
ABSTRACT
Tailoring extracellular vesicles (EVs) as targeted drug delivery systems to enhance the therapeutic efficacy showed superior advantage over liposomal therapies. Herein, we developed a novel nanotool for targeting B16.F10 murine melanoma, based on EVs stabilized with Polyethylene glycol (PEG) and loaded with doxorubicin (DOX). Small EVs were efficiently enriched from melanoma cells cultured under metabolic stress by ultrafiltration coupled with size exclusion chromatography (UF-SEC) and characterized by size, morphology, and proteome. To reduce their clearance in vivo, EVs were PEGylated and passively loaded with DOX (PEG-EV-DOX). Our data suggested that the low PEG coverage of EVs might still favor EV surface protein interactions with target proteins from intratumor cells, ensuring their use as “Trojan horses” to deliver DOX to the tumor tissue. Moreover, our results showed a superior antitumor activity of PEG-EV-DOX in B16.F10 murine melanoma models in vivo compared to that exerted by clinically applied liposomal DOX in the same tumor model. The PEG-EV-DOX administration in vivo reduced NF-κB activation and increased BAX expression, suggesting better prognosis of EV-based therapy than liposomal DOX treatment. Collectively, our results highlight the promising potential of EVs as optimal tools for systemic delivery of DOX to solid tumors.
Introduction
Extracellular vesicles (EVs) are nanosized bilayered natural structures produced by nearly all types of cells that emerged as main mediators of intercellular communication via conveying molecular constituents (proteins, RNA, DNA, and lipids) to recipient cells that can change their behavior.Citation1–3 Extensive findings highlighted the role of cancer-derived EVs as main mediators of tumor-stromal crosstalk in orchestrating the therapeutic outcome.Citation4–6 Moreover, EVs released from tumor tissues exert systemic effects that collectively contribute to the reinforcement of malignant progressionCitation7,Citation8 by promoting angiogenesis, metastasis, immunosuppression, and chemoresistance.
EVs are being studied for the past decades to understand their biological roles and are considered valuable biomarkers and prognostic tools for clinical diagnosis and response monitoring to therapies, but also as therapeutic targets (e.g., exosomes interfering with immunotherapiesCitation9 or promoting pre-metastatic niche formation in distant tissues).Citation10,Citation11 Besides EVs potential to revolutionize our understanding of the communication circuitry in cancer, there is increasing evidence regarding EVs use as cancer delivery systemsCitation12–16 due to their proteomic “signature” which could ensure the specific tumor targeting potential of EVs, but also because these communication tools are very attractive for drug delivery purposes through their analogy to liposomes. Thus, the solid research in the field of liposomes provided the grounds for acquiring fundamental knowledge about the physicochemical properties of EVs, their drug loading capacity, drug release, targeting, and stability.Citation17 Nevertheless, EVs possess the ability to be endocytosed by cancer cells more efficiently than liposomes.Citation17,Citation18 Therefore, one major focus of current EV research is their use as natural carrier systems for the delivery of therapeutics, which stems from the drawbacks observed with synthetic nanoparticulate delivery systems (e.g., liposomes, nanoparticles, and micelles) for cancer therapy. Although these therapeutic strategies have proven efficient in the past decades compared to conventional therapies (i.e., Doxil®, the first Food and Drug Administration-approved liposomal drug formulation in 1995Citation19), due to findings that associate their uptake by immune cells (e.g., macrophages) with systemic immunosuppression or the initiation of allergic reactions,Citation20,Citation21 current liposomal therapies need to be improved based on findings learned from their natural counterparts. Tailoring EVs for targeted drug delivery represents a promising strategy due to several advantages of EVs over liposomes, such as intrinsic capacity to package and deliver functional molecules across physical and biological barriers,Citation22,Citation23 their biocompatibility, as well as the feasibility of upscaling the EV production due to the optimization and development of novel technologies.Citation21
Several studies attempted to incorporate small drugs in EVs/exosomes and reported the efficient use of these natural drug delivery vehicles to convey cargo to distant sites and to increase therapeutic efficacy.Citation23–28 Thus, several studies reported the exosome-mediated delivery of doxorubicin (DOX),Citation23,Citation24 curcumin,Citation25 and paclitaxelCitation23 into tumor tissues. However, the previous in vivo studies indicated that most of the EVs administered intravenously were rapidly cleared by innate immune system cells, likely via complement system activation.Citation15 To avoid this major drawback, in this study, melanoma cell-derived EVs were decorated with a hydrophilic polymer, poly(ethylene glycol) (PEG),Citation18 due to its ability to inhibit both hydrophobic and electrostatic interactions of a variety of blood components with the EVs surface and to confer increased systemic circulation time while partially avoiding nonspecific uptake. Importantly, stealth coating the surface of nanoparticles with PEG was reported to generate an anti-PEG immune response, which could be reduced by the presence of specific surface proteins on EV membranes,Citation29,Citation30 which highlights the advantage of using biogenic particles for drug delivery.
Therefore, the aim of this study was to develop a novel EV-based nanoformulation based on PEG-functionalized EVs (PEG-EVs) to prolong their systemic circulation and avoid uptake by immune cells. These “sterically stabilized” EVs were loaded with DOX for targeting B16.F10 murine melanoma in vivo and their antitumor effects were compared with those induced by conventional liposomal DOX clinically applied (e.g., DOX encapsulated in long-circulating liposomes (LCL)). Our findings highlighted the preferential uptake of PEG-EVs by melanoma cells compared to the uptake of LCL, improved cytotoxic effects on these cells in vitro in the presence of M2 tumor-associated macrophages (TAM) and higher antitumor efficacy in vivo, reflected through the chemosensitization of tumors via interference with anti-apoptotic pathways and reduction of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) transcription factor activation and overexpression of BAX pro-apoptotic protein. Hence, this encouraging preclinical proof-of-concept study demonstrates the increased efficacy of functionalized EVs for drug delivery to solid tumors and further studies could contribute to the improvement of current liposome-based antitumor therapies.
Results
Efficient isolation and enrichment of small EVs (sEVs) via UF-SEC
Based on the elution graphic presented in , fractions 11–20 consisting of the first eluted peaks were pooled up and subjected to DLS analysis to verify the presence of nanosized particles (, supplementary figure S1 from supplementary file S1). DLS analysis showed a monodisperse population of EVs with an average size of 150 ± 28 and a polydispersity index (PDI) of 0.23 ± 0.06, highlighting the efficient isolation of EVs with a small size range (i.e. under 200 nm)Citation31,Citation32 to which we will further refer as small EVs (sEVs), as well as the lack of aggregates in these fractions. For a better delineation of EV morphological characteristics, UF-SEC-isolated EVs were subjected to TEM analysis, which also confirmed the presence of nanosized EVs, with the typical ‚cup shaped’ morphology of EVs and sizes averaging 60 ± 10 nm (). These sizes are smaller than the DLS reported sizes, as the vesicles are no longer in solution, which determines them to shrink. Protein content was on average 2.11 ± 0.42 mg/ml in a volume of approximately 500 μl. Moreover, Western blot analysis of commonly used EV markers () further confirmed the presence of EVs enriched in CD9 and TSG101 after UF-SEC isolation in accordance with previous reports.Citation33,Citation34 The absence or weak presence of cytochrome c (negative EV marker) in sEVs compared to cell lysates highlighted the efficient isolation of sEVs using the S-200-HR column, as described in the methods section.
Figure 1. (a) Elution graphic obtained after gel-filtration through the Sepharose 200-HR column. The graph is showing the absorption values at 280 nm for each fraction. Dark circles indicate the fractions that were further selected for the enrichment and characterization of nanosized EVs. (b) Western blot analysis for different EV markers in EV-enriched pooled fractions compared to parent cells. The Western blot figure shows results for CD9 and TSG101 as markers highlighting EV presence. Cytochrome c was used as a negative marker for EVs and β-actin was included as a loading control. MW (kDa) – the molecular weight of the proteins in kDa.
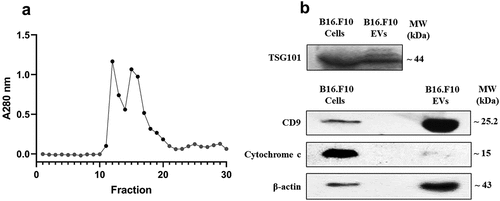
Figure 2. Transmission electron microscopy (TEM) of metabolic stress condition B16.F10 melanoma cells-derived nanosized EVs isolated through the UF-SEC technique. Exosomes were negatively stained with uranyl acetate. Dark arrows indicate the EVs, imaged as ‚cup-shaped’ structures with sizes averaging 60 nm. A – 1000 nm scale bar; B – 500 nm scale bar; C – 200 nm scale bar; D – 100 nm scale bar.
PEG-coated EVs as efficient DOX delivery systems (PEG-EV-DOX)
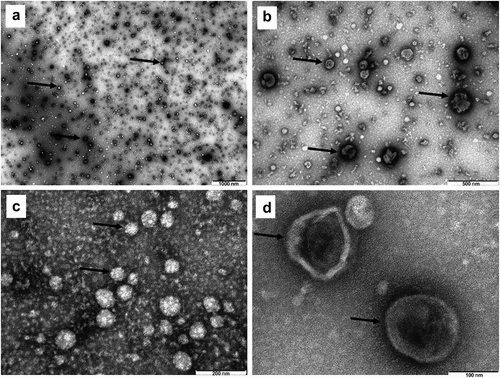
To shield UF-SEC-enriched EVs from opsonization or recognition by immune cells in the blood stream, they were decorated with PEG2000 by the post-insertion technique as described in the methods section. DLS analysis indicated that PEG-EVs had an average size of 164 ± 5 nm and an average PDI of 0.165 ± 0.07 (supplementary figure S2 from supplementary file S1), with a PEGylation efficiency of 0.1 mol % PEG concentration from total phospholipid mass of EVs, with an average concentration of 90 nmol/ml (μM). Quantitative spectrofluorimetric uptake measurements of PEG-EVs by B16.F10 cells were compared to the uptake of EVs and LCL by the same cells. The results suggested a higher uptake efficiency for EVs when compared to the uptake of PEG-EVs (by 3-fold, P = .0356) and LCL (by 14-fold, P = 0.0363), likely via exosome- and microvesicle-dependent uptake mechanisms (). Importantly, an increased uptake of PEG-EVs compared to LCL (by about 4-fold, P = .0266) was noted, being related with the tumor targeting potential of the surface proteins of EVs (). After DOX passive loading into PEG-EVs and efficient removal of the unencapsulated drug by UF-SEC, the vesicles had an average size of 117 ± 10.5 nm and DOX concentration in PEG-EV-DOX samples was around 455 µg/ml with EE% of 45.5 ± 15.4%, which suggested a high drug loading capacity with a relatively low interexperimental variation and the potential for systemic administration in vivo.
Figure 3. Spectrofluorimetric assessment of functionalized EVs (PEG-EVs) uptake by B16.F10 cells compared to natural (EV) or artificial vehicle uptake (LCL). Uptake studies were performed after 4 h incubation of B16.F10 cells with a concentration of 7.25 μM phospholipids of rhodamine (excitation at 540 nm, emission at 580 nm) fluorescently labeled long-circulating liposomes (LCL-Rhod), Cell Tracker Deep Red Dye-LabeleD EVs (EV-CTDR), and PEG functionalized EVs labeled with CTDR (excitation at 640 nm, emission at 680 nm). Results were expressed as mean ± SD of triplicate measurements and represented as Relative Fluorescence Units (RFU). Untreated B16.F10 cells were used to correct for cell autofluorescence; ns – not significant; P > .05; *, P < .05.
Proteomic signature of EVs isolated from cellular stress conditions
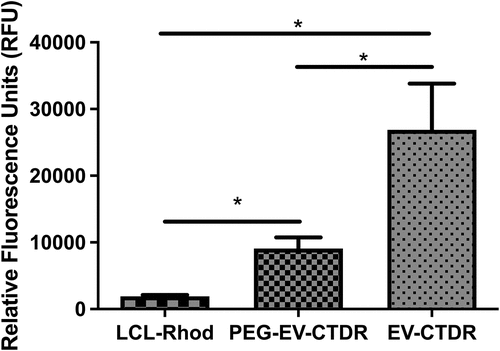
The data obtained by mass spectrometry analyses were presented in the supplementary material (S3_B16F10EVs_2uniqPept, and S4_B16F10cells_2uniqPept). A total of 1447 proteins were detected in the sEV-enriched sample, and 2666 proteins were detected in cell lysates of parent B16.F10 murine melanoma cells subjected to metabolic stress conditions. A total of 139 proteins were not detected in the EV databases for the target species and are likely uncharacterized proteins or proteins that were not previously detected in EV samples due to the metabolic stress experimental conditions (). Firstly, Venn diagram was used to confirm the presence of exosome- or microvesicle-associated proteins. For this, we inferred from the Venn diagram the intersection of the proteins detected in our samples with other dedicated murine databases used, and the results showed that 87.6% of proteins were characteristic for microvesicles and 37.66% of the proteins were specific for exosomes, thus confirming the enrichment of both exosomes and microvesicles by UF-SEC.
Figure 4. Global characterization of EVs enriched by UF-SEC that were detected by Mass spectrometry and bioinformatic analysis of EV membrane proteins. (a) Venn diagram showing the intersection of EVs released by B16.F10 murine melanoma cells subjected to metabolic stress (1% FBS) with the compendium of proteins detected in other studies involving EV research for the target species (ExoCarta, EVpedia, and Vesiclepedia); B16.F10 EVs = proteomic data from the current study. The diagram was obtained using the FunRich tool (http://www.funrich.org/). (b) Venn diagram showing the intersection of membrane proteins of B16.F10 EVs compared to membrane proteins of parent murine melanoma cells subjected to metabolic stress (1% FBS). Functional enrichment for the most frequently identified (27) membrane proteins from this study by Gene Ontology. Graphs represent the assigned classification of (c) the reactome pathway and (d) molecular function. Data were analyzed and represented using FunRich (http://funrich.org/download) tool.
Proteins associated with the membrane cell compartment detected by BUSCA were also validated by TMHMM web server and total B16.F10 cells membrane proteins were compared with the membrane proteins in sEV-enriched samples. The results showed that about half of these membrane proteins were differentially enriched in EVs (). Afterwards, frequently identified membrane proteins enriched in the EV samples that were detected by both BUSCA and TMHMM tools (27 proteins out of 214 membrane proteins), were subjected to ToppGene and FunRich analysis, and the results showed that the main roles of these proteins were associated with specific cellular uptake mediated by receptor or co-receptor activity, delineating their importance for EV internalization via exogenous protein binding (, ). Namely, membrane proteins such as tetraspanins 3, -4, -6, −9, and −14, CD9, CD63, CD82, CD109, CD151, the integrins β1, α −4, −5, -V, −6x1A, −9, and other surface proteins pivotal for EV internalization and responsible for specific interaction with recipient cells (CSPG4, CD109, L1CAM, and GPNMB) were identified via the MS analysis (Supplementary file S3). Moreover, the 27 membrane proteins were screened for interactors with IntAct and the identified interactors (n = 121) were subjected to reactome analysis to obtain an overview over the main types of immune cells that could be targeted by these EVs. These results highlighted the potential of EVs to interact with proteins belonging to the immune cell counterparts such as LAT, ITGB3, PGRMC, LTB, APP, APL2, EXT2, and LEPROT. Meanwhile, at the tumor site, EV uptake by immune cells capable of phagocytosis, such as intratumor macrophages, may depend mainly on the presence of dynamin, clathrin, galectins, and proteoglycans also present in our MS data (Supplementary information S3).Citation35,Citation36
Table 1. Membrane proteins involved in EV internalization and intercellular signaling processes
Evaluation of the antiproliferative effects of the PEG-EV-DOX on B16.F10 cells in monoculture and in co-culture with TAM
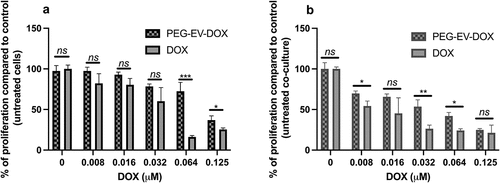
Based on the interactions found between EV surface proteins and other immune cells, we evaluated the effects of PEG-EV-DOX treatment compared to DOX treatment on the proliferation of B16.F10 cells in monoculture as well as in co-culture with M2-differentiated macrophages, which are the most abundant stromal cells at the tumor site. The results were expressed as % of proliferation compared to control (untreated cells) () and as IC50 values for each treatment administered (). The results suggested that PEG-EV-DOX exerted much higher inhibitory effects on the proliferation of B16.F10 melanoma cells co-cultured with macrophages than those exerted on the cancer cells cultured alone (IC50 of DOX = 0.123 µM in monoculture compared with IC50 of DOX = 0.0401 µM in co-culture) (). However, the same pattern was noted when free DOX was administered on monoculture and co-culture but with stronger efficacy likely due to DOX availability in vitro (, ).
Table 2. Determined IC50 values of DOX after 24 h treatment with PEG-EV-DOX or free DOX on B16.F10 cells in monoculture and in co-culture with M2 TAM
Figure 5. Anti-proliferative effects of PEG-EV-DOX and DOX on B16.F10 cells in monoculture and co-culture with M2 TAM. (a) after 24 h incubation of B16.F10 cells in monoculture with different concentrations of PEG-EV-DOX and DOX; (b) after 24 h incubation of B16.F10 cells in co-culture with M2 TAM with different concentrations of PEG-EV-DOX and DOX. Data represent mean ± SD of triplicate measurements. The unpaired t test was used to compare the effects of PEG-EV-DOX treatment to the effects of the same concentration of free DOX; ns – not significant; P > .05; *, P < .05; **, P < .01; ***, P < .001.
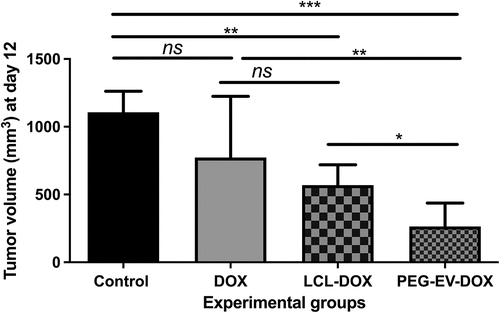
The antitumor efficacy of PEG-EV-DOX in B16.F10 melanoma-bearing mice was superior to that exerted by clinically applied liposomal DOX formulation
To test our in vitro findings regarding the preferential uptake of PEG-EVs, we assessed whether the administration of PEG-EV-DOX could exert stronger antitumor effects on the growth of B16.F10 melanoma compared to free drug (DOX) and liposomal drug (LCL-DOX). For this, syngeneic C57Bl/6 melanoma-bearing mice were i.v. injected on days 8 and 11 with a dose of 2 mg/kg DOX administered in either the free form or the designed approaches (incorporated in PEG-EVs versus encapsulated in LCL). The results were represented as tumor volumes (mm3) at day of sacrifice (day 12) and were presented in as mean ± SD from data acquired from 5 mice/experimental group. Our data revealed that the treatment with 2 mg/kg PEG-EV-DOX exerted a stronger suppression of the B16.F10 melanoma tumor growth than that induced by the administration of 2 mg/kg LCL-DOX (by 76% versus 51% inhibition compared to control tumors, P = .0369) (). The twofold increased efficacy of PEG-EV-DOX therapy versus LCL-DOX on murine melanoma might be consistent with the increased uptake observed in vitro () which could ensure a prolonged systemic circulation time due to hydrophilic PEG coating and the small size of EVs that led to a higher accumulation of DOX intratumorally and stronger tumor targeting potential of PEG-EV-DOX.Citation37
Figure 6. Antitumor effects of PEG-EV-DOX therapy on B16.F10 melanoma in vivo. For each experimental group, a dose of 2 mg/kg DOX was administered at days 8 and 11 after s.c. tumor cell inoculation, either as free drug (DOX), via artificial drug delivery vehicles (LCL-DOX) or via stabilized natural drug delivery particles (PEG-EV-DOX). Tumor volumes at sacrification day (12) were represented as mean ± SD of tumor volumes of five mice and were compared with control group (untreated tumors) or with the other experimental groups. ns – not significant; P > .05; *, P < .05; **, P < .01; ***, P < .001.
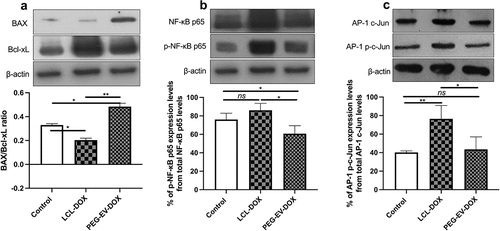
To preliminarily investigate the prognosis of PEG-EV-DOX treatment in comparison with LCL-DOX on melanoma, the intratumor apoptosis markers (such as pro-apoptotic BAX and anti-apoptotic protein Bcl-xL) as well as essential transcription factors (such as NF-κB and AP-1) involved in tumor inflammation and angiogenesis were screened. Our data suggested that PEG-EV-DOX favored pro-apoptotic and anti-inflammatory phenotype of the tumors, while LCL-DOX enhanced significantly the opposite phenotypes (e.g., anti-apoptotic and pro-inflammatory) of the same melanoma model (). Thus, PEG-EV-DOX treatment increased the intratumor production ratio of BAX/Bcl-xL compared to the same ratio in control (untreated tumors) (P = .0193), whereas LCL-DOX administration determined a reduction of the same ratio compared to control (P = .012) (). This was mainly due to the fourfold increase of BAX expression levels after PEG-EV-DOX therapy (P = .0219) compared with a significant increase by 2.5-fold of Bcl-XL when LCL-DOX was administered (P = .0318) (, graphics not shown). Moreover, our data suggested that PEG-EV-DOX exerted a slight reduction of NF-κB p65 activation (P = .0467) () and no significant changes in the activation of AP-1 c-Jun (P = .6482) () compared to control tumors. It seemed that LCL-DOX exerted pro-inflammatory action as NF-κB p65 activation was increased by 30% (P = .0443) and AP-1 c-Jun transcription factor was highly activated (by 65%, P = .0036) compared to untreated tumors ().
Figure 7. The effects of PEG-EV-DOX treatment on the intratumor production or activation of proteins associated with apoptosis (BAX, Bcl-xL), proliferation (c-Jun), and inflammation and angiogenesis (NF-κB p65). Cropped Western blot images and their representative graphs displaying the intratumor levels of proteins at day 12 when mice were sacrificed show the (a) pro-apoptotic BAX/ anti-apoptotic Bcl-xL ratio from samples run on the same blot; (b) The percentage of p-NF-κB p65 levels from total NF-κB p65 protein levels; (c) The percentage of AP-1 p-c-Jun activation from total AP-1 c-Jun protein levels; β-actin was used as loading control. The results were expressed as mean ± SD of two independent measurements; unpaired t-test was used for statistical analysis of the data; ns – not significant; P > .05; *, P < .05; **, P < .01; ***, P < .001.
Discussion
EVs are among the most attractive candidates for the development of novel drug delivery systems for therapeutic use against a variety of pathological conditions due to their intrinsic capacity to transfer functional cargo between cells and their increased specificity, biocompatibility, low immunogenicity, and toxicity profile.Citation21,Citation38 Moreover, several previous studies highlighted the feasibility of using EVs as natural cargo delivery tools for small antitumor molecules.Citation18,Citation23–25 Thus, all these characteristics could provide superior advantages over conventional drug delivery therapies based on nanoparticles, liposomes, micelles, and enhance chemotherapeutic efficacy.Citation20,Citation21 Therefore, in this study, we aimed to improve the therapeutic outcome of the cytotoxic drug DOX, by increasing its specificity for melanoma tissue using an EV-based therapeutic strategy.
Melanoma cells were used in this study based on the decreased responsiveness of human melanomas to the antitumor agent DOX, leading to therapeutic inefficacy, as shown in clinical trials.Citation39,Citation40 However, even though the commercially available Doxil®Citation19 was reported to show a lower clinical toxicity profile compared to the free drug, the antitumor efficacy of this formulation did not improve significantly.Citation41 For this purpose, we tailored an EV-based therapeutic strategy using PEG-functionalized EVs loaded with DOX for targeting B16.F10 murine melanoma, and we tested the efficacy of this approach both in vitro and in vivo.
Thus, to stimulate the EV production by B16.F10 murine melanoma cells, metabolic stress culturing conditions in vitro (1% FBS) were induced. Furthermore, to preserve EV functional characteristics for subsequent studies in vivo, UF-SEC isolation technique was used. As shown previously, this conventional EV enrichment method is fast, reliable, and optimal for our subsequent studies because it ensures the efficient isolation of small EVs with minimal contamination from the cell culture media and less vesicle aggregation due to the use of a low pressure system, which helps maintaining an EV intact biophysical surface and makes them suitable for post-isolation modification and systemic administration.Citation42–44
Physicochemical characterization of the sEVs enriched by UF-SEC was performed to achieve the minimal experimental requirements for extracellular vesicles suggested in the MISEV2018.Citation32 Collectively, our data confirmed a quick and efficient enrichment of EVs from cell culture media with an average size of 150 nm and a typical EV morphology, containing specific EV markers for both exosomes and microvesicles ( and 4a). Despite the biogenic properties of EVs and their inherent capacity to avoid the phagocytosis, melanoma, and other EV types administered in high amounts for therapeutic purposes have been shown, similarly to liposomes, to have a short half-life in circulation and to undergo rapid clearance after systemic administration either through uptake by macrophages or their accumulation in liver, spleen, and lungs early after injection (about 2 h),Citation15,Citation18,Citation45 which results in reduced targeting and drug delivery capacity to specific sites and is currently one of the major challenges ascribed to the use of EVs as drug delivery systems. To reduce the clearance of EVs after i.v. injection, they were functionalized with PEG,Citation18,Citation38,Citation46 which could potentially reduce their uptake by the reticuloendothelial system and increase EV systemic circulation time.Citation18 From this standpoint, we used the post-micellar insertion technique to directly engineer these EVs with DSPE-PEG2000 moieties that could confer them increased circulation longevity to an extent that, as far as current studies can confirm, minimally affects their structural properties, composition, and does not majorly interfere with their uptake by target cells.Citation18,Citation47,Citation48 Thus, we show that although PEGylation of EVs significantly reduced their uptake by B16.F10 cells compared to uncoated EVs () which is consistent with other studies,Citation18,Citation49 the preferential uptake of PEG-functionalized EVs by fourfold () compared to the uptake of PEG-coated liposomes demonstrates their increased tumor cell specificity and uptake mediated by lipid and protein (e.g., integrins, tetraspanins, and glycoproteins) interactions, which may likely play a major role in EV intratumor biodistribution and uptake in vivo.Citation50,Citation51
To identify EV membrane proteins potentially involved in the preferential EV uptake as well as specific uptake by recipient cells, ToppGene and FunRich functional enrichment analysis highlighted that a majority of these proteins were associated with specific cellular uptake mediated by receptor or co-receptor activity delineating their importance for EV internalization via exogenous protein binding (, ). Tetraspanins and integrins are ubiquitous surface molecules associated with EV uptake and exosome homing to specific tissues.Citation10,Citation51 In MS data we identified several such proteins as, for example, the tetraspanins 3, −4, −6, −9, and −14, CD9, CD63, CD82, CD109, CD151, and the integrins β1, α −4, −5, -V, −6x1A, −9 (Supplementary file S1). Besides the major roles of membrane EV proteins for their internalization by recipient cells, other roles are mainly associated with the regulation of tumor processes in the tumor microenvironment, such as proliferation, inflammation, apoptosis, angiogenesis, invasion, and metastasis (CSPG4, CD109, L1CAM, GPNMB, IGF2R, Plexin D1). The presence of these membrane proteins (tetraspanins, integrins, and other surface proteins) on the EV surface could explain their increased uptake by recipient cells, as shown by the uptake assay (), but also their potential to alter the behavior of recipient cells via activation of signaling pathways. Among these proteins, CD109 is a glycosylphosphatidylinositol‐anchored glycoprotein acting as a multifunctional receptor associated with aberrant cancer cell proliferation,Citation52 integrin subunit beta 1 (ITGB1) and integrin subunit alpha V (ITGAV) both bind CX3C chemokine, attracting leukocytes as well as guiding EVs toward distinct target tissues,Citation53,Citation54 transferrin receptor (TFRC) is a membrane glycoprotein that facilitates the cellular uptake,Citation55 sortilin 1 (SORT1) is involved in exosome release and transfer,Citation56 insulin-like growth factor 2 receptor (IGF2R) is a tumor suppressor and a positive regulator of T-cell coactivation, facilitating immune cell responses and tumor invasion.Citation24 Another identified protein, was the specific melanoma glycoprotein non-metastatic b (GPNMB), which is a prometastatic and immunosuppressive molecule, previously reported to be present on melanoma exosomes.Citation57,Citation58 Meanwhile, a required molecule for migration, Plexin D1, was also detected on EVs, and this protein is the receptor of the secreted protein semaphorin and together, they activate the Notch-PlexinD1 signaling axis that regulates cell migration and cancer cell metastatic potential.Citation59 Although these proteins mediate protumor processes, it is likely that PEG presence on the EV surface, as well as DOX presence as a cargo, could interfere with their tumor-promoting roles. Additionally, the low PEG coverage of EVs might interfere with the generation of anti-PEG immune response in the circulation while still allowing the interaction of EV surface proteins with target proteins from other cells as well as the phagocytosis of EVs by intratumor macrophages, ensuring the use of these EVs as “Trojan horses” to enter cancer or stromal cells and to orchestrate the therapeutic outcome.Citation29,Citation30
To test the functional efficacy of EVs as drug delivery systems, DOX was exogenously loaded into EVs by incubation with freshly isolated EVs that were functionalized with PEG by the post-insertion technique, which confers longer systemic circulation times, reduced clearance by macrophages of the reticuloendothelial system and tumor targeting potential.Citation18,Citation38,Citation60 PEG-EV-DOX samples had a drug EE% of about 45% suggesting that passive DOX loading into PEG-EVs could yield drug concentrations suitable for further in vivo studies and likely affected the least the EV membrane. However, other active drug loading methods such as electroporation or a pH gradient-dependent method could prove higher DOX entrapping efficiency into EVs. Subjecting the samples through sequential SEC steps for PEG grafting on the surface and DOX loading, led to the enrichment of sEVs with a size range around 110 nm that strongly facilitates their accumulation at the tumor site through the “Enhanced Permeability and Retention (EPR) Effect” that is characteristic for the leaky solid tumor vasculature.Citation61
To assert the potential of this novel EV-based nanoformulation for DOX delivery, which to our knowledge has not been previously described in functional studies in vivo, we screened for the antiproliferative effects of PEG-EV-DOX on B16.F10 cells as well as in co-cultures of melanoma cells with M2 TAM, which are pivotal players in mediating melanoma cells chemoresistance.Citation62,Citation63 Our findings highlighted an increased antiproliferative effect of PEG-EV-DOX on melanoma cells and M2 TAM in coculture compared to the effects on melanoma cells in monoculture, as shown by the threefold decrease in the IC50 value in coculture (IC50 = 0.0401 µM versus IC50 = 0.1272 µM) inferred from the proliferation assay. This would suggest that PEG-EV-DOX could interfere with the tumor promoting role of M2 TAM and predict an increased therapeutic outcome in vivo. Moreover, the abovementioned specific EV-M2 TAM protein interactions could account for the increased antiproliferative effect displayed by PEG-EV-DOX administration in co-cultures (). Although a stronger antiproliferative capacity was observed with free DOX treatment in vitro, likely due to the readily availability of the drug as compared to PEG-EV-DOX, which have a delayed uptake by cells,Citation64 the same effect was not observed in vivo, as free DOX administration did not significaltly inhibit tumor growth compared to control (untreated tumors) (). Importantly, our results showed a significant suppression of B16.F10 melanoma tumor growth (by twofold, P = .0369) when a 2 mg/kg DOX dose was administered via PEG-functionalized EVs (PEG-EV-DOX) compared to the administration of the same drug dose under LCL form (LCL-DOX) (). As these data suggested an increased antitumor advantage of EVs as drug delivery systems, we further screened for the effects of functionalized PEG-EV-DOX versus the effects of LCL-DOX on the expression levels of key proteins involved in apoptosis and on the activation of pivotal transcription factors associated with tumor proliferation, inflammation, and angiogenesis ().
Literature findings suggested that melanoma progression displays a phenotype with a decreased pro-apoptotic BAX and an increased anti-apoptotic Bcl-xL.Citation65 Western blot results indicated that PEG-EV-DOX administration altered the BAX/Bcl-xL intratumor ratio toward a more pro-apoptotic phenotype compared to the effects of LCL-DOX, which highly increased Bcl-xL levels () and consistently with other studies, is an indicator of melanoma stemness, aggressiveness, resistance to apoptosis, and poor therapeutic outcome.Citation66,Citation67 Notably, PEG-EV-DOX significantly increased BAX expression levels, an effect reported in the literature to increase tumor susceptibility to chemotherapy, as this protein is considered to be a pivotal regulator of apoptosis and inducer of programmed cell death.Citation66 Additionally, our results show that LCL-DOX induced a strong intratumor activation of AP-1 c-Jun (by 65%, P = .0036) and increased the activation levels of NF-κB p65 (by 30%, P = .0443) (). Since the activation of both of these transcription factors is tightly linked with tumor progression and metastasis, as well as resistance to apoptosis,Citation68–70 our results suggest that LCL-DOX induced a more aggressive tumor phenotype. Contrarily, PEG-EV-DOX administration did not induce the activation of AP-1 c-Jun and moderately reduced the NF-κB p65 activation (P = .0467), which could account for a less aggressive melanoma phenotype and susceptibility to DOX-induced apoptosis () reinforced by BAX overexpression.Citation62,Citation66
Conclusions
Altogether, our study showed confirmatory data for a rapid, cost-effective, reproducible, and repeatable UF-SEC approach for the isolation and enrichment of small EVs from B16.F10 melanoma cells subjected to metabolic stress conditions. This procedure minimally affects EV physical and functional characteristics, making them valuable tools for tailoring novel drug delivery systems that could be exploited like Trojan Horses. After grafting with PEG and loading with DOX, this PEG-EV-DOX formulation displayed an encapsulation efficacy of ~45% and increased antitumor effects in vivo, reflecting their suitable use for DOX delivery to B16.F10 murine melanoma. Our results highlighted the feasibility of using PEG-EV-DOX as melanoma-targeted therapy in vivo, which was highly superior to clinically applied liposomal DOX in terms of antitumor efficacy and intratumor molecular mechanisms of action. Future investigations should also focus on testing the effects of chronic therapy of melanoma tumors with PEG-EV-DOX and the assessment of long-term exposure of mice to the proposed drug delivery systems. However, for further clinical applications, we would need to investigate which of the bioactive molecules conveyed are pivotal for their targeting potential, and we envision the use of reconfigured EVs (exosome mimetics field) derived from non-tumor or immune cells (e.g., red blood cells, MSCs, and T cells),Citation71 functionalized with molecules that shield them from nonspecific uptake in systemic circulation and enhance their tumor targeting potential. Another important aspect is the feasibility of EVs large-scale production, which is pivotal for further clinical applications of these natural reconfigured drug delivery systems. The methodology proposed in this paper is suitable for upscaling. Importantly, the culturing conditions under metabolic stress stimulate the EV productionand the SEC technique, which would ensure the enrichment of biologically functional EVs from cell culture media while better conserving their characteristics, is currently being used for biotechnological applications and large-scale purification/enrichment of biomolecules. Nevertheless, we consider that these preliminary results are promising, and further validation of the methodology used according to the guidelines in this field,Citation72 as well as the optimization of the drug loading efficiency and identification of EV pivotal surface proteins involved in specific tumor targeting, and some of the perspective studies, which will better highlight the potential use of these natural drug delivery systems.
Material and methods
Cells
Murine melanoma B16.F10 (ATCC, CRL-6475) cancer cells were cultured in Dulbecco’s Modified Eagle medium (DMEM, Lonza), supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 IU/ml penicillin and 100 µg/ml streptomycin (DE17-602E, Lonza), and 4 mM L-Glutamine (BE17-605E, Lonza). Cancer cells were maintained as a monolayer at 37°C in a 5% CO2 humidified atmosphere.
Murine tumor model
Male C57Bl/6 mice (6–8 weeks of age) were obtained from the Cantacuzino Institute (Bucharest, Romania) and animals were kept in standard housing with standard rodent chow and water available ad libitum under a 12-h light/dark cycle. Experiments were performed according to the national regulations and were approved by the local animal experiments ethical committee (registration no. 31444/27.03.2017). For tumor induction, 106 B16.F10 cells were inoculated subcutaneously (s.c.) in the right flank of mice. The B16.F10 tumors became palpable at day 7 after cell inoculation. Tumor size was measured regularly with a caliper starting with day 7 and the tumor volume was calculated using the formula V = 0.52×a2×b, where a is the smallest and b is the largest superficial diameter (in mm). Body weight of mice was monitored regularly during treatments. At the end of the experiments, mice were sacrificed by CO2 asphyxiation, and tumors were collected for post-mortem analysis.
All methods used for obtaining the PEG-EV-DOX were summarized and shown in supplementary material, S6_Study-design.
In vitro metabolic stress conditions for enhancing EV production
The production of EVs was described to be enhanced by cellular stress conditions, which render cells more aggressive, such as nutrient deprivation.Citation73,Citation74 Thus, to harvest extracellular vesicles, cells were cultured to reach 60–70% confluency under normal culturing conditions. Afterward, cells were subjected to metabolic stress consisting of complete media containing 1% Exosome Depleted FBS (Thermo Scientific, A2720801), supplemented with 100 IU/ml penicillin, 100 μg/ml streptomycin, and 4 mM L-Glutamine, until cells reached 95% confluency.
Extracellular vesicles isolation and purification
The culture media was collected for isolation and purification of EVs using ultrafiltration coupled with size-exclusion chromatography (UF-SEC), as this combination of techniques ensures the efficient enrichment of small-sized EVs suitable for compositional and functional studies.Citation42,Citation43 First, the media was centrifuged for 10 min at 300xg to remove dead cells, for 10 min at 2500xg to remove cell debris and apoptotic bodies, and then filtered through a 0.2 μm membrane to remove large vesicles and aggregates. The media was concentrated using 100 kDa centrifugal ultrafiltration unit Amicon Ultra (UFC9100, Millipore/Sigma Aldrich) according to the instructions of the manufacturer. For EV enrichment, 1 ml of concentrated cell culture media from approximately six T150 flasks was subjected to SEC on a 30 cm length and 1.5 cm diameter Sephacryl S-200 HR (Sigma, GE17-0584-01) column at 4°C using sterile phosphate-buffered saline (PBS) as mobile phase. The absorbance of the collected fractions (approximately 30 fractions of 1 ml each) was measured at 280 nm to determine the EV-containing fractions, which were then further concentrated to a smaller volume, characterized, and used for subsequent studies.
Dynamic Light Scattering (DLS)
The size of EVs was determined by DLS analysis, also known as photon correlation spectroscopy, using the Zetasizer Nano ZS analyzer (Malvern Instruments, Malvern, UK). This technique measures the fluctuations in the intensity of scattered light as a function of time when light strikes particles in suspension and is reliable for the evaluation of monodisperse particle populations. The same instrument was also used for the measurement of the zeta potential of these particles throughout the experiments. For sample analysis, EVs were diluted 100-fold in PBS and analyzed at an angle of 90°, at 25°C. All measurements were carried out in triplicate and were reported at mean ± standard deviation (SD).
Transmission Electron Microscopy (TEM)
To confirm EV presence and their morphological characterization, TEM was used. This is a reliable technique for the examination of the EV quality and size, with sizes smaller than those detected by DLS due to the EV dehydration. For this, a drop of enriched EVs was added for 1 minute on a formvar-coated carbon grid followed by negative staining with uranyl acetate. After drying, the grids were imaged with a Jeol JEM 1010 instrument coupled with a Mega View III CCD camera for image capturing.
Western blot analysis for EV biomarker validation
EV protein concentration was determined by Bradford assay (Sigma-Aldrich). For validation of EV isolation and purification, 20 µg of EV proteins were subjected to denaturing electrophoresis (SDS-PAGE) and Western blot technique for identification of specific EV markers (CD9 Antigen (CD9) and Tumor susceptibility gene 101 (TSG101)),Citation33 as well as a negative control for EVs (cytochrome c)Citation34 compared to the presence of the same markers in B16.F10 cell lysates. Cell lysis buffer consisted of 10 mM HEPES, 200 mM NaCl, 1% Triton X, 10 mM MgCl2, 1 mM DTT and Complete Protease Inhibitor Cocktail tablets (11697498001, Roche Applied Science) were added to the lysis buffer. For Western blot, the nitrocellulose membranes were incubated overnight at 4°C with primary antibodies for CD9 (mouse monoclonal IgG anti-mouse, 1:1000 dilution, sc-13118, Santa Cruz Biotechnology, Santa Cruz, CA, USA), for TSG101 (mouse monoclonal IgG anti-mouse, 1:1000 dilution, MA5-32463, Thermo Fisher Scientific), for cytochrome c (mouse monoclonal IgG anti-mouse, dilution 1:200, JA5204, Calbiochem), and for β-actin (rabbit polyclonal IgG anti-mouse, 1:1000 dilution, sc-130656, Santa Cruz Biotechnology) as a loading control. Secondary antibodies were horseradish peroxidase (HRP)-labeled IgG goat anti-rabbit (sc-2004) or goat anti-mouse (sc-2005) (1 h incubation, 1:2500 dilution, Santa Cruz Biotechnology). All antibodies were diluted in 5% nonfat dry milk (Bio-Rad Laboratories, Hercules, CA, USA) prepared in Tris-buffered saline with 0.1% Tween-20 (Honeywell Atlas Ltd., London, UK). The immunocomplexes were developed using Clarity Western ECL (Bio-Rad, 170–5061) and the blots were exposed to a Kodak X-ray film (Z358487, Eastman Kodak, Rochester, NY, USA) for about 1–5 min. Films were imaged using a ChemiDoc Touch Imaging System (Bio-Rad). Uncropped images from the Western blot analysis are presented in supplementary figure S3 from supplementary file S2.
Nanoscale liquid chromatography coupled to tandem mass spectrometry (nano LC-MS/MS)
For mass spectrometry, we used liquid chromatography-mass spectrometry (LC-MS) chromasolv solvents (water, acetonitrile), ammonium bicarbonate, iodoacetamide (I6125), and dithiothreitol (DTT, 43815) from Sigma-Aldrich (St Louis, MO, USA), formic acid, eluent additive for LC-MS (56302, Sigma-Aldrich, Merck Sigma, UK), sequencing grade modified Trypsin (Promega, Madison, WI, USA). For sample preparation and nano-liquid chromatography tandem mass spectrometry (nanoLC-MS/MS) analysis, UF-SEC enriched sEVs were pooled from two independent isolations and lysed. The same procedure was applied for EV donor murine melanoma B16.F10 cells cultured under the metabolic stress condition (1% FBS). 50 µg of protein were simultaneously separated by SDS-PAGE and stained with Coomassie Brilliant Blue. Ten gel slices were excised from each lane/sample, cut into ~ 1 mm3 pieces, destained with a solution of 50 mM ammonium bicarbonate in 50% acetonitrile and subjected to an in-gel digestion protocol adapted from previously described protocols,Citation75,Citation76 using sequencing grade modified trypsin as protease. The extracted peptides were dried in a vacuum concentrator (Speed-Vac). All the samples were analyzed by nanoLC-MS/MS using an EASY nLC II (Thermo Fisher Scientific, Germany) online coupled to an LTQ™ – Orbitrap Velos Pro™ mass spectrometer (Thermo Fisher Scientific). The peptides were resuspended in 0.1% formic acid and 2% acetonitrile solution (solvent A). NanoLC analysis involved first trapping and desalting of the peptides on a C18 trap column (2 cm × 100 µm) (Thermo Fisher Scientific), followed by the chromatographic separation of the peptides on a C18 analytical column (10 cm × 75 µm), which was connected online to the mass spectrometer using a stainless steel emitter) (Thermo Fisher Scientific). The peptides were separated using a 90 min gradient of 2 to 30% solvent B (0.1% formic acid and 98% acetonitrile solution). A data-dependent acquisition method was implemented: a survey/precursor ion scan (300– 1650 m/z interval, resolution of 60,000 at 400 m/z) with Orbitrap detection, followed by five consecutive collision-induced dissociation fragmentation scans (performed in the linear ion trap) for the first five most intense ions from the survey scan, with +2, +3, or higher charge states. Two technical replicates were performed for each of the ten gel slices obtained from a sample.
The nanoLC-MS/MS data analysis was performed as follows. For peptide identification, the raw files were searched with the Sequest HT algorithm integrated into Proteome Discoverer v1.4, against the murine proteome (manually reviewed and annotated Mus musculus database downloaded from Swiss-Prot), using the following settings: trypsin (full) as enzyme, with maximum 2 missed cleavages, 10 ppm for precursor mass tolerance, 0.6 Da for fragment mass tolerance, carbamidomethylation (+57.021 Da) of cysteine residues as static modification and oxidation of methionine residues (+15.995 Da) as variable modification. A decoy database (which contained the reversed protein sequences from the mentioned murine proteome) was used to validate the identified peptide spectrum matches (PSMs), using the Target Decoy PSM Validator node. Only PSMs at 1% FDR and with a precursor mass tolerance of maximum 5 ppm and high confidence (minimum 99%) were kept in the final report. At least two unique peptides were required for each protein group to assess the identification of the proteins. Supplementary details regarding the MS analysis and the proteomic data are presented in the Supplementary file S5.
Bioinformatic qualitative analysis of proteomic data
To characterize the sEVs enriched by the UF-SEC technique, proteomic data were searched against the main EV databases: ExoCarta for published exosomal proteins (http://exocarta.org/), EVpedia (evpedia.info/) to verify the EV enrichment, and, respectively, Vesiclepedia (http://www.microvesicles.org/). Venn diagram was used to determine the prevalence of exosome versus microvesicles marker proteins in the obtained samples. Computational tools such as BUSCA (http://busca.biocomp.unibo.it)Citation77, and TMHMM Server v.2.0 (http://www.cbs.dtu.dk/services/TMHMM/) were used for predicting protein subcellular localization or to detect EV membrane proteins. The web-based bioinformatics functional tools such as FunRich (http://funrich.org/download)Citation78 and ToppGene (https://toppgene.cchmc.org/enrichment.jsp), and reactome analysis (https://reactome.org)Citation79 were used for Gene ontology enrichment analysis to functionally characterize different proteins (membrane, cargo) from the EVs obtained under metabolic stress culturing conditions. The statistical analyses of all the obtained data were performed using the standard settings of the tools used, and only interrogated proteins highlighted by the provided statistical indices (pValue, FDR B&H, FDR B&Y, respectively, Bonferroni), reflecting the degree of credibility for each individually analyzed protein were taken into consideration. Furthermore, IntAct molecular interaction database (http://www.ebi.ac.uk/intact)Citation80 was used to identify potential interactors of EV surface proteins, which could support findings regarding their preferential uptake by recipient cells.
PEG stabilization of EVs
Based on previous findings that showed an increase in the in vivo circulation time of PEGylated EVs (PEG-EVs), phenomenon known as stabilization (stealthiness) with analogy to liposomes, UF-SEC-isolated sEVs were decorated with polyethylene glycol-2000 (PEG2000) by the post-insertion method, which is the main preformed liposome synthetic post-modification strategy and has increased stability advantages over the classical pre-insertion method.Citation18,Citation81,Citation82 This technique relies on the incubation of EVs with DSPE-PEG2000 micelles at a temperature that increases membrane fluidity and flip-flop lipid movements contributing to the spontaneous transfer of DSPE-PEG from micelles to EVs.Citation18 These stable PEG anchors on the EV surface create a hydrophilic protective layer that prevents their opsonization or interaction with mononuclear phagocytes in the bloodstream that are responsible for their nonspecific uptake and accelerated systemic clearance.Citation81,Citation82 A micellar suspension of 2.75 mg/ml DSPE-PEG2000 (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] ammonium salt) (Lipoid GmbH, Ludwigshafen, Germany) was prepared in sterile PBS, at a concentration above the critical micelle concentration (0.5– 1 µm) that ensures the formation of micelles in suspension.Citation83 The suspension was heated at 60°C under agitation for 10 minutes and for reducing the micellar dimension, the suspension was sonicated for 5 minutes (2 seconds sonication and 5 seconds break) at 10% amplitude, room temperature. Afterward, EV samples were mixed in a 1:1 (v/v %) ratio with the micellar suspension for 2 h at 40°C with agitation, which is the optimal temperature which maintains EV characteristics.Citation18 PEG-EVs were purified from micelles by separation on a Sephacryl S-200 HR column using PBS as a mobile phase, and the fractions showing absorbance at 280 nm were concentrated to a smaller volume by ultrafiltration and subjected to DLS analysis to confirm EV presence.
Measurement of PEG functionalization of EVs
PEGylation of EVs for their use as therapeutic drug delivery systems interferes with the particle clearance from the systemic circulation and increases drug accumulation to the tumor tissue.Citation18 To determine the PEGylation efficiency of UF-SEC enriched sEVs, a Methoxy-Polyethylene Glycol (mPEG) ELISA kit was used (MPEG, Life Diagnostics) according to the manufacturer’s instructions. Briefly, this assay consists of 96-well plates coated with a mouse monoclonal antibody for capturing the polyoxyethylene backbone of PEG (catalog# 9B5-6-25-7) and uses an anti-mPEG mouse monoclonal HRP-antibody for detection. First, HRP anti-mPEG was added to the wells, and then PEG-EV or standards were also added and incubated for 1 h on a plate shaker. Afterward, wells were washed, and TMB reagent was added for 20 minutes to allow the development of a blue color. The reaction was stopped by the addition of HCl which changed the color to yellow for which the absorbance was measured at 450 nm. The extent of PEGylation was expressed as mol% of mPEG-conjugated phospholipid (incorporated into the EV bilayer) from total EVs phospholipids.
DOX incorporation into PEG-EVs
Drug loading was achieved by passive loading into PEG-EVs via incubation with a solution of 4 mg/ml DOX (Sigma-Aldrich, cat. no. D2975000) in sterile PBS in a volumetric ratio of 1:1 for 2 h at 37°C with agitation. To remove unincorporated DOX, the mix was run on a Sephadex G-25 (G2580, Sigma Aldrich) gel-filtration column (60 cm) using sterile PBS as mobile phase, at room temperature, and 80 fractions of 1 ml were collected. The fractions 37–51 containing PEG-EV-DOX that presented high absorbance at 280 nm were concentrated by ultrafiltration and used fresh for further experiments.
Physico-chemical characterization of PEG-EV-DOX
The size of the prepared PEG-EV-DOX was measured by DLS. The drug concentration of PEG-EV-DOX was determined in triplicate by diluting the samples 50-fold in ultrapure H2O and measuring the absorbance at a wavelength of 480 nm. DOX concentration was calculated from a standard curve of free DOX using serial dilutions between 1.00 and 1.5 µg/ml and reported as mean ± SD of two independent experiments. DOX encapsulation efficiency (EE) was calculated as % of the entrapped drug using the formula EE (%) = (Entrapped DOX/Total DOX) x100, where entrapped DOX was the quantity of DOX determined spectrophotometrically from PEG-EV-DOX and the total DOX represented the quantity of DOX initially used for incubation with the PEG-EVs. The EE% was calculated as mean ± SD of two independent experiments.
Uptake studies
To assess the quantitative uptake of PEG-EVs in comparison with LCLs, we performed spectrofluorimetric and fluorescence microscopy studies. A fluorescent LCL formulation was obtained using 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (ammonium salt) (810150 C, Avanti Polar Lipids) to confer fluorescent properties to the LCLs (LCL-Rhod) which were prepared by lipid film hydration method and characterized for a manuscript in preparation (data not shown). EVs or PEG-EVs were stained using the Cell Tracker™ Deep Red (CTDR) (C34565, Molecular probes by Life Technologies) dye, which is nontoxic, does not affect membrane lipids and displays fluorescence for a long time. The dye becomes fluorescent upon permeating the lipid membrane and subsequent transformation to a cell-impermeant product.Citation84 For staining, freshly obtained EVs or PEG-EVs, as described in the methods section above, were incubated in a 1:1 volumetric ratio with a solution of 10 µM CTDR for 4 hours at 37°C, then purified on a CL-4B (CL4B200, Sigma Aldrich) column at 4°C for removing the unincorporated dye. Fractions containing the EVs were identified spectrophotometrically at 280 nm and concentrated through ultrafiltration using 100 kDa filtration units. Lipids were extracted from EVs, PEG-EVs and LCL-Rhod with the Bligh and DyerCitation85 method, and total lipid concentration was determined using the RouserCitation86 method to further ensure the administration of the same lipid concentration for uptake studies.
To evaluate the efficiency of PEG-EVs uptake by B16.F10 cells compared to the uptake of EVs and LCL-Rhod, quantitative spectrofluorimetric measurements were performed. For this, B16.F10 cells were seeded at a 5 × 103 cells/well in a 96-well microplate for fluorescence and allowed to attach for 24 h. Afterward, cells were treated with 7.25 µM of phosholipids from either LCL-Rhod, EV-CTDR, PEG-EV-CTDR for 4h at 37°C, then washed with 100 μl sterile PBS and covered with another 100 μl of sterile PBS. For rhodamine, the excitation wavelength of 540 nm was used, and emission was monitored at 580 nm. CTDR fluorescence was monitored by excitation at a wavelenght of 640 nm and the measurement of the emitted light at 680 nm. Fluorescence intensity was measured as Relative Fluorescence Units (RFU) using the FLUOstar Omega plate reader (BMG Labtech). Cell autofluorescence (untreated B16.F10 cells) was subtracted from all other measurements and final results were expressed as mean ± SD of triplicate measurements.
Proliferation assay
The in vitro antiproliferative effects of PEG-EV-DOX treatment was assessed on B16.F10 melanoma cells in monoculture as well as in co-culture with bone marrow differentiated M2 TAM with 10 ng/ml granulocyte-macrophage colony-stimulating factor (GM-CSF, Cell Signaling Technology, MA, USA) and 20 ng/ml interleukin-4 (IL-4, Cell Signaling Technology, MA, USA), as previously described by Rauca et al., 2018.Citation87 For this, ELISA BrdU-colorimetric immunoassay (Roche Applied Science, Penzberg, Germany) was used, as previously described and according to the manufacturer’s instructions.Citation62 Thus, to test the efficacy of PEG-EV-DOX as compared to free DOX, B16.F10 melanoma cells were seeded in a 96-well at a ratio of 5000 cells/well for monocultures, while for co-cultures a ratio of 4000 B16.F10 cells to 1000 M2 TAM/well was used. This cell density ratio (4:1) was reported to approximate the in vivo physiological conditions of murine melanoma development.Citation88 After cells were allowed to attach for 24 h, serial concentrations of PEG-EV-DOX or DOX (ranging between 0.008 and 0.125 µM DOX) were tested in triplicate to assess the IC50 values after 24 h incubation with the treatment. The results were expressed as % of proliferation compared to control (untreated cells in monoculture and, respectively, in co-culture).
In vivo antitumor efficacy of PEG-EV-DOX in B16.F10 murine melanoma-bearing mice
The antitumor effects of PEG-EV-DOX on melanoma growth were compared with the effects of clinically applied PEG-coated liposomal DOX (LCL-DOX) on the same in vivo tumor model. LCL-DOX was prepared and characterized as previously described by Licarete et al., 2020.Citation62 To assess the effects of the stabilized PEG-EV-DOX on murine melanoma tumor growth doses of 2 mg/kg DOX, 2 mg/kg LCL-DOX and 2 mg/kg PEG-EV-DOX were i.v. injected at days 8 and 11 after s.c. tumor induction in syngeneic C57BL/6 mice, according to the treatment scheme previously reported.Citation89 Tumor size and body weights were measured daily. Each experimental group consisted of 5–6 mice. At day 12 after tumor cell inoculation, mice were sacrificed, and tumors were collected for post-mortem analysis.
Western blot analysis
Isolated tumors were weighed and then pooled to obtain tumor tissue lysates for each group. The protein content of the tumor tissue homogenates was assessed by biuret method.Citation90 To determine the effects of functionalized PEG-EV-DOX compared to the effects of LCL-DOX on the levels of key transcription factors for tumor inflammation, angiogenesis, and apoptosis, Western blot analysis was performed as described previously.Citation2 20 µg of protein was loaded per lane for each sample. Primary antibodies were incubated overnight at 4°C for p65 subunit of the NF-κB (NF-κB p65; mouse monoclonal IgG anti-mouse, 1:500 dilution, sc-56735, Santa Cruz Biotechnology, Santa Cruz, CA, USA), phosphorylated NF-κB p65 (p-NF-κB p65; mouse monoclonal IgG anti-mouse, 1:500 dilution, sc-33039, Santa Cruz Biotechnology), c-Jun subunit of activator protein 1 (AP-1 c-Jun; rabbit polyclonal IgG anti-mouse, 1:1000 dilution, sc-45, Santa Cruz Biotechnology), phosphorylated AP-1 c-Jun (AP-1 p-c-Jun; monoclonal IgG anti-mouse 1:1000, sc-7891-R, Santa Cruz Biotechnology), B-cell lymphoma–extra-large anti-apoptotic protein (Bcl-xL; rabbit monoclonal IgG anti-mouse, 1:500 dilution, 2764, Cell Signaling), Bcl-2-associated X protein (BAX; rabbit polyclonal IgG anti-mouse, 1:500 dilution, 2772S, Cell Signaling), and β-actin (rabbit polyclonal IgG anti-mouse, 1:1000 dilution, sc-130656, Santa Cruz Biotechnology). Secondary antibodies were HRP-labeled IgG goat anti-rabbit (sc-2004) or goat anti-mouse (sc-2005) secondary antibodies (1 h incubation, 1:2500 dilution, Santa Cruz Biotechnology). Uncropped images from the Western blot analysis are presented in supplementary figure S4 from supplementary file S2.
Statistical analysis
For the statistical analysis, we used GraphPad Prism software version 6. To assess significant differences between two experimental conditions we used the unpaired t-test. To determine significant differences between more experimental conditions we used one-way ANOVA with Bonferroni correction for multiple comparisons. For the calculation of the IC50 values, we used non-linear regression to obtain dose–response curves, from which the values were calculated. A P value lower than 0.05 was considered statistically significant.
Abbreviations
EV(s), Extracellular vesicle(s); PEG, Polyethylene glycol; DOX, Doxorubicin; UF-SEC, Ultrafiltration coupled with size-exclusion chromatography; LCL(s), Long-circulating liposome(s); TAM, Tumor-associated macrophages; NF-κB, Nuclear factor kappa-light-chain-enhancer of activated B cells; BAX, Bcl-2-associated X protein; DMEM, Dulbecco’s Modified Eagle medium DMEM; FBS, Fetal bovine serum; PBS, Phosphate-buffered saline; DLS, Dynamic Light Scattering; TEM, Transmission Electron Microscopy; SD, Standard deviation; SDS-PAGE, Sodium dodecyl sulphate-polyacrylamide gel electrophoresis; CD9, CD9 antigen; TSG101, Tumor susceptibility gene 101; HEPES, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; IgG, Immunoglobulin G; HRP, Horseradish peroxidase; LC-MS, Liquid chromatography coupled with mass spectrometry; DTT, Dithiothreitol; nanoLC-MS/MS, Nano-liquid chromatography tandem mass spectrometry; PSMs, Peptide spectrum matches; DSPE-PEG2000, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000; mPEG, Methoxy-Polyethylene Glycol; EE, Encapsulation efficiency; HBS, HEPES-buffered saline; Rhod, Rhodamine; CTDR, Cell Tracker™ Deep Red; RFU, Relative Fluorescence Units; AP-1 c-Jun, c-Jun subunit of activator protein-1; Bcl-xL, B-cell lymphoma-extra-large anti-apoptotic protein; ANOVA, Analysis of variance; IC50, Half-maximal inhibitory concentration; PDI, Polydispersity index; CSPG4, Chondroitin sulfate proteoglycan 4; L1CAM, L1 Cell Adhesion Molecule; GPNMB, Melanoma glycoprotein non-metastatic b; LAT, Linker for activation of T-cells family member 1; ITGB3, Integrin beta-3; PGRMC, Sigma-2 receptor and progesterone receptor membrane component 1; LTB, Lymphotoxin Beta; APP, Amyloid precursor protein; APL2, AP-1 complex subunit beta-1; EXT2, Exostosin glycosyltransferase-2; LEPROT, Leptin receptor gene-related protein; SORT1, Sortilin 1; LRP, Lipoprotein receptor-related protein; IGF2R, Insulin-like growth factor 2 receptor; ATP1A1, Sodium/potassium-transporting ATPase subunit alpha-1; HYOU1, Hypoxia Up-Regulated 1; ITGB1, Integrin beta-1; ENPP2, Ectonucleotide Pyrophosphatase; SLC3A2, 4F2 cell-surface antigen heavy chain; TFRC, Transferrin receptor.
Supplemental Material
Download Zip (4.4 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
Additional information
Funding
References
- Yuana Y, Sturk A, Nieuwland R. Extracellular vesicles in physiological and pathological conditions. Blood Rev. 2013;27(1):31–39. doi: 10.1016/j.blre.2012.12.002.
- Patras L, Fens MHAM, Vader P, Barendrecht A, Sesarman A, Banciu M, Schiffelers R. Normoxic tumour extracellular vesicles modulate the response of hypoxic cancer and stromal cells to doxorubicin in vitro. Int J Mol Sci. 2020;21(17):5951. doi: 10.3390/ijms21175951.
- Saber SH, Ali HEA, Gaballa R, Gaballah M, Ali HI, Zerfaoui M, Abd Elmageed ZY. Exosomes are the driving force in preparing the soil for the metastatic seeds: lessons from the prostate cancer. Cells. 2020;9(3):564. doi: 10.3390/cells9030564.
- Patras L, Banciu M. Intercellular crosstalk via extracellular vesicles in tumor milieu as emerging therapies for cancer progression. Curr Pharm Des. 2019;25(17):1980–2006. doi: 10.2174/1381612825666190701143845.
- Steinbichler TB, Dudás J, Skvortsov S, Ganswindt U, Riechelmann H, Skvortsova -I-I. Therapy resistance mediated by exosomes. Mol Cancer. 2019;18(1):58. doi: 10.1186/s12943-019-0970-x.
- Guo Q, Wang H, Yan Y, Liu Y, Su C, Chen H, Yan Y, Adhikari R, Wu Q, Zhang J, et al. The role of exosomal microRNA in cancer drug resistance. Front Oncol. 2020;10. doi:10.3389/fonc.2020.00472.
- Whiteside TL. Tumor-derived exosomes and their role in cancer progression. Adv Clin Chem. 2016;74:103–141. doi: 10.1016/bs.acc.2015.12.005.
- Bandari SK, Purushothaman A, Ramani VC, Brinkley GJ, Chandrashekar DS, Varambally S, Mobley JA, Zhang Y, Brown EE, Vlodavsky I, et al. Chemotherapy induces secretion of exosomes loaded with heparanase that degrades extracellular matrix and impacts tumor and host cell behavior. Matrix Biol. 2018;65(2018):104–118. DOI:10.1016/j.matbio.2017.09.001.
- Olejarz W, Dominiak A, Żołnierzak A, Kubiak-Tomaszewska G, Lorenc T. Tumor-derived exosomes in immunosuppression and immunotherapy. J Immunol Res. 2020;2020:1–11. doi: 10.1155/2020/6272498.
- Peinado H, Alečković M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, Hergueta-Redondo M, Williams C, García-Santos G, Ghajar CM, et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med. 2012;18(6):883–891. DOI:10.1038/nm.2753.
- Rodrigues G, Hoshino A, Kenific CM, Matei IR, Steiner L, Freitas D, Kim HS, Oxley PR, Scandariato I, Casanova-Salas I, et al. Tumour exosomal CEMIP protein promotes cancer cell colonization in brain metastasis. Nat Cell Biol. 2019;21(11):1403–1412. DOI:10.1038/s41556-019-0404-4.
- Vader P, Breakefield XO, Wood MJA. Extracellular vesicles: emerging targets for cancer therapy. Trends Mol Med. 2014;20(7):385–393. doi: 10.1016/j.molmed.2014.03.002.
- Henderson MC, Azorsa DO. The genomic and proteomic content of cancer cell-derived exosomes. Front Oncol. 2012;2. doi:10.3389/fonc.2012.00038.
- Simona F, Laura S, Simona T, Riccardo A. Contribution of proteomics to understanding the role of tumor-derived exosomes in cancer progression: state of the art and new perspectives. PROTEOMICS. 2013;13(10–11):1581–1594. doi: 10.1002/pmic.201200398.
- Smyth T, Kullberg M, Malik N, Smith-Jones P, Graner MW, Anchordoquy TJ. Biodistribution and delivery efficiency of unmodified tumor-derived exosomes. J Controlled Release. 2015;199:145–155. doi: 10.1016/j.jconrel.2014.12.013.
- Tkach M, Théry C. Communication by extracellular vesicles: where we are and where we need to go. Cell. 2016;164(6):1226–1232. doi: 10.1016/j.cell.2016.01.043.
- Kotmakçı M, Bozok Çetintaş V. Extracellular vesicles as natural nanosized delivery systems for small-molecule drugs and genetic material: steps towards the future nanomedicines. J Pharm Pharm Sci. 2015;18(3):396. doi: 10.18433/j36w3x.
- Kooijmans SAA, Fliervoet LAL, van der Meel R, Fens MHAM, Heijnen HFG, van Bergen En Henegouwen P.M.P., Vader P, Schiffelers RM. PEGylated and targeted extracellular vesicles display enhanced cell specificity and circulation time. J Controlled Release. 2016;224: 7PMP7–85. doi: 10.1016/j.jconrel.2016.01.009.
- Barenholz Y. Doxil®–the first FDA-approved nano-drug: lessons learned. J Control Release Off J Control Release Soc. 2012;160(2):117–134. doi: 10.1016/j.jconrel.2012.03.020.
- Sercombe L, Veerati T, Moheimani F, Wu SY, Sood AK, Hua S. A dvances and challenges of liposome assisted drug delivery. Front Pharmacol. 2015;6:286. doi: 10.3389/fphar.2015.00286.
- Elsharkasy OM, Nordin JZ, Hagey DW, de Jong OG, Schiffelers RM, Andaloussi SE, Vader P. Extracellular vesicles as drug delivery systems: why and how? Adv Drug Deliv Rev. 2020;159:332–343. doi: 10.1016/j.addr.2020.04.004.
- Haney MJ, Klyachko NL, Zhao Y, Gupta R, Plotnikova EG, He Z, Patel T, Piroyan A, Sokolsky M, Kabanov AV, et al. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J Controlled Release. 2015;207:18–30. doi: 10.1016/j.jconrel.2015.03.033.
- Yang Y, Chen Y, Zhang F, Zhao Q, Zhong H. Increased anti-tumour activity by exosomes derived from doxorubicin-treated tumour cells via heat stress. Int J Hyperthermia. 2015;31(5):498–506. doi: 10.3109/02656736.2015.1036384.
- Tian Y, Li S, Song J, Ji T, Zhu M, Anderson GJ, Wei J, Nie G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials. 2014;35(7):2383–2390. doi: 10.1016/j.biomaterials.2013.11.083.
- Sun D, Zhuang X, Xiang X, Liu Y, Zhang S, Liu C, Barnes S, Grizzle W, Miller D, Zhang H-G, et al. A novel nanoparticle drug delivery system: the anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol Ther. 2010;18(9):1606–1614. DOI:10.1038/mt.2010.105.
- Yang E, Wang X, Gong Z, Yu M, Wu H, Zhang D. Exosome-mediated metabolic reprogramming: the emerging role in tumor microenvironment remodeling and its influence on cancer progression. Signal Transduct Target Ther. 2020;5(1):1–13. doi: 10.1038/s41392-020-00359-5.
- Zhang Z, Dombroski JA, King MR. Engineering of exosomes to target cancer metastasis. Cell Mol Bioeng. 2019;13(1):1–16. doi: 10.1007/s12195-019-00607-x.
- Wang Y, Zhang Y, Cai G, Li Q. Exosomes as actively targeted nanocarriers for cancer therapy. Int J Nanomedicine. 2020;15:4257–4273. doi: 10.2147/IJN.S239548.
- Thangaraju K, Neerukonda SN, Katneni U, Buehler PW. Extracellular vesicles from red blood cells and their evolving roles in health, coagulopathy and therapy. Int J Mol Sci. 2020;22(1):153. doi: 10.3390/ijms22010153.
- Hu C-MJ, Zhang L, Aryal S, Cheung C, Fang RH, Zhang L. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc Natl Acad Sci. 2011;108(27):10980–10985. doi: 10.1073/pnas.1106634108.
- Möller A, Lobb RJ. The evolving translational potential of small extracellular vesicles in cancer. Nat Rev Cancer. 2020;20(12):697–709. doi: 10.1038/s41568-020-00299-w.
- Théry C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, Antoniou A, Arab T, Archer F, Atkin-Smith GK, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the international society for extracellular vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7(1):1535750. DOI:10.1080/20013078.2018.1535750.
- EL Andaloussi S, Mäger I, Breakefield XO, Wood, MJA. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov. 2013;12(5):347–357. doi: 10.1038/nrd3978.
- Lötvall J, Hill AF, Hochberg F, Buzás EI, Di Vizio D, Gardiner C, Gho YS, Kurochkin IV, Mathivanan S, Quesenberry P, et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the international society for extracellular vesicles. J Extracell Vesicles. 2014;3(1):26913. DOI:10.3402/jev.v3.26913.
- Mulcahy LA, Pink RC, Carter DRF. Routes and mechanisms of extracellular vesicle uptake. J Extracell Vesicles. 2014;3(1):24641. doi: 10.3402/jev.v3.24641.
- Feng D, Zhao W-L, Ye -Y-Y, Bai X-C, Liu R-Q, Chang L-F, Zhou Q, Sui S-F. Cellular internalization of exosomes occurs through phagocytosis. Traffic. 2010;11(5):675–687. doi: 10.1111/j.1600-0854.2010.01041.x.
- Kumar Khanna V. 2012. Targeted delivery of nanomedicines. ISRN Pharmacol. 2012:571394. doi: 10.5402/2012/571394.
- Sutaria DS, Badawi M, Phelps MA, Schmittgen TD. Achieving the promise of therapeutic extracellular vesicles: the devil is in details of therapeutic loading. Pharm Res.2017;34(5):1053–1066. doi: 10.1007/s11095-017-2123-5.
- Smylie MG, Wong R, Mihalcioiu C, Lee C, Pouliot J-F. A phase II, open label, monotherapy study of liposomal doxorubicin in patients with metastatic malignant melanoma. Invest New Drugs. 2007;25(2):155–159. doi: 10.1007/s10637-006-9002-y.
- Ugurel M, Schadendorf D, Fink W, Zimpfer-Rechner C, Thoelke A, Figl R, Kaatz M. Clinical phase II study of pegylated liposomal doxorubicin as second-line treatment in disseminated melanoma. Oncol Res Treat. 2004;27(6):540–544. doi: 10.1159/000081335.
- Xing M, Yan F, Yu S, Shen P, Lee JW. Efficacy and cardiotoxicity of liposomal doxorubicin-based chemotherapy in advanced breast cancer: a meta-analysis of ten randomized controlled trials. PloS One. 2015;10(7):e0133569. doi: 10.1371/journal.pone.0133569.
- Lai RC, Yeo RWY, Tan KH, Lim SK. Exosomes for drug delivery - a novel application for the mesenchymal stem cell. Biotechnol Adv. 2013;31(5):543–551. doi: 10.1016/j.biotechadv.2012.08.008.
- Benedikter BJ, Bouwman FG, Vajen T, Heinzmann ACA, Grauls G, Mariman EC, Wouters EFM, Savelkoul PH, Lopez-Iglesias C, Koenen RR, et al. Ultrafiltration combined with size exclusion chromatography efficiently isolates extracellular vesicles from cell culture media for compositional and functional studies. Sci Rep. 2017;7(1):15297. doi:10.1038/s41598-017-15717-7.
- Gámez-Valero A, Monguió-Tortajada M, Carreras-Planella L, Franquesa M, Beyer K, Borràs FE. Size-exclusion chromatography-based isolation minimally alters extracellular vesicles’ characteristics compared to precipitating agents. Sci Rep. 2016;6(1):33641. doi: 10.1038/srep33641.
- Imai T, Takahashi Y, Nishikawa M, Kato K, Morishita M, Yamashita T, Matsumoto A., Charoenviriyakul C, Takakura Y. Macrophage-dependent clearance of systemically administered B16BL6-derived exosomes from the blood circulation in mice. J Extracell Vesicles. 2015;4(1):26238. doi: 10.3402/jev.v4.26238.
- Deng B, Wang Z, Song J, Xiao Y, Chen D, Huang J. Analysis of doxorubicin uptake in single human leukemia K562 cells using capillary electrophoresis coupled with laser-induced fluorescence detection. Anal Bioanal Chem. 2011;401(7):2143–2152.doi: 10.1007/s00216-011-5315-6.
- Zhu L, Dong D, Yu Z-L, Zhao Y-F, Pang D-W, Zhang Z-L. Folate-engineered microvesicles for enhanced target and synergistic therapy toward breast cancer. ACS Appl Mater Interfaces. 2017;9(6):5100–5108. doi: 10.1021/acsami.6b14633.
- Jiang L, Luirink J, Kooijmans SAA, van Kessel, KPM, Jong, W, van Essen, M, Seinen, CW, de Maat, S, de Jong , OG, Gitz-François, JFF, et al. A post-insertion strategy for surface functionalization of bacterial and mammalian cell-derived extracellular vesicles. Biochim Biophys Acta BBA - Gen Subj. 2020;1865(4): doi: 10.1016/j.bbagen.2020.129763.
- Schindler C, Collinson A, Matthews C, Pointon A, Jenkinson L, Minter RR, Vaughan TJ, Tigue NJ. Exosomal delivery of doxorubicin enables rapid cell entry and enhanced in vitro potency. PLoS ONE. 2019;14(3):e0214545. doi: 10.1371/journal.pone.0214545.
- McKelvey KJ, Powell KL, Ashton AW, Morris JM, McCracken SA. Exosomes: mechanisms of Uptake. J Circ Biomark. 2015;4:7. doi: 10.5772/61186.
- Soekmadji C, Li B, Huang Y, Wang H, An T, Liu C, Pan W, Chen J, Cheung L, Falcon‐Perez JM, et al. The future of extracellular vesicles as theranostics - an ISEV meeting report. J Extracell Vesicles. 2020;9(1):1809766. doi:10.1080/20013078.2020.1809766.
- Mii S, Enomoto A, Shiraki Y, Taki T, Murakumo Y, Takahashi M. CD109: a multifunctional GPI-anchored protein with key roles in tumor progression and physiological homeostasis. Pathol Int. 2019;69(5):249–259. doi: 10.1111/pin.12798.
- Fujita M, Takada YK, Takada Y. Integrins αvβ3 and α4β1 act as coreceptors for fractalkine, and the integrin-binding defective mutant of fractalkine is an antagonist of CX3CR1. J Immunol Baltim Md 1950. 2012;189(12):5809–5819. doi: 10.4049/jimmunol.1200889.
- Hoshino A, Costa-Silva B, Shen T-L, Rodrigues G, Hashimoto A, Tesic Mark M, Molina H, Kohsaka S, Di Giannatale A, Ceder S, et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527(7578):329–335. DOI:10.1038/nature15756.
- Risha Y, Minic Z, Ghobadloo SM, Berezovski MV. The proteomic analysis of breast cell line exosomes reveals disease patterns and potential biomarkers. Sci Rep. 2020;10(1):13572. doi: 10.1038/s41598-020-70393-4.
- Wilson CM, Naves T, Vincent F, Melloni B, Bonnaud F, Lalloué F, Jauberteau M-O. Sortilin mediates the release and transfer of exosomes in concert with two tyrosine kinase receptors. J Cell Sci. 2014;127(Pt 18):3983–3997. doi: 10.1242/jcs.149336.
- Tomihari M, Chung J-S, Akiyoshi H, Cruz PD, Ariizumi K. DC-HIL/glycoprotein Nmb promotes growth of melanoma in mice by inhibiting the activation of tumor-reactive T cells. Cancer Res. 2010;70(14):5778–5787. doi: 10.1158/0008-5472.CAN-09-2538.
- Maric G, Rose AA, Annis MG, Siegel PM. Glycoprotein non-metastatic b (GPNMB): a metastatic mediator and emerging therapeutic target in cancer. OncoTargets Ther. 2013;6:839–852. doi: 10.2147/OTT.S44906.
- Rehman M, Gurrapu S, Cagnoni G, Capparuccia L, Tamagnone L. PlexinD1 is a novel transcriptional target and effector of notch signaling in cancer cells. PloS One. 2016;11(10):e0164660. doi: 10.1371/journal.pone.0164660.
- Deng H, Zhou Z, Yang W, Lin L-S, Wang S, Niu G, Song J, Chen X. Endoplasmic reticulum targeting to amplify immunogenic cell death for cancer immunotherapy. Nano Lett. 2020;20(3):1928–1933. doi: 10.1021/acs.nanolett.9b05210.
- Gabizon A, Papahadjopoulos D. Liposome formulations with prolonged circulation time in blood and enhanced uptake by tumors. Proc Natl Acad Sci U S A. 1988;85(18):6949–6953. doi:10.1073/pnas.85.18.6949.
- Licarete E, Rauca VF, Luput L, Drotar D, Stejerean I, Patras L, Dume B, Toma VA, Porfire A, Gherman C, et al. Overcoming intrinsic doxorubicin resistance in melanoma by anti-angiogenic and anti-metastatic effects of liposomal prednisolone phosphate on tumor microenvironment. Int J Mol Sci. 2020;21(8):2968. DOI:10.3390/ijms21082968.
- Castells M, Thibault B, Delord JP, Couderc B. 2012. Implication of tumor microenvironment in chemoresistance: tumor-associated stromal cells protect tumor cells from cell death . Int J Mol Sci. 13(8): 9645–9571 . doi:10.3390/ijms13089545.
- Sanchez L, Yi Y, Yu Y. Effect of partial PEGylation on particle uptake by macrophages. Nanoscale. 2017. 9(1):288–297. doi: 10.1039/c6nr07353k.
- Bush JA, Li G. The role of Bcl-2 family members in the progression of cutaneous melanoma. Clin Exp Metastasis. 2003;20(6):531–539. doi: 10.1023/A:1025874502181.
- Eberle J, Hossini AM. Expression and function of Bcl-2 proteins in Melanoma. Curr Genomics. 2008;9(6):409–419. doi: 10.2174/138920208785699571.
- Trisciuoglio D, Tupone MG, Desideri M, Di Martile M, Gabellini C, Buglioni S, Pallocca M, Alessandrini G, D’Aguanno S, Del Bufalo D, et al. BCL-XL overexpression promotes tumor progression-associated properties article. Cell Death Dis. 2017;8(12): doi:10.1038/s41419-017-0055-y.
- Kashani-Sabet M, Shaikh L, Miller JR, Nosrati, M, Ferreira, CMM, Debs, RJ, Sagebiel, RW, et al. NF-kappa B in the vascular progression of melanoma. J Clin Oncol. 2004;22(4):617–623. DOI:10.1200/JCO.2004.06.047.
- Ueda Y, Richmond A. NF-κB activation in melanoma. Pigment Cell Res. 2006;19(2):112–124. doi: 10.1111/j.1600-0749.2006.00304.x.
- Madonna G, Ullman CD, Gentilcore G, Palmieri G, Ascierto PA. NF-κB as potential target in the treatment of melanoma. J Transl Med. 2012;10(1):53. doi: 10.1186/1479-5876-10-53.
- Crivelli B, Chlapanidas T, Perteghella S, Lucarelli E, Pascucci L, Brini AT, Ferrero I, Marazzi M, Pessina A, Torre ML, et al. Mesenchymal stem/stromal cell extracellular vesicles: from active principle to next generation drug delivery system. J Control Release Off J Control Release Soc. 2017;262:104–117. doi: 10.1016/j.jconrel.2017.07.023.
- Rankin-Turner S, Vader P, O’Driscoll L, Giebel B, Heaney LM, Davies OG. A call for the standardised reporting of factors affecting the exogenous loading of extracellular vesicles with therapeutic cargos. Adv Drug Deliv Rev. 2021;173:479–491. doi: 10.1016/j.addr.2021.04.012.
- Mitrus I, Bryndza E, Kazura M, Smagur A, Sochanik A, Cichon T, Szala S. Properties of B16-F10 murine melanoma cells subjected to metabolic stress conditions. Acta Biochim Pol. 2012;59(3):363–366. doi:10.18388/abp.2012_2122.
- de Jong OG, Verhaar MC, Chen Y, Vader P, Gremmels H, Posthuma G, Schiffelers RM, Gucek M, van Balkom BWM. Cellular stress conditions are reflected in the protein and RNA content of endothelial cell-derived exosomes. J Extracell Vesicles. 2012;1(1):18396. doi: 10.3402/jev.v1i0.18396.
- Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc. 2006;1(6):2856–2860. doi: 10.1038/nprot.2006.468.
- Chiritoiu GN, Jandus C, Munteanu CVA, Ghenea S, Gannon PO, Romero P, Petrescu SM. Epitope located N-glycans impair the MHC-I epitope generation and presentation. Electrophoresis. 2016;37(11):1448–1460. doi: 10.1002/elps.201500449.
- Savojardo C, Martelli PL, Fariselli P, Profiti G, Casadio R. BUSCA: an integrative web server to predict subcellular localization of proteins. Nucleic Acids Res. 2018;46(W1):W459–W466. doi: 10.1093/narA/gky320.
- Pathan M, Keerthikumar S, Chisanga D, Alessandro R, Ang C-S, Askenase P, Batagov, AO, Benito-Martin , A, Camussi, G, and Clayton, A, et al. A novel community driven software for functional enrichment analysis of extracellular vesicles data. J Extracell Vesicles. 2017;6(1):1321455. DOI:10.1080/20013078.2017.1321455.
- Jassal B, Matthews L, Viteri G, Gong, C, Lorente P, Fabregat A., Sidiropoulos K, Cook J, Gillespie M, Haw R, et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020;48(D1):D498–D503. DOI:10.1093/nar/gkz1031.
- Orchard S, Ammari M, Aranda B, Breuza L, Briganti L, Broackes-Carter F, Campbell NH, Chavali G, Chen C, del Toro N, et al. The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014;42(6):D358–363. DOI:10.1093/nar/gkt1115.
- Nakamura K, Yamashita K, Itoh Y, Yoshino K, Nozawa S, Kasukawa H. Comparative studies of polyethylene glycol-modified liposomes prepared using different PEG-modification methods. Biochim Biophys Acta. 2012;1818(11):2801–2807. doi: 10.1016/j.bbamem.2012.06.019.
- Nosova AS, Koloskova OO, Nikonova AA, Simonova V A., Smirnov VV, Kudlay D, Khaitov MR. Diversity of PEGylation methods of liposomes and their influence on RNA delivery. MedChemComm. 2019;10(3):369–377. doi: 10.1039/c8md00515j.
- Li WM, Xue L, Mayer LD, Bally MB. Intermembrane transfer of polyethylene glycol-modified phosphatidylethanolamine as a means to reveal surface-associated binding ligands on liposomes. Biochim Biophys Acta. 2001;1513(2):193–206. doi: 10.1016/s0005-2736(01)00351-0.
- Lim CY, Owens NA, Wampler RD, Ying Y, Granger JH, Porter MD, Takahashi M, Shimazu K. Succinimidyl ester surface chemistry: implications of the competition between aminolysis and hydrolysis on covalent protein immobilization. Langmuir ACS J Surf Colloids. 2014;30(43):12868–12878. doi: 10.1021/la503439g.
- Bligh EG, Dyer WJ. 1959. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 37(8):911–917. doi: 10.1139/o59-099.
- Rouser G, Fkeischer S, Yamamoto A. Two dimensional thin layer chromatographic separation of polar lipids and determination of phospholipids by phosphorus analysis of spots. Lipids. 1970;5(5):494–496. doi: 10.1007/BF02531316.
- Rauca V-F, Licarete E, Luput L, Sesarman A, Patras L, Bulzu P, Rakosy-Tican E, Banciu M. Combination therapy of simvastatin and 5, 6-dimethylxanthenone-4-acetic acid synergistically suppresses the aggressiveness of B16.F10 melanoma cells. PloS One. 2018;13(8):e0202827. doi: 10.1371/journal.pone.0202827.
- Haase-Kohn C, Wolf S, Herwig N, Mosch B, Pietzsch J. Metastatic potential of B16-F10 melanoma cells is enhanced by extracellular S100A4 derived from RAW264.7 macrophages. Biochem Biophys Res Commun. 2014;446(1):143–148. doi: 10.1016/j.bbrc.2014.02.126.
- Banciu M, Schiffelers RM, Storm G. Investigation into the role of tumor-associated macrophages in the antitumor activity of Doxil. Pharm Res. 2008;25(8):1948–1955. doi: 10.1007/s11095-008-9629-9.
- Gornall AG, Bardawill CJ, David MM. Determination of serum proteins by means of the biuret reaction. J Biol Chem. 1949;177(2):751–766. doi:10.1016/S0021-9258(18)57021-6.