
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
p53-R249S (p53-RS) is frequently detected in human hepatocellular carcinoma (HCC) that is highly associated with hepatitis B infection and aflatoxin B1 exposure. Our previous study showed that CDK4/Cyclin D1 phosphorylates p53-RS at the cancer-derived Ser249 and promotes its interaction with c-Myc in the nucleus, consequently enhancing c-Myc-dependent ribosomal biogenesis and HCC cell proliferation. Here we explored the possibility of co-targeting CDK4 and p53-RS with available small molecule inhibitors as a potential combined therapy for HCC that harbor p53-RS. Indeed, co-treatment of p53-RS-containing, but not wild-type p53 or p53-null, HCC cells with PD-0332991 (PD), a CDK4/6 inhibitor, and CP-31398 (CP), a compound that can restore the intrinsic conformation and transcriptional activity of mutant p53, drastically repressed the c-Myc activation function of p53-RS. This combination of PD with CP exhibited a synergistic effect on the inhibition of HCC cell growth in a p53-RS dependent manner, especially at a lower dose. These results suggest that co-targeting CDK4 and p53-RS can serve as a potential approach for the development of an effective therapy for HCC that harbor p53-RS.
1. Introduction
Liver cancer is the fifth most common cancer and the second deadliest cancer in the world with about 700,000 deaths per year in recent years,Citation1 which accounts for about 7% of all cancersCitation2 and over 80% of primary liver cancer.Citation3,Citation4 A recent estimate showed that the male-female ratio is 2.4 in its worldwide distribution,Citation5 especially in developing countries.Citation6,Citation7 Hepatocellular carcinoma (HCC) is a major form of liver cancer and has many known risk factors, including AFB1 exposure, chronic hepatitis B virus (HBV) and hepatitis C virus (HCV) infection, alcoholism, autoimmune liver disease, diabetes, obesity, and other metabolic diseases.Citation8
Several treatments have been used for intermediate-stage HCC treatment in clinics, such as transarterial chemoembolization (TACE), radiotherapy, locoregional therapy, and chemotherapy.Citation9 However, those with advanced-stage disease can only benefit from molecule-targeted therapy.Citation10 One example is the development of the tyrosine kinase inhibitor sorafenib for patients with HCC, who do not have an effective treatment.Citation11 Since sorafenib showed a survival advantage in patients with advanced-stage HCC, a number of new target-based drugs have been tested in first-line and second-line randomized phase III trials.Citation12 However, with the exception of regorafenib,Citation13 none has been shown to be able to prolong survival. Also, no relevant data has been produced so far in combination therapies for HCC. Therefore, there is an urgent need for developing a more effective therapy for this type of cancer.
Like other cancers, HCC provides one of the most striking examples of a mutation “fingerprint” in the human genome left by a carcinogen.Citation14–Citation16 The molecular hallmark of HCC is a mutation at codon 249 in p53, resulting in the substitution of Arg by Ser (p53-R249S, p53-RS),Citation17 which was found in more than 50% of the cases in high incidence areas associated with hepatitis B virus (HBV) infection and AFB1 exposure.Citation18 This mutation accounts for 26% of all p53 mutations described to date in HCC, but is rare (less than 2%) in other cancers.Citation19 Therefore, p53-RS is a promising target for developing an anti-HCC therapy.
Remarkably, this mutant p53 could gain its oncogenic function via a cell cycle-regulated mechanism.Citation20 Specifically, we recently showed that CDK4/Cyclin D1 can phosphorylate the HCC-derived Ser249 of p53-RS. Once phosphorylated, it binds to PIN1 that facilitates the nuclear import of this mutant. In the nucleus, phosphorylated p53-RS interacts with c-Myc and stabilizes it by preventing FBW4A-mediated ubiquitination and degradation of this oncoprotein, consequently activating c-Myc-dependent transcription in HCC cells and their proliferation.Citation21,Citation22 Amazingly, this signaling pathway has also been confirmed in some primary human HCC tissues.Citation21,Citation22 The levels of c-Myc, CDK4, Pin1, and p53-RS phosphorylation were relatively higher than that in HCC tissues without the p53-RS mutation. Also, the p53-RS-CDK4-c-Myc complex was also detected in these p53-RS-harboring HCC tissues. These results suggest that co-targeting any of the two molecules in this pathway, such as p53-RS and CDK4, might be a better approach for developing an effective anti-HCC therapy for p53-RS-containing HCCs.
Fortunately, several CDK4 inhibitors have been developed into attractive therapeutic options.Citation23 These selective CDK4/6 inhibitors include palbociclib (PD0332991, PD), abemaciclib (LY2835219), and ribociclib (LEE011).Citation24 Among them, PD combined with letrozole or fulvestrant has led to the significant improvement of the progression-free survival in stage II trials of advanced breast cancer with estrogen receptor (ER) positive and HER2-negative. This combination was recently approved by the U.S. Food and Drug Administration (FDA) for breast cancer treatment.Citation25,Citation26 Also, PD shows tolerable toxicity and is predicted to be effective in other tumor types, such as HCC that often harbors an intact Rb.Citation27
Also, small molecules have been identified to restore the wild-type conformation of mutant p53 in tumor cells.Citation28 One exemplified compound is CP-31398 (CP) that could directly interact with a mutant p53 molecule and mediate its folding into the wild-type conformation during biosynthesis.Citation28 As such, it can restore the intrinsic conformation and transcriptional activity of mutant p53 in cancer cells, causing the induction of p53 target genes, such as p21.Citation29,Citation30 Remarkably, CP inhibited tumor growth by 75% in mice that harbor human melanoma xenograft A375.S2 with a mutation at amino acid 249 of p53, but with little toxicity at the therapeutic doses in vivo.Citation29 It has been shown that CP combination with cisplatin, doxorubicin, or VP-16 has an additive effect on cell culture.Citation31 Therefore, we decided to determine if combining this compound with the CDK4 inhibitor PD as mentioned above could show a synergistic inhibition of HCC cell growth and proliferation.
As detailed below, we found that indeed combination of the mutant p53 converter compound CP with the CDK4 inhibitor PD could synergistically inhibit HCC survival and colony formation particularly at lower doses by more effectively suppressing c-Myc activity in mutant p53-, but not wild-type p53- or p53-null, HCC cells. Our results suggest that co-targeting CDK4 and p53-RS in HCC might serve as a more effective therapeutic approach for HCC that harbor this mutant p53.
2. Materials and methods
2.1. Cell lines, reagents, and antibodies
HCC cell lines including PLC/PRF/5 (p53-RS), HepG2 (wild-type p53), and Hep3B (p53-null) were kindly gifted from Dr. Tong Wu (Tulane University) and used in our previous study.Citation21 Stable Hep3B cells expressing p53-RS (Hep3B-p53-RS) were infected by lenti-virus that harbors pLentif6-p53-RS, which was purchased from Addgene created by Dr. Bernard Futscher), and then established by Blasticidin. The cells stated above were cultured in Dulbecco′s Modified Eagle′s Medium (DMEM) (GIBCO/Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) (GIBCO/Invitrogen, Carlsbad, CA) and 1% penicillin (50 U/ml)/streptomycin (0.1 mg/ml) (Invitrogen, Carlsbad, CA) at 37°C in a humidified atmosphere of 5% CO2. These cells were treated with PD (99.66% purity, dissolved in water) (Selleckchem, USA) and CP (99.10% purity, dissolved in water) (Tocris, USA), as well as PD/CP combination. Anti-c-Myc (ab32072, Abcam), anti-p53 (DO-1, FL-393 Santa Cruz Biotechnology), anti-p21 (CP74, Neomarkers), anti-β-actin (C4, Santa Cruz Biotechnology) antibodies, and HRP-labeled goat anti-mouse IgG (Santa Cruz Biotechnology) were commercially purchased.
2.2. Cell colony formation assay
HCC cancer cells as mentioned above were seeded into the flat-bottomed six-well culture plates (Wuxi NEST Biotechnology Co., Ltd, Wuxi, Jiangsu, China) in triplicate in 2 ml of medium containing 10% FBS (PLC/PRF/5: 1000 cells per well; HepG2: 2000 cells per well; Hep3B: 1000 cells per well; Hep3B-p53-RS: 1500 cells per well). After overnight incubation, culture media were replaced with a fresh one with 10% FBS or same medium containing PD, CP, or their mixture as shown in in a humidified atmosphere of 5% CO2 at 37°C for additional 2 weeks. Then, the cells were washed one time with cold PBS, and visible cell colonies were stained and fixed with a solution containing 0.5% crystal violet (Sigma-Aldrich, Inc) in methanol for an hour, followed by washing with tap water until excess dye removed. HCC colonies were counted by imageJ software that is designed by the National Institutes of Health.
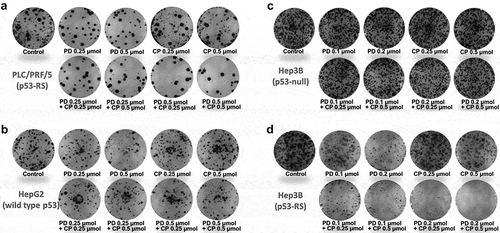
Figure 1. Combination of PD with CP effectively inhibits colony formation of HCC cells.
The indicated cell lines were seeded into six-well culture plates at a desired density. Then, the cells were treated with PD, CP or their concurrent combination the next day. After 2 weeks, the plates were stained for the formation of cell colonies with crystal violet dye in methanol. The photograph of one well in a representative experiment is shown for each treatment in the colony formation assay. (A) PLC/PRF/5 cells (p53-RS); (B) HepG2 cells (wild-type p53); (C) Hep3B cells (p53 null); (D) Hep3B cells (p53-RS).
2.3. Cell proliferation inhibition assay
HCC cells were seeded into the 96-well culture plates (Wuxi NEST Biotechnology Co., Ltd, Wuxi, Jiangsu, China) in 0.2 ml of medium containing 10% FBS (PLC/PRF/5: 2500 cells per well; HepG2: 3000 cells per well; Hep3B: 2000 cells per well; Hep3B-p53-RS: 2500 cells per well) and grown over night. Then, cells were cultured in IncuCyte® S3 Live-Cell Analysis System (Essen BioScience, MI USA) for additional 5 days with or without various treatment groups (five wells per group), and cell growth was monitored by IncuCyte every 12 h. Data analysis was conducted using the IncuCyte® Cell Count Proliferation Assay methodology. Cell proliferation inhibition rate = (1-Cell confluence Treatment group/Cell confluence Control group) × 100%. Synergetic effect of the combination of PD and CP was analyzed by Jin′s formula.Citation32–Citation34 The formula is where
and
are the average effects (inhibition rate) of the combination treatment, PD only, and CP only, respectively. In this method, Q ≥ 1.15 shows synergism; 0.85 ≤ Q < 1.15 shows additive effects, and Q < 0.85 shows antagonism.
2.4. Western blot analysis
After treatment for 18 h under desired conditions, HCC cells were harvested and then were washed once with ice-cold PBS and protein was obtained using 80–100 μl lysis buffer consisting of 50 mM Tris/HCl (pH7.5), 0.5% Nonidet P-40 (NP-40), 1 mM EDTA, 150 mM NaCl, 1 mM dithiothreitol (DTT), 0.2 mM phenylmethylsulfonyl fluoride (PMSF), 10 μM pepstatin A and 1 mM leupeptin for 30 min on ice, and microcentrifuged at 13 × 103 rpm for at least 15 min. Supernatants were collected for protein concentration determination using the Quick Start™ Bradford Protein Assay Kit (Bio-Rad Laboratories, Inc). Extracted protein (30 μg) with 5× loading buffer containing 2-mercaptoethanol were separated by size on a gradient SDS gel and transferred to polyvinylidene fluoride (PVDF) membranes (Bio-Rad Laboratories, Inc). Membranes were blocked for half an hour in blocking buffer (1× TBST with 5% free-fat dry milk) at room temperature and placed in primary antibodies overnight at 4°C. The primary antibodies for c-Myc, p53, p21, and β-actin were reacted. After that, the membranes were washed three times in 1× TBST and were incubated with corresponding HRP-conjugated secondary antibody for an hour at room temperature. The blots were washed thrice with 1× TBST and the signals were detected and analyzed with the Clarity Western ECL Substrate (Bio-Rad Laboratories, Inc) and ChemiDoc XRS+ system (Bio-Rad Laboratories, Inc).
2.5. RNA isolation and quantitative RT-PCR (qRT-PCR) analysis
Total RNA was isolated from HCC cells with Trizol agent (Invitrogen, Carlsbad, CA, USA) following the instructions of the manufacturer. Three hundred nanograms of total RNA were used as templates for reverse transcription using poly-(T)20 primers and M-MLV reverse transcriptase (Promega, Madison, WI, USA). qRT-PCR reactions used SYBR green mix with the Real-Time PCR Detection systems (Bio-Rad, Hercules, CA, USA) using a GAPDH probe as an internal control, whose expression remains relatively consistent across the test samples. All reactions were carried out in triplicate. The relative quantitation of gene expression in terms of fold change was analyzed by using the comparative ΔΔCt method. Relative expression levels of the target genes in each treatment group were normalized to the endogenous control. The following oligonucleotide primers for qRT-PCR were used: 5ʹ-TCACCCCTCTGCCATTAAAGG-3ʹ and 5ʹ-AGCAGTGTATTCCCCAGGCC-3ʹ for human E2F2; 5ʹ-CCTTCGATAGCTCAGCTGGTAG-3ʹ and 5ʹ-GGAATCGGAACCAGCGACCTAAG-3ʹ for human tRNA-Tyr; 5ʹ-TAATACGACTCACTA- TAGGG-3ʹ and 5ʹ-TAGAAGGCACAGTCGAGG-3ʹ for human CDK4; 5ʹ-TTGGCCGAGCGGTCTAT-3ʹ and 5ʹ-ATTCGAACCCTCGCATCT-3ʹ for human tRNA-Leu; 5ʹ-GAGTCAACGGATTTGGTCGT-3ʹ and 5ʹ-GACAAGCTTCCCGTTCTCAG-3ʹ for human GAPDH.
2.6. Statistical analysis
All of the statistical analyses were performed using the SPSS 13.0 software. The quantitative data were obtained from at least three independent experiments and expressed as means ± standard deviation (SD). Statistical comparisons were performed using Student’s t-tests (comparisons between two groups) and ANOVA (statistical analysis for three or more groups). P < .05 was considered to be a statistically significant difference.
3. Results
3.1. Combination of PD with CP more effectively inhibits p53-RS-dependent HCC cell growth
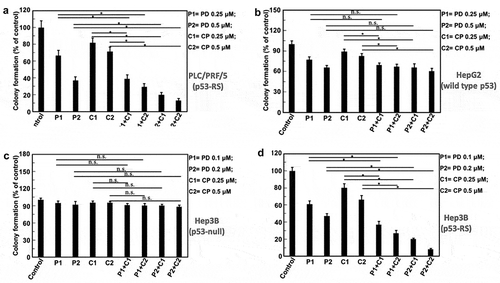
We first determined the effects of PD or CP alone and their combination treatment on the proliferation of human PLC/PRF/5 (p53-RS), HepG2 (wild type p53), and Hep3B (p53-null) HCC cells in colony formation assays. PD and CP at a lower dose (0.25 μM) alone either moderately suppressed or had minimal effect on colony formation of any of these HCC cells (– and ). However, the combination of the two drugs drastically reduced the colony numbers of PLC/PRF/5 cells in a dose-dependent fashion ( and ). This suppressive effect was not seen in either HepG2 ( and ) or Hep3B cells ( and ), compared to control or single-agent treatments. These results suggest that PD and CP might cooperate with each other to inhibit HCC cell growth by targeting CDK4/6 and mutant p53 (p53-RS), respectively, as either wild-type p53-containing or p53 null HCC cells did not respond to their combination treatment ( and ,). This statement was further supported by the following experiment. Remarkably, when p53-RS was introduced into p53-null Hep3B cells, the p53-RS-containing Hep3B cells became significantly more sensitive to the combined treatment of PD with CP than were their parental cells, compared to either PD or CP alone ( and ), which was consistent with that shown in PLC/PRF/5 cells ( and ). Thus, these results demonstrate that the combination of PD with CP is much more effective than either of the single agent in inhibiting the colony formation and growth of HCC cells in a p53-RS dependent manner (P < .05, ).
Figure 2. The quantification of the results from the HCC colony formation assays.
The indicated cells were treated with PD and CP either alone or in combination treatment as described in legends. Values represent the mean±SD (n = 3, * P < .05; n.s. P> .05). (A) PLC/PRF/5 cells (p53-RS); (B) HepG2 cells (wild-type p53); (C) Hep3B cells (p53 null); (D) Hep3B cells (p53-RS).
3.2. PD and CP exert a strong synergistic effect on cell proliferation
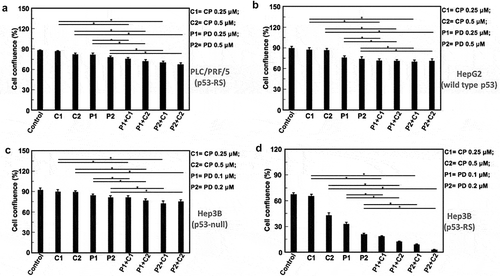
Next, we determined if the PD/CP combination might have a synergistic effect on HCC cell proliferation by employing the same set of HCC cells as mentioned above. To do so, cells were first treated for consecutive 5 days with these two drugs, either alone or in combination as indicated in . The cell confluence after treatment with different combinations was compared with that for each individual compound alone as shown in . Notably, cell proliferation assays showed that the HCC cell viability in the p53-RS-containing Hep3B group was more significantly decreased than any of the other treatment groups including PLC/PRF/5, HepG2, and Hep3B cells after 5-days treatment (P < .05, and ). Using the Jin’s formula, we found that PD and CP in combination yielded synergism across a wide range of concentrations in p53-RS Hep3B cells. Even though the suppression of PLC/PRF/5 cell proliferation by the PD/CP combination ( and ) was not as apparent as that of p53-RS Hep3B cells ( and ), this combination did significantly impair their cell viability with a synergistic effect on PLC/PRF/5 cells (Q ≥ 1.15) (). By contrast, four combination treatments to HepG2 and Hep3B cells only showed additive effects (0.85 ≤ Q < 1.15) (). Consistently, the PD/CP combination treatment with different doses also more synergistically suppressed the proliferation (Q ≥ 1.15) of p53-RS Hep3B cells (). These results again demonstrate that the synergistic suppression of HCC cell growth by these two compounds is dependent on p53-RS. Interestingly, the largest degree of synergism was observed when PD and CP were administered at a lower dose (PD: 0.25 μM; CP: 0.25 μM in PLC/PRF/5 cells; PD: 0.1 μM; CP: 0.25 μM in p53-RS Hep3B cell lines). As such, this combination treatment was utilized for the following experiments for further investigation of their combined effects and mechanisms of action.
Table 1. Synergetic effect of PD and CP combination on HCC growth as analyzed by using Jin′s formula*.
Figure 3. Analysis of cell survival using the IncuCyte ZOOM system.
The indicated cell lines were seeded in 96-well plates at a given density. The growth of the human HCC cells treated with PD and CP alone or in combination was measured for 5 days using the IncuCyte Live Cell Imaging system that monitors cell confluency in real time. The growth curves for these cells are plotted as the mean±SD confluence from n = 5 wells in an experiment representative of the three independent experiments. (A) PLC/PRF/5 cells (p53-RS); (B) HepG2 cells (wild-type p53); (C) Hep3B cells (p53 null); (D) Hep3B cells (p53-RS).
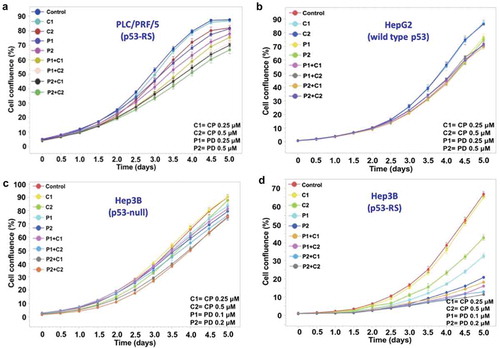
Figure 4. Combination of PD with CP more significantly inhibits proliferation and survival of HCC cells that harbor p53-RS.
Cell growth of HCC cells treated with PD or CP alone or in combination was monitored as indicated in legends with the IncuCyte Live Cell Imaging system. Mean±SD confluence at the fifth day in the analysis of cell confluence (n = 5, * P < .05). (A) PLC/PRF/5 cells (p53-RS); (B) HepG2 cells (wild-type p53); (C) Hep3B cells (p53 null); (D) Hep3B cells (p53-RS).
3.3. CP synergizes the potential of PD in c-Myc downregulation
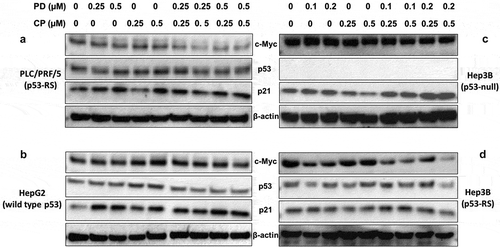
c-Myc is a nuclear transcription factor that is critical to the proliferation and renewal of stem cells,Citation35,Citation36 as well as to the survival of cancer stem cellsCitation37,Citation38 by activating gene expression that is essential to ribosomal biogenesis and protein synthesis.Citation39 It is reported that c-Myc is highly expressed in nearly 80% of human cancers.Citation40 Our recent study showed that p53-RS acquires its GOF by stabilizing and activating c-Myc in response to CDK4 activity in HCC cells.Citation21 Hence, we wanted to check if the aforementioned synergistic effect of PD and CP on HCC cell survival and colony formation might be due to the down-regulation of c-Myc by the CDK4 inhibitor PD and activation of mutant p53-RS by CP. To gain insight into the mechanism of the observed synergism, HCC cells with p53-RS or wild-type p53 or p53 null were treated with different doses of PD or CP alone or their combination for 18 h, and then cells were collected for Western blot analysis. As shown in and Supplementary Figure 1, PD by itself modestly reduced c-Myc levels, while CP alone increased p53 and p21 levels in a dose-dependent manner in PLC/PRF/5 cells. However, the level of c-Myc was more dramatically decreased when combining these two compounds at the lower doses (PD: 0.25 μM; CP: 0.25 μM). Also, this combination at this dose induced p53 and p21 levels in PLC/PRF/5 cells. Interestingly, co-treatment of wild-type p53 HepG2 or p53 null Hep3B cells with the same combination of the compounds did not show any apparent effect on c-Myc and p53 protein levels, but with the induction of p21 (). The induction of p21 in these HCC cells might be due to the activation of p73 or p63 by these agents independent of p53.Citation41,Citation42 When p53-RS Hep3B cell lines were used, the effects were similar to that of PLC/PRF/5 cells (), further demonstrating that this synergistic effect is specific to p53-RS. Taken together, these results show that simultaneous inhibition of CDK4/CyclinD1 by PD and function rescue of mutant p53 by CP can synergistically decrease the c-Myc level and activate p53 in HCC cells at the lower doses.
Figure 5. Combination with low doses of PD and CP shows a significant synergy on c-Myc reduction with elevated p53 activity in a p53-RS-dependent manner.
The indicated cell lines were plated in 6-well culture plates and treated with PD or CP alone or in combination at the indicated doses for 18 h. The cells were then harvested for preparation of whole cell lysates and subsequent Western blot analysis with antibodies for c-Myc, p53, and p21, respectively. Protein (30 μg) was loaded in each lane, and an anti-β-actin antibody was used to be a loading control. (A) PLC/PRF/5 cells (p53-RS); (B) HepG2 cells (wild-type p53); (C) Hep3B cells (p53 null); (D) Hep3B cells (p53-RS).
3.4. PD and CP synergistically inhibit c-Myc activity
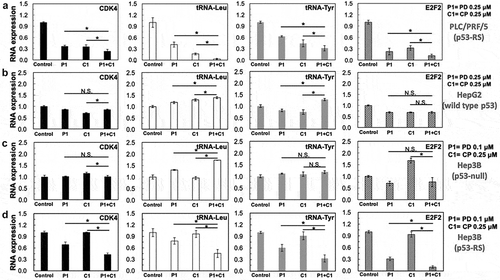
We previously showed that p53-RS after being phosphorylated by CDK4/Cyclin D1 can enhance c-Myc transcriptional activity and boost up c-Myc-driven ribosomal biogenesis in HCC cells.Citation21 Next, we decided to determine if PD and CP can synergistically inhibit the expression of selected c-Myc target genes (CDK4, tRNA-Leu, tRNA-Tyr, and E2F2) by qPCR analysis of RNAs isolated from HCC cell lines with expressed p53-RS, wild-type p53 and p53 null. Indeed, the expression of genes stated above was decreased in PLC/PRF/5 and p53-RS Hep3B after PD/CP combination treatment compared to the single treatment either PD or CP alone (). Statistically significant differences (P < .05) between combination treatment and drugs alone were observed for CDK4, tRNA-Leu, tRNA-Tyr, and E2F2 according to Student’s t-tests (). By contrast, these inhibitory effects were not observed in wild-type p53-containing HepG2 or p53 null Hep3B cells regardless of a single or combination treatment or not (). Of note, the mRNA expression of some c-Myc target genes was increased significantly (e.g., tRNA-Leu or tRNA-Tyr) in these HCC cells with unknown mechanisms (). Statistical analysis using ANOVA model showed that the mRNA level alterations of c-Myc target genes in Hep3B cells were opposite to that in p53-RS Hep3B cells (), suggesting that the reduction of c-Myc target genes’ expression by PD/CP combination treatment is likely to be p53-RS-dependent. Also, these results demonstrate that synergistic inhibitory effect of PD with CP on HCC survival and colony formation is at least in part attributed to the suppression of c-Myc transcription activity and the consequent inhibition of its target gene expression, such as CDK4, tRNA-Leu, tRNA-Tyr, and E2F2 (Supplementary Figure 2).
Figure 6. Combination of PD with CP more significantly suppresses the expression of c-Myc target genes at RNA levels.
Cells as indicated below were treated with either PD or CP alone or in combination for 16 h and harvested for RNA extraction and q-PCR analyses for specific c-Myc target genes as indicated in the figure. GAPDH was used as negative control. Three independent experiments were performed in duplicates. Differences between the combination treatment and PD or CP alone for each target were analyzed using either Student’s t-tests (comparisons between two groups) or ANOVA (statistical analysis for 3 groups) (n = 3, * P < .05; n.s. P> .05). (A) PLC/PRF/5 cells (p53-RS); (B) HepG2 cells (wild-type p53); (C) Hep3B cells (p53 null); (D) Hep3B cells (p53-RS).
4. Discussion
Cancer cases increased by 33% between 2005 and 2015 and it is expected to increase to 22 million in the next 20 years.Citation43 As the most common primary malignant tumor in the liver, HCC is the fifth most common cancer in men, worldwide, and seventh in women.Citation44 It is the second leading cause of cancer-related mortality with over half a million of new cases diagnosed annually in the world and it accounts for nearly 70% of cancer deaths in part of developing countries.Citation44 Therefore, there is an urgent need for effective therapies against this type of cancer, particularly the malignant ones. As part of the effort in addressing this issue, we tested if the combination of a CDK4 inhibitor PD with a mutant p53 converter compound CP could serve as a possible combinatory treatment for human HCCs that harbor p53-RS.
This combination makes sense for the following reasons. First, HCCs in Asia and Africa are often highly associated with HBV infection and AFB1 exposure.Citation45 These two risk factors are highly related to the mutation of p53 with substitution of Arg249 with Ser and often accompanied by activated CDK4/Cyclin D1 and c-Myc.Citation21,Citation22 Also, our recent study unveiled that CDK4/Cyclin D1 can phosphorylate Ser249 of p53-RS and this phosphorylation leads to its nuclear import mediated by PIN1. As a result, phosphorylated p53-RS binds to c-Myc and stabilizes its protein stability by inhibiting FBW7A ubiquitin ligase activity, consequently activating the c-Myc transcription pathway and promoting HCC cell growth and survival. It is through this CDK4/Cyclin D1-c-Myc pathway that p53-RS acquires its new GOF critically important for HCC cell proliferation and growth. Finally, there are available CDK4 and mutant p53 inhibitors, which are either in clinic use for breast cancer, such as PD,Citation46 or on clinical trials, such as CP analogs,Citation47 for AML. Therefore, co-targeting CDK4 and mutant p53 could be an effective approach for the development of anti-HCC therapy specifically for the cancers that harbor p53-RS. The availability of these two compounds that have been approved to be safe in the clinic made our preliminary testing in cultured HCC cells possible.
Indeed, our results as reported here simply support this idea with the following lines of evidence. First, we showed that PD/CP combination treatment leads to more significant inhibition on colony formation and cell proliferation or survival of HCC cells that contain endogenous p53-RS (PLC/PRF/5 cells) or exogenous p53-RS (p53-RS Hep3B cells), but not of HCC cells (HepG2 or Hep3B) that harbor either wild-type p53 or no p53 (–). Also, this combination displayed a synergistic effect on the former two cell lines, but not the latter two cell lines (). Mechanistically, we found that PD and CP combination leads to a drastic reduction of c-Myc level and activity as well as induction of wild-type p53 activity as represented by p21 induction, particularly at their low doses ( and ). However, the changes of p53, p21, and PUMA levels were not in a dose-dependent manner in human HCCs that harbor p53-RS, and other mechanisms underlying the synergistic effect on cell proliferation may be involved in PD/CP combination treatment at their higher doses in addition to the reduction of c-Myc level and activity by this combination. Moreover, this synergistic effect was only observed in p53-RS-containing PLC/PRF/5 or Hep3B cells, but not in wild-type p53-containing or p53 null HCC cells ( and ). Hence, in light of these results, we believe that co-targeting CDK4 and p53-RS could be an effective approach for the development of a future anti-HCC therapy against those HCCs that harbor p53-RS (). Since both of the small molecules have been either used or on trails in the clinic, we hope that these results can offer useful information for clinical oncologists to design their clinical trials for patients with HBV-positive and p53-RS-positive HCC. In the future, we will perform more studies using future available HCC cells or hepatomas that harbor p53-R249S to further investigate if co-targeting this mutant p53 and CDK4 could present a more effective therapy for HCCs that harbor this mutant p53. However, in consideration of the different impacts of CP+PD on cell proliferation of PLC/PRF/5 and Hep3B (R249S) cells, we think that further research using experimental animal models in vivo is also warranted. Moreover, further analyses using RNA seq, proteomics, and epigenetic tools would allow us to mechanistically dissect and to provide molecule insights into the different responses of these two cell lines to this combination of the two drugs. These lines of information would be more instrumental for the future design of a combination therapy with these compounds for HCCs that harbor p53-R249S
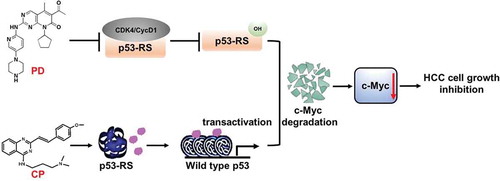
Figure 7. The model for the mechanisms underlying the p53-RS-dependent synergistic antitumor effect of PD and CP in combination.
As a CDK4 inhibitor, PD inhibits the phosphorylation of p53-RS by CDK4/Cyclin D1 as previously shown.Citation21 In doing so, this inhibitor can overcome the activation of c-Mcy by p53-RS via promoting its degradation. Also, the mutant p53 converter compound CP rescues native conformation and transcriptional transactivation activity of p53-RS. The sum outcome of the co-treatment of p53-RS-bearing HCC cells with the two compounds is the more significant reduction of c-Myc level and activity, consequently suppressing HCC cell proliferation and growth in a p53-RS-dependent manner.
Conflict of interests
The authors declare that they have no conflict of interest.
Supplemental Material
Download MS Word (235 KB)Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- The Cancer Genome Atlas Research Network. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. 2017;169:1327–1341.
- Aghemo A, Colombo M. Hepatocellular carcinoma in chronic hepatitis C: from bench to bedside. Semin Immunopathol. 2013;35:111–120.
- Sangiovanni A, Del Ninno E, Fasani P, De Fazio C, Ronchi G, Romeo R, Morabito A, De Franchis R, Colombo M. Increased survival of cirrhotic patients with a hepatocellular carcinoma detected during surveillance. Gastroenterol. 2004;126:1005–1014.
- Cheng P, Cheng Y, Su MX, Li D, Zhao GZ, Gao H, Li Y, Zhu JY, Li H, Zhang T. Bicluster and pathway enrichment analysis of HCV-induced cirrhosis and hepatocellular carcinoma. Asian Pac J Canc Prev. 2012;13:3741–3745.
- Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108.
- Mori M, Hara M, Wada I, Hara T, Yamamoto K, Honda M, Naramoto J. Prospective study of hepatitis B and C viral infections, cigarette smoking, alcohol consumption, and other factors associated with hepatocellular carcinoma risk in Japan. Am J Epidemiol. 2000;151:131–139.
- Levrero M. Viral hepatitis and liver cancer: the case of hepatitis C. Oncogene. 2006;25:3834–3847.
- Yang JD, Roberts LR. Hepatocellular carcinoma: a global view. Nat Rev Gastroenterol Hepatol. 2010;7:448–458.
- Dutta R, Mahato RI. Recent advances in hepatocellular carcinoma therapy. Pharmacol Ther. 2017;173:106–117.
- Llovet JM, Montal R, Sia D, Finn RS. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat Rev Clin Oncol. 2018;15:599–616.
- Llovet JM, Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology. 2008;48:1312–1327.
- Terashima T, Yamashita T, Toyama T, Arai K, Kawaguchi K, Kitamura K, Yamashita T, Sakai Y, Mizukoshi E, Honda M, et al. 2018. Surrogacy of time to progression for overall survival in advanced hepatocellular carcinoma treated with systemic therapy: a systematic review and meta-analysis of randomized controlled trials. Liver Cancer. DOI:10.1159/000489505
- Bruix J, Qin S, Merle P, Granito A, Huang YH, Bodoky G, Pracht M, Yokosuka O, Rosmorduc O, Breder V, et al. Resource Investigators. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389:56–66.
- Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–799.
- Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–428.
- Ozen C, Yildiz G, Dagcan AT, Cevik D, Ors A, Keles U, Topel H, Ozturk M. Genetics and epigenetics of liver cancer. New Biotechnol. 2013;30:381–384.
- Gouas D, Shi H, Hainaut P. The aflatoxin-induced TP53 mutation at codon 249 (R249S): biomarker of exposure, early detection and target for therapy. Cancer Lett. 2009;286:29–37.
- Szymańska K, Hainaut P. TP53 and mutations in human cancer. Acta Biochimica Polonica. 2003;50:231–238.
- Szymańska K, Lesi OA, Kirk GD, Sam O, Taniere P, Scoazec JY, Mendy M, Friesen MD, Whittle H, Montesano R, et al. Ser-249TP53 mutation in tumour and plasma DNA of hepatocellular carcinoma patients from a high incidence area in the Gambia, West Africa. Int J Cancer. 2004;110:374–379.
- Di Agostino S, Strano S, Emiliozzi V, Zerbini V, Mottolese M, Sacchi A, Blandino G, Piaggio G. Gain of function of mutant p53: the mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell. 2006;10:191–202.
- Liao P, Zeng SX, Zhou X, Chen TJ, Zhou F, Cao B, Jung JH, Del Sal G, Luo SW, Lu H. Mutant p53 gains its function via c-Myc activation upon CDK4 phosphorylation at serine 249 and consequent PIN1 binding. Mol Cell. 2017;68:1134–1146.
- Wang H, Liao P, Zeng SX, Lu H. 2019. It takes a team: a gain-of-function story of p53-R249S. J Mol Cell Biol. 2019;11:277–283.
- Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015;14:130–146.
- Sherr CJ, Beach D, Shapiro GI. Targeting CDK4 and CDK6: from discovery to therapy. Cancer Discov. 2016;6:353–367.
- Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, Ettl J, Patel R, Pinter T, Schmidt M, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 2015;16:25–35.
- Turner NC, Ro J, André F, Loi S, Verma S, Iwata H, Harbeck N, Loibl S, Huang Bartlett C, Zhang K, et al. Palbociclib in hormone-receptor-positive advanced breast cancer. N Engl J Med. 2015;373:209–219.
- Bollard J, Miguela V, Ruiz de Galarreta M, Venkatesh A, Bian CB, Roberto MP, Tovar V, Sia D, Molina-Sánchez P, Nguyen CB, et al. Palbociclib (PD-0332991), a selective CDK4/6 inhibitor, restricts tumour growth in preclinical models of hepatocellular carcinoma. Gut. 2017;66:1286–1296.
- Rippin TM, Bykov VJ, Freund SM, Selivanova G, Wiman KG, Fersht AR. Characterization of the p53-rescue drug cp-31398 in vitro and in living cells. Oncogene. 2002;21:2119–2129.
- Foster BA, Coffey HA, Morin MJ, Rastinejad F. Pharmacological rescue of mutant p53 conformation and function. Science. 1999;286:2507–2510.
- Demma MJ, Wong S, Maxwell E, Dasmahapatra B. Cp-31398 restores DNA-binding activity to mutant p53 in vitro but does not affect p53 homologs p63 and p73. J Biol Chem. 2004;279:45887–45896.
- Takimoto R, Wang W, Dicker DT, Rastinejad F, Lyssikatos J, El-Deiry WS. The mutant p53-conformation modifying drug, CP-31398, can induce apoptosis of human cancer cells and can stabilize wild-type p53 protein. Cancer Biol Ther. 2002;1:47–55.
- Zhu H, Huang M, Ren D, He J, Zhao F, Yi C, Huang Y. 2013. The synergistic effects of low dose fluorouracil and TRAIL on TRAIL-resistant human gastric adenocarcinoma AGS cells. Biomed Res Int. 2013:293874. doi:10.1155/2013/293874.
- Wu J, Li X, Fang H, Yi Y, Chen D, Long Y, Gao X, Wei X, Chen CY. 2016. Investigation of synergistic mechanism and identification of interaction site of aldose reductase with the combination of gigantol and syringic acid for prevention of diabetic cataract. BMC Complement Altern Med. 16:286. doi:10.1186/s12906-016-1251-5.
- Liu Y, Maccarini P, Palmer GM, Etienne W, Zhao Y, Lee CT, Ma X, Inman BA, Vo-Dinh T. 2017. Synergistic immuno photothermal nanotherapy (SYMPHONY) for the treatment of unresectable and metastatic cancers. Sci Rep. 7:8606. doi:10.1038/s41598-017-09116-1.
- Bouchard C, Staller P, Eilers M. Control of cell proliferation by Myc. Trends Cell Biol. 1998;8:202–206.
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676.
- Gordan JD, Thompson CB, Simon MC. HIF and c-Myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell. 2007;12:108–113.
- Kim J, Woo AJ, Chu J, Snow JW, Fujiwara Y, Kim CG, Cantor AB, Orkin SH. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell. 2010;143:313–324.
- Grandori C, Gomez-Roman N, Felton-Edkins ZA, Ngouenet C, Galloway DA, Eisenman RN, White RJ. c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat Cell Biol. 2005;7:311–318.
- Dang CV. MYC on the path to cancer. Cell. 2012;149:22–35.
- Lee CW, La Thangue NB. Promoter specificity and stability control of the p53-related protein p73. Oncogene. 1999;18:4171–4181.
- Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, Dötsch V, Andrews NC, Caput D, McKeon F. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell. 1998;2:305–316.
- Fitzmaurice C. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: a systematic analysis for the global burden of disease study. JAMA Oncol. 2017;3:524–548.
- Mittal S, El-Serag HB. Epidemiology of HCC: consider the population. J Clin Gastroenterol. 2013;47:S2–S6.
- El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology. 2012;142:1264–1273.
- Murphy CG, Dickler MN. The role of CDK4/6 inhibition in breast cancer. Oncologist. 2015;20:483–490.
- Bykov VJ, Wiman KG. Mutant p53 reactivation by small molecules makes its way to the clinic. FEBS Lett. 2014;588:2622–2627.