic signaling through nicotinic acetylcholine receptors stimulates the proliferation of cervical cancer cells: An explanation for the molecular role of tobacco smoking in cervical carcinogenesis? Int J Cancer 2009; 124: 1090-6; PMID:19048619; http://dx.doi.org/10.1002/ijc.24053
- van Koppen CJ, Kaiser B. Regulation of muscarinic acetylcholine receptor signaling. Pharmacol Ther 2003; 98:197-220; PMID:12725869; http://dx.doi.org/10.1016/S0163-7258(03)00032-9
- Wessler I, Kirkpatrick CJ. Acetylcholine beyond neurons: the non-neuronal cholinergic system in humans. Br J Pharmacol 2008; 154:1558-71; PMID:18500366; http://dx.doi.org/10.1038/bjp.2008.185
- Cheng K, Zimniak P, Raufman JP. Transactivation of the epidermal growth factor receptor mediates cholinergic agonist-induced proliferation of H508 human colon cancer cells. Cancer Res 2003; 63:6744-50; PMID:14583469
- Jimenez E, Montiel M. Activation of MAP kinase by muscarinic cholinergic receptors induces cell proliferation and protein synthesis in human breast cancer cells. J Cell Physiol 2005; 204:678-86; PMID:15744749; http://dx.doi.org/10.1002/jcp.20326
- Rayford W, Noble MJ, Austenfeld MA, Weigel J, Mebust WK, Shah GV. Muscarinic cholinergic receptors promote growth of human prostate cancer cells. Prostate 1997; 30:160-6; PMID:9122040; http://dx.doi.org/10.1002/(SICI)1097-0045(19970215)30:3%3c160::AID-PROS3%3e3.0.CO;2-Q
- Song P, Sekhon HS, Lu A, Arredondo J, Sauer D, Gravett C, Mark GP, Grando SA, Spindel ER. M3 muscarinic receptor antagonists inhibit small cell lung carcinoma growth and mitogen-activated protein kinase phosphorylation induced by acetylcholine secretion. Cancer Res 2007; 67:3936-44; PMID:17440109; http://dx.doi.org/10.1158/0008-5472.CAN-06-2484
- Hua N, Wei X, Liu X, Ma X, He X, Zhuo R, Zhao Z, Wang L, Yan H, Zhong B, Zheng J. A novel muscarinic antagonist R2HBJJ inhibits non-small cell lung cancer cell growth and arrests the cell cycle in G0/G1. PLoS One 2012; 7:e53170; PMID:23285263; http://dx.doi.org/10.1371/journal.pone.0053170
- Kaniaris P, Fassoulaki A, Liarmakopoulou K, Dermitzakis E. Serum cholinesterase levels in patients with cancer. Anesth Analg 1979; 58:82-4; PMID:571250; http://dx.doi.org/10.1213/00000539-197903000-00004
- Martínez-Moreno P, Nieto-Cerón S, Torres-Lanzas J, Ruiz-Espejo F, Tovar-Zapata I, Martínez-Hernández P, Rodríguez-López JN, Vidal CJ, Cabezas-Herrera J. Cholinesterase activity of human lung tumors varies according to their histological classification. Carcinogenesis 2006; 27:429-36; PMID:16272577; http://dx.doi.org/10.1093/carcin/bgi250
- Ami N, Koga K, Fushiki H, Ueno Y, Ogino Y, Ohta H. Selective M3 muscarinic receptor antagonist inhibits small-cell lung carcinoma growth in a mouse orthotopic xenograft model. J Pharmacol Sci 2011; 116:81-8; PMID:21512307; http://dx.doi.org/10.1254/jphs.10308FP
- Stork PJ, Schmitt JM. Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol 2002; 12:258-66; PMID:12074885; http://dx.doi.org/10.1016/S0962-8924(02)02294-8
- Carlisle DL, Liu X, Hopkins TM, Swick MC, Dhir R, Siegfried JM. Nicotine activates cell-signaling pathways through muscle-type and neuronal nicotinic acetylcholine receptors in non-small cell lung cancer cells. Pulm Pharmacol Ther 2007; 20:629-41; PMID:17015027; http://dx.doi.org/10.1016/j.pupt.2006.07.001
- Dasgupta P, Rizwani W, Pillai S, Kinkade R, Kovacs M, Rastogi S, Banerjee S, Carless M, Kim E, Coppola D, Haura E, Chellappan S. Nicotine induces cell proliferation, invasion and epithelial-mesenchymal transition in a variety of human cancer cell lines. Int J Cancer 2009; 124:36-45; PMID:18844224; http://dx.doi.org/10.1002/ijc.23894
- Davis R, Rizwani W, Banerjee S, Kovacs M, Haura E, Coppola D, Chellappan S. Nicotine promotes tumor growth and metastasis in mouse models of lung cancer. PLoS One 2009; 4:e7524; PMID:19841737
- Wu SQ, Lv YE, Lin BH, Luo LM, Lv SL, Bi AH, Jia YS. Silencing of periostin inhibits nicotine-mediated tumor cell growth and epithelial-mesenchymal transition in lung cancer cells. Mol Med Rep 2013; 7:875-80; PMID:23314871
- Wess J, Angeli P, Melchiorre C, Moser U, Mutschler E, Lambrecht G. Methoctramine selectively blocks cardiac muscarinic M2 receptors in vivo. Naunyn Schmiedebergs Arch Pharmacol 1988; 338:246-9; PMID:3057387; http://dx.doi.org/10.1007/BF00173395