
ABSTRACT
Chromosomal duplication is targeted by various chemotherapeutic agents for the treatment of cancer. However, there is no specific inhibitor of DNA polymerases that is viable for cancer management. Through structure-based in silico screening of the ZINC database, we identified a specific inhibitor of DNA polymerase δ. The discovered inhibitor, Zelpolib, is projected to bind to the active site of Pol δ when it is actively engaged in DNA replication through interactions with DNA template and primer. Zelpolib shows robust inhibition of Pol δ activity in reconstituted DNA replication assays. Under cellular conditions, Zelpolib is taken up readily by cancer cells and inhibits DNA replication in assays to assess global DNA synthesis or single-molecule bases by DNA fiber fluorography. In addition, we show that Zelpolib displays superior antiproliferative properties to methotrexate, 5-flourouracil, and cisplatin in triple-negative breast cancer cell line, pancreatic cancer cell line and platinum-resistant pancreatic cancer cell line. Pol δ is not only involved in DNA replication, it is also a key component in many DNA repair pathways. Pol δ is the key enzyme responsible for D-loop extension during homologous recombination. Indeed, Zelpolib shows robust inhibition of homologous recombination repair of DNA double-strand breaks and induces “BRCAness” in HR-proficient cancer cells and enhances their sensitivity to PARP inhibitors.
Introduction
The majority of successful chemotherapeutics are those that interfere with DNA replicationCitation1,Citation2. The success of these drugs is explicable given that the defining characteristic of neoplastic cells, regardless of histology, is unregulated and persistent cell division. Various components of DNA metabolism serve as targets for cancer therapy including chromosomal template for replication, required deoxynucleotides (dNTPs) for replicating the daughter strand, and necessary enzymes. The most diverse category of chemotherapeutic strategies act to directly damage the templateCitation3 to prevent its replication. These strategies involve various covalent modifications of DNA strands including crosslinking, alkylation, and lysis, as elicited by both chemical and physical means. Strategies that damage the DNA, however, have a narrow therapeutic window because of the propensity to damage all cells1,Citation3.
The second class of compounds, broadly termed antimetabolitesCitation4, target the synthesis of dNTPs. Thus, these compounds act to deplete the substrates for DNA replication. This strategy is – in general – less toxic than the aforementioned strategies that damage DNA because it has minimal impact on non-dividing cells. Consequently, the relevant compounds offer a better side-effect profile and can be taken for longer duration than the DNA damaging agents. Methotrexate and its analogsCitation5, which competitively inhibit dihydrofolate reductase, constitute important examples of antimetabolite drugs. Indeed, methotrexate is prescribed for numerous disorders that exhibit aberrant cell proliferation, such as arthritis and psoriasis. The long course of these diseases has meant that, in some cases, methotrexate has been prescribed and well-tolerated for decades. Even so, the targets of antimetabolites, such as dihydrofolate reductaseCitation5, thymidylate synthaseCitation2, or ribonucleotide reductaseCitation6 lie at the intersection of numerous metabolic pathways, which increases the propensity for off-target side effects.
The required enzymes that can be targeted for chemotherapeutic intervention are topoisomerasesCitation7-Citation9 which act to prepare chromosomes for replication, helicases which act to unwind the duplex DNA, and polymerases which synthesize the daughter strand during replication. Of these, topoisomerasesCitation5 have been successfully targeted. A number of topoisomerase inhibitors have gained FDA approval, such as the topotecan analogs and etoposide. Topoisomerases, however, are indispensable for the resolution of DNA topology issues during transcriptionCitation10 and topoisomerase inhibitors also have the propensity to induce toxic single- and double-DNA strand breaksCitation11.
Ostensibly missing are chemical entities that target the most important enzymes in DNA replication – DNA polymerases, even though DNA or RNA polymerases are the targets for many antiviral therapeutics. Reversible inhibitors of DNA polymerases should inhibit cancer cell proliferation and possess some advantages over DNA damaging agents and topoisomerase inhibitors. First, due to the specific function of replicative polymerases, reversible DNA polymerase inhibitors will inhibit DNA replication alone, and thus cell proliferation without affecting non-proliferating cells. Secondly, these inhibitors will not damage the chromosomes of non-dividing cells and should have minimal impact on transcription. Consequently, the side-effect profile of polymerase inhibitors should be at least as good as antimetabolites exemplified by methotrexate. There are three essential replicative DNA polymerases, Pol α, Pol δ and Pol ε. Cells will not replicate DNA without one of the three and inhibiting one is sufficient to disable chromosome duplication and cancer cell proliferation. On the other hand, the three replicative Pols share a common fold and near identical active site structures as shown by x-ray crystallographyCitation12-Citation14. Inhibitors that bind to the active sites most likely will inhibit all three Pols simultaneously.
Here we report the discovery of a novel Pol δ inhibitor, Zelpolib through structural based in silico screen of the ZINC databaseCitation15. We further show that Zelpolib causes robust inhibition of Pol δ in vitro and in vivo and exhibits superior antiproliferative activities as compared to a number of currently-used chemotherapeutics.
Results
Identification of Zelpolib through in silico screen
Through two cycles of screening () top 300 compounds (supplemental movie 1) were selected for detailed analysis. We aimed to select compounds that will inhibit Pol δ when it is actively engaged in DNA replication. At the minimum, three key requirements are necessary: interaction with template strand, hydrophobic stacking with the preceding nucleotide base of the primer, and extensive interaction with Pol δ. We selected 10 compounds that satisfy our requirements. All selected compounds showed inhibition of Pol δ activity when assayed at 100 μM with the poly(dA)/oligo(dT) assay (data not shown) and only one compound showed significant inhibition when assayed at 10 μM. This compound, subsequently named Zelpolib (), Zilch Polymerase activity), is predicted to have reasonable pharmacological properties (). Zelpolib exhibits a unique binding mode in the model structure ()). The catechol moiety occupies the base position of the incoming nucleotide (,b), and supplemental movie 2), in a position to form hydrophobic stacking with previous base of the primer (cytosine). The catechol groups are in position to form two hydrogen bonds with guanine on the template strand ()). In fact, half of the inhibitor molecule (from catechol to thiourea segment of the inhibitor, )) almost overlaps with the incoming nucleotide in the crystal structure (3IAY.PDB, dCTP, ) vs 2B). In the final model, Zelpolib is also in a position to form 5 potential hydrogen bonds with Pol δ()). The three residues forming hydrogen bonds with Zelpolib, N698, D602 and D757, also play important roles in interaction with incoming nucleotide (3IAY). Potentially, more hydrogen bonds are formed between amide of Y607 and guanidine side chain of R667 with Zelpolib. Overall, Zelpolib occupies significantly more space within the Pol δ active site () than the incoming nucleotide and is projected to be a strong inhibitor that binds to Pol δ active site when it is in complex with template/primer, i.e. when actively synthesizing DNA during replication or repair.
Figure 1. Flow-chart of the in silico screening protocol by Dock 6.4 of the ChemoBridge library.
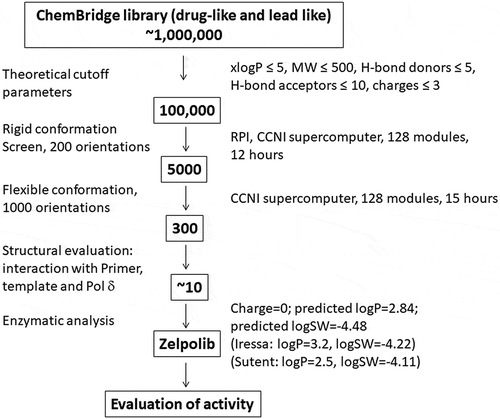
Figure 2. Interactions of Zelpolib at the active site of Pol δ. A, Zelpolib forms two hydrogen bonds with dG on the template strand. The catechol moiety stacks against dOC (dideoxycytosine) on the primer. B, comparative position of dCTP at the active site of Pol δ. C, chemical structure of Zelpolib. D, 5 potential hydrogen-bonds can be formed between Zelpolib and Pol δ.
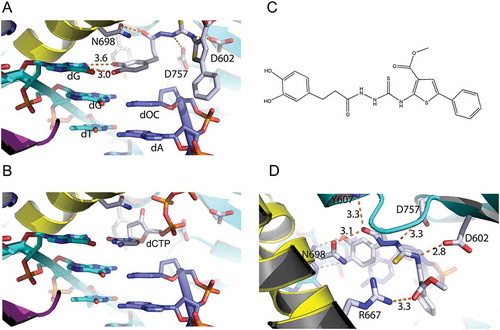
Figure 3. Comparison of Zelpolib with dCTP in binding to the active site of Pol δ. Zelpolib in stick (A) and ball (B) representation with dCTP (C and D) in comparison.
Inhibition of Pol δ activity by Zelpolib in enzymatic assays
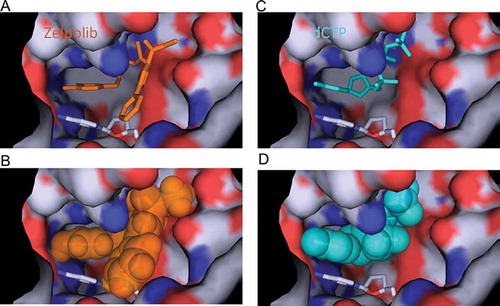
In order to validate the predicted inhibitory properties of Zelpolib, we used the well-established DNA polymerase assay, poly(dA)/oligo(dT) methodCitation1,Citation16, to quantify the inhibition constants. In this assay, a poly(dA) template (up to 4 kilobases) was randomly primed with oligo(dT) primers (40mer). In the presence of proliferating cell nuclear antigen (PCNA), Pol δ will initiate the elongation of oligo dT primer with the incorporation of radio-labeled deoxythymidine Citation3H3H. The polymerase activity can then be precisely quantified by counting the isotope incorporated. In this assay, Zelpolib showed robust inhibition of Pol δ activity in a non-competitive manner ()) against dTTP with an inhibition constant Ki of 4.3 ± 0.3 μM (RCitation2= 0.99).
Figure 4. Zelpolb inhibits Pol δ activity. A, enzymatic assay by poly(dA)/oligo(dT) method demonstrates noncompetitive inhibition with Ki of 4.3 μM. B, Zelpolib is likely unique in inhibiting Pol δ activity in comparison to current FDA approved small molecule oncology drugs. C, quantification of the full-length products (40mer, integration by ImageJ). Lower peak height indicates less product. D, quantification of unextended primer. Higher peaks correspond to higher amounts of primer left.
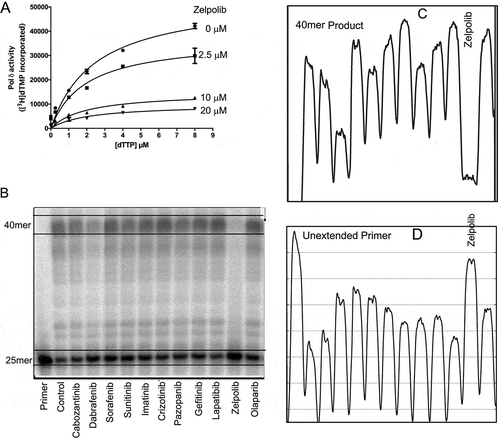
Next, we investigated the inhibitory properties of Zelpolib in a primer extension assayCitation17,Citation18 with a template that consists of all four deoxynucleotide, which is more reflective of cellular DNA replication processes than the poly(dA) template. The substrate consisted of a 25mer primer hybridized to a 40mer template. In the presence of PCNA and dNTPs, the primers were extended upon addition of Pol δδand a full extension would generate a 40mer copy of the template. The primers were labeled at the 5’-end withCitation19 32P, and the products were separated on a sequencing gel for visualization and quantification. Consistent with results from poly(dA)/oligo(dT) assays, Zelpolib showed concentration dependent inhibition of the primer extension activities of Pol δ (Supplemental Figure S1). In the second assay, we aimed to investigate whether inhibition of DNA replication is exhibited by other existing cancer drugs, especially kinase inhibitors. The idea originated from the fact that most kinase inhibitors compete for binding with ATP. It is conceivable that they might compete with dATP for binding to polymerases. In addition, there are some structural similarities between Zelpolib and many kinase inhibitors currently in clinical application. We randomly selected 10 small-molecule-cancer drugs with nine being kinase inhibitorsCitation20 and one parp inhibitor (olaparib)Citation21. At 20 μM, Zelpolib was the only molecule shown to exhibit robust inhibition of Pol δ activity ()), with 94% of the primer unextended ()). In the presence of any other inhibitors, at least 30% of the primers were extended. If we analyze the full-length product formed, it is barely detectible in the presence of 20 μM Zelpolib, whereas in the presence of any other inhibitors, the full-length products are clearly detectible ()).
In our in silico screening, the active site cavity was created by the removal of dCTP in the original crystal structure. One of the questions we asked was if Zelpolib displays any preference in the inhibition of a specific dNTP among the four dNTPs. Therefore, we assayed the inhibition patterns of all four individual dNTP molecules in a single nucleotide incorporation assay. In this assay, only one nucleotide is present for the primer extension for the next appropriate base pairing to the template. Interestingly, Zelpolib inhibited the incorporation of all four nucleotides to similar extents in a noncompetitive manner (Supplemental Figure S2).
Our enzymatic analyses of Zelpolib on Pol δ activity have validated the in silico screen results. Zelpolib show strong inhibition of Pol δ activity in two different assays and inhibited the incorporation of all four nucleotide in a noncompetitive manner with Ki in the μM range. In addition, the mechanism of inhibiting DNA replication by Zelpolib is not likely shared by other small molecule drugs currently in clinical application in oncology.
Zelpolib inhibits DNA replication directly under cellular conditions
Upon establishing that Zelpolib inhibits Pol δ activities in enzymatic assays, we conducted experiments to establish that Zelpolib inhibits DNA synthesis in vivo. Extensive cellular studies have shown that kinase inhibitors can arrest cell cycle progression and induce apoptosisCitation22 upon prolonged exposure of 24 to 96 hours. The effects of prolonged exposure could be a combination of many cellular pathway disturbances that eventually results in halting DNA synthesis. In order to assess the direct effect on DNA replication, we avoided prolonged cellular exposure to Zelpolib so that any effects on DNA replication are not due to cell cycle regulation. In our first assay, we incubated cells with Zelpolib for 1 to 2 hours to ensure uptake. Then the cells were pulse labeled with 5-ethynyl-2’-deoxyuridine (EdU)Citation23 so that the majority of incorporation was due to ongoing DNA replication processes. The uridine analog, EdU, is incorporated into DNA as dTTP analogs and can be detected by “Click” chemistry with fluorescent azideCitation23,Citation24. We tested Zelpolib on a pancreatic cancer cell line, BxPC-3 cells, followed by EdU incorporation analysis using laser scanning cytometry. Zelpolib showed concentration dependent inhibition of EdU incorporation. The integrated fluorescence signal is shown in ), with significant reduction of EdU incorporation at 10 and 20 μM in comparison with sham treatment.
Figure 5. Zelpolib inhibits cellular DNA replication. A, EdU incorporation by whole cell population is inhibited by Zelpolib. Exponentially growing HCC1395 (TNBC) cells were treated with Zelpolib for 2 hours prior to pulse labeling with EdU for 30 minutes. Amount of EdU quantified with “click” chemistry and measured by laser scanning cytometry (LSC). Error bar shows mean value with SEM (triplicates) and P values were calculated using unpaired Student’s t-test. B, treatment scheme of DNA fiber fluorography assay. C, DNA fiber-length comparison between untreated and treated with 50 μM Zelpolib (HCC1395 cells, see Figure S4 for original images). D, quantification of DNA fiber length for BxPC-3 cells. E, quantification of DNA fiber length for HCC1395 cells. 75 fibers were analyzed per sample. Scattered dot plot shows ratio of green/red fiber lengths (ratios) with SEM. P values were calculated using unpaired T test. *** indicates p < 0.0001.
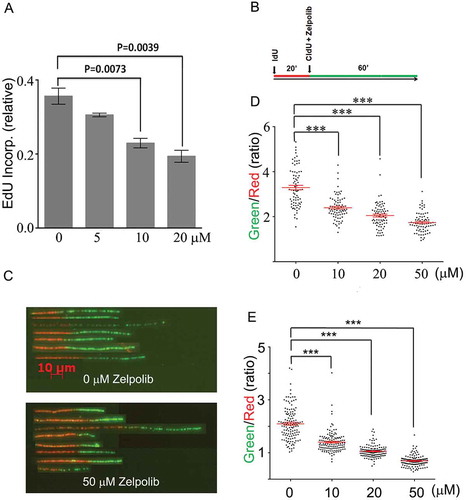
In our search for Pol δ inhibitors, we focused on the identification of compounds that can inhibit Pol δ when it is actively replicating DNA. DNA fiber fluorography with dual labeling techniqueCitation25 provides a possibility that we can analyze the effect of Zelpolib on ongoing DNA replication at a single-molecule level. Both IdU and CldU are halogenated thymidine analogs that can be incorporated into DNA. Subsequent DNA fiber spreading and antibody detection are used to visualize the replicated DNA fiber. The incorporation of IdU gives rise to red fibers and CldU produces green fibers with antibody detectionCitation25. Therefore, in this assay, the cells were first incubated with IdU for 20 minutes in the absence of Zelpolib ()) to establish ongoing DNA replication and the basal replication rates of each individual DNA fibers. Subsequently, CldU were added to cell culture either with or without simultaneous addition of Zelpolib. DNA replication were allowed for an additional hour. For any ongoing replication, the green fiber (60 minutes of replication) should be collectively about 3X the length of the red fiber (20 minutes of replication, )) either in the absence of Zelpolib or even in the presence of Zelpolib if the cells failed to take up Zelpolib fast enough in that short duration. Under this stringent condition, Zelpolib showed concentration dependent inhibition of ongoing cellular DNA replication (), BxPC-3 cells). In order to be certain that this inhibition was not cell line dependent, we conducted an identical experiment on a triple-negative breast cancer cell line as well. All three concentrations of Zelpolib showed significant inhibition of ongoing DNA replication (,e)). The DNA fibers ()) from a single field of view (Supplemental Figure S3) were realigned for visual comparison.
These cellular studies demonstrated that Zelpolib inhibits cellular DNA replication significantly with concentrations that inhibited Pol δ activities in enzymatic assays. The DNA fiber analysis also demonstrates that, as intended in the screening protocol, Zelpolib can be taken up readily by cancer cells and inhibits ongoing DNA replication. At 20 μM, Zelpolib inhibited the DNA fiber extension by about 40%, indicating that cellular uptake took less than 30 minutes as the total exposure time was 60 minutes.
Zelpolib displayed superior antiproliferative properties to widely used anti-cancer drugs
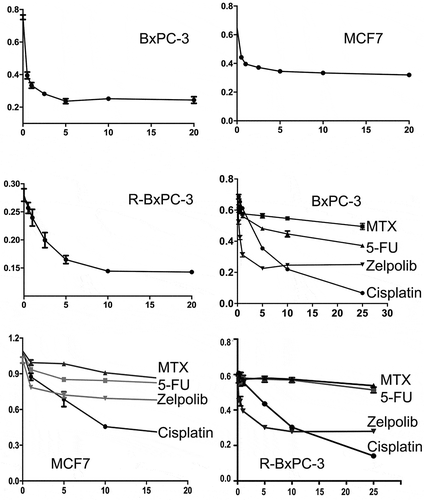
DNA replication precedes cell division. Thus, cancer cells cannot divide and proliferate without successful chromosomal duplication. A DNA polymerase inhibitor should inhibit cancer cell proliferation. We characterized the antiproliferative properties of Zelpolib on cancer cell lines originated from pancreatic cancer (BxPC-3) and breast cancer (MCF7 for ER+ breast cancer). In addition, we developed a cisplatin resistant subline of a pancreatic cancer cell line, R-BxPC3 for our studies. We used the MTT methodCitation26 for analyzing cell proliferation. This assay is based on the reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide by live cells to an insoluble formazon, which were subsequently dissolved in DMSO and quantified by UV absorbance. Therefore, the cell number is proportional to the optical density in the established linear region. Based on this assay, Zelpolib showed robust antiproliferative properties against BxPC-3, R-BxPC-3, and MCF7 cells (), all reaching a plateau at about 5 μM. The plateau phenomena indicate inhibition of proliferation without cell killing, similar to these observed from hydroxyureaCitation27, aphidicolinCitation28, and to a certain extent, methotrexate treatmentsCitation29. Direct inhibition of DNA polymerase activity provides a unique mechanism that is not employed by any of the current cancer therapeutics. We selected a number of most widely prescribed chemotherapeutics with distinct mechanisms to compare their antiproliferative potential in the two cancer cell lines. We included alkylating agent cisplatin, antimetabolites methotrexate and 5-flourouracil (5FU)Citation30–Citation32. When investigated on pancreatic cancer BxPC-3 cells, Zelpolib is superior to methotrexate and 5-FU at all concentrations; is superior to cisplatin up to 10 μM (). With MCF7 cells, Zelpolib is superior to methotrexate and 5-FU across the concentration range. Whereas the comparison with cisplatin is biphasic. Zelpolib is at least as effective as cisplatin in inhibiting MCF7 cell growth up to 5 μM, whereas cisplatin is more effective beyond 5 μM, as the cytotoxic effects of cisplatin start to dominate (). Similar advantages of Zelpolib were also observed with cisplatin-resistant Pancreatic cancer cells (). Within the time frames of our study of 48 to 72 hours, treatment with Zelpolib alone does not lead to significant increase in apoptosis or senescence (data not shown), consistent with its mechanism of action. Aphidicolin, a Pol δ inhibitor, is commonly used for cell cycle synchronization in laboratory practice without induction of cell apoptosis or senescence.
Figure 6. Antiproliferative activities of Zelpolib. Concentration dependent inhibition of cell proliferation by Zelpolib on three different cell lines and comparison with methotrexate, 5-FU, and cisplatin by MTT assays. All samples were in triplicates and presented as averages with standard deviation.
Indeed, Zelpolib provides effective antiproliferative activities with a unique mechanism of action, which has not been successfully employed in current cancer therapeutics.
Zelpolib inhibits DNA double strand break repair by homologous recombination (HR)
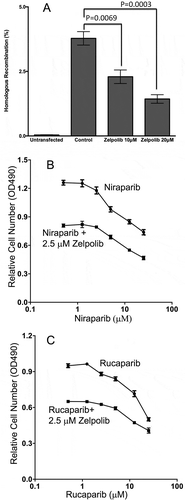
In addition to being indispensable for the duplication of chromosomes, Pol δ also plays critical roles in various DNA repair pathwaysCitation19. It has been shown that Pol δ is the preferred polymerase for the D-loop extension during homologous recombination repairCitation33. Thus, we investigated the effect of Zelpolib on HR. We used the two plasmid DR-GFP assay developed by Nakanishi et alCitation34, which is a modified version of the initial methodCitation35. The first plasmid contains a mutant GFP that has lost the ability to fluoresce. The second plasmid contains the coding region of a restriction enzyme, I-SceI. The co-transfection of the two plasmids will result in the expression of I-SceI, which can digest the DR-GFP plasmid at the mutation site leading to the formation of a double-strand break. Successful homologous recombination repair of the double strand break will restore the GFP fluorescent signal. Therefore, the percentage of fluorescent cells is the readout of the efficacy of HR. To have conditions for valid comparison, HEK 293T cells were transfected with both plasmids under identical conditions. The transfected cells were allowed to recover for 4 to 5 hours and then replaced with fresh media. Subsequently, Zelpolib was added at two different concentrations. The efficacy of HR was analyzed about 40 hours later by flow cytometry. Zelpolib showed dose-dependent inhibition of HR (). The reduction in HR efficacy at 20 μM Zelpolib is comparable to those induced by siRNA knockdown of BRCA1 or BRCA2 individually as reportedCitation36.
Figure 7. Zelpolib inhibits homologous recombination in cell-based assay. a, Dual plasmids (DR-GFP and I-Scel) reporter assay was used to measure the effect of Zelpolib on DSB repair in 293T cells as reportedCitation34. The error bar represents mean values of four repeats with SEM. P values were calculated using unpaired T test. b, Zelpolib enhances the sensitivity of triple negative breast cancer cells (HCC1395) to niraparib by MTT assay. c, Zelpolib enhanced the sensitivity of TNB cells (HCC1395) towards Rucaparib.
Zelpolib increases the sensitivity of HR proficient cells to PARP inhibitors
PARP inhibitors have been a breakthrough therapy for HR-deficient ovarian cancers due to BRCA1/2 mutationsCitation37-Citation41. However, there have been minimal clinical benefits for HR-proficient cancersCitation37-Citation41. Since Zelpolib can inhibit HR, we explored the effect of Zelpolib in combination with PARP inhibitors on HR-proficient cancer cells. The hypothesis is that Zelpolib will lead to an HR-defect phenotype through inhibition of Pol δ and render HR-proficient cells sensitive to PARP inhibition. We tested the combination on a triple-negative breast cancer cell line, HCC1395, which is insensitive to PARP inhibitors NiraparibCitation42 or RucaparibCitation43. The cells were treated with increasing concentrations of PARP inhibitor Niraparib ()) or Rucaparib ()) in the absence or presence of 2.5 μM Zelpolib for 48hrs. The number of cells were then analyzed by MTT assay. Zelpolib increased the sensitivity of HCC1395 cells to Niraparib across the whole range of concentrations, with the differences being significant up to 20 μM. The effect of 25 μM of Niraparib on the proliferation of HCC1395 cells can be achieved by 5 μM in the presence of 2.5 μM Zelpolib. The combination of Zelpolib with Rucaparib behaved similarly to Zelpolib and Niraparib combination, with the difference being statistically significant up to 25 μM.
Inhibition of the polymerase activity required for homologous homologous recombination by Zelpolib can render HR proficient cells more sensitive to PARP inhibition.
Discussion
Advantages of a reversible polymerase inhibitor
Current cancer therapies take advantage of the quantitative differences in biological pathways between cancer and non-cancer cells. These differences have been summarized by the landmark publication of “Hallmarks of Cancer”Citation44 by Hanahan and Weinberg and a subsequent updateCitation45. Among these, the most prominent difference is the rate of proliferation. The high proliferating rates of cancer cells demand expedited nutritional supply through accelerated neoangiogenesis, and rapid chromosome duplication at the sacrifice of genomic stability through inactivation of checkpoint and DNA repair pathways. In fact, these differences and vulnerabilities have been the targets of majority of current therapeutic regimen. Kinase inhibitors, especially VEGFR inhibitors, disrupt neoangiogenesisCitation46,Citation47 and inhibit cancer cell proliferation. These inhibitors are an essential part of our current anticancer arsenal.
On the disruption of chromosome duplication front, the most successful chemotherapy reagents target DNA replication by direct DNA damage. These drugs include platinum derivatives that form DNA interstrand crosslinks, and alkylating agents that modify DNA bases. These reagents can modify DNA of all cell types and impact normal cellular function on many fronts. One of the unintended consequences of direct DNA damage is the effect on gene transcription inhibitionCitation10,Citation48, a necessary cellular activity shared by all cells, proliferating or quiescent. Some of the side effects and toxicities of DNA damaging agents likely originate from inhibition of transcription in normal tissuesCitation49. The consequences of transcription inhibition are somewhat under-appreciated, even though the toxicity from α-amanitin, the toxin from some poisonous mushrooms, is well knownCitation50,Citation51. The initial effects of α-amanitin exposure share significant similarities with those from chemotherapy, such as abdominal pain, vomiting, and diarrheaCitation51,Citation52. Evidently, for cancer treatment, the challenge is to inhibit DNA replication with minimal impact on other nucleotide metabolism pathways such as transcription.
The three replicative polymerases, Pol α, δ, and ε, share a relatively conserved active-site structure and an inhibitor of one most likely will inhibit the others as well. A reversible inhibitor of replicative DNA polymerases could accomplish the goal of inhibiting DNA replication without significant impact on other cellular processes such as transcription or translation. This is partially confirmed by results from this study, as Zelpolib alone showed minimal cell cytotoxicity with pronounced antiproliferative activities. Additional support is from studies on one of the well-characterized replicative polymerase inhibitor, aphidicolin. A number of publications have shown the robust anticancer activities of aphidicolin in cell line and animal modelsCitation53-Citation56. More importantly, the safety profile in miceCitation55 and phase 1 clinical studyCitation57 further validated the tolerability of polymerase inhibitors. However, aphidicolin progressed no further in clinical development due to limitations in biological availability and stabilityCitation55,Citation57.
Inhibition of cellular polymerases requires much lower concentrations of inhibitors in comparison with tyrosine kinase inhibitors, if the inhibition constants were comparable. There are over 30 kinase inhibitors which have been widely used to treat various cancers. The difficulty in inhibiting kinases arises from the fact that cellular ATP concentrations are relatively high (2–10 mM) and its Km values of ATP towards kinases are generally in the 10 to 50 μM range. Therefore, it requires a high [inhibitor]/Ki ratio for the inhibitors to overcome the ATP saturation of kinase active sites. On the other hand, cellular deoxyribonucleotide concentrations (dNTPs) are much lower and generally in the micromolar rangeCitation58,Citation59, therefore it requires lower inhibitor [inhibitor]/Ki ratio to compete with dNTPs for the polymerase active sites. These analyses are supported by the fact that nucleoside analogs such as cytosine arabinosideCitation60,Citation61 (ara-C), which has similar affinities towards polymerases after conversion to triphosphate to those of dNTPs, is effective in cancer therapy.
The antiproliferative potential and unique mechanism of action Zelpolib displays could make it a component of cancer therapy to minimize the toxicities associated with current cancer chemotherapeutics.
Inhibition of DNA repair
The majority of current cancer therapeutics has been based on the quantitative differences in biochemical pathways between cancer and non-cancer cells. More recent advances take advantage of the qualitative differences between cancer and non-cancer cells to minimize the side effects or to induce more durable responses. The breakthroughs come from investigations on the deficiencies in cancer cells, notably DNA repair pathwaysCitation62. The universally increased genomic instability of cancers most likely originates from loss of certain DNA repair functions, some have been identified and others remain to be characterized. Two identified pathways, homologous repair deficiency and mismatch repair defect, have been characterized across many cancer types, with HR deficiency due to BRCA1/2 mutations mainly in female reproductive cancersCitation63 and MMR deficiencyCitation64 in colorectal cancersCitation65.
The first major advance in a long time in ovarian cancer treatment comes from the discovery that nearly half of epithelial ovarian cancers (EOCs) are HR deficient due to genetic or epigenetic alterations and roughly 40 percent of these are due to BRCA1/2 mutationsCitation66. Cancers with HR defects have exquisite sensitivity to PARP inhibition as a consequence of synthetic lethality. In 2012, results from a pivotal clinical trial with olaparib for the maintenance therapy in patients with platinum-sensitive relapsed disease attained a hazard ratio of 0.18Citation21 (82% reduction in the risk of progression or death) in progression-free survival for the BRCA1/2 mutant population. Subsequent clinical trials with other PARP inhibitors, niraparibCitation67 and rucaparibCitation43, showed similar efficacy to olaparibCitation38. These results are unprecedented, considering the challenges throughout the years in treating ovarian cancers. In addition, PARP inhibitors have been well tolerated and patients have been on olaparib continuously for many years under maintenance settings.
The loss of MMR function, which results in microsatellite instabilityCitation65 (MSI), was first observed in colorectal cancer. Later, it was found in a small percentage of cancers of all types. Clinical trials have shown that cancers with MSI, regardless of histology, respond favorably to immunotherapy with pembrolizumab, a monoclonal antibody that blocks PD-1 induced immune suppressionCitation68,Citation69. Sensitivity to immunotherapy represents a breakthrough in cancer therapy, as these responses are generally durable and significantly prolong patient survival.
However, both breakthrough treatments are limited by the facts that only a small percentage of all cancers have either HR or MMR defects. The question is whether these defects can be induced to cancers without genetic mutations in HR or MMR proteins. Both HR and MMR pathways involve many proteins that coordinate the repair process and could provide targets for pharmaceutical interventions. One common enzymatic activity required for both pathways is DNA polymerization, through a polymerase. In HR, the required DNA polymerase has been suggested to be either Pol δ or Pol ε, with reported preference for Pol δCitation33. SiRNA knockdown of either protein reduces the HR efficacy by about 50%. Other polymerases, such as Pol η and Pol ξ, have been implicated by a limited number of publicationsCitation70. In MMR, a polymerase is required to refill the gap post excision and most studies suggest the role is filled by Pol δCitation71.
Zelpolib displays robust inhibition of the homologous recombination process and renders HR proficient triple negative breast cancer cells more sensitive to PARP inhibition. These results suggest possible clinical applications to widen the scope of applicability of PARP inhibitors. More broadly, the participation of Pol δ in a wide range of DNA repair pathways implies that Zelpolib will impact many DNA repair pathways in cancer cells. The disruption of these DNA repair pathways could render various cancers more responsive to advanced immune therapies available today and tomorrow.
Materials and methods
In silico screening of compound library
A model structure of the catalytic domain of human Pol δ was generated based on the structure of yeast pol δCitation72 by SWISSPDBCitation73. The primary sequences of both proteins are conserved, and the active sites structures were virtually identical between the model structure and that of yeast pol δ. The bound primer/template was kept in the complex whereas the incoming nucleotide was removed in order to identify molecules that occupy the active site. Residues within a radius of 10 Å of the ε-amino group of residue 694K were defined as the active site to construct a grid for the virtual screening. The compound library from ChemBridge (~1,000,000 compounds) from ZINC database was used for screening. Initial cutoff loosely based on Lipinski’s Rule of FiveCitation74 (xlogP<5, MW<500, H-bond donors<5, H-bond acceptors<10, and charges<3) reduced the library size to 100,000. A three-step protocol () was used with DOCK (6.4)Citation75 to complete the screening process. The most probable conformation of the compound was initially used as a rigid molecule and maximum orientation of 200 was used in the first step. The 5,000 compounds with the highest scores were selected for a second cycle of screening. In this cycle, the conformations of these compounds were regarded as flexible and the maximum number of orientations was increased to 1,000. Subsequently, the 300 compounds (supplemental movie 1) with the highest scores were selected and analyzed by Pymol. The individual structural analysis with PyMol to select compounds that could inhibit Pol δ when it is actively engaged in DNA replication. We set three key requirements for the selection process: 1) the compound can interact with the template strand, especially the nucleoside base that incoming nucleotide will pair with; 2) the compound can form meaningful hydrophobic interactions with the preceding nucleotide base of the primer, preferably through π-π electron interactions; 3) extensive interaction with Pol δ. 10 compounds satisfied our initial requirements and were purchased for initial enzymatic analysis by poly(dA)/oligo(dT) with inhibitor concentrations of 100 μM. Further assay with 10 μM concentration narrowed the choice to a single compound.
Cell culture
MCF7, BxPC-3, HCC1395, and 293T cells were purchased from American Type Culture Collection (ATCC, Manassas, VA) and maintained in media recommended by ATCC. Cisplatin resistant pancreatic cancer cell line, R-BxPC-3, was derived from parental BxPC-3 cells with sequential increases in the concentration of cisplatin with the final concentration of 1.25 μM according to protocol published previouslyCitation76,Citation77.
Protein expression and purification
Recombinant Pol δ was expressed in insect cells and purified as reported previouslyCitation18. PCNA was expressed in E coli and purified as reportedCitation18.
DNA polymerase assay
Three different assays are used to analyze DNA polymerase activities and the impact of Zelpolib. The poly(dA)/oligo(dT) assayCitation1,Citation16 was used for precise quantification as reported. Briefly,the standard reaction mixture for DNA polymerase assay contained 0.375 μM sparsely primed poly(dA)/oligo(dT) (Supertechs, MD), 150 μg/ml BSA, 7.5% glycerol, 7.5 mM MgCl2, 75 mM HEPES (pH 6.5), 0.75 μM Citation3HdTTP (100 cpm/pmol). The reaction was initiated by the addition of pol δ and PCNA. After 10 to 30 minutes, the reaction was terminated by the addition of 20 mM EDTA. Aliquots of reaction mixtures were spotted on DE81 ion exchange membranes followed by washing three times with 0.3 M ammonium formate (pH 7.8) and once with 95% ethanol. Citation3HdTMP incorporation was quantified on liquid scintillation counter. Experiment was performed in triplicates.
The primer extension assays were conducted as previously reportedCitation18. Sequences for 25-mer primer oligonucleotide and four different 40-mer oligonucleotide templates are as follows:
25-mer: 5’-GCC ACT ACA GCA CCT TGA CAG CCA G-3’
40-mer (G): 5’-TCA TCG GTC G CA TCG CTG GCT GTC AAG GTG CTG TAG TGG C-3’
40-mer (C): 5’-TCA TCG GTC G CA TCC CTG GCT GTC AAG GTG CTG TAG TGG C-3’
40-mer (A): 5’-TCA TCG GTC G CA TCA CTG GCT GTC AAG GTG CTG TAG TGG C-3’
40-mer (T): 5’-TCA TCG GTC G CA TCT CTG GCT GTC AAG GTG CTG TAG TGG C-3’
Briefly, the standard reaction mixture contained 5 μM dNTP, 5 mM MgCl2, 50 mM Tris–HCl (pH 7.5), 2 mM dithiothreitol, 100 mg/ml BSA, 50 mM NaCl, 20 nM primer/template, 50 nM PCNA, 10 nM Pol δ and variable concentrations of inhibitors. The reactions were initiated by addition of dNTPs and MgCl2. Reaction was allowed to proceed at 25°C for 2 min before addition of stopping buffer (95% formamide/25 mM EDTA). The products were separated on a 20% polyacrylamide urea gel. Products were visualized on phosphorimager and quantitated using Image-J software. For single nucleotide primer extension assayCitation18, the protocol is identical to primer extension assay except a single dNTP is used.
Cell proliferation assay-MTT method
Human cancer cells (5 × 103 – 7.5 × 103) were initially seeded in 96-well plates (Cellstar) containing 0.2 ml media per well. After 24 hrs, these cells were treated with variable concentrations of drug combinations for 48–72 hrs. Subsequently, 4,5-dimethylthiazol-2-yl-2,5-diphenyltetrazolium bromide (MTT) (20 μl of 5 mg/ml) was added to each well for 3 hrs. Formazan crystals were then dissolved in 150 μl DMSO and optical density was read at 490 nmCitation26. All experiments were performed in triplicates. Results were analyzed by GraphPad Prism. Data represent mean value ± SEM for each concentration of inhibitor combinations.
DNA fiber fluorography dual labeling
To analyze the effect of Zelpolib, DNA fiber fluorography was used with dual labeling technique as reported. Cells were seeded in 6-well plates (Cellstar) and allowed to attach for 24 hours before pulse labeled with 25 μM IdU for 20 min. Subsequently, cells were washed three times with PBS and pulse labeled with 250 μM of CldU (Sigma) for 60 min in the presence or absence of Zelpolib. Cell lysis and fiber spreading were conducted according to Schwab & NiedzwiedzCitation25. IdU and CldU were detected by primary mouse anti-BrdU antibody (BD biosciences, 1:25) and rat anti-BrdU antibody (Abcam, 1:200) and secondary fluorescent antibodies Alexa anti-mouse 594 (ThermoFisher scientific, 1:625) and Alexa anti-rat 488 (ThermoFisher scientific, 1:200). DNA fibers were imaged on fluorescence microscope and fiber lengths were measured using Image-J software. Statistics were calculated using GraphPad prism software.
Fluorescence based double strand break repair assay
The efficacy of homologous recombination was analyzed by a two-plasmids system according to published protocolCitation34,Citation35. HEK 293T cells (0.15 × 106 cells/ml) were seeded in 6-well plate allowed to recover for 24 hours before being transfected with 1.5 μg of pDRGFP and pCBASceI (1:1) plasmids using Lipofectamine 3000 (Invitrogen). After 4 hours, separate wells were treated with different concentrations of Zelpolib or DMSO alone for 44 hours. Resulting cells were washed three times in ice cold 1X PBS and trypsinized. Cellular fluorescence for GFP was measured by flow cytometry (Beckman coulter, MoFlo XDP). About 32,000 cells were analyzed per sample. Transfection efficacy was estimated with a GFP plasmid (Invitrogen) under identical conditions. The experiment was repeated three more times and statistics were calculated using GraphPad prism software.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Download Zip (54.1 MB)Acknowledgments
The authors thank Thomas Jeitner for proofreading the manuscript.
Supplementary material
Supplementary data can be accessed here
Additional information
Funding
References
- Karran P. Mechanisms of tolerance to DNA damaging therapeutic drugs. Carcinogenesis. 2001;22:1931–1937.
- Wilson PM, Danenberg PV, Johnston PG, Lenz HJ, Ladner RD. 2014. Standing the test of time: targeting thymidylate biosynthesis in cancer therapy. Nat Reviews Clin Oncology. 11:282–298. doi:10.1038/nrclinonc.2014.51.
- Cheung-Ong K, Giaever G, Nislow C. 2013. DNA-damaging agents in cancer chemotherapy: serendipity and chemical biology. Chem Biol. 20:648–659. doi:10.1016/j.chembiol.2013.04.007.
- Tiwari M. 2012. Antimetabolites: established cancer therapy. J Cancer Res Ther. 8:510–519. doi:10.4103/0973-1482.106526.
- Hagner N, Joerger M. 2010. Cancer chemotherapy: targeting folic acid synthesis. Cancer Manag Res. 2:293–301. doi:10.2147/CMR.S10043.
- Shao J, Liu X, Zhu L, Yen Y. 2013. Targeting ribonucleotide reductase for cancer therapy. Expert Opin Ther Targets. 17:1423–1437. doi:10.1517/14728222.2013.840293.
- Delgado JL, Hsieh C-M, Chan N-L, Hiasa H. 2018. Topoisomerases as anticancer targets. Biochem J. 475:373–398. doi:10.1042/BCJ20160583.
- Nitiss JL. DNA topoisomerases in cancer chemotherapy: using enzymes to generate selective DNA damage. Current Opinion Investigational Drugs (London, England: 2000). 2002;3:1512–1516.
- Nitiss JL. 2009. Targeting DNA topoisomerase II in cancer chemotherapy. Nat Reviews Canc. 9:338–350. doi:10.1038/nrc2607.
- Collins I, Weber A, Levens D. 2001. Transcriptional consequences of topoisomerase inhibition. Mol Cell Biol. 21:8437–8451. doi:10.1128/MCB.21.24.8437-8451.2001.
- Pommier Y. 2013. Drugging topoisomerases: lessons and challenges. ACS Chem Biol. 8:82–95. doi:10.1021/cb300648v.
- Doublie S, Zahn KE. 2014. Structural insights into eukaryotic DNA replication. Front Microbiol. 5:444. doi:10.3389/fmicb.2014.00547.
- Zhang D, O’Donnell M. 2016. The eukaryotic replication machine. Enzym. 39:191–229. doi:10.1016/bs.enz.2016.03.004.
- Johansson E, Macneill SA. 2010. The eukaryotic replicative DNA polymerases take shape. Trends Biochem Sci. 35:339–347. doi:10.1016/j.tibs.2010.01.004.
- Irwin JJ Using ZINC to acquire a virtual screening library. Current protocols in bioinformatics/editoral board, Andreas D Baxevanis [et al] 2008; Chapter 14:Unit 14 6.
- Zhang P, Frugulhetti I, Jiang Y, Holt GL, Condit RC, Lee MY. Expression of the catalytic subunit of human DNA polymerase delta in mammalian cells using a vaccinia virus vector system. J Biol Chem. 1995;270:7993–7998.
- Meng X, Zhou Y, Lee EYC, Lee MYWT, Frick DN. 2010. The p12 subunit of human polymerase delta modulates the rate and fidelity of DNA synthesis. Biochemistry. 49:3545–3554. doi:10.1021/bi100042b.
- Meng X, Zhou Y, Zhang S, Lee EY, Frick DN, Lee MY. 2009. DNA damage alters DNA polymerase delta to a form that exhibits increased discrimination against modified template bases and mismatched primers. Nucleic Acids Res. 37:647–657. doi:10.1093/nar/gkn1000.
- Lee M, Wang X, Zhang S, Zhang Z, Lee EYC. Regulation and modulation of human DNA polymerase delta activity and function. Genes. 2017;8(7):190.
- Wu P, Nielsen TE, Clausen MH. 2015. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol Sci. 36:422–439. doi:10.1016/j.tips.2015.04.005.
- Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott CL, Meier W, Shapira-Frommer R, Safra T, et al. 2014. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 15:852–861. doi:10.1016/S1470-2045(14)70228-1.
- George Paul A, Sharma-Walia N, Kerur N, White C, Chandran B. 2010. Piracy of prostaglandin E2/EP receptor-mediated signaling by Kaposi’s sarcoma-associated herpes virus (HHV-8) for latency gene expression: strategy of a successful pathogen. Cancer Res. 70:3697–3708. doi:10.1158/0008-5472.CAN-09-3934.
- Salic A, Mitchison TJ. 2008. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc Natl Acad Sci U S A. 105:2415–2420. doi:10.1073/pnas.0712168105.
- Buck SB, Bradford J, Gee KR, Agnew BJ, Clarke ST, Salic A. 2008. Detection of S-phase cell cycle progression using 5-ethynyl-2’-deoxyuridine incorporation with click chemistry, an alternative to using 5-bromo-2’-deoxyuridine antibodies. Biotechniques. 44:927–929. doi:10.2144/000112812.
- Schwab RA, Niedzwiedz W. Visualization of DNA replication in the vertebrate model system DT40 using the DNA fiber technique. J Visualized Experiments: JoVE. 2011;56:e3255.
- Twentyman PR, Luscombe M. A study of some variables in a tetrazolium dye (MTT) based assay for cell growth and chemosensitivity. Br J Cancer. 1987;56:279–285.
- Wong SJ, Myette MS, Wereley JP, Chitambar CR. Increased sensitivity of hydroxyurea-resistant leukemic cells to gemcitabine. Clinil Cancer Res. 1999;5:439–443.
- Kuriyama I, Mizuno T, Fukudome K, Kuramochi K, Tsubaki K, Usui T, Imamoto N, Sakaguchi K, Sugawara F, Yoshida H, et al. 2008. Effect of dehydroaltenusin-C12 derivative, a selective DNA polymerase alpha inhibitor, on DNA replication in cultured cells. Molecules. 13:2948–2961. doi:10.3390/molecules13122948.
- McBurney MW, Whitmore GF. Mechanism of growth inhibition by methotrexate. Cancer Res. 1975;35:586–590.
- Mitchell EP. Oxaliplatin with 5-FU or as a single agent in advanced/metastatic colorectal cancer. Oncology (Williston Park, NY). 2000;14:30–32.
- Brennan MJ, Vaitkevicius VK. 5-Fluorouracil in clinical cancer experience with 155 patients. Cancer Chem Rep. 1960;6:8–11.
- Gold GL, Hall TC, Shnider BJ, Selawry O, Colsky J, Owens AH, Jr., Dederick MM, Holland JF,Brindley CO, Jones R. A clinical study of 5-fluorouracil. Cancer Res. 1959;19:935–939.
- Maloisel L, Fabre F, Gangloff S. 2008. DNA polymerase delta is preferentially recruited during homologous recombination to promote heteroduplex DNA extension. Mol Cell Biol. 28:1373–1382. doi:10.1128/MCB.01651-07.
- Nakanishi K, Cavallo F, Brunet E, Jasin M. 2011. Homologous recombination assay for interstrand cross-link repair. Methods Mol Biol. 745:283–291. doi:10.1007/978-1-61779-129-1_16.
- Weinstock DM, Nakanishi K, Helgadottir HR, Jasin M. 2006. Assaying double-strand break repair pathway choice in mammalian cells using a targeted endonuclease or the RAG recombinase. Methods Enzymol. 409:524–540. doi:10.1016/S0076-6879(05)09031-2.
- Ahrabi S, Sarkar S, Pfister SX, Pirovano G, Higgins GS, Porter ACG, Humphrey TC. 2016. A role for human homologous recombination factors in suppressing microhomology-mediated end joining. Nucleic Acids Res. 44:5743–5757. doi:10.1093/nar/gkw326.
- De Picciotto N, Cacheux W, Roth A, Chappuis PO, Labidi-Galy SI. 2016. Ovarian cancer: status of homologous recombination pathway as a predictor of drug response. Crit Rev Oncol Hematol. 101:50–59. doi:10.1016/j.critrevonc.2016.02.014.
- Kim G, Ison G, McKee AE, Zhang H, Tang S, Gwise T, Sridhara R, Lee E, Tzou A, Philip R, et al. 2015. FDA approval summary: olaparib monotherapy in patients with deleterious germline BRCA-mutated advanced ovarian cancer treated with three or more lines of chemotherapy. Clinil Cancer Res. 21:4257–4261. doi:10.1158/1078-0432.CCR-15-0887.
- Meehan RS, Chen AP. 2016. New treatment option for ovarian cancer: PARP inhibitors. Gynecologic Onc Res Practice. 3:3. doi:10.1186/s40661-016-0024-7.
- Moore DC, Ringley JT, Patel J. Rucaparib: A Poly(ADP-Ribose) polymerase inhibitor for BRCA-mutated relapsed ovarian cancer. J Pharm Pract. 2017;30:897190017743131.
- Taylor KN, Eskander RN. 2017. PARP inhibitors in epithelial ovarian cancer. Recent Pat Anticancer Drug Discov. 2018;13:145–158.
- Scott LJ. 2017. Niraparib: first global approval. Drugs. 77:1029–1034. doi:10.1007/s40265-017-0752-y.
- Syed YY. 2017. Rucaparib: first global approval. Drugs. 77:585–592. doi:10.1007/s40265-017-0716-2.
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70.
- Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell. 144:646–674. doi:10.1016/j.cell.2011.02.013.
- Arora A, Scholar EM. 2005. Role of tyrosine kinase inhibitors in cancer therapy. J Pharmacol Exp Ther. 315:971–979. doi:10.1124/jpet.105.084145.
- Gotink KJ, Verheul HMW. 2010. Anti-angiogenic tyrosine kinase inhibitors: what is their mechanism of action? Angiogenesis. 13:1–14. doi:10.1007/s10456-009-9160-6.
- Todd RC, Lippard SJ. 2009. Inhibition of transcription by platinum antitumor compounds. Metallomics: Integrated Biometal Science. 1:280–291. doi:10.1039/b907567d.
- Sonohara Y, Iwai S, Kuraoka I. 2015. An in vitro method for detecting genetic toxicity based on inhibition of RNA synthesis by DNA lesions. Genes env. 37:8. doi:10.1186/s41021-015-0014-8.
- Ward J, Kapadia K, Brush E, Salhanick SD. 2013. Amatoxin poisoning: case reports and review of current therapies. J Emerg Med. 44:116–121. doi:10.1016/j.jemermed.2012.02.020.
- Allen B, Desai B, Lisenbee N. 2012. Amatoxin: A Review. ISRN Emerg Med. 2012:1–4. doi:10.5402/2012/190869.
- Boussios S, Pentheroudakis G, Katsanos K, Pavlidis N. Systemic treatment-induced gastrointestinal toxicity: incidence, clinical presentation and management. Ann gastroenterology. 2012;25:106–118.
- Damia G, Tagliabue G, Zucchetti M, Davoli E, Sessa C, Cavalli F, D’Incalci M. Activity of aphidicolin glycinate alone or in combination with cisplatin in a murine ovarian tumor resistant to cisplatin. Cancer Chemother Pharmacol. 1992;30:459–464.
- Moreland NJ, Illand M, Kim YT, Paul J, Brown R. Modulation of drug resistance mediated by loss of mismatch repair by the DNA polymerase inhibitor aphidicolin. Cancer Res. 1999;59:2102–2106.
- O’Dwyer PJ, Moyer JD, Suffness M, Harrison SD Jr., Cysyk R, Hamilton TC, Plowman J. Antitumor activity and biochemical effects of aphidicolin glycinate (NSC 303812) alone and in combination with cisplatin in vivo. Cancer Res. 1994;54:724–729.
- Sargent JM, Elgie AW, Williamson CJ, Taylor CG. Aphidicolin markedly increases the platinum sensitivity of cells from primary ovarian tumours. Br J Cancer. 1996;74:1730–1733.
- Sessa C, Zucchetti M, Davoli E, Califano R, Cavalli F, Frustaci S, Gumbrell L, Sulkes A, Winograd B, D’Incalci M. Phase I and clinical pharmacological evaluation of aphidicolin glycinate. J Natl Cancer Inst. 1991;83:1160–1164.
- Leeds JM, Mathews CK. Cell cycle-dependent effects on deoxyribonucleotide and DNA labeling by nucleoside precursors in mammalian cells. Mol Cell Biol. 1987;7:532–534.
- Leeds JM, Slabaugh MB, Mathews CK. DNA precursor pools and ribonucleotide reductase activity: distribution between the nucleus and cytoplasm of mammalian cells. Mol Cell Biol. 1985;5:3443–3450.
- Momparler RL. Biochemical pharmacology of cytosine arabinoside. Med Pediatr Oncol. 1982;10(Suppl 1):45–48.
- Perrino FW, Mekosh HL. Incorporation of cytosine arabinoside monophosphate into DNA at internucleotide linkages by human DNA polymerase alpha. J Biol Chem. 1992;267:23043–23051.
- Dietlein F, Thelen L, Reinhardt HC. 2014. Cancer-specific defects in DNA repair pathways as targets for personalized therapeutic approaches. Trends in Genetics: TIG. 30:326–339. doi:10.1016/j.tig.2014.06.003.
- Prakash R, Zhang Y, Feng W, Jasin M. 2015. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb Perspect Biol. 7:a016600. doi:10.1101/cshperspect.a016600.
- Eshleman JR, Markowitz SD. Mismatch repair defects in human carcinogenesis. Hum Mol Genet. 1996;5 :1489-1494.
- Boland CR, Goel A. 2010. Microsatellite instability in colorectal cancer. Gastroenterology. 138:2073–87 e3. doi:10.1053/j.gastro.2009.12.064.
- Konstantinopoulos PA, Ceccaldi R, Shapiro GI, D’Andrea AD. 2015. Homologous recombination deficiency: exploiting the fundamental vulnerability of ovarian cancer. Cancer Discov. 5:1137–1154. doi:10.1158/2159-8290.CD-15-0714.
- Caruso D, Papa A, Tomao S, Vici P, Panici PB, Tomao F. 2017. Niraparib in ovarian cancer: results to date and clinical potential. Ther Adv Med Oncol. 9:579–588. doi:10.1177/1758834017718775.
- Chang L, Chang M, Chang HM, Chang F. Microsatellite instability: a predictive biomarker for cancer immunotherapy. Applied Imm Chem Mol Morphology: AIMM. 2018;26:e15–e21.
- First tissue-agnostic drug approval issued. Cancer Discov. 2017;7:656.
- McVey M, Khodaverdian VY, Meyer D, Cerqueira PG, Heyer W-D. 2016. Eukaryotic DNA Polymerases in homologous recombination. Annu Rev Genet. 50:393–421. doi:10.1146/annurev-genet-120215-035243.
- Longley MJ, Pierce AJ, Modrich P. DNA polymerase delta is required for human mismatch repair in vitro. J Biol Chem. 1997;272:10917–10921.
- Swan MK, Johnson RE, Prakash L, Prakash S, Aggarwal AK. 2009. Structural basis of high-fidelity DNA synthesis by yeast DNA polymerase delta. Nat Struct Mol Biol. 16:979–986. doi:10.1038/nsmb.1663.
- Guex N, Peitsch MC. 1997. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 18:2714–2723. doi:10.1002/elps.1150181505.
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26.
- Allen WJ, Balius TE, Mukherjee S, Brozell SR, Moustakas DT, Lang PT, Case DA, Kuntz ID, Rizzo RC. 2015. DOCK 6: impact of new features and current docking performance. J Comput Chem. 36:1132–1156. doi:10.1002/jcc.23905.
- Mezencev R, Matyunina LV, Wagner GT, McDonald JF. 2016. Acquired resistance of pancreatic cancer cells to cisplatin is multifactorial with cell context-dependent involvement of resistance genes. Cancer Gene Ther. 23:446–453. doi:10.1038/cgt.2016.71.
- Coley HM. Development of drug-resistant models. Methods Mol Med. 2004;88:267–273.