
ABSTRACT
The five-year survival rate for pancreatic ductal adenocarcinoma (PDAC) has remained a dismal 9% for approximately 40 years with an urgent need for novel therapeutic interventions. ONC201 is the founding member of the imipridone class, comprised of orally bioavailable small molecules that have shown efficacy in multiple tumor types both in animal models and in Phase I/II clinical trials. ONC201 is a potent inducer of the tumor necrosis factor related apoptosis inducing ligand (TRAIL) pathway. TRAIL is an innate immune mechanism which induces programmed cell death of cancer cells. We observed that PDAC cells upregulated ATF4, CHOP, and DR5 after treatment with ONC201. This occurred in cell lines that are susceptible to ONC201-induced apoptosis and in ones that are not. In response to ONC201, PDAC cells downregulated anti-apoptotic proteins including c-FLIP, BclXL, XIAP, cIAP1, and survivin. We hypothesized that TRAIL receptor agonists might induce selective, synergistic apoptosis in pancreatic cancer cell lines treated with ONC201. We screened 7 pancreatic cancer cell lines and found synergy with ONC201 and rhTRAIL or the novel TRAIL receptor agonist TLY012 in 6 of the 7 cell lines tested. In vivo experiments using BxPC3 and HPAFII xenograft models showed that the combination of ONC201 plus TLY012 significantly delays tumor growth as compared to controls. Immunohistochemical analysis of the tumors after three doses of the combination showed significantly increased cleavage of caspase 3 in vivo as compared to controls. Taken together, the preclinical efficacy of ONC201 and TLY012 represents a novel therapeutic option for further testing in pancreatic cancer patients. This combination showed marked efficacy in tumor cells that are both sensitive and resistant to the pro-apoptotic effects of ONC201, providing rationale to further investigate the combination of ONC201 plus TLY012 in patients with pancreatic cancer.
Introduction
The five-year survival rate for pancreatic cancer has remained very low at 9%, across all Surveillance, Epidemiology and End Results (SEER) stages combined, for nearly the past 40 years.Citation1 This highlights the urgent need for novel therapeutic options. One such option is ONC201, a novel TNF-Related Apoptosis Inducing Ligand (TRAIL)-inducing compound that has shown marked efficacy in pre-clinical and clinical trials across various tumor typesCitation2 Though ONC201 has yet to be clinically evaluated in the context of pancreatic cancer, there is significant preclinical data from our lab showing efficacy in pancreatic cancer xenograft models.Citation3
TRAIL is an innate immune ligand that induces selective apoptosis in cancer cells through binding to Death Receptor 4 (DR4) and Death Receptor 5 (DR5).Citation4,Citation5 Given this impressive phenotype, much excitement was generated regarding the therapeutic potential of rhTRAIL in the clinic. However, due to poor pharmacokinetics, low bioavailability, and poor clinical activity as monotherapy in common tumor types, efforts to develop short-lived rhTRAIL have not been successful. This lack of success ultimately led to a search for small molecules and TRAIL receptor agonists that could induce apoptotic cell death via the TRAIL pathway.Citation6 This led to the eventual discovery of ONC201/TIC10.
ONC201 (originally named TIC10 for TRAIL-inducing compound 10) is a novel, first-in-class, orally bioavailable small molecule that was identified in a phenotypic reporter screen for its ability to induce the TRAIL pathway in a p53-independent manner.Citation7 It is currently being tested in approximately 20 clinical trials as monotherapy or in combination with other chemotherapies and is well-tolerated in humans. It has shown potent efficacy in subsets of patients with glioblastoma.Citation8 ONC201 can selectively induce apoptosis in cancer cells by blocking the phosphorylation of Akt and ERK, downstream effectors of the canonical MAPK growth pathway. Blocking the phosphorylation of Akt and ERK prevents the phosphorylation of FOXO3a. Thus, FOXO3a can translocate to the nucleus, and this combined with activation of the integrated stress response, upregulates ATF4, TRAIL, and DR5.Citation7 In addition to this novel mechanism, ONC201 has a promising pharmacokinetic/pharmacodynamic split; the half-life of ONC201 is 6–11 hours, but its intracellular effects can be observed up to a week later. This is a helpful property of the drug for combination therapies, as the short half-life prevents drug-drug interactions.Citation2,Citation9
The clinical failure of rhTRAIL also set off a search for TRAIL mimetics – ligands that were modified to increase the short half-life of rhTRAIL and address some of its other biochemical shortcomings. One such TRAIL mimetic is TLY012 that can target both DR4 and DR5. This is in contrast to other approaches, like AMG655, which solely target DR5.Citation10 Chae et al.Citation11 previously established that TLY012 has a markedly improved PK and PD profile compared to rhTRAIL in mouse models in vivo. They also showed that TLY012 induces a more potent, anti-tumor effect in colorectal cancer mouse models in vivo, suggesting that TLY012 has promising anti-cancer prospects.Citation11 Further research also demonstrated that TLY012 has marked activity against fibrotic cells, which are known to upregulate DR5.Citation12 Thus, TLY012 is entering clinical trials for pancreatitis and other fibrotic diseases. TLY012 also has good prospects as an anti-cancer agent, especially for fibrosis-driven cancers such as pancreatic cancer.
Pancreatic cancer cells are notoriously resistant to extrinsic TRAIL-induced apoptosis and all pancreatic cancer cells are known to undergo type II extrinsic apoptosis.Citation13,Citation14 Type II extrinsic apoptosis amplifies cell death signaling through the mitochondria. By contrast, type I apoptosis does not involve mitochondrial signaling.Citation15 TRAIL-resistance in pancreatic cancer cells is partially due to the overexpression of various IAP family proteins, including cIAP1, XIAP, and survivin. cIAP-1 and XIAP inhibit TRAIL-induced apoptosis by blocking the cleavage of caspase 3, 7, or 9 and thereby preventing downstream apoptotic events.Citation16
Another key protein involved in mediating TRAIL resistance is cellular, FLICE-like inhibitory protein (cFLIP). High levels of cFLIP, as often observed in pancreatic cancer cells, can block TRAIL-induced apoptosis by competing with caspase-8 for binding to the Fas-associated Death Domain (FADD) adaptor protein. Blocking the binding of caspase-8 to FADD prevents the proper formation of the Death-Inducing Signaling Complex (DISC) and prevents activating cleavage of caspase-8. Further downstream from cFLIP, cIAP1, and XIAP, members of the Bcl-2 family of proteins – especially Bcl-XL, Bcl-2, and Mcl-1 – also modulate sensitivity to TRAIL-induced apoptosis. These proteins block the activation of Bax and Bak, other Bcl-2 family members. Activated Bax and Bak are responsible for cell death through increased mitochondrial outer membrane permeability (MOMP). Thus, Bcl-XL, Bcl-2, and Mcl-1 inhibit apoptosis by preventing permeabilization of the mitochondrial membrane.Citation17
ONC201 is known to induce either an apoptotic or anti-proliferative phenotype in a variety of tumor types.Citation18,Citation19 Interestingly, the apoptotic or anti-proliferative phenotype does not depend on the endogenous TRAIL sensitivity of the PDAC tumor cell lines. Further analysis of PDAC tumor cell lines and their response to ONC201 showed that the anti-proliferative response is more common than the full, apoptotic response. Thus, we present a novel strategy to convert the response of pancreatic cancer cells to ONC201 from anti-proliferative to apoptotic.
In this study, we analyze the preclinical potential of a novel combination of ONC201 and TRAIL receptor agonists, rhTRAIL or TLY012. Initially, we explored the concept through the use of rhTRAIL. However, given rhTRAIL’s clinical shortcomings, we also explored the potential for combination of ONC201 with TLY012. Here, we report that the combination of ONC201 and TLY012 can induce selective, synergistic apoptosis in vitro and significantly delays tumor xenograft growth in vivo. This combination treatment shows marked efficacy in models that are both sensitive and resistant to ONC201-induced apoptosis providing a promising outlook for patients whose tumors do not respond to ONC201 and providing potential for regression in patients’ tumors that do respond to ONC201.
Results
ONC201 induces potent anti-proliferative and pro-apoptotic effects in multiple pancreatic cancer cell lines
ONC201 has been previously reported to induce apoptosis in several different tumor types through activation of the TRAIL pathway. In the context of pancreatic cancer, ONC201 shows two major phenotypes: anti-proliferative and/or apoptotic. Cells that undergo ONC201-induced apoptosis are referred to as ‘sensitive,’ and cells that do not undergo ONC201-induced apoptosis are referred to as ‘resistant.’ Table S1 depicts the cell lines used for this study, their major mutations, and their response to ONC201.Citation20
To test the anti-proliferative effects of ONC201, we used a clonogenicity assay, a widely used methodology that determines the ability of a single cell to form a colony unit. The number of colonies is then used as a measure of proliferative capacity of the cancer cells. We hypothesized that ONC201 decreases cellular clonogenicity in both ‘sensitive’ and ‘resistant’ pancreatic cancer cell lines. Cells were pre-treated with ONC201 for 3 days and grown in drug-free media for 7 days after. As shown in , cell proliferation was significantly reduced amongst AsPC-1, BxPC3, HPAFII, and PANC-1 cells after treatment with 2 or 4 μM ONC201. BxPC3, HPAFII, and PANC-1 also showed significantly decreased plate coverage after treatment with 1 μM ONC201 ().
Figure 1. ONC201 induces potent anti-proliferative and apoptotic effects across multiple pancreatic cancer cell lines. A) Clonogenicity assays of AsPC-1, BxPC3, HPAFII, and PANC-1 cell lines after treatment with ONC201. Cells were pre-treated for 3 days and grown in drug-free media for 7 days after. B) Quantification of clonogenicity assays shown in Figure 1a. C) Sub-G1 analysis of various pancreatic cancer cells treated with ONC201 for 72 hours. D) Time-course western blot of AsPC-1 cells treated with ONC201 for 12, 24, 48, or 72 hours at 3 μM (+) or 6 μM (++). Quantification of western blot is displayed below. * p < .05, ** p < .01, ** p < .001
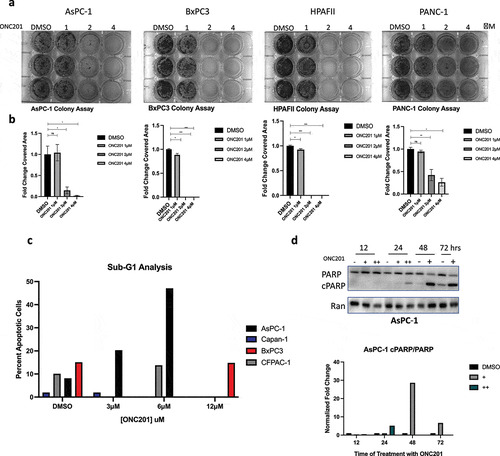
To confirm sensitivity to ONC201-induced apoptosis, we conducted a flow cytometric sub-G1 assay and western blots for PARP cleavage. As expected, AsPC-1 cells underwent an apoptotic-response showing high-levels of sub-G1 DNA particles (). AsPC-1 cells also showed increased PARP cleavage at the 48- and 72-hour time-points in a dose-dependent manner confirming that cells were undergoing apoptosis (). Furthermore, there was no increase in sub-G1 DNA as compared to vehicle in BxPC3 cells treated with 12 μM ONC201 (). Similar, non-apoptotic effects were observed in CFPAC-1 and Capan-1 cell lines using sub-G1 analysis (). Taken together, these data suggest that ONC201 can induce an anti-proliferative effect in cell lines that are sensitive and resistant to ONC201-induced apoptosis.
ONC201 upregulates the integrated stress response including ATF4, CHOP, and DR5
Our previous data has shown that ONC201 can induce its potent anti-tumor effects through activation of the Integrated Stress Response (ISR), a mammalian stress pathway known to regulate homeostasis.Citation19 Among the key molecules in the ISR is activating transcription factor 4 (ATF4). ATF4 is known to induce transcription of various ISR molecules, including C/EBP homologous protein (CHOP).
To confirm activation of the ISR in pancreatic cancer cell lines, we conducted a time-course experiment and analyzed ATF4 expression in HPAFII, AsPC-1, and CFPAC-1 cell lines (). As expected, ONC201 induced transient expression of ATF4 at 24, 48, and 72 hours in all cell lines tested. We then analyzed the expression of CHOP and DR5 in HPAFII cells. We observed that CHOP is upregulated at both 24 and 48 hours after treatment of HPAFII cells with ONC201 (3 μM and 6 μM; ). At 72 hours, CHOP was only upregulated after treatment of HPAFII cells with 6 μM ONC201 (). Similarly, DR5 was upregulated in HPAFII cells at 24 and 48 hours after treatment with both 3 μM and 6 μM ONC201 (). In AsPC-1 cell lines, ATF4 was upregulated after treatment with 6 μM ONC201 at both 48 and 72 hours (). Similarly, CFPAC-1 cells showed upregulation of ATF4 after treatment with ONC201 at 24, 48, and 72 hours ().
Figure 2. ONC201 induces potent activation of the integrated stress response in a panel of pancreatic tumor cell lines. A) Time-course western blot of HPAFII cells treated with ONC201 (+, 3 μM; ++, 6 μM) for 24, 48, or 72 hours. B) Time-course western blot of AsPC-1 cells treated with ONC201 (+, 3 μM) showing ATF4 upregulation at 48 and 72 hours. Time-course western blot of CFPAC-1 Cells treated with ONC201 (+, 6 μM; ++, 12 μM) D) Time-course western blot assessing DR5 expression in BxPC3 cells after treatment with ONC201 for 48 and 72 hours (+, 12 μM; ++, 20 μM). Quantification of all blots is shown
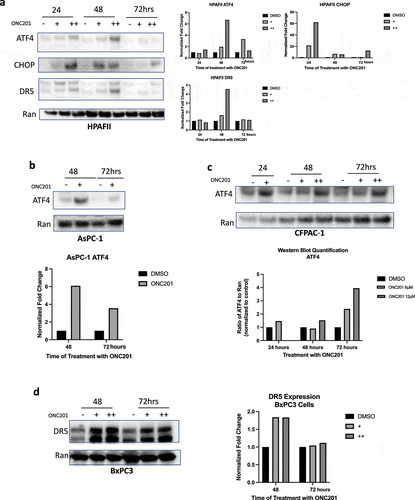
In BxPC3 cells, ONC201-induced potent upregulation of DR5 at 48 and 72 hours confirming ISR activation similar to that observed in HPAFII cells (). These data indicate that ONC201 upregulates the ISR in multiple pancreatic cancer cell lines. Furthermore, activation of the ISR appears to be independent of innate sensitivity of cell lines to ONC201-induced apoptosis. This is evidenced by activation of ATF4 in HPAFII and AsPC-1, ONC201 sensitive cell lines, and CFPAC-1, a cell line that is resistant to ONC201-induced apoptosis.
ONC201 primes pancreatic cancer cell lines for TRAIL-induced cell death through downregulation of key anti-apoptotic proteins
Sensitivity to TRAIL-induced apoptosis in pancreatic cancer is mediated by the expression of certain anti-apoptotic proteins. It is ultimately the intracellular balance of pro-apoptotic and anti-apoptotic proteins that determines cell fate. Thus, in addition to DR5 expression, we examined whether or not ONC201 can modulate the expression of these key anti-apoptotic proteins to further sensitize cells to TRAIL-induced apoptosis.
Previously, Lev et al.Citation3 established that ONC201 can reduce expression of XIAP, Mcl-1, and Bcl-XL in PANC-1, HPAFII, and BxPC3 cell lines. Thus, to further examine the downregulation of anti-apoptotic proteins, western blots were used to analyze the expression of cIAP-1, survivin, cFLIP, Bcl-2, and Bcl-XL across multiple PDAC tumor cell lines. Each of these proteins inhibits various components of the extrinsic apoptosis pathway, posing unique challenges in anti-tumor therapy.
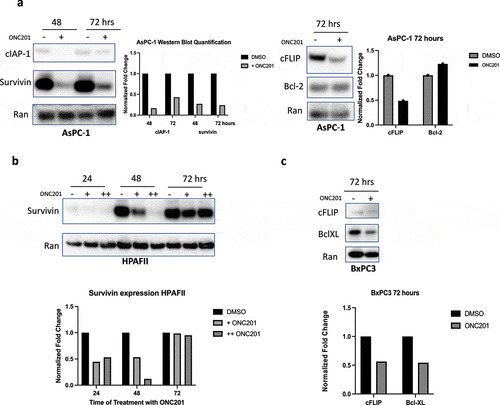
Analysis of cIAP-1 and survivin showed potent downregulation after treatment with 3 μM and 6 μM ONC201 in AsPC-1 cells (). Similarly, potent downregulation of survivin was observed in HPAFII cells after treatment with ONC201 (). To further analyze the downregulation of anti-apoptotic proteins, samples were probed for cellular-flice like inhibitory protein (cFLIP), Bcl-2, and Bcl-XL. In AsPC-1 and BxPC3 cells, significant downregulation of cFLIP after treatment with ONC201 was observed, suggesting that downregulation of anti-apoptotic proteins occurs in cells that are both sensitive and resistant to ONC201-induced apoptosis (, ). Interestingly, analysis of Bcl-2 in AsPC-1 cells showed a small increase (). However, analysis of Bcl-XL in BxPC3 revealed downregulation after treatment with 12 μM ONC201 for 72 hours (). Taken together, our data show that ONC201 primes pancreatic cancer cells for TRAIL-induced apoptosis through high levels of death receptor expression and low levels of anti-apoptotic proteins.
Figure 3. ONC201 primes pancreatic tumor cell lines for TRAIL-induced apoptosis through downregulation of key anti-apoptotic proteins. A) Western blots in AsPC-1 cell lines treated with ONC201 (+, 3 μM) B) HPAFII cells treated with ONC201 (+, 3 μM; ++, 6 μM). C) BxPC3 cells treated with ONC201 for 72 hours (+, 12 μM)
Addition of TLY012 or rhTRAIL after pre-treatment with ONC201 induces synergistic apoptosis in pancreatic tumor cell lines
The potent upregulation of DR5 and downregulation of various anti-apoptotic proteins after treatment with ONC201 posed an interesting, albeit counterintuitive, question: could addition of rhTRAIL (or rhTRAIL mimetics) induce synergistic apoptosis in cells pre-treated with ONC201? This question is particularly relevant and meaningful because activation of the ISR and apoptotic priming is observed in cell lines that are both sensitive and resistant to ONC201-induced apoptosis. Thus, a potential combination treatment would be applicable to cells that are resistant to ONC201-induced apoptosis.
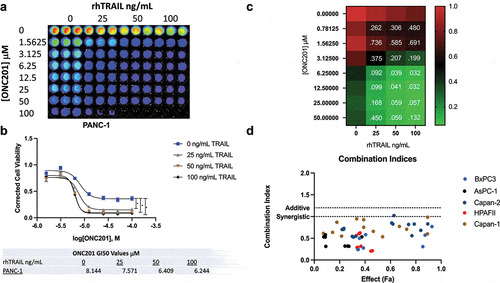
To test this hypothesis, seven pancreatic cancer cell lines were screened for potential synergy using cell viability assays. As a proof of concept, the first set of experiments was conducted using rhTRAIL. Consistent with the hypothesis, the combination of ONC201 and rhTRAIL induced potent, synergistic loss of cell viability in six of seven cell lines tested. PANC-1 cells responded extremely well to the combination treatment showing a statistically significant decrease in cell viability after adding only 25 ng/mL rhTRAIL (). The combination indices for PANC-1 cells treated with ONC201 and rhTRAIL are shown in a heatmap in . Cell-Titer Glo Images and cell viability curves for the other cell lines tested are in Figure S1 and S2. Interestingly, this response was not limited to PDAC tumor cell lines. Sub-G1 flow cytometry analysis of SW480 colon cancer cell line treated with ONC201 and rhTRAIL showed potent, synergistic apoptosis after treatment with both drugs (Figure S3). For cell viability assays, synergy was assessed using the Chou-Talalay method. Combination indices for six of the test cell lines are shown in No synergistic loss of cell viability was observed in the CFPAC-1 cell line (Figure S4A, S4B).
Figure 4. Combination of ONC201 and TRAIL receptor agonists induces synergistic apoptosis across PDAC tumor cell lines in vitro. A) Cell viability assay of PANC-1 cells treated with varying doses of ONC201 and rhTRAIL. Cells were pre-treated with ONC201 for 72 hours. rhTRAIL was added for 4 hours before imaging. B) Dose response curve for image shown in Panel 4A. C) Heat map of PANC-1 cells treated with ONC201 and TLY012. The color of the wells represents fraction alive according to the scale bar. Combination index values are listed inside the wells. Values less than 1 indicate synergy. C) Combination indices for PDAC tumor cell lines pre-treated with ONC201 for 72 hours and TLY012 for 4 hours
To determine if this response was apoptotic, western blots were conducted and probed for expression of cleaved PARP. Synergistic PARP cleavage was observed in HPAFII, BxPC3, and Capan-1 cell lines after treatment with the combination of ONC201 and rhTRAIL ().
Figure 5. Combination of ONC201 and TRAIL induces selective, synergistic apoptosis across multiple PDAC tumor cell line in vitro with minimal toxicity to non-transformed cells. A) PARP cleavage western blots of HPAFII, BxPC3, and Capan-1 cells after 72 hours treatment with ONC201 (3 μM, 12 μM, or 6 μM, respectively) and 4 hours treatment with either 50 or 100 ng/mL rhTRAIL. B) PARP cleavage western blot of PANC-1 cells treated with 1 μM ONC201 for 72 hours. TLY012 subsequently added for 4 hours. C) CellTiter Glo assay assessing cell viability of non-transformed HFF-1 cells treated with ONC201 for 72 hours and subsequent addition of TLY012 for 4 hours. D) PARP cleavage western blot of HFF-1 cells treated with 2 μM ONC201 for 72 hours and of TLY012 for 4 hours
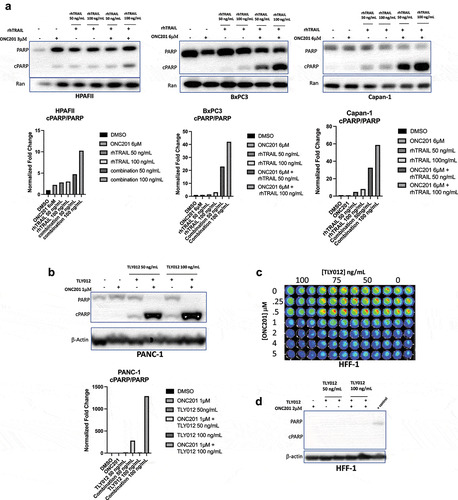
Given the clinical failure of rhTRAIL as a mono-agent, the potential for a combination of ONC201 and TLY012, a novel TRAIL receptor agonist, was also examined. Western blots of PANC-1 cells treated with the combination of ONC201 and TLY012 also showed potent, synergistic PARP cleavage, suggesting that TLY012 could be used to replace rhTRAIL for combination with ONC201 (). Cell viability analysis was also conducted in HPAFII and AsPC-1 cells with the combination ONC201 and TLY012 (Figure S5A). The HPAFII cell line showed potent synergistic loss of cell viability (Figure S5B, S5C). Taken together, these data suggest that TLY012 can be used in combination of ONC201 to synergistically induce apoptosis in PDAC tumor cell lines in vitro.
The combination of ONC201 and TLY012 does not induce synergistic cell death in human foreskin fibroblasts
The in vitro efficacy of the combination treatment of ONC201 and TLY012 raised some interesting prospects for translation. Thus, to explore in vitro toxicity Human Foreskin Fibroblasts (HFF-1) were treated with the combination of ONC201 and TLY012 and analyzed via cell viability assays and western blots. There was a minimal decrease in cell viability in HFF-1 cells after treatment with ONC201 and TLY012 (), and no PARP cleavage was observed after treating HFF-1 cells with the combination of ONC201 and TLY012 ().
Combination of ONC201 and TLY012 significantly delays pancreatic xenograft tumor growth
The selectivity of this ONC201 plus TLY012 apoptotic response to cancer cells further increased prospects for translation. Thus, the in vivo efficacy of ONC201 and TLY012 was examined using a pancreatic cancer xenograft model. BxPC3 and HPAFII cell lines were used for the in vivo xenograft studies.
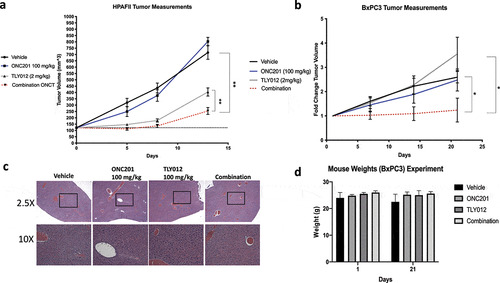
For both experiments, mice were dosed once weekly with ONC201 and TLY012. ONC201 was administered orally and TLY012 was administered via intraperitoneal injections. In the HPAFII model, two doses of the combination treatment showed a statistically significant difference in tumor volume between all four groups (). HPAFII xenografts treated with the combination of ONC201 were nearly four times smaller than the vehicle control group. In the BxPC3 model, tumor growth was significantly slower in the combination group as compared to both the vehicle and TLY012. No significant difference was observed between ONC201 and the combination group. Pairwise comparisons for all of the groups included in both of these xenograft experiments are shown in Figure S6. Liver sections from mice bearing HPAFII xenografts treated with three, weekly doses of ONC201 and TLY012 were stained with Hematoxylin & Eosin. Minimal differences and no liver toxicity were observed between any of the four groups (). Animal weights were tracked for mice bearing BxPC3 xenograft tumors. No appreciable weight loss was observed from Day 1 to Day 21, after three, once weekly doses of the combination of ONC201 and TLY012. These data indicate the combination treatment of ONC201 and TLY012 is nontoxic in vivo.
Figure 6. Combination of ONC201 and TLY012 significantly slows PDAC tumor growth in HPAFII and BxPC3 tumor models in vivo. A) Mice bearing HPAFII xenograft tumors were treated with two doses of the combination of ONC201 and TLY012. ONC201 (100 mg/kg) was administered first and TLY012 (2 mg/kg) was injected three days later. The second dose was initiated one week after initial treatment with ONC201. Treatment was initiated once tumors reached the optimal size of 100–150 mm3. Vehicle (n = 5), ONC201 (n = 5), TLY012 (n = 6), Combination (n = 8). B) Mice bearing BxPC3 xenograft tumors were treated with three doses of the combination of ONC201 and TLY1012. ONC201 (100 mg/kg) was administered first and TLY012 (2 mg/kg) was injected three days later. Mice were dosed once per week with the combination treatment. To combat heterogeneity in tumor formation, mice were dosed in a ‘rolling format’ and separated into two cohorts, allowing tumors to reach the optimal size of ~150mm3 before treatment. Vehicle (n = 4), ONC201 (n = 5), TLY012 (n = 5), Combination (n = 5). C) H&E liver sections from mice bearing HPAFII xenografts. Mice were treated with three, weekly doses ONC201 (100 mg/kg) and TLY012 (2 mg/kg). D) Mouse weights from mice bearing BxPC3. Mice were dosed with three, weekly doses of ONC201 (100 mg/kg) and TLY012 (2 mg/kg). * p < .05, ** p < .01
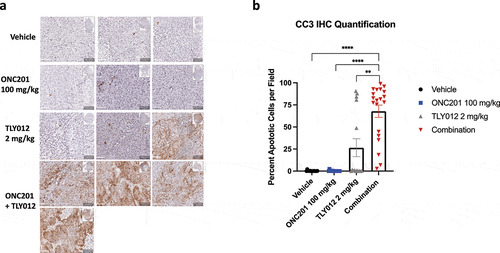
Immunohistochemical analysis of cleaved caspase 3 (CC3) in mice bearing HPAFII tumors treated with the three doses of the combination treatment showed a marked increase in CC3 after treatment with the combination. Minimal CC3 was observed in the vehicle and ONC201 conditions. Interestingly, one tumor in the TLY012 group showed a marked increase in CC3. All four tumors treated with the combination of ONC201 and TLY012 showed a marked increase in CC3 after treatment with three, weekly doses. (). Quantification of five high power fields per tumor showed a marked, statistically significant increase in CC3 positive cells in the combination group as compared to all other remaining groups (). Whole tumor scans stained with CC3 can be found in Figure S7. HPAFII xenografts tumors were also analyzed for ISR activation and Ki67 positivity, but we did not observe any robust, statistically significant differences between the four groups (Figure S8A, S8B). Similarly, BxPC3 xenograft tumors were analyzed for Ki67, ATF4 activation, and cell death. No robust, qualitative differences were observed between any of the groups for any of the markers probed in the BxPC3 tumor xenograft model. (Figure S9A, S9B, S9C).
Figure 7. Combination of ONC201 and TLY012 induces potent apoptosis in an HPAFII tumor model in vivo. A) Immunohistochemical analysis of cleaved caspase 3 (CC3) in HPAFII tumor xenograft models treated with three, once weekly doses of ONC201 (100 mg/kg) and TLY012 (2 mg/kg). ONC201 was administered first, and TLY012 was administered 3 days later. Each dose of the combination was administered one week apart. Treatment was initiated once tumors reached between 100–150mm3. Mice were euthanized 1 day after administration of the third dose of TLY012. All scale bars indicate 50 μM. B) Graph showing quantification of 5 20X fields per tumor. * p < .05, ** p < .01, *** p < .001, **** p < . 0001
Taken together, the in vivo data from these experiments shows that the combination of ONC201 and TLY012 induces a potent, apoptotic anti-tumor effect in PDAC tumor models. Furthermore, this drug combination is minimally toxic suggesting a promising outlook for the clinical translation of ONC201 and TLY012.
Discussion
ONC201 is a novel, orally bioavailable small molecule with promising therapeutic potential for use in patients with pancreatic cancer as a mono-agent and in combination. Here, we demonstrate that ONC201 can induce both anti-proliferative and pro-apoptotic effects in multiple PDAC cell lines. Interestingly, the pro-apoptotic effects of ONC201 do not depend on the endogenous TRAIL sensitivity of these cell lines. As seen in other tumor cell types, ONC201 induced potent activation of the ISR including ATF4, CHOP, and DR5. Upon more in depth examination of the effects of ONC201, we observed that it induces potent downregulation of anti-apoptotic proteins – even in cell lines that do not display the pro-apoptotic phenotype. The depth and breadth of this ‘priming’ response is promising, as it may provide additional therapeutic options for patients with tumors that are resistant to ONC201-induced apoptosis.Citation21 When combined with rhTRAIL, ONC201 induced an apoptotic response that is selective to cancer cells in 6 of the 7 cell lines tested. The apoptotic response was not observed in the CFPAC-1 cell line, but we did observe potent activation of the ISR. Future experiments will involve testing whether or not the CFPAC-1 cell line downregulates cFLIP, BclXL, Mcl-1, and other key anti-apoptotic proteins.
As expected, combining ONC201 with TLY012, a novel TRAIL receptor agonist, yielded a similar, selective response to that observed with rhTRAIL. This is an exciting result, as TLY012 is currently entering clinical trials for use in targeting fibrotic cells and has a promising safety profile.
Furthermore, the in vivo xenograft studies for ONC201 plus TLY012 combination therapy-treated mice showed significant differences from the vehicle-treated mice in both tumor models tested. While the BxPC3 model does not show a statistically significant difference between the ONC201 and combination groups, the observed trend was approaching statistical significance. The data from the HPAFII model is especially promising, as statistically significant regression can be observed after the first dose. Though the tumors continued growing after the first dose, the combination tumors were significantly different – nearly half the size – of all other groups. Future iterations of this experiment could involve a more aggressive dosing regimen to examine the potential for sustained regression. Toxicity was examined using mouse weights and Hematoxylin & Eosin-stained liver sections. No appreciable changes were observed across groups in both mouse weights and the liver morphology suggesting that this combination treatment is well-tolerated.
Immunohistochemical analysis of CC3 in the HPAFII tumor xenograft model showed promising results. Here, we observed robust staining of CC3 in all four tumors treated with the combination of ONC201 and TLY012 and statistically significant differences from all other groups. These data corroborated the change in tumor volume observed when the HPAFII xenografted tumors were treated with the combination of ONC201 and TLY012. Further analysis of ATF4 and Ki67 in the HPAFII tumor cell line xenograft model did not show a significant change between any of the four groups. The reason for this disparity remains unclear, but one potential reason could be cell selection at the later time point of three, weekly doses. IHC analyses were performed on samples from the BxPC3 xenograft model. Here, we did not observe any marked differences in tumors probed for Ki67, ATF4, and CC3. However, the long-term tumor growth data did show a significant reduction in tumor growth between the vehicle, TLY012, and combination of ONC201 plus TLY012. No statistically significant difference was observed between the combination and ONC201. Additional experiments can use varied dosing regimens and time-point variations to better assess the strength of this response in vivo. Further analysis can also focus on the effects of the combination treatment on the long-term survival of tumor-bearing mice.
Our data provide a promising outlook for the clinical translation of the combination of ONC201 and TLY012 for use in PDAC patients. To gain a better understanding of the role of the PDAC tumor microenvironment, future work could involve the use of a genetically engineered mouse model (GEMM), such as the KPC mouse model.Citation22 Using such models will enable us to analyze the efficacy of this combination treatment in an orthotopic, syngeneic setting. These experiments would address some of the clinical challenges faced when caring for patients with PDAC – such as the role of the tumor microenvironment – and would address some limitations of the subcutaneous xenograft tumor models used for this study.
Other future directions for this study include using a syngeneic model with Ncr1-GFP mice to assess the intra-tumoral recruitment of NK cells in PDAC tumor models. Previous data from our lab has shown that ONC201 can induce the intra-tumoral recruitment of NK cells in colorectal cancer.Citation23 However, this has yet to be explored in pancreatic cancer. As TRAIL is expressed on the surface of NK cells, understanding the potency of immune recruitment in PDAC is integral to assessing the efficacy of ONC201 and this combination treatment.Citation24 Furthermore, using an immunocompetent model will also allow us to examine the immune effects of intravenous administration of exogenous TRAIL receptor agonists.
The combination treatment of ONC201 and TLY012 presents a novel, promising therapeutic option for PDAC patients. This combination shows selective, synergistic apoptosis in PDAC cell lines that are both sensitive and resistant to the pro-apoptotic effects of ONC201. We hope that this and resulting future work will lead to successful translation of this combination treatment to the clinic, providing hope and options for treatment of patients with PDAC.
Materials and methods
Cell culture
Cell lines used for this study were acquired from the American Type Culture Collection (ATCC), unless otherwise indicated. All pancreatic cancer cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) with 10% Fetal Bovine Serum (FBS). Human Foreskin Fibroblasts (HFF) were grown in DMEM with 15% FBS. ONC201 was provided by Oncoceutics, Inc. DMSO was used as a vehicle for dissolving ONC201 for in vitro experiments. rhTRAIL was generated in-house using a protocol previously developed by our lab and detailed in Kim et al. TLY012 was provided by Theraly Fibrosis, Inc. TLY012 was diluted in sterile phosphate buffered saline (PBS) for in vitro experiments.
Clonogenicity (Colony) assay
Colony assays were conducted to assess the proliferative capacity of cells after treatment with ONC201. 500 cells/well were plated overnight into 6-well plates. Cells were grown in media containing ONC201 for 3 days. After 3 days, the media was changed to fresh media and cells were grown for a week. After one week, cells were fixed and stained using a crystal violet solution. Subsequently, plates were imaged.
Cell viability assay
To assess cell viability, cells were plated overnight in 96-well plates at a density of 1.0 × 104 cells/well. All cells were plated in triplicates. Cells were first pre-treated with ONC201 for 72 hours. After 72 hours, the media in the wells was replaced with either fresh media (controls) or with media containing various doses of rhTRAIL or TLY012. After 4 hours of incubation, cells were treated with Cell-Titer Glo (Promega) and imaged to assess cell viability. Synergy and combination indices were determined using Compusyn, which uses the Chou-Talalay method for determining synergy.
Flow cytometric analysis
Sub-G1 analysis was conducted following a well-established lab protocol. Cells were plated overnight before treating with drugs. After the experiment was completed, all floating and adherent cells were harvested. Once cells were harvested, they were gently centrifuged and washed with PBS to remove the phenol red found in the culture medium. Once thoroughly washed, cells were resuspended and fixed in 70% cold ethanol and stored at 4°C overnight. Once cells were fixed, they were stained with propidium-iodide to bind DNA. Cells were then run on an Epics Elite (Beckman Coulter) flow cytometer. FlowJo was used to analyze the percentage of apoptotic cells.
Western blotting
Cells were plated overnight before any drug treatment.Citation25 Approximately 5 × 105 cells/well were plated in 6-well plates. Upon completion of the experiment, 1 mL of media containing floating cells was harvested. Adherent cells were crushed and collected into the remaining 1 mL of media, and this total solution was pelleted for 5 minutes at 400 Rcf. The pellets were then washed by resuspending with PBS once. Cells were then lysed using RIPA buffer (Sigma-Aldrich) with 1X protease inhibitor and 1X phosphatase inhibitor (Roche). Upon lysis, cells were centrifuged at 13,000 RPM at 4°C for approximately 20 minutes. The supernatants were collected. Proteins were estimated using a BCA Assay (Thermo Scientific). Gels used for analysis were pre-cast NuPAGE 4–12% Bis-Tris (Thermo Scientific). Bands were quantified using an established ImageJ protocol from the ImageJ user guide.
In vivo tumor xenograft studies
All in vivo studies conducted for this manuscript were approved by the Brown University IACUC. For in vivo tumor xenograft studies, we used female, athymic nude mice acquired from Taconic Biosciences (NCR-Nu-F, genotype: sp/sp). Mice were aged 5–8 weeks at the time of tumor inoculation. Cells were mixed in a 50:50 Matrigel (Corning):PBS solution and mixed at various dilutions. Total inoculation volume was 200 μL, irrespective of tumor model or number of cells inoculated. ONC201 was always administered via oral gavage, and doses were titrated such that mice only received 100 μL of solution. The vehicle is a solution of 20% Cremophor EL (Sigma-Aldrich), 70% PBS, and 10% DMSO. rhTRAIL was administered through intravenous tail vein injections. TLY012 was administered via intraperitoneal injections.
Tumor volume calculations
Measurements were taken using Vernier calipers. Thus, the equation for calculating tumor volume is: Volume = (WidthCitation2 * Length)/2.
BxPC3 long-term tumor growth experiment
Approximately 4 million cells were inoculated on the right flank. Mice were given one tumor per mouse. To minimize the variability of tumor size at starting time, mice were separated into two cohorts. As each cohort reached the optimal size range of 100–150 mm3, treatment was initiated.
HPAFII long-term tumor growth experiment:
Mice were inoculated with approximately 4 million cells on the right flank. Treatment was initiated once the entire cohort of tumors reached an optimal volume between 100–150mm3.
Immunohistochemical staining
Tumors were fixed in formalin immediately after harvesting in cassettes. After fixation, cassettes were paraffin embedded. Slides were cut 5 μm thick. Immunohistochemistry was initiated by deparaffinizing slides using xylene. Slides were dehydrated through sequential dilutions of ethanol. The antigen retrieval step was conducted by heating slides for 10 minutes in pH 6.0 citrate acid buffer. Ki67 (MIB-1) antibody was obtained from Cell Signaling Technologies, used at 1:200 dilution. CC3 Antibodies obtained from BD Biosciences, used at 1:100 dilution. Slides were incubated in primary antibodies overnight; respective secondary antibodies were added the following day. Slides were developed using DAB Staining Kit (Vector Labs) and mounted using a xylene-based mount, Cytoseal XYL.
Immunohistochemical quantification
Slides were quantified using QuPath, an open-source, automated program for immunohistochemistry quantification.Citation26 Ki67 staining was analyzed in a binary fashion with only positive and negative nuclei. CC3 staining was analyzed by taking 5 representative high power (20X) fields per tumor and using QuPath to quantify cells that stained positive.
Statistical analysis
All statistical analyses were conducted using Microsoft Excel and figures were designed using Prism GraphPad. P-values were determined using the Student’s T-test and significance was determined as P < .05.
Disclosures
W.S.E-D. is a co-founder of Oncoceutics Inc., a subsidiary of Chimerix, and is fully compliant with NIH and institutional disclosure guidelines. V.V.P. is an employee of Oncoceutics, Inc., a subsidiary of Chimerix. S.L. is an employee and shareholder of Theraly Fibrosis, Inc., a subsidiary of D&D Pharmatech.
Supplemental Material
Download MS Word (20.2 KB)Acknowledgments
This work was presented in an earlier form as part of the Annual Meeting of the American Association for Cancer Research (AACR) in April 2020. This work was supported in part by NCI grants CA173453 (W.S.E-D.), P30CA006927, and a grant from D&D Pharmatech (to W.S.E-D.). W.S.E-D. is an American Cancer Society Research Professor and is supported by the Mencoff Family University Professorship at Brown University. The contents of this manuscript are solely the responsibility of the authors and do not necessarily represent the official views of the National Cancer Institute, the National Institutes of Health, or the American Cancer Society.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
Additional information
Funding
References
- Yabar CS, Winter JM. Pancreatic cancer: a review. Gastroenterol Clin North Am. 2016 Sep;45(3):429–445.https://doi.org/10.1016/j.gtc.2016.04.003. PMID: 27546841
- Prabhu VV, Morrow S, Rahman Kawakibi A, Zhou L, Ralff M, Ray J, Jhaveri A, Ferrarini I, Lee Y, Parker C, et al. ONC201 and imipridones: anti-cancer compounds with clinical efficacy. Neoplasia. 2020Dec;22(12):725–744. https://doi.org/10.1016/j.neo.2020.09.005. Epub 2020 Oct 23. PMID: 33142238
- Lev A, Lulla AR, Wagner J, Ralff Md, Kiehl JB, Zhou Y, Benes CH, Prabhu VV, Oster W, Astsaturov I, et al. Anti-pancreatic cancer activity of ONC212 involves the unfolded protein response (UPR) and is reduced by IGF1-R and GRP78/BIP. Oncotarget. 2017 Sep 12;8(47):81776–81793. https://doi.org/10.18632/oncotarget.20819. PMID: 29137221; PMCID: PMC5669847
- von Karstedt S, Montinaro A, Walczak H. Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nat Rev Cancer. 2017 May 24;17(6):352–366. https://doi.org/10.1038/nrc.2017.28. PMID: 28536452
- Itoh N, Yonehara S, Ishii A, Yonehara M, Mizushima S, Sameshima M, Hase A, Seto Y, Nagata S. The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell. 1991 Jul 26;66(2):233–243. https://doi.org/10.1016/0092-8674(91)90614-5. PMID: 1713127
- Kelley SK, Harris LA, Xie D, Deforge L, Totpal K, Bussiere J, Fox JA. Preclinical studies to predict the disposition of Apo2L/tumor necrosis factor-related apoptosis-inducing ligand in humans: characterization of in vivo efficacy, pharmacokinetics, and safety. J Pharmacol Exp Ther. 2001 Oct;299(1):31–38. PMID: 11561060.
- Allen JE, Krigsfeld G, Mayes Pa, Patel L, Dicker DT, Patel AS, Dolloff NG, Messaris E, Scata KA, Wang W, et al. Dual inactivation of Akt and ERK by TIC10 signals Foxo3a nuclear translocation, TRAIL gene induction, and potent antitumor effects. Sci Transl Med. 2013 Feb 6;5(171):171ra17. https://doi.org/10.1126/scitranslmed.3004828. PMID: 23390247
- Hall MD, Odia Y, Allen JE, Tarapore R, Khatib Z, Niazi TN, Daghistani D, Schalop L, Chi AS, Oster W, et al. First clinical experience with DRD2/3 antagonist ONC201 in H3 K27M-mutant pediatric diffuse intrinsic pontine glioma: a case report. J Neurosurg Pediatr. 2019 Apr 5;1–7. https://doi.org/10.3171/2019.2.PEDS18480. Epub ahead of print. PMID: 30952114.
- Allen JE, Kline CL, Prabhu VV, Wagner J, Ishizawa J, Madhukar N, Lev A, Baumeister M, Zhou L, Lulla A, et al. Discovery and clinical introduction of first-in-class imipridone ONC201. Oncotarget. 2016 Nov 8;7(45):74380–74392. https://doi.org/10.18632/oncotarget.11814. PMID: 27602582
- Graves JD, Kordich JJ, Huang TH, Piasecki J, Bush TL, Sullivan T, Foltz IN, Chang W, Douangpanya H, Dang T, et al. Apo2L/TRAIL and the death receptor 5 agonist antibody AMG 655 cooperate to promote receptor clustering and antitumor activity. Cancer Cell. 2014 Aug 11;26(2):177–189. https://doi.org/10.1016/j.ccr.2014.04.028. Epub 2014 Jul 17. PMID: 25043603
- Chae SY, Kim TH, Park K, Jin CH, Son S, Lee S, Youn YS, Kim K, Jo DG, Kwon IC, et al. Improved antitumor activity and tumor targeting of NH(2)-terminal-specific PEGylated tumor necrosis factor-related apoptosis-inducing ligand. Mol Cancer Ther. 2010 Jun 9;(6):1719–1729. https://doi.org/10.1158/1535-7163.MCT-09-1076. Epub 2010 Jun 1. PMID: 20515949
- Park JS, Oh Y, Park YJ, Park O, Yang H, Slania S, Hummers LK, Shah AA, An HT, Jang J, et al. Targeting of dermal myofibroblasts through death receptor 5 arrests fibrosis in mouse models of scleroderma. Nat Commun. 2019 Mar 8;10(1):1128. https://doi.org/10.1038/s41467-019-09101-4. PMID: 30850660
- Hamacher R, Schmid RM, Saur D, Schneider G. Apoptotic pathways in pancreatic ductal adenocarcinoma. Mol Cancer. 2008 Jul 24;7:64. https://doi.org/10.1186/1476-4598-7-64. Pmid: 18652674
- Hinz S, Trauzold A, Boenicke L, Sandberg C, Beckmann S, Bayer E, Walczak H, Kalthoff H, Ungefroren H. Bcl-XL protects pancreatic adenocarcinoma cells against CD95- and TRAIL-receptor-mediated apoptosis. Oncogene. 2000 Nov 16;19(48):5477–5486. https://doi.org/10.1038/sj.onc.1203936. PMID: 11114725
- Ozören N, El-Deiry WS. Defining characteristics of Types I and II apoptotic cells in response to TRAIL. Neoplasia. 2002 Nov-Dec;4(6):551–557.https://doi.org/10.1038/sj.neo.7900270. PMID: 12407450
- Deveraux QL, Reed JC. IAP family proteins–suppressors of apoptosis. Genes Dev. 1999 Feb 1;13(3):239–252. https://doi.org/10.1101/gad.13.3.239. PMID: 9990849
- Carneiro BA, El-Deiry WS. Targeting apoptosis in cancer therapy. Nat Rev Clin Oncol. 2020 Jul;17(7):395–417.https://doi.org/10.1038/s41571-020-0341-y. Epub 2020 Mar 23. PMID: 32203277
- Ralff MD, Kline CLB, Küçükkase OC, Wagner J, Lim B, Dicker DT, Prabhu VV, Oster W, El-Deiry WS. ONC201 demonstrates antitumor effects in both triple-negative and non-triple-negative breast cancers through TRAIL-dependent and TRAIL-independent mechanisms. Mol Cancer Ther. 2017 Jul;16(7):1290–1298.https://doi.org/10.1158/1535-7163.MCT-17-0121. Epub 2017 Apr 19. PMID: 28424227
- Kline CL, Van den Heuvel AP, Allen JE, Prabhu VV, Dicker DT, El-Deiry WS. ONC201 kills solid tumor cells by triggering an integrated stress response dependent on ATF4 activation by specific eIF2α kinases. Sci Signal. 2016 Feb 16;9(415):ra18. https://doi.org/10.1126/scisignal.aac4374. PMID: 26884600
- Deer EL, González-Hernández J, Coursen JD, Shea JE, Ngatia J, Scaife CL, Firpo MA, Mulvihill SJ. Phenotype and genotype of pancreatic cancer cell lines. Pancreas. 2010 May;39(4):425–435.https://doi.org/10.1097/MPA.0b013e3181c15963. Erratum in: Pancreas. 2018 Jul;47(6):e37.PMID: 20418756; PMCID: PMC2860631
- Ralff MD, Jhaveri A, Ray JE, Zhou L, Lev A, Campbell KS, Dicker DT, Ross EA, El-Deiry WS. TRAIL receptor agonists convert the response of breast cancer cells to ONC201 from anti-proliferative to apoptotic. Oncotarget. 2020 Oct 20;11(42):3753–3769. https://doi.org/10.18632/oncotarget.27773. PMID: 33144917
- Lee JW, Komar CA, Bengsch F, Graham K, GL B. Genetically engineered mouse models of pancreatic cancer: the KPC model (LSL-Kras(G12D/+);LSL-Trp53(R172H/+);Pdx-1-Cre), Its variants, and their application in immuno-oncology drug discovery. Curr Protoc Pharmacol. 2016 Jun 1;73:14.39.1–14.39.20. https://doi.org/10.1002/cpph.2. PMID: 27248578
- Wagner J, Kline CL, Zhou L, Campbell KS, MacFarlane AW, Olszanski AJ, Cai KQ, Hensley HH, Ross EA, Ralff MD, et al. Dose intensification of TRAIL-inducing ONC201 inhibits metastasis and promotes intratumoral NK cell recruitment. J Clin Invest. 2018 Jun 1;128(6):2325–2338. doi: https://doi.org/10.1172/JCI96711. Epub 2018 Apr 30. PMID: 29533922
- Mirandola P, Ponti C, Gobbi G, Sponzilli I, Vaccarezza M, Cocco L, Zauli G, Secchiero P, Manzoli FA, Vitale M. Activated human NK and CD8+ T cells express both TNF-related apoptosis-inducing ligand (TRAIL) and TRAIL receptors but are resistant to TRAIL-mediated cytotoxicity. Blood. 2004 Oct 15;104(8):2418–2424. doi: https://doi.org/10.1182/blood-2004-04-1294. Epub 2004 Jun 17. PMID: 15205263
- Hernandez-Borrero LJ, Zhang S, Lulla A, Dicker DT, El-Deiry WS. CB002, a novel p53 tumor suppressor pathway-restoring small molecule induces tumor cell death through the pro-apoptotic protein NOXA. Cell Cycle. 2018;17(5):557–567. doi:https://doi.org/10.1080/15384101.2017.1346762. Epub 2018 Feb 19. PMID: 28749203.
- Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, McQuaid S, Gray RT, Murray LJ, Coleman HG, et al. QuPath: open source software for digital pathology image analysis. Sci Rep. 2017 Dec 4;7(1):16878. doi: https://doi.org/10.1038/s41598-017-17204-5. PMID: 29203879; PMCID: PMC5715110