
Abstract
EGFR and EGFRvIII are overexpressed in various types of cancer, serving as optimal targets for cancer therapy. Capitalizing on the high specificity of humanized antibody 806 (mAb806) to the EGFR and EGFRvIII overexpressed in cancer, we designed and generated a bivalent recombinant immunotoxin (RIT, DT390-BiscFv806) by fusing the mAb806-derived bivalent single-chain variable fragment with a diphtheria toxin fragment, DT390. In vitro, DT390-BiscFv806 efficiently internalized into the cells and exhibited high cytotoxicity against the U87 glioblastoma cells and the EGFRvIII-transfected U87 (U87-EGFRvIII) cells with a half maximal inhibition concentration (IC50) of 1.47 nM and 2.26 × 10−4 nM, respectively. Notably, DT390-BiscFv806 was 4 orders of magnitude more potent against the U87-EGFRvIII cells than against the parent U87 cells. The cytotoxicity against a group of 6 head and neck squamous cell carcinoma cell lines were further analyzed, showing an IC50 ranging from 0.24 nM to 156 nM, depending on the expression level of EGFR/EGFRvIII. In animals, the U87-EGFRvIII tumor xenografts grew extremely faster than the parental U87, and systemic administration of DT390-BiscFv806 significantly inhibited the growth of established U87-EGFRvIII and U87 tumor xenografts, showing a growth inhibition rate of 76.3% (59.82–96.2%) and 59.4% (31.5–76.0%), respectively. In pathology, the RIT-treated tumors exhibited a low mitotic activity and a large number of degenerative tumor cells, compared with the control tumors. The results indicate that DT390-BiscFv806 is promising for treatment of various types of cancer, especially for those with high EGFR expression or with EGFR and EGFRvIII co-expression.
Abbreviations
| EGFR | = | epidermal growth factor receptor |
| EGFRvIII | = | epidermal growth factor receptor variant III |
| GBM | = | glioblastoma multiforme |
| mAbs | = | monoclonal antibodies |
| TKIs | = | tyrosine kinase inhibitors |
| RIT | = | recombinant immunotoxin |
| biscFv | = | bivalent single-chain variable fragment |
| DT | = | diphtheria toxin |
| DT390 | = | an engineered diphtheria toxin fragment |
| P. pastoris | = | Pichia pastoris |
| VL | = | light chain variable domain |
| VH | = | heavy chain variable domain |
| scFv | = | single-chain variable fragment |
| SDS-PAGE | = | sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| HPLC | = | high-performance liquid chromatography |
| IC50 | = | half maximal inhibition concentration |
| RTV | = | individual relative tumor volume |
| %TGI | = | percentage tumor growth inhibition. |
Introduction
Epidermal growth factor receptor (EGFR) is a transmembrane tyrosine kinase belonging to the HER/erbB family. Activation of EGFR by its ligands results in homo- or heterodimerization of EGFR and autophosphorylation of tyrosine residues in its C-terminal domain, leading to cell proliferation and migration.Citation1 Various types of human cancer have been shown to overexpress the EGFR and possess the cancer-specific EGFR variant III (EGFRvIII).Citation2 One example is the glioblastoma multiforme (GBM), which shows EGFR overexpression in approximately 60–90% and amplification in 40–50% of tumors, and of the EGFR-amplified GBM, up to 60–70% possess EGFRvIII.Citation3,4 Another example is the head and neck squamous cell carcinoma (HNSCC), which shows EGFR overexpression in up to 90% and gene amplification in 10–58%.Citation5,6 A subset of HNSCC also possesses the EGFRvIII, although the EGFRvIII frequency varies from 0 to 42% in literature.Citation7,8 Other types of cancer overexpressing EGFR/EGFRvIII include non-small cell lung carcinoma, breast cancer, colorectal carcinoma, and ovarian cancer. The high prevalence of EGFR overexpression and EGFRvIII-type mutation in various types of cancer has sparked the development of anti-EGFR monoclonal antibodies (mAbs) and tyrosine kinase inhibitors (TKIs).Citation9-11 Indeed, a large group of mAbs and TKIs have been generated and tested for their anti-tumor efficacy and benefits in treatment of patients.Citation12,13 However, only mAbs have to date demonstrated a certain degree of survival improvement for a small subset of patients with GBM, HNSCC, and other types of cancer.Citation9-11 Results with reversible and irreversible TKIs are generally disappointing.Citation12 Continued development of mAbs is facing formidable challenges, especially with the dose-limiting side effects induced by their binding with the EGFR expressed in normal tissues, and with the innate and acquired resistance associated with up-regulation of ligands to compete with mAbs for receptor binding, persistent activation of downstream signaling through multiple interacting pathways, and EGFR mutations.Citation13
Recombinant immunotoxins (RITs) are a group of protein-based therapeutics comprising 2 functional moieties: one is the antibody fragment that allows the RITs to bind specifically to target cells and another is the bacterial toxin fragment that kills the cells upon cellular internalization.Citation14,15 Compared to mAbs and antibody conjugates, RITs have an improved penetration capability and a greater anti-tumor efficacy. Because of the unique features of RITs such as high specificity, extraordinary potency and lack of drug resistance, EGFR-targeted RITs have been designed and constructed with different mAbs as the template such as the scFv(225)-ETA, scFv(14E1)-ETA, 425(scFv)-ETA', D2C7-IT, MR1scFvPE38KDEL, and scFv/rGel (E/rG).Citation16-20 RITs have also been generated by using epidermal growth factor (EGF) or transforming growth factor-α peptide as a template such as TP-38.Citation21 Several RITs have been advanced from laboratory to the stage of Phase I and Phase II clinical trials. In general, several disadvantages of the current RITs limit their overall anti-tumor efficacy in clinical applications. Majority of these RITs have a relatively low binding affinity with EGFR due to their monovalency and most react with the EGFR expressed in cancer as well as in normal tissues, resulting in some dose-limiting side effects. Furthermore, only the RIT, D2C7-IT, has been reported to recognize both the wild-type EGFR and the EGFRvIII.Citation17-19
The high specificity of mAb806 to the overexpressed EGFR and EGFRvIII in cancer, but not to the EGFR in normal tissue highlights its advantage over other mAbs for generation of RITs.Citation22-24 The mAb806 was raised against mouse fibroblast cells expressing the EGFRvIII, and its unique specificity contributes to the fact that mAb806 binds to an epitope exposed only in the transitional untethered form of EGFR when it is overexpressed in cancer.Citation25 Preclinical and Phase I first-in-man clinical studies have confirmed the specificity as well as the high therapeutic index of mAb806, showing lack of normal tissue uptake in cancer patients.Citation26 Taking advantage of the unique specificity and humanization-derived benefits of the mAb806, we generated a bivalent RIT, designated as DT390-BiscFv806, by fusing an engineered diphtheria toxin (DT) fragment (DT390) with the humanized mAb806-derived bivalent single-chain variable fragment (biscFv) via amino acid linkers. DT390 is an engineered fragment consisting of the catalytic and translocation domains of DT with a size of 390 amino acids. This bivalent RIT was expressed in a DT-resistant Pichia pastoris (P. pastoris) system (Patent No.: US7892786) and exhibited extraordinary potency against cancer cells with overexpressed EGFR only and those with co-expressed EGFR and EGFRvIII. Herein, we report the design and generation of the DT390-BiscFv806, and its cytotoxicity and anti-tumor efficacy.
Results
Generation and characterization of DT390-BiscFv806
DT390-BiscFv806 was constructed by cloning and integrating the light (VL) and heavy (VH) variable domain sequences into plasmid vectors and linked with the peptide (G4S)3 sequentially.Citation27-30 The full linear sequence of DT390-BiscFv806 was DT390-VL-(G4S)3-VH-(G4S)3-VL-(G4S)3-VH, which was confirmed by DNA sequencing. The cartoon structure of DT390-BiscFv806 was shown in , presenting a bivalent tandem scFv format. The product was expressed with the DT-resistant P. pastoris system. Purification of the product was achieved with a 4-step scheme as described in the Materials and Methods. The raw and purified yields of DT390-BiscFv806 were ∼15 mg/L and ∼12 mg/L, respectively. The final product was adjusted to 0.1 mg/ml, filter-sterilized and stored at −80 °C in the buffer containing 10 mM Tris (pH, 7.2), 1 mM EDTA, 150 mM NaCl, and 5% glycerol.
Figure 1. Schematic description and characterization of the EGFR- and EGFRvIII-specific bivalent recombinant immunotoxin, DT390-BiscFv806. 1A shows the linear sequence (upper panel) and cartoon structure (lower panel) of DT390-BiscFv806. 1B and IC show the SDS-PAGE gel electrophoresis and HPLC analysis of DT390-BiscFv806, respectively. The loading volume of DT390-BiscFv806 was 6 µl (0.768 µg) and 90 µl (11.52 µg) for gel electrophoresis and HPLC analysis, respectively. Lanes 1 and 2 on the SDS-PAGE gel in 1B are DT390-BiscFv806 with different preparations and having different purity. The product in lane 1 was used in the present studies. Superdex 200 size-exclusion column analysis showed a major and a minor peak at the elution times of 28.323 min and 25.041 min, representing the purified and the aggregated product, respectively (1C). The third peak appeared in the HPLC profile was used as a reference, which was confirmed to be due to the EDTA added in the sample buffer in our previous studies. Both SDS-PAGE electrophoresis and HPLC analysis indicate a high purity of the final product.
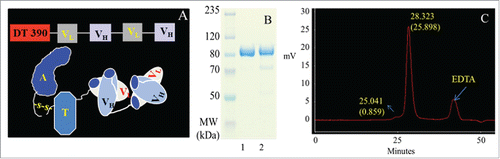
The purity of the final product was >95% as estimated under non-reducing condition, presenting a single band in the 4–12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gel (). The product shown in lane 1 of was used for the present studies. High-performance liquid chromatography (HPLC) with superdex 200 size-exclusion column analysis showed a major and a minor peak at the elution times of 28.323 min and 25.041 min, respectively (). The major peak at 28.323 min represented the purified and the minor peak at 25.041 min might be the aggregated product. The third peak appeared in the HPLC profile was used as a reference, which was confirmed to be due to the EDTA added in the sample buffer in our previous studies. These results indicate that the final product of DT390-BiscFv806 was in a high purity with little aggregation.
Enhanced proliferation of U87 cells and growth of tumor xenografts by EGFRvIII expression
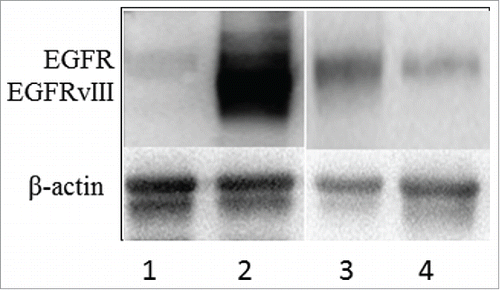
U87 cells are known to express EGFR, but do not possess EGFRvIII. The U87-EGFRvIII subline was established by stable transfection of EGFRvIII. High expression of EGFRvIII in the U87-EGFRvIII cells was confirmed with Western blotting (). To better understand the therapeutic efficacy of DT390-BiscFv806, we analyzed the effect of enforced expression of EGFRvIII on the growth of U87 cells as well as U87 tumor xenografts.
Figure 2. Western blot analysis of EGFR and EGFRvIII protein expression in cultured cells. Lanes 1 and 2 show the U87 and the U87-EGFRvIII cells without and with high expression of EGFRvIII, respectively. Lanes 3 and 4 are JHU-13 and JHU-22 cells as a representative of EGFR expression in head and neck squamous cell carcinoma (HNSCC) cell lines. No EGFRvIII expression was detected in the 6 HNSCC cell lines analyzed.
Cell availability assay showed that the proliferation of U87-EGFRvIII cells was significantly faster than that of the parental U87 cells with the cell doubling time of 11.18 h and 15.20 h, respectively. Interestingly, the stationary phase in the growth curve of U87-EGFRvIII cells delayed significantly. When 5 × 103 of U87 cells were seeded in the wells of 96-well plates, the stationary phase in the cell growth curve reached along with 100% confluence after 48 h. On the contrary, the stationary phase for U87-EGFRvIII cells became prominent only on day 6 after seeding of the cells, showing persistent proliferation.
In animals, enforced expression of EGFRvIII in U87-EGFRvIII cells resulted in the formation and growth of tumors significantly earlier than that of the parental U87 cells. The tumor nodules of U87-EGFRvIII were palpable as early as 10 days, while the tumor nodules of U87 were palpable at 20–30 d after 1 × 106 cell inoculation. The mean volume reached 117.1 ± 30.9 mm3 and 2341.1 ± 523.5 mm3 on day 13 and day 30, respectively for U87-EGFRvIII tumors, while the mean volume of U87 tumor xenografts was 18.8 ± 2.5 mm3 and 1048.7 ± 111.2 mm3 on day 30 and day 51, respectively, after cell inoculation (mean ± SEM, n = 5 mice/group). The latent phase of U87-EGFRvIII tumor formation was 10–20 d shorter than that of the U87 tumor formation. However, no significant difference in the tumor volume doubling time was observed between the U87-EGFRvIII and U87 tumor xenografts (3.32 vs. 3.38 days, P > 0.05) as calculated based on the log phase of tumor growth curve.
High cytotoxicity of DT390-BiscFv806 against GBM and HNSCC cells
The cytotoxicity of DT390-BiscFv806 against the cultured cancer cells was determined after the cells were exposed to graded concentrations of DT390-BiscFv806. shows the survival curves of different cancer cells plotted by the cell viability vs. the drug concentration, and the cell morphology under different concentrations of DT390-BiscFv806. The half maximal inhibitory concentration (IC50) of DT390-BiscFv806 was measured to be 1.47 nM and 2.26 × 10−4 nM for U87 and U87-EGFRvIII cells, respectively (). Notably, DT390-BiscFv806 had more than 4 orders of magnitude more potency against the U87-EGFRvIII cells than against the parental U87 cells. In morphology, cell death was obvious after exposure to DT390-BiscFv806 ().
Figure 3. The high cytotoxicity of DT390-BiscFv806 against cancer cells. The panels 1 and 2 show the cell viability curves of glioblastoma cells and head and neck squamous cell carcinoma cells after exposure to graded concentrations of DT390-BiscFv806. The panels 3 and 4 show the representative morphology of the U87 (A to D) and U87-EGFRvIII (E to H) cells, respectively, following exposure of DT390-BiscFv806. The concentrations of DT390-BiscFv806 in the panels 3 and 4 were 0, 3 × 10−11, 1 × 10−10, and 1 × 10−8 M for A to D, and 0, 1 × 10−15, 1 × 10−13, and 1 × 10−11 for E to H, respectively.
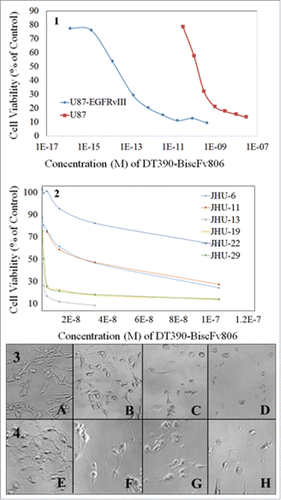
Table 1. Cytotoxicity of DT390-BiscFv806 against cancer cells
We further analyzed the cytotoxicity of DT390-BiscFv806 against a group of 6 HNSCC cell lines (). The cytotoxicity (IC50) of DT390-BiscFv806 varied among the HNSCC cell lines with the highest against the JHU-13 (0.24 nM) and JHU-19 cells (0.89 nM), followed by JHU-29 (1.33 nM), JHU-6 (14.6 nM) and JHU-11 (21.7 nM), and the lowest against the JHU-22 cells (156 nM). To understand the EGFR expression status in the 6 HNSCC cell lines, we analyzed the presence of EGFRvIII using reverse transcription PCR (RT-PCR), the copy numbers of EGFR gene using real-time PCR, and the EGFR protein expression levels using Western blotting.Citation31,32 In the analysis, U87-EGFRvIII cells were used as a positive control for the presence of EGFRvIII. Neither presence of EGFRvIII nor amplification of EGFR gene was detected in all of the 6 HNSCC cell lines (data not shown).Citation31 The protein expression level of EGFR varied among the 6 cell lines with the highest in JHU-13 and the lowest in JHU-22 ().Citation32 Comparison between the cytotoxicity and EGFR protein level showed a positive relationship between them in the HNSCC cell lines.
To understand the potential non-specific toxicity from DT390-BiscFv806, we treated the 4 most sensitive cell lines including U87-EGFRvIII, U87, JHU-13 and JHU-29 with a prostate-specific membrane antigen (PSMA)-targeted bivalent RIT, A-dmDT390-scfbDb(PSMA). The results showed that no significant cytotoxicity of A-dmDT390-scfbDb(PSMA) to either of the 4 cell lines were observed at the concentration of >100 nM. A-dmDT390-scfbDb(PSMA) was constructed by fusing a biscFv from antibody J591 with DT390, similar to the procedures to construct the DT390-BiscFv806. In our previous studies, A-dmDT390-scfbDb(PSMA) was confirmed to be highly cytotoxic to PSMA-expressing LNCaP prostate cancer cells (IC50, 0.57 nM), but not to PSMA-negative PC-3 cells (IC50, >100 nM).Citation29
High anti-tumor efficacy of DT390-BiscFv806 against established tumor xenografts
The efficacy of DT390-BiscFv806 was evaluated against the growth of established U87-EGFRvIII and U87 tumor xenografts. Because of the effect of enforced EGFRvIII expression, the growth patterns of U87 and U87-EGFRvIII tumor xenografts were significantly different. Therefore, different treatment regimens were designed to test the anti-tumor efficacy of DT390-BiscFv806 against the established U87 and U87-EGFRvIII tumor xenografts. Irrespective of the regimens, DT390-BiscFv806 significantly inhibited the growth of both U87-EGFRvIII and U87 tumor xenografts ().
Figure 4. Growth inhibition of established tumor xenografts by DT390-BiscFv806. 4A and 4C represent the changes of the relative tumor volume (RTV) of U87-EGFRvIII and U87 tumor xenografts, respectively, at different times after beginning of treatment. 4B and 4D show the changes of the percentage tumor growth inhibition (%TGI) of U87-EGFRvIII and U87 xenografts, respectively, at different times after beginning of treatment. The data were expressed as mean ± SD of 6 mice per group. RTV and %TGI were calculated as indicated in the Section of Materials and Methods.
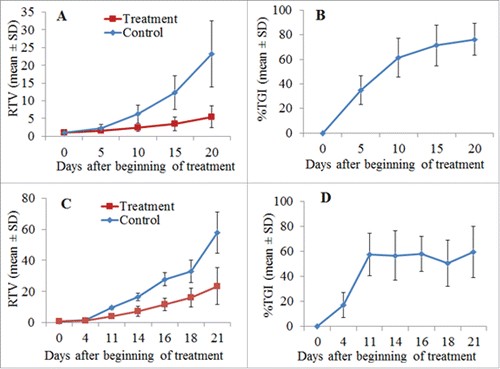
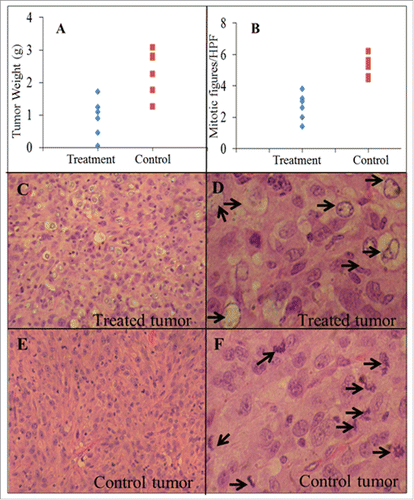
show the changes of mean individual relative tumor volume (RTV) and percentage tumor growth inhibition (%TGI) for U87-EGFRvIII tumors after DT390-BiscFv806 administration. The RTV changes over the time were significantly different between the treatment and control groups (n = 6 mice/group). At the end point of experiment (day 18 after beginning of treatment), the RTV was 5.5 ± 3.0 vs. 23.1 ± 9.2; the tumor weight was 0.9 ± 0.6 g vs. 2.2 ± 0.9 g; and the tumor volume was 836.7 ± 555.7 mm3 vs. 2523.7 ± 1136.2 mm3 for the treatment vs. control groups (mean ± SD, all P<0.05) (). DT390-BiscFv806 led to a %TGI of 76.3% (ranging from 59.8%-96.2%) on the day 18, the last day after beginning of treatment. The cumulative %TGI increased over time, showing 34.7% ± 11.7, 61.5% ± 15.9, 71.3% ± 16.3, and 76.3% ± 13.1 on days 4, 10, 14, and 18, respectively ().
Figure 5. Comparative analysis between DT390-BiscFv806-treated U87-EGFRvIII tumors and control tumors. Compared with the control tumors, the treated tumors had a much lower tumor weight (2.2 ± 0.9 vs. 0.9 ± 0.6 g, P<0.05), a lower number of mitosis (5.23 ± 0.66 vs. 2.67 ± 0.86 per high power field, P < 0.05), and many degenerative tumor cells throughout the entire tumors. These degenerative tumors cells were rarely seen in the control tumors. Arrows indicate degenerative tumor cells in 5D and mitosis in 5F. The original magnification was 100x for 5C and 5E, and 400x for 5D and 5F. Tumors were dissected on day 18 after beginning with or without DT390-BiscFv806 treatment.
show the changes of mean RTV and %TGI over time for U87 tumors following DT390-BiscFv806 administration. Similarly, the RTV at each defined time point was also significantly different between the treatment and control groups (n = 5 mice/group). At the end point of experiment (day 21 after beginning of treatment), the RTV was 23.5 ± 11.8 vs. 57.9 ± 13.2 (P<0.05) for the treatment vs. control groups and a %TGI of 59.4% (ranging from 31.5%-76.0%) was obtained. As shown in , the cumulative %TGI was relatively stable from day 11 to day 21 after an initially rapid increase of tumor growth inhibition (day 4 to day 11) following treatment, showing 57.6% ± 17.1, 56.7% ± 19.8, 58.0% ± 13.9, 50.6% ± 18.4, and 59.4% ± 20.4 on days 11, 14 16, 18 and 21, respectively, which was different from that of the inhibitory growth of U87-EGFRvIII tumors; the latter showed an increased cumulative %TGI over time ().
With the present dosage regimen, DT390-BiscFv806 was in general well tolerated by mice. However, a body weight loss of >10% was observed in 3 of the 12 mice bearing U87-EGFRvIII tumor xenografts (one control and 2 RIT-treated mice) and 3 of the 10 mice bearing U87 tumor xenografts (one control and 2 RIT-treated mice). The mouse body weight loss in the control group might be associated with the rapid tumor growth, while in the treatment group it might be associated with the tumor growth as well as the DT390-BiscFv806 toxicity. There was a significant difference for the body weight loss/gain between the control and RIT-treated mice bearing the U87-EGFRvIII tumor xenografts (P = 0.021), but there was no significant difference for mice bearing U87 tumor xenografts (P = 0.524). During the experiments, no significant clinical signs of toxicity were observed such as sickness, diarrhea and lethargy.
Pathologic findings
We comparatively analyzed the pathology between the RIT-treated and the control tumor xenografts. Under microscopy, both of the RIT-treated and control tumors were composed of densely arranged tumor cells with heteromorphic and large nuclei. One striking change was the lower mitotic activity in the RIT-treated tumors than in the control tumors (). The mitotic figures were 5.23 ± 0.66 per high-powered fields (HPF, 400x) in the control group vs. 2.67 ± 0.86 per HPF in the RIT-treated group (2-tailed t-test, P=0.0002) (). Another striking change that was observed in all RIT-treated tumors was the presence of a large number of degenerative tumor cells. As shown in , these degenerative cells had an empty cytoplasm and the outer boundary of nucleus with loss of the nuclear details. These cells distributed throughout the entire tumors with more in the central region than in the peripheral region of tumors. These degenerative cells were rarely seen in the tumors of control group (). The mechanism and significance for the presence of a large number of degenerative cells in the RIT-treated tumors remain to be studied.
Necrosis was observed in tumors from both groups, showing more necrosis in larger tumors, but there was no obvious difference in the extent of necrosis among the tumors with similar size from the 2 groups. Inflammatory cell infiltration was not obvious in the stroma of both RIT-treated and control tumors.
Discussion
The DT390-BiscFv806 we generated possesses some desirable properties as an EGFR- and EGFRvIII-targeted therapeutic agent. This RIT was constructed with a humanized biscFv that was derived from the mAb806 and its toxin component was DT390, different from other RITs that are Pseudomonas exotoxin A (PE) fragment-based. DT390 is a truncated form of DT, which retains its enzyme activity and membrane translocation function, while deleting the binding domain to prevent its binding with normal cells, thereby diminishing its systemic toxicity. Engineered DT and PE fragments have their own advantages and disadvantages.Citation33-35 In general, PE fragments are more resistant to genetic manipulation, but induce more side effects (especially liver toxicity) and stronger immunogenicity than DT fragments. However, previous immunization of patients with DT vaccine could be of concern when DT fragment is used. Fortunately, no significant neutralizing effect by the existing circulating anti-DT antibodies has been observed in clinical trials with DT390-based RITs.Citation35,36 This may be explained by the assumption that the epitopes targeted by the DT vaccine-induced neutralizing antibodies are mostly located in the deleted binding domain of DT. A DT390-based anti-CD3 monospecific RIT, OntakTM, was approved in 1999 by the U.S. FDA for treatment of cutaneous T-cell lymphoma and other diseases.Citation37 Because immunogenicity of RITs that is induced by the toxin component as well as the murine antibody fragments is a major issue for their clinical application, use of the humanized antibody fragment and DT390 in construction of DT390-BiscFv806 is expected to reduce its immunogenicity and side effects, making it more suitable for clinical use.
A challenge in RIT development is the difficulty in generating highly functional, multi-domain RITs, requiring an efficient expression system that should not only be resistant to the toxin component, but also able to properly fold proteins with multiple domains.Citation33 Bacteria are the most frequently used expression systems because of their resistance to toxins, but they lack the ability to efficiently fold complex proteins. Following expression in bacteria, multi-domain RITs must be re-folded chemically to recover their binding capability and bioactivity, thus the function of multi-domain RITs cannot be fully recovered in most situations. The majority, if not all of the current EGFR-targeted RITs, were monovalent and generated with bacterial expression systems. Toxin-resistant cell lines such as CHO and HEK293T are also used to produce RITs, but the production yield in most cases is very limited. To efficiently express DT-based RITs, Dr. Liu et al. the coauthor of this manuscript, have invented a DT-resistant P. pastoris expression system (Patent No.: US7892786). P. pastoris is a methylotrophic yeast with 2 alcohol oxidase genes. Its strong inducible promoter AOX1 allows P. pastoris to express a high level of proteins in a simple, inexpensive medium with a high growth rate in either a shake flask or a fermenter, making it suitable for both small and large scale production. Importantly, P. pastoris is capable of folding multi-domain RITs properly by forming disulfide bonds.Citation27-30 The expression and purification parameters of this system have been further optimized for production scale up of RITs according to the U.S. FDA GMP guidelines. An example is the anti-CD3 RIT, Resimmune (A-dmDT390-bisFv(UCHT1)), which is under Phase II clinical trials and has been produced at a raw yield of ∼207 mg/L and a purified yield of ∼144.2 mg/L after a 163-hour induction period in a single batch of 120 L bioreactor culture.Citation38 For the DT390-BiscFv806 in the present study, its expression level was ∼15 mg/L in shake-flasks, higher than that obtained for Resimmune under the same expression conditions.
Valency, molecular size, and circulation time are 3 key factors affecting the anti-tumor efficacy of a RIT.Citation29,39,Citation40 Use of biscFv to construct RIT can significantly improve its functional affinity. Under most conditions for bivalent binding, 2 measurable KD exist, one for monovalent and the other for bivalent. The overall binding affinity is determined by the fraction of bivalent binding. Increasing the bivalent binding fraction is one approach to enhance the binding affinity. Our previous studies on different formats of RITs have confirmed the higher binding affinity of bivalent RITs than that of monovalent RITs.Citation28,29,Citation40 The studies with RITs targeting PSMA (A-dmDT390-scfbDb(PSMA)) or CD3 (A-dmDT390-bisFv(UCHT1)) have shown that the binding affinity and cytoxicity of the bivalent format are >2.5-fold and >10-fold higher, respectively, than that of a monovalent format.Citation28,29,Citation40 We expected that the bivalent DT390-BiscFv806 would have a high binding affinity and cytotoxicity to the targeted cells. Indeed, our data demonstrated that the cytotoxicity (IC50) of DT390-BiscFv806 was comparable to that of A-dmDT390-bisFv(UCHT1) (1.7 × 10−14 on Jurkat T cells) and A-dmDT390-scfbDb(PSMA) (5.7 × 10−12 on LNCaP prostate cancer cells).Citation29,38 Regarding the molecular size, RITs with a small molecular size usually have a better tumor-penetrating capability than those with larger molecular size. DT390-BiscFv806 has a moderate molecular size of ∼97 kDa, much smaller than full antibodies (∼150 kDa), suggesting a better penetration capability than mAbs and a longer circulation time than monovalent RITs, increasing the chance to accumulate in tumors. Increasing the penetration capability and binding affinity while maintaining a proper circulating time by optimizing the primary and secondary structure of a RIT is critical to achieve an optimal anti-tumor efficacy.
To better understand the anti-tumor efficacy of our newly generated RIT, we first established a subline U87-EGFRvIII with stable co-expression of EGFR and EGFRvIII. Enforced expression of EGFRvIII led to significant changes of the U87 cell behaviors. Under the experimental conditions presented in the Materials and Methods, the U87 cells reached the stationary phase of growth after 100% confluence at ∼48 h from seeding of the cells. On the contrary, U87-EGFRvIII cells kept proliferating up to day 6 when we completed the studies. This phenomenon was also reflected in the tumor formation in animals. The latent phase of U87-EGFRvIII tumor formation was much shorter than that of U87 tumor formation, although the doubling times of their tumor volumes were similar once the tumor growth entered the log growth phase. A possible explanation for this phenomenon is that enforced expression of EGFRvIII makes the U87-EGFRvIII cells resistant to apoptosis induced after 100% confluence, and when they are inoculated in mice, most cells stay alive and grow to a tumor mass with a shorter latent phase than the U87 cells. The results are consistent with other reports, although further detailed studies are necessary to elucidate the mechanisms underlying this phenomenon.Citation41-43
We first tested the cytotoxicity of DT390-BiscFv806 against the U87 and U87-EGFRvIII cells. The results showed that DT390-BiscFv806 was highly toxic against both U87 and U87-EGFRvIII cells. Interestingly, DT390-BiscFv806 had more than 4 orders of magnitude more potency against the U87-EGFRvIII cells than against the parental U87 cells. This result could be explained by the targeting effect of DT390-BiscFv806 on both EGFR and EGFRvIII, and indicates that DT390-BiscFv806 recognizes both EGFR and EGFRvIII. We further analyzed a group of 6 HNSCC cell lines to confirm the cytotoxic effect of DT390-BiscFv806. Although these HNSCC cell lines were not detected to possess EGFRvIII, they also showed a high sensitivity to DT390-BiscFv806, which was associated with the protein expression levels of EGFR.
We tested the specificity of DT390-BiscFv806 against EGFR/EGFRvIII by treating the 4 most sensitive cell lines (U87-EGFRvIII, U87, JHU-13 and JHU-29) with a prostate-specific membrane antigen (PSMA)-targeted bivalent RIT, A-dmDT390-scfbDb(PSMA). We did not detect significant cytotoxicity of A-dmDT390-scfbDb(PSMA) to either of the 4 cell lines at the concentration of >100 nM. A-dmDT390-scfbDb(PSMA) was constructed by fusing a biscFv from the PSMA-specific antibody J591 with DT390 using similar procedures in constructing the DT390-BiscFv806. We have confirmed that A-dmDT390-scfbDb(PSMA) is highly cytotoxic and exhibits a high potency to PSMA-expressing LNCaP cultured cells and tumor xenografts, but not to PSMA-negative PC-3 cultured cells and tumor xenografts in our previous studies.Citation29
We then tested the efficacy of DT390-BiscFv806 against the established U87-EGFRvIII and U87 tumor xenografts. Keeping the different growth features of U87-EGFRvIII and U87 cells in mind, we designed different protocols showing that the tumor growth was inhibited significantly by systemic administration of DT390-BiscFv806 with a mean %TGI of 76.3% (ranging from 59.8%-96.2%) and 59.4% (ranging from 31.5%-76.0%), respectively. These results indicate that DT390-BiscFv806 is efficacious to tumors with EGFR expression alone or with EGFR and EGFRvIII co-expression; and more efficacious against the latter. It is worth noting that most existing RITs react not only with the EGFR overexpressed in cancer, but also react with the EGFR expressed in normal tissue, resulting in unwanted toxicity to key organs, and to date, only the monovalent RIT, D2C7-IT, has been reported to recognize both the wild type EGFR and EGFRvIII.Citation17-19 Because EGFRvIII is specifically expressed in cancer and plays a significant role in tumor progression and treatment failure, a RIT that recognizes the EGFRvIII is expected to enhance its anti-tumor benefits.
To better understand the anti-tumor mechanisms of DT390-BiscFv806, we comparatively analyzed the pathological differences between RIT-treated tumors and control tumors. Except for the decreased mitotic activity, a significant finding in the treated tumors is the appearance of a large number of degenerative tumor cells, but such cells were rarely observed in the control tumors. Interestingly, there was no obvious difference in the extent of necrosis among the tumors with similar size from the 2 groups. We hypothesized that the tumor cell death induced by RITs may be a slow process following the inhibition of protein synthesis by RITs and the appearance of degenerative tumor cells may be a representing phenomenon of this slow process, although further studies are necessary.
In summary, we generated a humanized bivalent scFv-derived, DT390-based RIT, DT390-BiscFv806, by capitalizing upon the unique specificity of mAb806 against EGFR and EGFRvIII overexpressed in cancer but not the EGFR in normal tissue, and taking the mAb806 humanization-derived benefits. This novel RIT was expressed with a DT-resistant Pichia expression system in a high yield and purity. DT390-BiscFv806 exhibited high cytotoxicity and anti-tumor efficacy against the cancer cells either with EGFR expression alone or with EGFR and EGFRvIII co-expression. It is notable that DT390-BiscFv806 was 4 orders of magnitude more potent against EGFR and EGFRvIII co-expressing U87-EGFRvIII cells than against the parental U87 cells with EGFR expression alone. The high anti-tumor efficacy and the unique properties of DT390-BiscFv806 make it promising for treatment of cancers such as GBM, HNSCC, and cancers of lung, breast, prostate, and ovary.
Materials and Methods
Design and generation of the bivalent RIT, DT390-BiscFv806
Design and cloning of DT390-BiscFv806
DT390-BiscFv806 was designed to have a linear sequence arrangement of DT390-VL-(G4S)3-VH-(G4S)3-VL-(G4S)3-VH, where (G4S)3 is the peptide linker, and VL/VH are the VL and VH variable domain sequences derived from the humanized mAb806 (). DT390-BiscFv806 was constructed following the strategy we used to construct other RITs.Citation27-30 Briefly, a set of primers were designed based on the sequences of VL and VH variable domains of the humanized mAb806, and the VL and VH domains were amplified with PCR. The amplified products were analyzed by electrophoresis in 1% agarose gels and purified using QIAquik Gel Extraction Kit (Qiagen, Valencia, CA). The BamHI digested VL PCR fragment was ligated to Bgl II digested VH fragment using T4 ligase to generate the scFv. After confirming the scFv sequence with DNA sequencing, the first and second scFv fragments were subcloned into the XhoI and EcoRI sites of the plasmid vector pwPICZα-DT390, which contains the DT390 fragment. The (G4S)3 was used as the linker to connect the 2 scFv to obtain the biscFv fragment.
DT390-BiscFv806 expression and purification
The linearized DT390-BiscFv806 construct in pwPICZα-DT390 vector was transformed into the DT-resistant P. pastoris strain (Patent No.: US7892786). The transformants were selected on YPD (1% yeast extract, 2% peptone and 2% dextrose) plates containing zeocin (100 μg/ml). Six colonies were randomly picked and cultivated in test tubes containing 5 mL YPD at 30 °C at 250 rpm for 24 h as growth phase I, then in YPG (1% yeast extract, 2% peptone, 1% glycerol) for another 24 h as growth phase II. The cultures were induced with methanol for 48 h at 25 °C at 225 rpm. Antifoam (0.02%) was added in all of the growth and induction medium, and phenylmethanesulfonyl fluoride (1 mM) was added with methanol to inhibit the protein degradation during the induction phase. The culture supernatants were analyzed under non-reducing condition with SDS-PAGE (Life Technologies, Grand Island, NY). The optimal clone was selected based on the gel electrophoresis and cultivated in 1000 mL shake-flasks for 48 h for growth and induction phase, respectively. The supernatant containing the desired product of DT390-BiscFv806 was purified following a 4-step scheme: diafiltration, capture by hydrophobic chromatography, borate anion exchange chromatography, and anion exchange chromatography according to our established protocols.Citation27-30
SDS-PAGE and HPLC analysis
The product of DT390-BiscFv806 was characterized with 4–12% SDS-PAGE and HPLC. Proteins fractionated by running the precast SDS-PAGE gels were stained with Coomassie blue staining reagent (Life Technologies) after washing the gels with distilled water. HPLC analysis was performed with a Shimadzu HPLC system using Superdex 200 size-exclusion column, 10/300 GL (GE healthcare, Pittsburgh, PA). The sample volume was 90 μl using 100 μl loop. The flow rate was 0.5 ml/min. The running time was 120 min and the running buffer was composed of 50 mM phosphate buffer (pH, 7.0) and 150 mM NaCl.
In vitro experimental design
Cell lines and cell culture
The human glioblastoma cell line U87 and its subline U87-EGFRvIII and 6 HNSCC cell lines were used in the present studies. The U87 cell line was purchased from the American Type Culture Collection (ATCC, Rockville, MD) and classified as grade IV glioblastoma by the ATCC. U87-EGFRvIII subline was established by stable transfection of EGFRvIII to U87 cells. EGFR expression in U87 cells and EGFR/EGFRvIII co-expression in U87-EGFRvIII cells were confirmed using RT-PCR and Western blotting with the antibody Ab-5 (Thermo Scientific, Cambridge, MA). The antibody Ab-5 reacts with the extracellular domains of both EGFR and EGFRvIII. The effect of EGFRvIII expression on the U87 cell proliferation in vitro and in animals was also characterized (see Results section). The six HNSCC cell lines including JHU-6, JHU-11, JHU-13, JHU-19, JHU-22, and JHU-29, were originally established at Johns Hopkins University. JHU-6, JHU-19 and JHU-29 were from HNSCC arising from the base of tongue; JHU-11 and JHU-22 were from the larynx; and JHU-13 was from the neck node metastasis. Cells were routinely cultured in Dulbecco's Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F12) supplemented with 10% fetal bovine serum and 50 mg/mL penicillin/streptomycin (Life Technologies). All cultures were conducted at 37 °C in a humidified atmosphere containing 5% CO2 in air.
Western blot analysis of EGFR and EGFRvIII expression
Cultured cells at the confluence of 70–80% were washed twice with phosphate buffered saline (PBS) and collected in RIPA lysis buffer (Santa Cruz Biotechnology, Santa Cruz, CA). The protein concentration was quantified with the Bio-Rad protein assay (Bio-Rad, Hercules, CA). Whole-cell proteins (30 µg) were separated on 8% SDS-PAGE gels and transferred to polyvinylidene difluoride membranes (Amersham Corp., Arlington Heights, IL). The membranes were blocked with 5% nonfat dry milk for 1 h and then hybridized overnight at 4 °C with the primary antibody Ab-5. After washing with TBS-T buffer (20 mM Tris (pH 8.0), 150 mM NaCl, and 0.1% Tween 20), the membrane was rehybridized with horseradish peroxidase-conjugated anti-mouse antibody (Santa Cruz Biotechnology) for 1 h. Specific protein signals were detected with the chemiluminescence detection system (Bio-Rad). Same membrane was also probed with the antibody against β-actin (Thermo Scientific).
Determination of the DT390-BiscFv806 cytotoxicity
We first determined the cell proliferation of U87-EGFRvIII cells following stable transfection of EGFRvIII. U87-EGFRvIII and U87 cells (5 × 103) were seeded, respectively, in 96-well plates and the cell growth was determined at different time points from 3 h to 6 d with PrestoBlue cell viability reagent according to manufacturer's instruction (Life Technologies). The absorbance values at 570 nm and 600 nm (reference wavelength for normalization) were measured, respectively, with an OPTImaxTM Tunable Microplate Reader (Molecular Devices, Sunnyvale, CA). The normalized absorbance values were converted to the cell numbers based on the standard cell number-absorbance curve. Cell doubling time was used as the indicator of cell growth, which was calculated with the formula of T In2/In(Xe/Xi), where T is the cell incubation time in the unit of h; and Xi and Xe are the cell numbers at the beginning and end of exponential phase, respectively, in the cell growth curve.
To determine the cytotoxicity of DT390-BiscFv806 against U87, U87-EGFRvIII, and HNSCC cells, 5 × 103 to 5 × 104 cells were seeded in wells of 96-well plates and allowed to grow overnight. Cells were then exposed to DT390-BiscFv806 at graded concentrations from 0 to 1 × 10−7 M for 48 h. The medium was replaced with fresh medium without DT390-BiscFv806 and the cells were allowed to grow for further 24 h. The cell viability was measured with the PrestoBlue cell viability reagent. The IC50 of DT390-BiscFv806 against the cells was calculated with the Microsoft Excel software. The cell morphology was recorded with an inverted microscope connected to an image processing computer.
To test the potential non-specific cytotoxicity from DT390-BiscFv806, a PSMA-targeted bivalent RIT, A-dmDT390-scfbDb(PSMA), was used as a control. A-dmDT390-scfbDb(PSMA) was confirmed to be highly cytotoxic to PSMA-expressing LNCaP prostate cancer cells (IC50, 0.57 nM), but not to PSMA-negative PC-3 cells (IC50, >100 nM).Citation29 The GBM and HNSCC cell lines are absent of PSMA expression.
In vivo experimental design
Evaluation of the effect of enforced EGFRvIII expression on tumor xenograft growth
To evaluate the effect of enforced expression of EGFRvIII on the growth of U87 tumor xenografts, 1 × 107 subconfluent U87-EGFRvIII and U87 cells in 50 µl medium were inoculated, respectively, into the lower back of female athymic nude mice (ages ∼6 weeks, Harlan, Indianapolis, IN). The mice were maintained in a ventilated rack system, and food and water were provided ad libitum. Animals were observed closely and the tumor size was measured with a caliper. The tumor volume was calculated with the formula of Vt = (L x W2)/2, where Vt is the volume in mm3 at the defined time point, and L and W are the length (large diameter) and width (small diameter) of a tumor, respectively.
The mouse body weight was also weighed at each time point. The mouse body weight loss was calculated with the formula of % = [(We-Wi)/Wi] x 100%, where Wi is the mouse body weight in gram at the beginning of treatment and We is the body weight at the end time point of experiment. The Wi was adjusted to remove the tumor weight, while the tumor weight at the beginning of treatment was calculated based on the tumor volume and tumor density (1.05 g/mL).
Evaluation of anti-tumor efficacy against established tumor xenografts
The U87-EGFRvIII and U87 tumor xenografts were established by inoculating 1 × 107 subconfluent cells in the lower back of female athymic nude mice. When the tumor volume reached >50 mm3, the mice were divided into treatment and control groups with similar size and size distribution between the 2 groups. The treatment groups were given DT390-BiscFv806 and the control groups were given PBS. The anti-tumor efficacy was evaluated with the following parameters: tumor volume, tumor weight, RTV, and %TGI. The tumor weight was obtained at the end point of the experiment. The RTV was calculated as Vt/Vi, where Vi is the volume in mm3 at the beginning of treatment and Vt is the volume at a defined time point. The %TGI was calculated with the equation of %TGI=100-(T/Cx100), where T is the individual or mean RTV in the treatment group and C is the mean RTV in the control group at a defined time point.
Therapeutic protocols
Protocol 1: Therapeutic regimen for U87-EGFRvIII tumor xenografts: Two intense short courses of treatment were designed considering the extremely rapid growth of U87-EGFRvIII tumor xenografts. DT390-BiscFv806 was administrated intravenously via tail vein injection. Each course included one dose of 50 µg/kg body weight (∼1 µg per mouse) per day for 5 consecutive days, with an interval of 2 d in between the 2 courses. The activity of mice was monitored closely and their body weights were measured.
Protocol 2: Therapeutic regimen for U87 tumor xenografts: Because of the relatively slow growth of U87 tumor xenografts compared with that of the U87-EGFRvIII tumor xenografts, a different therapeutic regimen was designed including one intense short course (one dose per day for 5 consecutive days), followed by one dose every other day for an additional 6 doses. Each dose was 50 µg/kg body weight. DT390-BiscFv806 was administered intravenously via tail vein injection.
All animal studies were carried out in accordance with the guidelines of the Howard University Institutional Animal Care and Use Committees.
Pathological analysis
At the completion of experiments, mice were sacrificed with overdose of isoflurane followed by neck dislocation. Tumors were sectioned in the maximum diameter, fixed in 10% neutral buffered formalin, and embedded in paraffin. Hematoxylin-eosin stained sections with 5 µm thickness were used for pathological analysis. The mitotic activity was assessed by counting the mitotic figures in 10 HPF (400x) from the most proliferative areas of tumor.
Statistical analysis
Statistical analyses were conducted with the statistical software OriginPro 7.0 (OriginLab, Northampton, MA). The treatment and control groups were compared using Student's t-test. Statistical significance was defined as P < 0.05.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported in part by the NIH/NCRR/RCMI/4, G12 RR003048 at Howard University, the US Army Medical Research and Material Command (W81XWH-10–1–0767),NIH 1R15DE025138-01 and Howard University College of Medicine Bridge Fund (U400067).
References
- Hatanpaa KJ, Burma S, Zhao D, Habib AA. Epidermal growth factor receptor in glioma: signal transduction, neuropathology, imaging, and radioresistance. Neoplasia 2010; 9:675-84; http://dx.doi.org/10.1593/neo.10688
- Wikstrand CJ, Sampson JH, Bigner DD. EGFRvIII: an oncogene deletion mutant cell surface receptor target expressed by multiple tumour types. Expert Opin Ther Targets 2000; 4:497-514; http://dx.doi.org/10.1517/14728222.4.4.497
- Kuan CT, Wikstrand CJ, Bigner DD. EGFRvIII as a promising target for antibody-based brain tumor therapy. Brain Tumor Pathol 2000; 17:71-8; PMID:11210174; http://dx.doi.org/10.1007/BF02482738
- Lo HW. EGFR-targeted therapy in malignant glioma: novel aspects and mechanisms of drug resistance. Curr Mol Pharmacol 2010; 3:37-52; PMID:20030624; http://dx.doi.org/10.2174/1874467211003010037
- Chung CH, Ely K, McGavran L, Varella-Garcia M, Parker J, Parker N, Jarrett C, Carter J, Murphy BA, Netterville J et al. Increased epidermal growth factor receptor gene copy number is associated with poor prognosis in head and neck squamous cell carcinomas. J Clin Oncol 2006; 24:4170-6; PMID:16943533; http://dx.doi.org/10.1200/JCO.2006.07.2587
- Temam S, Kawaguchi H, El-Naggar AK, Jelinek J, Tang H, Liu DD, Lang W, Issa JP, Lee JJ, Mao L. Epidermal growth factor receptor copy number alterations correlate with poor clinical outcome in patients with head and neck squamous cancer. J Clin Oncol 2007; 25:2164-70; PMID:17538160; http://dx.doi.org/10.1200/JCO.2006.06.6605
- Sok JC, Coppelli FM, Thomas SM, Lango MN, Xi S, Hunt JL, Freilino ML, Graner MW, Wikstrand CJ, Bigner DD, et al. Mutant epidermal growth factor receptor (EGFRvIII) contributes to head and neck cancer growth and resistance to EGFR targeting. Clin Cancer Res 2006; 12:5064-73; PMID:16951222; http://dx.doi.org/10.1158/1078-0432.CCR-06-0913
- Keller J, Shroyer KR, Batajoo SK, Zhao HL, Dong LM, Hayman MJ, Chan EL. Combination of phosphorylated and truncated EGFR correlates with higher tumor and nodal stage in head and neck cancer. Cancer Invest 2010; 28:1054-62; PMID:20873989; http://dx.doi.org/10.3109/07357907.2010.512602
- Chen LF, Cohen EEW, Grandis JR. New Strategies in Head and Neck Cancer: epidermal growth factor receptor inhibition in head and neck cancer. Clin Cancer Res 2010; 16:2489-95; PMID:20406834; http://dx.doi.org/10.1158/1078-0432.CCR-09-2318
- Cohen RB. Current challenges and clinical investigations of epidermal growth factor receptor (EGFR)- and ErbB family-targeted agents in the treatment of head and neck squamous cell carcinoma (HNSCC). Cancer Treat Rev 2014; 40:567-77; PMID:24216225; http://dx.doi.org/10.1016/j.ctrv.2013.10.002
- Taylor TE, Furnari FB, Cavenee WK. Targeting EGFR for treatment of glioblastoma: molecular basis to overcome resistance. Curr Cancer Drug Targets 2012; 12:197-209; PMID:22268382; http://dx.doi.org/10.2174/156800912799277557
- De Witt Hamer PC. Small molecule kinase inhibitors in glioblastoma: a systematic review of clinical studies. Neuro Oncol 2010; 12:304-16; PMID:20167819; http://dx.doi.org/10.1093/neuonc/nop068
- Chong CR, Jänne PA. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med 2013; 19:1389-400; PMID:24202392; http://dx.doi.org/10.1038/nm.3388
- Pastan I, Hassan R, FitzGerald DJ, Kreitman RJ. Immunotoxin treatment of cancer. Annu Rev Med 2007; 58:221-37; PMID:17059365; http://dx.doi.org/10.1146/annurev.med.58.070605.115320
- Shan L, Wang PC. Recombinant immunotoxin therapy of solid tumors: challenges and strategies. J Basic Clin Med 2013; 2:1-6; PMID:25309827
- Schmidt M Vakalopoulou E, Schneider DW, Wels W. Construction and functional characterization of scFv(14E1)-ETA - a novel, highly potent antibody-toxin specific for the EGF receptor. Br J Cancer 1997; 75:1575-84; PMID:9184171; http://dx.doi.org/10.1038/bjc.1997.270
- Ochiai H, Archer GE, Herndon JE 2nd, Kuan CT, Mitchell DA, Bigner DD, Pastan IH, Sampson JH. EGFRvIII-targeted immunotoxin induces antitumor immunity that is inhibited in the absence of CD4+ and CD8+ T cells. Cancer Immunol Immunother 2008; 57:115-21; PMID:17634939; http://dx.doi.org/10.1007/s00262-007-0363-7
- Chandramohan V, Bigner DD. A novel recombinant immunotoxin-based therapy targeting wild-type and mutant EGFR improves survival in murine models of glioblastoma. Oncoimmunol 2013; 2:e26852; http://dx.doi.org/10.4161/onci.26852
- Chandramohan V, Bao X, Keir ST, Pegram CN, Szafranski SE, Piao H, Wikstrand CJ, McLendon RE, Kuan CT, Pastan IH, et al. Construction of an immunotoxin, D2C7-(scdsFv)-PE38KDEL, targeting EGFRwt and EGFRvIII for brain tumor therapy. Clin Cancer Res 2013; 19:4717-27; PMID:23857604; http://dx.doi.org/10.1158/1078-0432.CCR-12-3891
- Azemar M, Schmidt M, Arlt F, Kennel P, Brandt B, Papadimitriou A, Groner B, Wels W. Recombinant antibody toxins specific for ErbB2 and EGF receptor inhibit the in vitro growth of human head and neck cancer cells and cause rapid tumor regression in vivo. Int J Cancer 2000; 86:269-75; PMID:10738256; http://dx.doi.org/10.1002/(SICI)1097-0215(20000415)86:2%3c269::AID-IJC18%3e3.0.CO;2-8
- Sampson JH, Akabani G, Archer GE, Bigner DD, Berger MS, Friedman AH, Friedman HS, Herndon JE 2nd, Kunwar S, Marcus S, et al. Progress report of a Phase I study of the intracerebral microinfusion of a recombinant chimeric protein composed of transforming growth factor (TGF)-α and a mutated form of the Pseudomonas exotoxin termed PE-38 (TP-38) for the treatment of malignant brain tumors. J Neurooncol 2003; 65:27-35; PMID:14649883; http://dx.doi.org/10.1023/A:1026290315809
- Panousis C, Rayzman VM, Johns TG, Renner C, Liu Z, Cartwright G, Lee FT, Wang D, Gan H, Cao D, et al. Engineering and characterisation of chimeric monoclonal antibody 806 (ch806) for targeted immunotherapy of tumours expressing de2-7 EGFR or amplified EGFR. Br J Cancer 2005; 92:1069-77; PMID:15770208; http://dx.doi.org/10.1038/sj.bjc.6602470
- Luwor RB, Johns TG, Murone C, Huang HJ, Cavenee WK, Ritter G, Old LJ, Burgess AW, Scott AM. Monoclonal antibody 806 inhibits the growth of tumor xenografts expressing either the de2-7 or amplified epidermal growth factor receptor (EGFR) but not wild-type EGFR. Cancer Res 2001; 61:5355-61; PMID:11454674
- Johns TG, Mellman I, Cartwright GA, Ritter G, Old LJ, Burgess AW, Scott AM. The antitumor monoclonal antibody 806 recognizes a high-mannose form of the EGF receptor that reaches the cell surface when cells over-express the receptor. FASEB J 2005; 19:780-2; PMID:15774576
- Johns TG, Adams TE, Cochran JR, Hall NE, Hoyne PA, Olsen MJ, Kim YS, Rothacker J, Nice EC, Walker F, et al. Identification of the epitope for the epidermal growth factor receptor-specific monoclonal antibody 806 reveals that it preferentially recognizes an untethered form of the receptor. J Biol Chem 2004; 279:30375-84; PMID:15075331; http://dx.doi.org/10.1074/jbc.M401218200
- Scott AM, Lee FT, Tebbutt N, Herbertson R, Gill SS, Liu Z, Skrinos E, Murone C, Saunder TH, Chappell B, et al. A phase I clinical trial with monoclonal antibody ch806 targeting transitional state and mutant epidermal growth factor receptors. Proc Natl Acad Sci USA 2007; 104:4071-6; PMID:17360479; http://dx.doi.org/10.1073/pnas.0611693104
- Woo JH, Liu YY, Stavrou S, Neville DM. Increasing secretion of bivalent anti-T-cell immunotoxin by Pichia pastoris. Appl Environ Microbiol 2004; 70:3370-6; PMID:15184133; http://dx.doi.org/10.1128/AEM.70.6.3370-3376.2004
- Kim GB, Wang Z, Liu YY, Stavrou S, Mathias A, Goodwin KJ, Thomas JM, Neville DM. A fold-back single-chain diabody format enhances the bioactivity of an anti-monkey CD3 recombinant diphtheria toxin-based immunotoxin. Protein Eng Des Sel 2007; 20:425-32; PMID:17693455; http://dx.doi.org/10.1093/protein/gzm040
- Zhang F, Shan L, Liu Y, Neville D, Woo JH, Chen Y, Korotcov A, Lin S, Huang S, Sridhar R, et al. An anti-PSMA bivalent immunotoxin exhibits specificity and efficacy for prostate cancer imaging and therapy. Adv Healthc Mater 2013; 2:736-44; PMID:23184611; http://dx.doi.org/10.1002/adhm.201200254
- Liu YY, Woo JH, Neville DM. Overexpression of an anti-CD3 immunotoxin increases expression and secretion of molecular chaperone BiP/Kar2p by Pichia pastoris. Appl Environ Microbiol 2005; 71:5332-40; PMID:16151122; http://dx.doi.org/10.1128/AEM.71.9.5332-5340.2005
- Hauser BR, Zhu X, Califano J, Gu X. Detection of gene copy number of epidermal growth factor receptor in head and neck squamous cell carcinomas cell lines. Cancer Res 2008; 68: 3413; PMID:18451169; http://dx.doi.org/10.1158/0008-5472.CAN-07-1919
- Hauser BR. Epidermal growth factor receptor (EGFR) gene expression and its related biomarkers in head and neck squamous cell carcinoma [master's thesis]. [Washington DC]: Howard University; 2008. 58 p.
- Liu YY, Woo JH, Neville DM. Targeted introduction of a diphtheria toxin resistant mutation into the chromosomal EF-2 locus of Pichia pastoris and expression of immunotoxin in the EF-2 mutants. Protein Expr Purif 2003; 30:262-74; PMID:12880776; http://dx.doi.org/10.1016/S1046-5928(03)00129-3
- Li YM, Vallera DA, Hall WA. Diphtheria toxin-based targeted toxin therapy for brain tumors. J Neurooncol 2013; 114:155-64; PMID:23695514; http://dx.doi.org/10.1007/s11060-013-1157-8
- Adkins I, Holubova J, Kosova M, Sadilkova L. Bacteria and their toxins tamed for immunotherapy. Curr Pharm Biotechnol 2012; 13:1446-73; PMID:22339216; http://dx.doi.org/10.2174/138920112800784835
- Matar AJ, Pathiraja V, Wang Z, Duran-Struuck R, Gusha A, Crepeau R, Tasaki M, Sachs DH, Huang CA. Effect of pre-existing anti-diphtheria toxin antibodies on T cell depletion levels following diphtheria toxin-based recombinant anti-monkey CD3 immunotoxin treatment. Transpl Immunol 2012; 27:52-4; PMID:22676970; http://dx.doi.org/10.1016/j.trim.2012.05.003
- Manoukian G, Hagemeister F. Denileukin diftitox: a novel immunotoxin. Expert Opin Biol Ther 2009; 9:1445-51; PMID:19817678; http://dx.doi.org/10.1517/14712590903348135
- Woo JH, Liu JS, Kang SH, Singh R, Park SK, Su Y, Ortiz J, Neville DM, Willingham MC, Frankel AE. GMP production and characterization of the bivalent anti-human T cell immunotoxin, A-dmDT390-bisFv(UCHT1), for phase I/II clinical trials. Protein Expr Purif 2008; 58:1-11; PMID:18160309; http://dx.doi.org/10.1016/j.pep.2007.11.006
- Bühler P, Wetterauer D, Gierschner D, Wetterauer U, Beile UE, Wolf P. Influence of structural variations on biological activity of anti-PSMA scFv and immunotoxins targeting prostate cancer. Anticancer Res 2010; 30:3373-9; PMID:20944111
- Thompson J, Stavrou S, Weetall M, Hexham JM, Digan ME, Wang Z, Woo JH, Yu Y, Mathias A, Liu YY, et al. Improved binding of a bivalent single-chain immunotoxin results in increased efficacy for in vivo T-cell depletion. Protein Eng 2001; 14:1035-41; PMID:11809934; http://dx.doi.org/10.1093/protein/14.12.1035
- Nagane M, Coufal F, Lin H, Bögler O, Cavenee WK, Huang HJ. A common mutant epidermal growth factor receptor confers enhanced tumorigenicity on human glioblastoma cells by increasing proliferation and reducing apoptosis. Cancer Res 1996; 56:5079-86; PMID:8895767
- Nishikawa R, Ji XD, Harmon RC, Lazar CS, Gill GN, Cavenee WK, Huang HJ. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc Natl Acad Sci USA 1994; 91:7727-31; PMID:8052651; http://dx.doi.org/10.1073/pnas.91.16.7727
- Inda MM, Bonavia R, Mukasa A, Narita Y, Sah DW, Vandenberg S, Brennan C, Johns TG, Bachoo R, Hadwiger P, et al. Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev 2010; 24:1731-45; PMID:20713517; http://dx.doi.org/10.1101/gad.1890510