
Abstract
It has been reported that persistent or excessive autophagy promotes cancer cell death during chemotherapy, either by enhancing the induction of apoptosis or mediating autophagic cell death. Here, we show that miR-15a and miR-16 are potent inducers of autophagy. Rictor, a component of mTORC2 complex, is directly targeted by miR-15a/16. Overexpression of miR-15a/16 or depletion of endogenous Rictor attenuates the phosphorylation of mTORC1 and p70S6K, inhibits cell proliferation and G1/S cell cycle transition in human cervical carcinoma HeLa cells. Moreover, miR-15a/16 dramatically enhances anticancer drug camptothecin (CPT)-induced autophagy and apoptotic cell death in HeLa cells. Collectively, these data demonstrate that miR-15a/16 induced autophagy contribute partly to their inhibition of cell proliferation and enhanced chemotherapeutic efficacy of CPT.
Abbreviations
| BAFA1 | = | bafilomycin A1 |
| CPT | = | Camptothecin |
| miRNA | = | microRNA |
| LC3 | = | microtubule-associated protein 1 light chain 3 |
| mTOR | = | mechanistic target of rapamycin |
| Rictor | = | Rptor independent companion of mTOR complex 2 |
| MUT | = | mutated |
| UTR | = | untranslated region |
| NC | = | negative control |
Introduction
Autophagy is an evolutionarily conserved cellular catabolic process, by which damaged or excessive organelles and cytoplasmic proteins are degraded through lysosome degradation system.Citation1 Recently, autophagy has been proposed to play important roles in many physiological and pathological processes, such as cell proliferation, differentiation and metabolism.Citation2–5 The mechanistic target of rapamycin (mTOR) signaling pathway senses and integrates a variety of environmental cues to regulate organismal growth and homeostasis, including autophagy.Citation6 It is well established that activation of rapamycin-sensitive mTORC1 inhibits autophagosome formation through phosphorylating and repressing Ulk1 kinase activity.Citation7 mTORC1 also indirectly regulates autophagy by controlling lysosome biogenesis through phosphorylation of transcription factor EB (TFEB), which drives the transcription of several lysosome- and autophagy-specific genes.Citation8 Unlike mTORC1, much less is known about rapamycin-insensitive mTORC2 pathway. After being activated by growth factors, mTORC2 regulates cell growth and proliferation via phosphorylation of Akt at Ser473 and SGK1.Citation9 Activation of Akt and SGK1 results in phosphorylation and nuclear export inactivation of the transcription factor FoxO3a,Citation9,10 which regulates the expression of autophagy-related genes, such as LC3 and BNIP3.Citation11,12 In this manner, mTORC2 also acts as a negative regulator of autophagy.
MicroRNAs (miRNAs) are a class of noncoding RNA that negatively regulate gene expression through binding to the 3′ untranslated region (3′UTR).Citation13,14 MiRNAs are important regulators in various fundamental biological and pathological processes, such as cell proliferation, apoptosis, differentiation, metabolism, and cancer, as well as autophagy.Citation15–20 MiRNAs have recently been identified as important regulators of autophagy.Citation19 Some miRNAs suppress autophagic activity via targeting autophagy-related proteins (ATGs) under stress conditions. It has been shown that miR-20a impedes autophagy through Atg16L1 during hypoxia induced osteoclast differentiation,Citation21 while miR-181a inhibits starvation and rapamycin induced autophagy through Atg5.Citation22 Some miRNAs promote autophagy by repressing important upstream signals of autophagy pathway. For example, hypoxia induced miR-155 promotes autophagic activity through targeting Rheb and other component in mTOR signaling.Citation23,24 miR-34a inhibits autophagy and enhances chemotherapy-induced apoptosis in the retinoblastoma cell through HMGB1.Citation25
The miR-15a/107 group of miRNA contains a series of miRNAs, including miR-15a, miR-15b, miR-16, miR-103 and miR-107 family. MiR-15a and miR-16 form 2 different clusters in mammals and these 2 miRNAs are found to be frequently deleted or downregulated in many types of cancers, including chronic lymphocytic leukemia (CLL), colorectal cancer, squamous-cell carcinoma, ovarian cancer and prostate cancer.Citation26 As tumor suppressors, miR-15a and miR-16 were shown to inhibit cancer cell proliferation by targeting various cyclins and CDKs and induce apoptosis through down-regulating the anti-apoptotic gene Bcl-2.Citation27–29 In the present study, we report miR-15a and miR-16 directly target Rictor, an important component in mTORC2 pathway. Overexpression of miR-15a/16 or knockdown Rictor downregulate mTORC1/p70S6K, increase autophagic activity. In addition, miR-15a/16 significantly enhances anticancer drug CPT-induced apoptotic cell death through excessive autophagy.
Results
MiR-15a/16 increases autophagic activity
To elucidate the effects of miR-15a and miR-16 on autophagy, we performed LC3 puncta formation and LC3 conversion assays. HeLa cells stably expressing GFP-LC3 fusion protein were transiently transfected with miR-15a or miR-16 mimics. GFP-LC3 puncta formation was visualized by confocal microscope. As shown in , there was a significant increase of GFP-LC3 puncta in cells transfected with miR-15a or miR-16 compared with the negative control (NC). Quantification of GFP-LC3 dots in each cell confirmed that autophagosomes accumulated when miR-15a and miR-16 were over-expressed ().
Figure 1. MiR-15a and miR-16 increase autophagic activity. (A, B) MiR-15a and miR-16 promote GFP-LC3 puncta formation. HeLa cells stably expression GFP-LC3 were transfected with miR-15a, miR-16 or negative control (NC). Cells were fixed at 48 h post transfection. (A) Representative images were captured by confocal microscope. Scale bar, 10 μm. (B) Quantitative analysis of GFP-LC3 puncta in (A). Data shown are means ± SD of three independent experiments. **P < 0.01. (C) Over-expression of miR-15a or miR-16 induces LC3-II conversion and p62 degradation. Densitometric analysis were calculated using Image J software. (D) Overexpression of miR-15a and miR-16 increase autophagic flux. HeLa cells transfected with NC, miR-15a or miR-16 were treated with Baf A1 (10 nM) for 2 h. Western blotting was performed to analyze the status of LC3, p62 and GAPDH. (E) Densitometric analysis of LC3-II/GAPDH or (F) p62/GAPDH ratios from immunoblots using Image J. Data shown are means ± SD of three independent experiments. *P < 0.05, **P < 0.01, student 2-tailed t test.
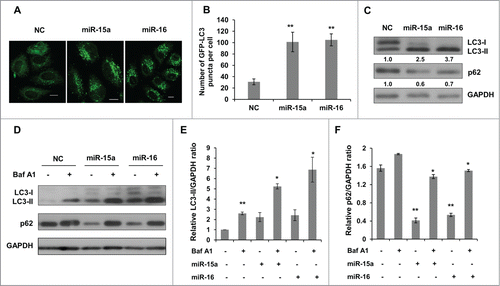
We then detected the conversion of LC3-I to LC3-II and p62 expression using western blot. Consistent with the results from the GFP-LC3 puncta assay, there was a significant increase of lipidated LC3-II in cells transfected with miR-15a or miR-16 (). p62/SQSTM1 is a selective substrate for autophagy-lysosome degradation, so total p62 protein levels reflect autophagic activity. Indeed, overexpression of miR-15a and miR-16 resulted in reduced p62 protein levels (). To test whether the increase in autophagosomes is due to increased autophagy activity or a block in downstream degradation, we performed an autophagic flux assay. Bafilomycin A1 (Baf A1) is a lysosomotropic reagent that blocks autophagosome degradation. As we expected, Baf A1 treatment caused elevated levels of LC3-II in the NC, miR-15a and miR-16 transfected cells (). In addition, p62 protein levels also increased upon Baf A1 treatment in miR-15a and miR-16 transfected cells (). Therefore, we conclude that miR-15a and miR-16 increase autophagic activity.
Inhibition of endogenous miR-15a and miR-16 repress autophagic activity
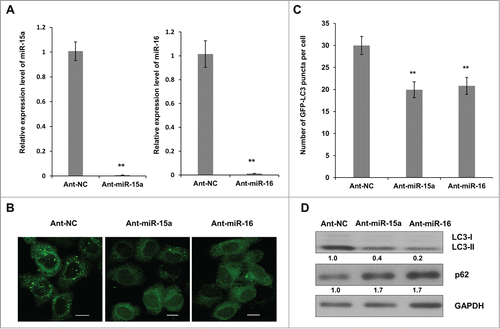
To further identify the connection between miR-15a, miR-16 and autophagy, we inhibited endogenous miR-15a and miR-16 expression and repeated the above assays. We used a single strand miRNA inhibitor (Ant-miR-15a and Ant-miR-16) to block endogenous miR-15a and miR-16 expression (). GFP-LC3 puncta accumulation was suppressed when endogenous miR-15a and miR-16 were inhibited (). A Western blot of LC3 conversion and p62 expression also showed that inhibition of miR-15a or miR-16 resulted in a decrease in LC3-II conversion and an increase in p62 expression (). These results demonstrate the relevance of endogenous miR-15a and miR-16 with autophagy.
Figure 2. Inhibition of endogenous miR-15a and miR-16 repress autophagic activity. (A) Blockage of endogenous miR-15a or miR-16 expression by transfection of single strand ant-miRNA. (B) Inhibition of endogenous miR-15a or miR-16 inhibits GFP-LC3 puncta formation. HeLa cells transfected with ant-miR-15a, ant-miR-16 or control (ant-NC) were fixed at 48 h after transfection. Representative images were captured by confocal microscope. Scale bar, 10 μm. (C) Quantitative analysis of GFP-LC3 puncta per cell. At least 200 cells were examined in each experimental group. Data shown are means ± SD of three independent experiments. **P < 0.01. (D) HeLa cells were transfected with ant-miR-15a, ant-miR-16 or ant-NC. Western blotting was performed to analyze the status of LC3, p62, and GAPDH.
Rictor is a direct target of miR-15a and miR-16
Having confirmed miR-15a and miR-16 function in the regulation of autophagy, we employed the miRNA prediction algorithm, Findtar and TargetScan, to clarify potential targets of miR-15a and miR-16.Citation30,31 Both of the algorithms predicted Rictor as a potential target of miR-15a and miR-16 (). We measured Rictor protein expression levels using immunoblot and results showed that Rictor protein expression was significantly lower when miR-15a and miR-16 were overexpressed and protein levels significantly increased when endogenous miR-15a and miR-16 were inhibited ().
Figure 3. Rictor is a direct target of miR-15a and miR-16. (A) Predicted binding sequence of miR-15a, miR-16 in Rictor 3′ UTR. (B) Western blot analysis of Rictor and GAPDH proteins in HeLa cells transfected with miR-15a, miR-16, NC, ant-miR-15a, ant-miR-16 or anti-NC. Protein ratios were determined by ImageJ densitometric analysis. (C) Luciferase reporter assay of wild type (WT) or mutated (MUT) Rictor 3′UTR vector co-transfected with NC, miR-15a or miR-16 respectively. Data shown are means ± SD of three independent experiments, **P < 0.01, student 2-tailed t test. (D) HeLa cells were transfected with Rictor siRNAs, miR-15a, miR-16 or NC. Western blotting was performed to analyze the status of LC3, p62, Rictor and GAPDH.
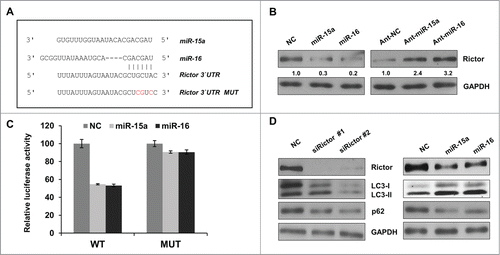
In order to confirm the direct binding of miR-15a and miR-16 to 3′UTR of Rictor mRNA, the 3′UTR fragments containing wild-type (WT) or mutated (MUT) seed regions of the miR-15a and miR-16 targets were cloned into a dual-luciferase reporter vector (). The WT reporter vector exhibited a significant reduction of luciferase activity when co-transfected with miR-15a and miR-16 compared to NC. In contrast, the seed region mutation vector abrogated this inhibitory effect of miR-15a and miR-16 (). Therefore, miR-15a and miR-16 suppress Rictor expression through direct binding to its 3′UTR.
Knockdown of Rictor has effects similar to miR-15a or miR-16
After we identified that Rictor is real target of miR-15a and miR-16, we wanted to test whether knockdown of Rictor has a similar effect on autophagy. We used Rictor siRNA to knock down its endogenous expression and explore its function on LC3 conversion or p62 protein degradation. As shown in , Rictor siRNA has the effect of increasing LC3 conversion and decreasing p62 protein levels, similar to miR-15a and miR-16. Therefore, miR-15a and miR-16 may induce autophagy by suppressing Rictor expression.
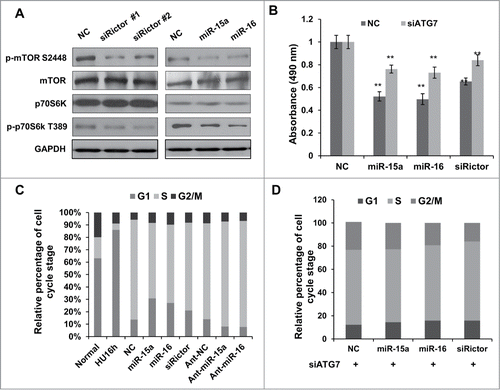
It has previously been reported that mTORC2 can activate mTORC1 through Akt phosphorylation.Citation32 Consistent with previous descriptions, mTORC1 activity was downregulated when we knocked down Rictor with siRNA. Phosphorylation of mTOR and downstream p70S6K were reduced in Rictor knocked-down cells. Overexpression of miR-15a or miR-16 had an effect on mTORC1 and p70S6K activity, similar to Rictor siRNA (). These data cumulatively reveal that miR-15a and miR-16 have an effect on autophagy and mTORC1 activity that is similar to Rictor siRNA.
Figure 4. MiR-15a and miR-16 inhibit cell proliferation partly through autophagy. (A) HeLa cells were transfected with Rictor siRNAs, miR-15a, miR-16 or NC. Western blotting were performed to analyze the status of total and phosphorylated mTOR and p70S6K. (B) MTT assay of HeLa cells transfected with miR-15a, miR-16, Rictor siRNA or NC, in the presence or absence of ATG7 siRNA. Cell viability (OD 490 nm absorbance) was examined at 48 h after transfection. Data shown are means ± SD of three independent experiments, **P < 0.01, student 2-tailed t test. (C) miR-15a and miR-16 induces G1/S cell cycle arrest. HeLa cells transfected with miR-15a, miR-16, Rictor siRNA or NC were synchronized at G1/S boundary by treatment with hydroxyurea (HU). Cells were released from HU block for 4 h, fixed and stained with with PI for Flow cytometry analysis. (D) Inhibition of autophagy partially rescues miR-15a and miR-16 induced G1/S cell cycle arrest. HeLa cells co-transfected with ATG7 siRNA and miR-15a, miR-16, Rictor siRNA or NC were synchronized at G1/S and released for 4 h. Percentage of G1, S and G2/M cells was quantified from flow cytometry analysis.
MiR-15a and miR-16 inhibit cell growth partly through autophagy
In order to test whether miR-15a and miR-16 exert their cell inhibitory function through autophagy, we performed MTT and flow cytometry analysis. As shown in , the MTT assay revealed that there was a significant reduction in cell growth at 48h in cells transfected with miR-15a, miR-16 or Rictor siRNA. A flow cytometry assay also showed G1 arrest in cells overexpressing miR-15a/16, as well as in cells with Rictor knockdown (). Atg7 is an E1-like enzyme and required for covalent conjugation of Atg12 to Atg5. Atg7 is an essential gene for autophagy, so we used Atg7 siRNA to block autophagy. Our results demonstrated that blocking autophagic activity abolished the G1/S arrest by miR-15a/16 or Rictor siRNA (). The cell proliferation assay also showed that depletion of Atg7 partly restored the inhibitory effect of miR-15a/16 and Rictor siRNA on cell proliferation (). These indicate that there is a potential link between miR-15a and miR-16 induced autophagy and inhibition of cell proliferation.
MiR-15a and miR-16 enhance chemotherapeutic efficacy of Camptothecin
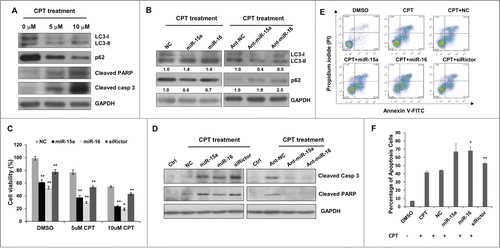
Camptothecin (CPT) is an anti-cancer drug used in cancer chemotherapy. At concentrations of both 5uM and 10uM, treatment with CPT dramatically induced autophagy and apoptotic cell death in HeLa cells, as revealed by LC3-II conversion, p62 degradation, cleavage of PARP and caspase-3 (). Interestingly, overexpression of miR-15a or miR-16 significantly increased CPT-induced autophagic activity, whereas knockdown endogenous miR-15a/16 produced the opposite effects (). Then we tested whether miR-15a and miR-16 would enhance the anti-cancer efficacy of CPT. Cells transfected with miR-15a or miR-16 exhibited more rigorously decrease in the cell proliferation assay (). Immunoblotting results also confirmed that CPT induced apoptosis was greatly enhanced by miR-15a or miR-16 compared with NC while knockdown endogenous miR-15a and miR-16 decreased CPT induced apoptosis as determined by cleaved PARP1 and caspase-3 (). Moreover, flow cytometry assay showed that miR-15a and miR-16 transfection led to more apoptotic cells than CPT only (Fig. 5E and F). Collectively, these data support the notion that miR-15a and miR-16 down-regulate cell proliferation partly through modulation of autophagy and enhance chemotherapeutic efficacy of the anticancer drug CPT.
Figure 5. MiR-15a and miR-16 enhance chemotherapeutic efficacy of Camptothecin. (A) Camptothecin (CPT) induced LC3 conversion, p62 degradation, and cleavage of caspase 3 and PARP. (B) Transfection of miR-15a or miR-16 increased CPT-induced autophagy and inhibition of endogenous miR-15a or miR-16 had the opposite effects on CPT-induced autophagy. Image J densitometric analysis of LC3-II/GAPDH and p62/GAPDH was shown. (C) Cell viability assay of HeLa cells transfected with miR-15a, miR-16 or Rictor siRNA in DMSO or CPT treated cells. Data shown are means ± SD of three independent experiments, **P < 0.01, student 2-tailed t test. (D) Western blot analysis of cleaved PARP1 and caspase 3 in HeLa cells transfected with miR-15a, miR-16, Rictor siRNA, NC or ant-miR-15a, ant-miR-16, ant-NC and treated with CPT for 24 h. (E) HeLa cells transfected with miR-15a, miR-16, Rictor siRNA, or NC were incubated with 10 uM CPT for 36 h. Cells were harvested and stained with PtdIns and annexin V-FITC for apoptotic analysis. (F) The percentage of apoptotic cells were the sum of the early apoptotic cells located in the lower right quadrant (annexin V–FITC +/PI - cells), as well as late apoptotic cells located in the upper right quadrant (annexin V–FITC +/PtdIns - cells). Data shown are means ± SD of three independent experiments, *P < 0.05, **P < 0.01, student 2-tailed t test.
Discussion
MiRNAs are important regulators in various biological processes by targeting genes in different signaling pathways. MiR-15a and miR-16 were reported as tumor suppressors and inhibited cancer cell proliferation.Citation26 MiR-15a and miR-16 are frequently deleted or down-regulated in cancer, especially in chronic lymphocytic leukemia (CLL), overexpression of miR-15a and miR-16 suppresses cancer cell proliferation.Citation33 In this study, we discovered that miR-15a and miR-16 inhibit cell proliferation partly through autophagy induction. MiR-15a/16 mediated repression of Rictor lead to downregualtion of mTORC1/p70S6K, induction of autophagy and inhibition of cell proliferation.
mTOR plays a central role in nutrient sensing and homeostasis of cellular biosynthesis and degradation. mTOR forms 2 complexes, designated as mTORC1 and mTORC2, respectively. Rictor is an essential component of mTORC2 and is activated by growth factors.Citation34,35 Once mTORC2 is activated, it induces cell proliferation through the phosphorylation of the AGC family of kinases, such as Akt, SGK and PKC.Citation9 Through phosphorylation of Akt, mTORC2 can positively regulate the activity of mTORC1.Citation9,32 It would be reasonable to speculate that mTORC2 acts as a negative regulator of autophagy. Consistent with this view, inhibition of mTORC2 activity induces autophagy and muscle atrophy through activation of FoxO3, a transcription factor downstream of Akt that induces expression of many autophagy-related genes.Citation11,12 However, mTORC2 has also been reported as a positive regulator of autophagy through mTORC2-Ypk signaling during amino acid starvation.Citation36,37 Therefore, the function of mTORC2 in autophagy regulation is dependent on cell context. Our results indicate that mTORC2 acts as a negative regulator and knockdown of Rictor induces autophagy.
Chemotherapy and radiotherapy play important roles in current cancer therapies and create an interplay between autophagy and cancer therapy.Citation38 Combination chemotherapy is widely used to treat cancer and is more effective than single chemotherapy because of resistance to single chemical treatments.Citation39 Several lines of evidence implicates a paradoxical role of autophagy following anticancer treatments. Autophagy induction can increase or decrease anticancer drug efficacy. On the one hand, autophagy is activated as a pro-survival mechanism in some cancer cells during chemotherapy. Inhibition of autophagy can re-sensitize drug resistant cancer cells and augment cytotoxicity of chemotherapeutic agents. On the other hand, persistent or excessive autophagy promotes cancer cell death during chemotherapy, either by enhancing the induction of apoptosis or mediating autophagic cell death.Citation40 Several lines of evidence suggest that miRNA sensitizes cancer cells to chemotherapeutic drugs through the regulation of autophagy signaling.Citation41-44 It has been reported that miR-30a suppresses Beclin 1-mediated autophagy and sensitizes tumor cells to cisplatinum.Citation45 However, miR-21 downregulation causes enhanced autophagic activity and increased chemosensitivity to etoposide or doxorubicin in K562 and KYO-1 cells, and this chemosensitivity was reverted by pre-treating cells with autophagy inhibitor 3-MA.Citation46 Inhibition of miR-25 leads to autophagic cell death and increases chemosensitivity to isoliquiritigenin in MCF-7/ADR cells.Citation47 Considering the paradoxical role of autophagy in cancer therapy, recognizing functional autophagy status in tumors will be required to direct the appropriate therapy.
Taken together, our study suggests that Rictor is one of the direct targets of miR-15a and miR-16 and this accounts for its function in autophagy induction. The connection between Rictor and autophagy contributes partly to the inhibition of cell proliferation caused by miR-15a and miR-16. In addition, Furthermore, miR-15a and miR-16-mediated autophagy induction increases the chemosensitivity of HeLa cells to CPT. Both miR-15a and miR-16 might be potential candidates for combination therapy of miRNA and CPT or other chemotherapeutic agents.
Materials and Methods
Cell culture
HeLa (human cervical cancer, ATCC, CCL-2) cells were cultured in Dulbeco's modified Eagle's medium (DMEM, Gibco, 12800–017) supplemented with 10% fetal bovine serum (FBS, PAA, A15–101), 10 U/mL pencillin-streptomycine (Gibco/Invitrogen, 15140–122) at 37°C in a humidified 5% CO2 incubator.
Target prediction
Two conventional online programs: Targetscan (http://www.targetscan.org) and Findtar (http://bio.sz.tsinghua.edu.cn) were used to predict the targets and precise binding sites of MIR15a and MIR16.
MiRNA and siRNA transfection
All miRNA mimics, miRNA inhibitors and siRNAs were purchased from Shanghai GenePharma Co. Ltd. The sequences of siRNAs were used as follows:
siRictor-1: 5′-GCAGCCUUGAACUGUUUAATT-3′, 5′-UUAAACAGUUCAAGGCUGCTT-3′
siRictor-2: 5′-CUUGUGAAGAAUCGUAUCUUTT-3′, 5′-AAGAUACGAUUCUUCACAAGTT-3′
A small RNA with a random sequence was used as a negative control (NC). For transfection, HeLa cells were suspended transfected with Lipofectamine 2000 (Invitrogen, 11668–019), according to the manufacture's protocol, 100 pmol siRNA or miRNA was added to each well of a 6-well plate.
RNA isolation and quantitative RT-PCR
Total RNA was isolated using RNAiso Plus (Takara, D9108B) according to the manufacturer's instructions. QRT-PCRs of mRNAs were performed using a reverse transcription kit (Takara, D6130) and SYBR Green Real-time PCR Master Mix (Toyobo, QPK-201). QRT-PCRs of miRNAs were performed according to the manufacturer's instructions using a TaqMan® MicroRNA Reverse Transcription Kit (ABI. 4366597) and Real-time PCR Master Mix (Toyobo, QPK-101). MiRNA probes were purchased from ABI: has-miR-15a: (ABI, 000390), has-miR-16 (ABI, 000391), RNU6 (ABI, 001093). MRNA and miRNA expression levels were normalized to GAPDH and RNU6 respectively.
Luciferase activity assay
The 3′ UTRs of RICTOR containing either wild type (WT) or mutated (MUT) miR-15a or miR-16 binding sites were inserted into pmirGLO Dual-Luciferase miRNA Target Expression Vectors (Promega, E1330). For luciferase activity assays, HeLa cell were co-transfected with WT or MUT UTR luciferase vectors and miRNAs using lipofectamine 2000 in 24-well plates. About 30h later, Luciferase activity was measured by Dual-Luciferase Reporter assay system (Promega, E1980) and normalized. Each experiment was repeated in triplicate.
Western blotting
HeLa cells were lysed with an ice-cold cell extract buffer (50mM Tris-HCl pH8.0, 4M Urea and 1% Triton X-100), containing a protease inhibitor cocktail (Roche, 04693132001). Total protein was equalized and separated by SDS-PAGE and then analyzed by western blotting. Primary antibodies used in this paper were as follows: SQSTM1 (MBL, PM045), LC3B (Sigma, L7543), GAPDH (Proteintech, 10494–1-AP), RICTOR (CST, 2114), phospho-MTOR (Ser2448) (CST, 5536), MTOR (CST, 2972), RPS6KB2/p70S6K (CST, 2708), phospho-RPS6KB2/p70S6K (Thr389) (CST, 9205), Cleaved PARP1 (CST, 5625), Cleaved Caspase-3 (CST, 9661). After incubation with horseradish peroxidase-coupled secondary anti-rabbit antibodies (KPL, 474–1506), protein bands were detected with ECL blotting detection reagents (KPL, 547100).
GFP-LC3 puncta assay
Hela GFP-LC3 stable cell were tansfected with miRNAs or anti-miRNAs. Two days after transfection, cells were fixed in 4% paraformaldehyde. GFP-LC3 puncta formation was determined by capturing images using an Olympus FV1000 confocal microscope (Olympus, Japan). The average number of GFP-LC3 doted per cell was counted from at least 200 cells.
Flow cytometry analysis for cell cycle and apoptosis
Cell were transfected with miRNA and/or treated with Camptothecin (CPT, KeyGen, KGA 8152) or Hydroxyurea (HU, Sigma, H8627). For cell cycle distribution assays, the cells were collected and fixed in 70% ethanol in PBS overnight and counter-stained with propidium iodide (PI, Sigma, P 4170) and analyzed for DNA content with a BD InfluxTM (BD Biosciences) flow cytometer. For apoptosis analysis, cells were trypsinized, washed with PBS 3 times and stained for 15 min in the dark with PtdIns and Annexin V (BD, 556547) at room temperature.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Additional information
Funding
References
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011; 147:728-41; PMID:22078875; http://dx.doi.org/10.1016/j.cell.2011.10.026
- Mizushima N. The pleiotropic role of autophagy: from protein metabolism to bactericide. Cell Death Differ 2005; 12(Suppl 2):1535-41; PMID:16247501; http://dx.doi.org/10.1038/sj.cdd.4401728
- Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 2010; 90:1383-435; PMID:20959619; http://dx.doi.org/10.1152/physrev.00030.2009
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069-75; PMID:18305538; http://dx.doi.org/10.1038/nature06639
- Mah LY, Ryan KM. Autophagy and cancer. Cold Spring Harb Perspect Biol 2012; 4:a008821; PMID:22166310; http://dx.doi.org/10.1101/cshperspect.a008821
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012; 149:274-93; PMID:22500797; http://dx.doi.org/10.1016/j.cell.2012.03.017
- Chan EY. mTORC1 phosphorylates the ULK1-mAtg13-FIP200 autophagy regulatory complex. Sci Signal 2009; 2:pe51; PMID:19690328; http://dx.doi.org/10.1126/scisignal.284pe51
- Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 2012; 31:1095-108; PMID:22343943; http://dx.doi.org/10.1038/emboj.2012.32
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005; 307:1098-101; PMID:15718470; http://dx.doi.org/10.1126/science.1106148
- Mori S, Nada S, Kimura H, Tajima S, Takahashi Y, Kitamura A, Oneyama C, Okada M. The mTOR pathway controls cell proliferation by regulating the FoxO3a transcription factor via SGK1 kinase. PloS One 2014; 9:e88891; PMID:24558442; http://dx.doi.org/10.1371/journal.pone.0088891
- Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 2007; 6:458-71; PMID:18054315; http://dx.doi.org/10.1016/j.cmet.2007.11.001
- Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH, Goldberg AL. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab 2007; 6:472-83; PMID:18054316; http://dx.doi.org/10.1016/j.cmet.2007.11.004
- Ambros V. The functions of animal microRNAs. Nature 2004; 431:350-5; PMID:15372042; http://dx.doi.org/10.1038/nature02871
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 2009; 136:215-33; PMID:19167326; http://dx.doi.org/10.1016/j.cell.2009.01.002
- Mendell JT. MicroRNAs: critical regulators of development, cellular physiology and malignancy. Cell Cycle 2005; 4:1179-84; PMID:16096373; http://dx.doi.org/10.4161/cc.4.9.2032
- Ventura A, Jacks T. MicroRNAs and cancer: short RNAs go a long way. Cell 2009; 136:586-91; PMID:19239879; http://dx.doi.org/10.1016/j.cell.2009.02.005
- Dumortier O, Hinault C, Van Obberghen E. MicroRNAs and metabolism crosstalk in energy homeostasis. Cell Metab 2013; 18:312-24; PMID:23850315; http://dx.doi.org/10.1016/j.cmet.2013.06.004
- Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet 2010; 11:597-610; PMID:20661255
- Frankel LB, Lund AH. MicroRNA regulation of autophagy. Carcinogenesis 2012; 33:2018-25; PMID:22902544; http://dx.doi.org/10.1093/carcin/bgs266
- Pourrajab F, Babaei Zarch M, BaghiYazdi M, Hekmatimoghaddam S, Zare-Khormizi MR. MicroRNA-based system in stem cell reprogramming; differentiation/dedifferentiation. Int J Biochem Cell Biol 2014; 55C:318-28; PMID:25150833; http://dx.doi.org/10.1016/j.biocel.2014.08.008
- Sun KT, Chen MY, Tu MG, Wang IK, Chang SS, Li CY. MicroRNA-20a regulates autophagy related protein-ATG16L1 in hypoxia-induced osteoclast differentiation. Bone 2014; 74:145-53.
- Tekirdag KA, Korkmaz G, Ozturk DG, Agami R, Gozuacik D. MIR181A regulates starvation- and rapamycin-induced autophagy through targeting of ATG5. Autophagy 2013; 9:374-85; PMID:23322078; http://dx.doi.org/10.4161/auto.23117
- Wan G, Xie W, Liu Z, Xu W, Lao Y, Huang N, Cui K, Liao M, He J, Jiang Y, et al. Hypoxia-induced MIR155 is a potent autophagy inducer by targeting multiple players in the MTOR pathway. Autophagy 2014; 10:70-9; PMID:24262949; http://dx.doi.org/10.4161/auto.26534
- Wang J, Yang K, Zhou L, Minhaowu Wu Y, Zhu M, Lai X, Chen T, Feng L, Li M, et al. MicroRNA-155 promotes autophagy to eliminate intracellular mycobacteria by targeting Rheb. PLoS Pathog 2013; 9:e1003697; PMID:24130493; http://dx.doi.org/10.1371/journal.ppat.1003697
- Liu K, Huang J, Xie M, Yu Y, Zhu S, Kang R, Cao L, Tang D, Duan X. MIR34A regulates autophagy and apoptosis by targeting HMGB1 in the retinoblastoma cell. Autophagy 2014; 10:442-52; PMID:24418846; http://dx.doi.org/10.4161/auto.27418
- Aqeilan RI, Calin GA, Croce CM. miR-15a and miR-16-1 in cancer: discovery, function and future perspectives. Cell Death Differ 2010; 17:215-20; PMID:19498445; http://dx.doi.org/10.1038/cdd.2009.69
- Liu Q, Fu H, Sun F, Zhang H, Tie Y, Zhu J, Xing R, Sun Z, Zheng X. miR-16 family induces cell cycle arrest by regulating multiple cell cycle genes. Nucleic Acids Res 2008; 36:5391-404; PMID:18701644; http://dx.doi.org/10.1093/nar/gkn522
- Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A 2005; 102:13944-9; PMID:16166262; http://dx.doi.org/10.1073/pnas.0506654102
- Cai CK, Zhao GY, Tian LY, Liu L, Yan K, Ma YL, Ji ZW, Li XX, Han K, Gao J, et al. miR-15a and miR-16-1 downregulate CCND1 and induce apoptosis and cell cycle arrest in osteosarcoma. Oncol Rep 2012; 28:1764-70; PMID:22922827
- Ye W, Lv Q, Wong CK, Hu S, Fu C, Hua Z, Cai G, Li G, Yang BB, Zhang Y. The effect of central loops in miRNA:MRE duplexes on the efficiency of miRNA-mediated gene regulation. PloS One 2008; 3:e1719; PMID:18320040; http://dx.doi.org/10.1371/journal.pone.0001719
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005; 120:15-20; PMID:15652477; http://dx.doi.org/10.1016/j.cell.2004.12.035
- Urbanska M, Gozdz A, Swiech LJ, Jaworski J. Mammalian target of rapamycin complex 1 (mTORC1) and 2 (mTORC2) control the dendritic arbor morphology of hippocampal neurons. J Biol Chem 2012; 287:30240-56; PMID:22810227; http://dx.doi.org/10.1074/jbc.M112.374405
- Veronese A, Pepe F, Chiacchia J, Pagotto S, Lanuti P, Veschi S, et al. Allele-specific loss and transcription of the miR-15a/16-1 cluster in chronic lymphocytic leukemia. Leukemia 2014; 29:86-95.
- Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, Hall MN. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol 2004; 6:1122-U30; PMID:15467718; http://dx.doi.org/10.1038/ncb1183
- Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol 2004; 14:1296-302; PMID:15268862; http://dx.doi.org/10.1016/j.cub.2004.06.054
- Vlahakis A, Graef M, Nunnari J, Powers T. TOR complex 2-Ypk1 signaling is an essential positive regulator of the general amino acid control response and autophagy. Proc Natl Acad Sci U S A 2014; 111:10586-91; PMID:25002487; http://dx.doi.org/10.1073/pnas.1406305111
- Vlahakis A, Powers T. A role for TOR complex 2 signaling in promoting autophagy. Autophagy 2014; 10:2085-6; PMID:25426890; http://dx.doi.org/10.4161/auto.36262
- Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W, DiPaola RS, Lotze MT, White E. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res 2011; 17:654-66; PMID:21325294; http://dx.doi.org/10.1158/1078-0432.CCR-10-2634
- Vu T, Sliwkowski MX, Claret FX. Personalized drug combinations to overcome trastuzumab resistance in HER2-positive breast cancer. Biochim Biophys Acta 2014; 1846:353-65; PMID:25065528
- Sui X, Chen R, Wang Z, Huang Z, Kong N, Zhang M, Han W, Lou F, Yang J, Zhang Q, et al. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis 2013; 4:e838; PMID:24113172; http://dx.doi.org/10.1038/cddis.2013.350
- Zhang H, Tang J, Li C, Kong J, Wang J, Wu Y, Xu E, Lai M. MiR-22 regulates 5-FU sensitivity by inhibiting autophagy and promoting apoptosis in colorectal cancer cells. Cancer Lett 2015; 356:781-90; PMID:25449431; http://dx.doi.org/10.1016/j.canlet.2014.10.029
- Dai F, Zhang Y, Chen Y. Involvement of miR-29b signaling in the sensitivity to chemotherapy in patients with ovarian carcinoma. Hum Pathol 2014; 45:1285-93; PMID:24767251; http://dx.doi.org/10.1016/j.humpath.2014.02.008
- Pan B, Yi J, Song H. MicroRNA-mediated autophagic signaling networks and cancer chemoresistance. Cancer Biother Radiopharm 2013; 28:573-8; PMID:23841710; http://dx.doi.org/10.1089/cbr.2012.1460
- Pennati M, Lopergolo A, Profumo V, De Cesare M, Sbarra S, Valdagni R, Zaffaroni N, Gandellini P, Folini M. miR-205 impairs the autophagic flux and enhances cisplatin cytotoxicity in castration-resistant prostate cancer cells. Biochem Pharmacol 2014; 87:579-97; PMID:24370341; http://dx.doi.org/10.1016/j.bcp.2013.12.009
- Zou Z, Wu L, Ding H, Wang Y, Zhang Y, Chen X, Zhang CY, Zhang Q, Zen K. MicroRNA-30a sensitizes tumor cells to cis-platinum via suppressing beclin 1-mediated autophagy. J Biol Chem 2012; 287:4148-56; PMID:22157765; http://dx.doi.org/10.1074/jbc.M111.307405
- Seca H, Lima RT, Lopes-Rodrigues V, Guimaraes JE, Almeida GM, Vasconcelos MH. Targeting miR-21 induces autophagy and chemosensitivity of leukemia cells. Curr Drug Targets 2013; 14:1135-43; PMID:23834154; http://dx.doi.org/10.2174/13894501113149990185
- Wang Z, Wang N, Liu P, Chen Q, Situ H, Xie T, Zhang J, Peng C, Lin Y, Chen J. MicroRNA-25 regulates chemoresistance-associated autophagy in breast cancer cells, a process modulated by the natural autophagy inducer isoliquiritigenin. Oncotarget 2014; 5:7013-26; PMID:25026296