
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Nimesulide is a nonsteroidal anti-inflammatory drug and a COX-2 inhibitor with antitumor and antiproliferative activities that induces apoptosis in oral, esophagus, breast, and pancreatic cancer cells. Despite being removed from the market due to hepatotoxicity, nimesulide is still an important research tool being used to develop new anticancer drugs. Multiple studies have been done to modify the nimesulide skeleton to develop more potent anticancer agents and related compounds are promising scaffolds for future development. As such, establishing a mechanism of action for nimesulide remains an important part of realizing its potential. Here, we show that nimesulide enhances TRAIL-induced apoptosis in resistant pancreatic cancer cells by promoting clustering of DR5 in the plasma membrane. In this way, nimesulide acts like a related compound, DuP-697, which sensitizes TRAIL-resistant colon cancer cells in a similar manner. Our approach applies a time-resolved FRET-based biosensor that monitors DR5 clustering and conformational states in the plasma membrane. We show that this tool can be used for future high-throughput screens to identify novel, nontoxic small molecule scaffolds to overcome TRAIL resistance in cancer cells.
Introduction
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) selectively induces apoptosis in cancer cells via engagement of its cognate receptor, death receptor 5 (DR5).Citation1 However, a significant number of cancer cells are resistant to TRAIL; particularly some highly malignant cancers such as melanoma and pancreatic cancer.Citation2,Citation3 Several novel drugs targeting TRAIL receptors are currently in clinical trials,Citation4 though TRAIL resistance limits their effectiveness. The current mechanistic understanding of TRAIL resistance remains muddied, with several overlapping factors likely conspiring to confer a resistant phenotype. These factors include, among others: altered surface expression of death receptors;Citation5,Citation6 the inability of the receptors to initiate death signal due to mutations;Citation7 defects in adaptor proteins which are essential for assembly of the death-inducing signaling complex;Citation8,Citation9 and upregulation of apoptosis inhibiting proteins.Citation10,Citation11 Regardless of its origins, there have been considerable efforts to develop therapeutic strategies that circumvent or overcome TRAIL-resistance.Citation12
One surprising advance came in the study of cyclooxygenase-2 (COX-2) inhibitors. COX-2 is a central enzyme in the production of proinflammatory prostaglandin from arachidonic acid, and is known to inhibit apoptosis and immune surveillance, stimulate angiogenesis, promote cancer invasion and metastasis.Citation13–15 Accordingly, COX-2 is considered a therapeutic target for prevention of several cancers including hepatocellular, colorectal, and pancreatic cancers.Citation16–19 Previous studies have shown that COX-2 inhibitors sensitize cancer cells to death receptor ligands including Fas, tumor necrosis factor and TRAIL.Citation20,Citation21 For example, DuP-697, a selective COX-2 inhibitor, sensitizes TRAIL resistant colon carcinoma cell to apoptosis by promoting the clustering of DR5 in the plasma membrane.Citation20 More recently, nimesulide, another COX-2 inhibitor and the subject of this study, has been associated with antitumor and an antiproliferative effect.Citation22,Citation23 Nimesulide is a nonsteroidal anti-inflammatory drug that induces apoptosis in breast cancer, oral and esophagus carcinoma cells, inhibits lung cancer cell proliferation and suppresses gastric carcinogenesis.Citation24–28 Recent studies also showed that nimesulide inhibits proliferation and induces apoptosis of pancreatic cancer cells by upregulating the tumor suppressor gene PTEN (phosphatase and tensin homolog deleted on chromosome 10).Citation29–31 Whether nimesulide also has the effect of sensitizing resistant pancreatic cancer cells to TRAIL is unknown.
Here, we investigated the effects of nimesulide on TRAIL sensitivity in human lymphoma Jurkat cells and two TRAIL-resistant pancreatic cancer cell lines, AsPC1 and Panc1, all of which express DR5 on their surface. Cell-based functional assays confirm that AsPC1 and Panc1 cancer cells are resistant to TRAIL-induced apoptosis, and show that co-treatment of TRAIL with nimesulide confers dose-dependent TRAIL sensitivity (as measured by caspase-8 activation). Using fluorescence microscopy, we illustrate that nimesulide’s enhancement of TRAIL-induced apoptosis, like DuP-697, correlates with clustering of DR5 in the plasma membrane. These clusters are thought to be essential to DR5 activation, and they reflect receptor association with lipid rafts and concomitant backbone conformational changes.Citation20,Citation32 Using nimesulide and a second positive control, bioymifi, we then show that small-molecule induced clustering of DR5 can be monitored through a FRET-based DR5 biosensor we previously developed.Citation33 Building on that technology, we provide a first proof-of-principle that this FRET platform can be extended for future high-throughput screening campaigns aimed at identifying novel scaffolds for enhancing TRAIL-sensitivity in resistant cells.
Materials and methods
Cell cultures and reagents
HEK293 cells were cultured in phenol red-free DMEM (Gibco) supplemented with 2 mM L-Glutamine (Invitrogen). Jurkat and AsPC1 cell line were cultured in RPMI 1640 with HEPES, sodium pyruvate, and L-glutamine (ATCC). Panc1 cells were cultured in Dulbecco’s Modified Eagle’s Medium with 4 mM L-glutamine, 4500 mg/L glucose, 1 mM sodium pyruvate, and 1500 mg/L sodium bicarbonate (ATCC). All media was supplemented with heat-inactivated 10% fetal bovine serum (Gibco), 100 U/mL penicillin and 100 mg/mL streptomycin (HyClone). Cell cultures were maintained in an incubator with 5% CO2 (Forma Series II Water Jacket CO2 Incubator: Thermo Fisher Scientific) at 37°C.
Molecular cloning
TagRFP and EGFP plasmids were prepared as described previously.Citation34 Genes encoding DR5ΔCD (1–240) were inserted at the N-terminus of the TagRFP and EGFP vectors using standard cloning techniques, as described previously.Citation35 To prevent the dimerization and aggregation of EGFP, alanine 206 was mutated to lysine (A206K).Citation36,Citation37
Detection of endogenous expression DR5 and DR4 on Jurkat and pancreatic cells
To examine the surface expression of DR5, cancer cells were washed 3x with cold PBS and incubated with allophycocyanin-conjugated anti-DR5 antibody for 1–2 hours on ice. Cells were washed three times with cold PBS and surface expression of DR5 was determined using flow cytometry. To test the effect of nimesulide on surface expression of DR5, cancer cells were seeded in a 10 ml flask and incubated for 24 hours at 37°C. Cells were treated with nimesulide (50 or 100 μM) and DMSO only controls and incubated for 24 hours at 37°C. Cells were washed three times with cold PBS and incubated with anti-DR5 antibody for 1–2 hours on ice. Cells were washed three times with cold PBS and surface expression of DR5 was determined using flow cytometry.
Western blot
Expression of DR5 and DR4 in cancer cells was also determined using a Western blot. Cancer cells were seeded in a 10 ml flask and incubated for 24 hours at 37°C. Cells were treated with (50 μM) and without nimesulide and incubated for 24 hours at 37°C. After incubation, cells were spun down and washed three times with cold PBS and lysed with native lysis buffer. Total protein concentration of lysates was determined by BCA assay, and equal amounts of total protein (20–80 μg) were mixed with 4x Bio-rad sample buffer and boiled for 3–5 min and resolved using 4–20% Tris–glycine SDS-PAGE gels (Bio-rad). Proteins were transferred to a PVDF membrane (EMD Millipore) and probed using antibodies against DR5, DR4, and β-Actin (Cell signaling technology and Biolegend). SHSY5Y cell lysate was used as a positive control for DR4.
Caspase-8 activity assay
The caspase-8 assay was used to assess the effects of TRAIL only, nimesulide only, and nimesulide+TRAIL on caspase-8 activity, and nimesulide effective concentration. Cancer cells were seeded in 96-well white opaque plates (Greiner Bio-One) at 7,500 cells/well and incubated for 24 hours at 37°C. Cells were incubated for 24 hours at 37°C with TRAIL alone (0.000001–10 μg/mL) or nimesulide alone (0.001–200 μM) or with nimesulide (50 μM) + TRAIL (0.000001–10 μg/mL) or DMSO only. An equal volume of Caspase-Glo 8 reagent (Promega) was added to each well, and the luminescence was measured after 45 minutes using a Cytation 3 Cell Imaging Multi-Mode Reader luminometer (BioTek). To determine the effective concentration (EC50) of nimesulide, Jurkat cells were seeded in 96-well white opaque plates at 7,500 cells/well and incubated for 24 h at 37°C. Cells were treated for 2 hours with nimesulide (0.001–200 μM) before being treated for 24 hours with TRAIL (0.01–0.1 μg/mL) at 37°C. An equal volume of Caspase-Glo 8 reagent (Promega) for each sample was added to each well, and the luminescence was measured after 45 min.
TRAIL binding assay
Jurkat cells were washed with PBS, and then cells (450,000/condition) were incubated with flag-tagged TRAIL (0.1 μg/mL) ± the nimesulide (EC50 concentration) for 2 hours on ice until equilibrated. After incubation, cells were washed two times with PBS to remove unbound TRAIL. Cells were labeled with rabbit anti-flag antibody, followed by AF647-conjugated anti-rabbit secondary antibody. TRAIL binding was measured using BD Accuri C6 flow cytometer.
DR5 clustering assay
To examine the effect of Nimesulide on DR5 clustering, HEK293 cells were transfected with the DR5ΔCD-RFP (7.5 μg) in a 10 cm plate using Lipofectamine 3000 (Invitrogen). After 24 hours of transfection, cells were lifted with TrypLE, resuspended in phenol red–free DMEM (Gibco) and centrifuged for 5 minutes at 200 g. Transfected cells were dispensed in a 24-well glass black clear bottom plate. The next day, transfected cells were treated separately with DMSO, bioymifi, TRAIL, nimesulide, or nimesulide+TRAIL and incubated overnight. Cells were fixed with 4% paraformaldehyde for 10–15 minutes and washed with PBS and imaged using a ImageXpress Pico imaging system. Panc1 cells, which have endogenous expression of DR5, were dispensed in a 24-well glass black clear bottom plate and treated separately with DMSO, bioymifi, TRAIL, nimesulide or nimesulide+TRAIL and incubated overnight. Next day cells were fixed with 4% paraformaldehyde for 10–15 minutes and washed with PBS, then labeled with APC-conjugated anti-DR5 antibody. Cells were mounted with antifade mounting medium with DAPI. Fluorescence images of cells were taken using ImageXpress Pico imaging system.
Pilot screening with LOPAC library
The LOPAC library, containing 1280 compounds, was purchased from Sigma-Aldrich. 384-well flat bottom polypropylene black plates were selected as the assay plates for their low autofluorescence and low inter-well cross talk (Greiner Bio-One). LOPAC compounds were formatted across four 384-well plates at 50 nL (10 μM final concentration/well) from 96-well mother plates using an Echo liquid dispenser. Plates were sealed and stored at −20°C until use. For screening, HEK293 cells were transfected using Lipofectamine 3000 with DR5ΔCD-GFP and DR5ΔCD-GFP:DR5ΔCD-RFP (1:6 ratio) for a total of 15.75 µg in 10 cm plates for 24 hours and transfection was confirmed using EVOS fluorescence microscopy. On the day of screening, drug plates were equilibrated at room temperature. The cells were harvested from the 10 cm plates by incubating for 2–4 minutes with TrypLE (Invitrogen), washed three times with PBS, filtered using 70 μm cell strainers (BD Falcon), and resuspended in PBS at a concentration of 1 million cells/mL. Next, cells were dispensed (50 μL/well) into the 384-well LOPAC drug plates (Greiner) by a Multidrop Combi Reagent Dispenser (Thermo Fisher Scientific) and allowed to incubate for 2 hours at room temperature before readings were acquired. Donor lifetime in the presence and absence of acceptor was measured by using a fluorescence lifetime plate reader (Fluorescence Innovations, Inc., Minneapolis, MN). GFP fluorescence was excited with a 473-nm microchip laser, and emission was filtered with 488-nm long-pass and 517/20-nm band-pass filters. Time-resolved fluorescence waveforms for each well were fitted to single-exponential decays using least-squares minimization global analysis software (Fluorescence Innovations, Inc.) to give donor lifetime (τD) and donor–acceptor lifetime (τDA). FRET efficiency (E) was then calculated based on EquationEq. (1(1)
(1) )
MTT assay
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was used to measure apoptosis and cell proliferation. For apoptosis measurements, pancreatic cancer cells were seeded in 96-well plates at a density of 7500 cells/well and incubated for 24 hours at 37°C and 5% CO2. After incubation, cells were treated with increasing concentrations of TRAIL (0.000001–10 μg/ml) + DMSO or nimesulide (50 μM) + TRAIL (0.000001–10 μg/ml), followed by 24 hours of incubation at 37°C. For cell proliferation, cancer cells were seeded in 96-well plates at a density of 7500 cells/well and incubated for 24 h at 37°C and 5% CO2. After incubation, cells were treated with increasing concentrations of nimesulide (0.001–200 μM), followed by 24 hours of incubation at 37°C. Cell viability and proliferation were assessed with a Cytation 3 Cell Imaging Multi-Mode Reader luminometer (BioTek).
Overexpression of FADD in HEK293 cells
Human-FADD plasmid was purchased from Origene technologies. HEK293 cells were transfected with the FADD plasmid (15 μg) in a 10 cm plate using Lipofectamine 3000 (Invitrogen). After 48 hours of transfection, cells were lifted with TrypLE, resuspended in phenol red–free DMEM (Gibco) and centrifuged for 5 minutes at 200 g. Transfected cells were washed three times with cold PBS and lysed with native lysis buffer (Abcam). Overexpression of FADD was confirmed by western blot with anti-FADD antibody (Cell Signaling).
Apoptosis induced by overexpression of FADD
To determine the effect of nimesulide on apoptosis induced by overexpression of FADD, HEK293 cells were transfected with the FADD plasmid (15 μg) in a 10 cm plate using Lipofectamine 3000 (Invitrogen). After 24 hours of transfection, cells were lifted with TrypLE and resuspended in phenol red–free DMEM (Gibco). Next, transfected cells were plated in 96-well white opaque plates (7,500 cells/well) and incubated for 24 hours at 37°C. Cells were then treated with nimesulide (0.001–200 μM) and incubated for 24 hours at 37°C. An equal volume of Caspase-Glo 8 reagent (Promega) to sample was added to each well, and the luminescence was measured after 45 min using a Cytation 3 Cell Imaging Multi-Mode Reader luminometer (BioTek).
DR5 knockdown using DR5 siRNA
For DR5 knockdown experiments, DR5 siRNA Gene Silencer was purchased from Santa Cruz Biotechnology (sc-40237, Santa Cruz Biotechnology). Cells were transfected with DR5 siRNA (25 nM) using lipofectamine 3000 (Invitrogen) according to the manufacturer’s recommendations. Transfected cells were incubated for 48 hours before further analysis. DR5 expression was determined using an anti-DR5 antibody and analyzed by flow cytometry.
Results
Varied TRAIL sensitivity of cancer cell lines
It is well documented that TRAIL induces apoptosis in various types of cancer cells via engaging its cognate receptor DR5.Citation38 However, not all cancer cells are sensitive to TRAIL, which could be due to the lack of DR5 expression. So, before testing the effect of nimesulide on TRAIL-induced apoptosis, we first examined the surface expression of DR5 in human lymphoma Jurkat cells and two pancreatic cancer cells, AsPC1 and Panc1. Flow cytometry data showed that all three cell lines were positive for DR5 expression (). However, DR5 expression was slightly higher in Jurkat cells () when compared with AsPC1 () and Panc1 ().
Figure 1. Surface expression of DR5 and TRAIL sensitivity of Jurkat and pancreatic cancer cells. FACS data demonstrate surface expression of DR5 on Jurkat (a), AsPC1(b) and Panc1 (c) cells. Cells were incubated with the APC-conjugated anti-DR5 antibody and analyzed by flow cytometry. The red line indicates cells labeled with anti-DR5 antibody and black line for unlabeled cells. (d) TRAIL sensitivity of AsPC1, Panc1 and Jurkat cancer cells. Caspase-8 activity was measured in Jurkat, and pancreatic cancer cells treated with increasing concentrations of TRAIL (0.000001–10 μg/ml). Data are presented as mean ± standard deviation (N = 3).
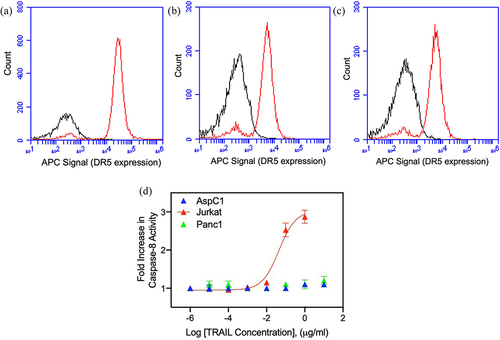
Next, we tested the sensitivity of Jurkat and pancreatic cancer cells to TRAIL using a caspase-8 activation assay. Studies have shown that upon TRAIL binding DR5 triggers apoptosis by recruiting the apoptosis initiator caspase-8 through the adaptor FADD.Citation39 Caspase-8 activity was increased in Jurkat cells in the presence of TRAIL in a dose-dependent manner, whereas the AsPC1 and Panc1 showed very minimal sensitivity to TRAIL (). These results confirm that the AsPC1 and Panc1 cells are resistant to TRAIL despite their intact expression of DR5. As previously mentioned, TRAIL resistance can be due to a wide variety of mechanisms, including defects in death inducing signaling complex proteins like FADD and caspase-8.Citation8–10
Nimesulide increases TRAIL-induced caspase-8 activity in Jurkat cells
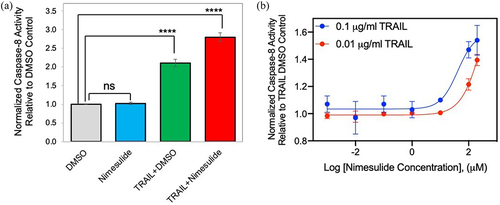
To test the biological effect of nimesulide on TRAIL-induced apoptosis, the caspase-8 activation assay was performed with Jurkat cells. These results show that nimesulide increases the TRAIL-induced caspase-8 activity by 1.4-fold compared to DMSO+TRAIL treated cells (). However, cell treated with nimesulide-only did not affect the caspase-8 activity compared to DMSO control (). These results suggest that nimesulide and TRAIL work synergistically to increase caspase-8 activity. Next, we determined the effective concentration (EC50) of nimesulide in the presence of TRAIL (0.01 and 0.1 μg/ml) using the caspase-8 assay. As shown in , nimesulide increased caspase-8 activity in a dose-dependent manner, with an EC50 of 57 μM at 0.1 μg/ml TRAIL and 915 μM at 0.01 μg/ml TRAIL. Taken together, nimesulide enhances TRAIL-induced caspase-8 activity in Jurkat cells.
Figure 2. Effect of nimesulide on TRAIL-induced caspase-8 activity. (a) Caspase-8 activity was measured in Jurkat cells treated with nimesulide-only (50 μM), TRAIL (0.1 μg/ml) +DMSO or TRAIL+nimesulide (50 μM) or DMSO only. (b) Determination of EC50 of nimesulide by caspase-8 assay. Jurkat cells were incubated with increasing concentrations of nimesulide (0.001–200 uM) and TRAIL (0.01–0.1 μg/ml). Data are presented as mean ± standard deviation (N = 3). ****P < .0001 compared to control by two-tailed unpaired t test.
Nimesulide sensitizes pancreatic cancer cells to TRAIL-induced caspase-8 activity
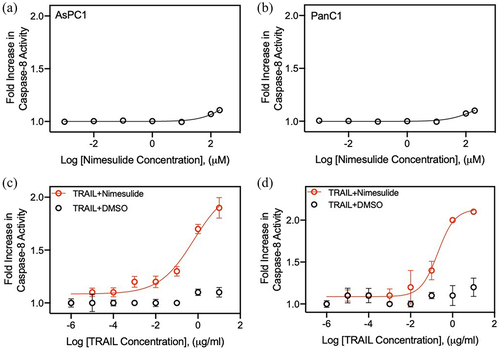
To determine the promise of nimesulide for clinical translation, we tested its ability to sensitize AsPC1 and Panc1 cells to TRAIL-induced caspase-8 activity. We first investigated whether nimesulide alone activates caspase-8 in these cell lines. As shown in , nimesulide-only treatment resulted in a 10% increase in caspase-8 activity at a high dose (200 M) in AsPC1 () and Panc1 (). However, co-treatment of TRAIL with nimesulide (50 μM) increased caspase-8 activity by 1.7-fold over DMSO in AsPC1 () and by 2-fold over DMSO in Panc1 cells (). These results confirm that nimesulide increases TRAIL-induced caspase-8 activity in pancreatic cancer cells.
Figure 3. Effect of nimesulide on caspase-8 activity in AsPC1 and Panc1 cancer cells. Caspase-8 activity was measured in AsPC1 (a) and Panc1 (b) cancer cells treated with increasing concentrations of nimesulide-only (0.001–200 μM). (c) AspC1 cells treated with increasing concentrations of TRAIL (0.000001–10 μg/ml) +DMSO or TRAIL+nimesulide (50 μM). (d) PanC1 cells treated with increasing concentrations of TRAIL (0.000001–10 μg/ml) +DMSO or TRAIL+nimesulide (50 μM). Data are presented as mean ± standard deviation (N = 3).
Nimesulide increases TRAIL-induced apoptosis in pancreatic cancer cells
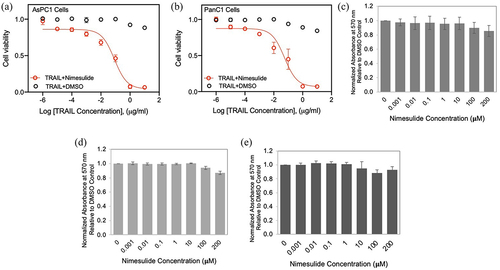
Previous studies have reported that nimesulide promotes apoptosis in several different cancer cells.Citation25,Citation26 So, we measured the effect of nimesulide on TRAIL-induced apoptosis in AsPC1 and Panc1 cells using MTT assay. While TRAIL-only treatment showed a 12% increase in cell death at a high concentration (10 μg/ml), co-treatment of TRAIL with nimesulide (50 μM) increased apoptosis by >90% in both AsPC1 () and Panc1 cells (). These results clearly confirm that nimesulide sensitizes pancreatic cancer cells to TRAIL-induced apoptosis.
Figure 4. Effect of nimesulide on TRAIL-induced apoptosis in pancreatic cancer cells and proliferation of all three cancer cells. Apoptosis was measured using MTT assay in AsPC1 (a) and Panc1 (b). Cancer cells were treated with increasing concentrations of TRAIL (0.000001–10 μg/ml) +DMSO or TRAIL+nimesulide (50 μM). Data are presented as mean ± standard deviation (N = 3). AsPC1 (c), Panc1 (d) and Jurkat (e) cells were treated separately with DMSO or nimesulide (0.001–200 μM) and incubated for 24 hours and cell proliferation was measured using MTT assay. Data are mean ± SD (N = 3).
Effect of nimesulide on cell proliferation
It has been reported that nimesulide inhibits cell proliferation and suppresses carcinogenesis in several different cancer cells.Citation29 So, we sought to investigate the effect of nimesulide on Jurkat and pancreatic cell proliferation using the MTT assay. MTT assay was developed as a non-radioactive alternative to tritiated thymidine incorporation into DNA for measuring cell proliferation.Citation40 These studies showed that low doses of nimesulide did not affect the proliferation of all three cancer cells (). However, 200 μM treatment decreased cell proliferation by 20%. In spite of this anti-proliferative effect at a high dose, we observed an increase in caspase-8 activity and apoptosis (). These findings demonstrate that cell proliferation is not the source of nimesulide proapoptotic activity.
Effect of nimesulide on TRAIL binding
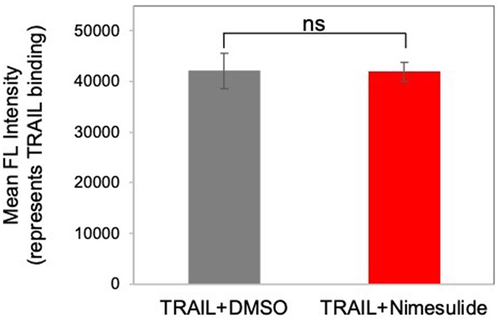
We tested whether antitumor effect of nimesulide is coming from an increase in TRAIL binding. So, we examined the effect of nimesulide on TRAIL binding using flow cytometry. This assay was performed at the effective dose of the nimesulide. No significant difference was observed in the binding of TRAIL to Jurkat cells in the presence versus absence of nimesulide (). These results confirm that nimesulide is not altering TRAIL binding.
Figure 5. Effect of nimesulide on TRAIL binding. Jurkat cells were incubated with TRAIL (0.1 μg/ml) +DMSO or TRAIL+nimesulide (effective dose concentration). TRAIL binding was detected with rabbit anti-Flag antibody, followed by AF647-conjugated anti-rabbit secondary antibody, as measured by flow cytometry. Data are presented as mean ± standard deviation (N = 3).
Nimesulide enhances TRAIL-induced capase-8 activity in pancreatic cancer cells by promoting DR5 clustering
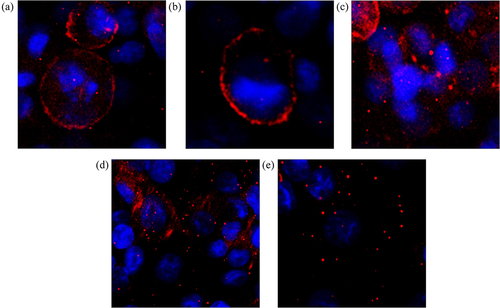
Previously studies reported that death receptor clustering at the cell membrane is required for the initiation of death signaling and is an important factor for TRAIL-induced apoptosis.Citation41–44 A recent study showed that DuP-697, a selective COX-2 inhibitor, sensitizes human colon carcinoma cells to TRAIL-mediated apoptosis via clustering of DR5 in cholesterol enriched and ceramide-enriched caveolae.Citation20 Also, a small molecule bioymifi increases apoptosis in several different cancer cells by promoting ligand-independent DR5 clustering.Citation45 So, to determine the mode of action of nimesulide, we first tested its effect on DR5 clustering in Panc1 cells using the ImageXpress Pico imaging system. Fluorescence microscopy data showed that DR5 clustering was significantly increased in cells treated with bioymifi (positive control, ) and nimesulide () when compared with DMSO control (). Interestingly, co-treatment of TRAIL with nimesulide further enhanced DR5 clustering ( and Supplementary Figure 2B). However, DR5 clustering was not increased in Panc1 cells treated with TRAIL alone ( and Supplementary Figure 2a) in comparison with control (). These results suggest that nimesulide increases the ligand-independent and -dependent clustering of DR5.
Figure 6. Effect of nimesulide on DR5 clustering. Panc1 cells were treated separately with DMSO (a), TRAIL+DMSO (b), bioymifi (c), nimesulide (d) and nimesulide+TRAIL (e). The next day cells were fixed with 4% paraformaldehyde and then labeled with APC-conjugated anti-DR5 antibody. Cells were mounted with antifade mounting medium with DAPI. Fluorescence images of cells were taken using ImageXpress Pico imaging system.
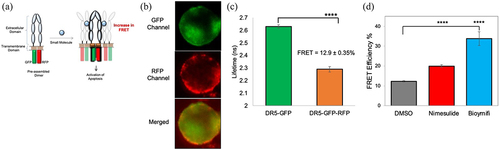
To further corroborate the effect of nimesulide on receptor–receptor interactions, we tested its effect on pre-ligand receptor–receptor interactions using TR-FRET method. We have previously used this method to monitor DR5-DR5 interactions.Citation33 TR-FRET experiments were performed in HEK293 cells transiently expressing DR5 without a cytoplasmic domain (DR5ΔCD) attached to GFP and coexpressing DR5ΔCD fused to GFP and RFP just downstream of the transmembrane domain of the receptors (). Fluorescence images of HEK293 cells co-expressing DR5ΔCD-GFP and DR5ΔCD-RFP (DR5ΔCD-FRET pair) showed an overlap of the GFP and RFP, indicating the colocalization of the receptors (). Lifetime measurements showed a substantial decrease in the DR5ΔCD-GFP (donor) fluorescence lifetime in the presence of the DR5ΔCD-RFP (acceptor) compared with the donor only, which confirms the efficient energy transfer between the FRET pairs (). This data recapitulates our previous finding that DR5ΔCD receptors are capable of homophilic interactions in the absence of cytoplasmic death domain and ligand.Citation46–48 FRET efficiency was significantly increased in cells treated with bioymifi (positive control) or nimesulide when compared with DMSO control ().
Figure 7. Effect of nimesulide on DR5-DR5 interactions. (a) Schematic of DR5ΔCD-FRET pair biosensor. Pre-ligand assembled DR5 dimers show FRET, which will be increased if small molecules are promoting DR5 clustering. (b) Fluorescence microscopy images showing the expression of DR5ΔCD-GFP and DR5ΔCD-RFP in cells transfected with DR5ΔCD-FRET pair (c) Fluorescence lifetime measurements with HEK293 cells transiently expressing DR5ΔCD-GFP only and DR5ΔCD-FRET pair. (d) FRET efficiency increased with nimesulide and bioymifi treatment compared to DMSO control. Data are mean ± SD (N = 3). ****P < .0001 compared to DMSO control by two-tailed unpaired t test.
We next investigated if increase in FRET efficiency is due to DR5 clustering. For this study, we used HEK293 cells transiently expressing DR5ΔCD-RFP. Fluorescence microscopy data showed that DR5 clustering was significantly increased in cells treated with bioymifi (positive control, Supplementary Figure 1C) and nimesulide (Supplementary Figure 1D) when compared with DMSO control (Supplementary Figure 1A). Unlike Panc1 cells, DR5 clustering was increased in HEK293 cells treated with TRAIL compared to DMSO control (Supplementary Figure 1A-B). In HEK293 cells also, co-treatment of TRAIL with nimesulide further increased DR5 clustering (Supplementary Figure 1E). Taken together, these data confirm that nimesulide sensitizes pancreatic cancer cells to TRAIL-induced apoptosis by promoting DR5 clustering.
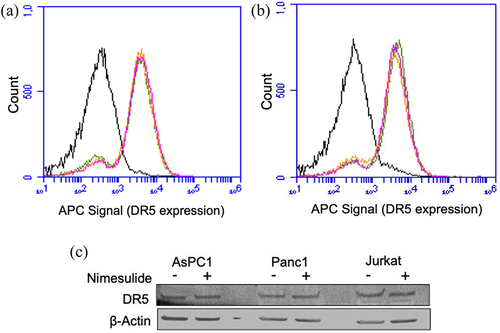
We next sought to examine if increase in DR5 clustering is due to upregulation of DR5 expression. Studies have shown that small molecules like Chloroquine and 3,3’-diindolylmethane enhances TRAIL-mediated apoptosis through upregulation of DR5.Citation49,Citation50 Surface expression of DR5 was examined in the presence and absence of nimesulide using flow cytometry () and western blot ( and supplementary Figure 3A). As shown in , nimesulide treatment did not affect the expression of DR5 in cancer cell lines. These results confirm that the expression level of DR5 was not involved in sensitization of these cancer cells to TRAIL.
Figure 8. Effect of nimesulide on DR5 expression. FACS data demonstrate surface expression of DR5 on AsPC1 (a) or Panc1 (b) cells. Cells were treated separately with DMSO or nimesulide and incubated for 24 hours. After incubation cells were washed with PBS and incubated with APC-conjugated anti-DR5 antibody and analyzed by flow cytometry. The black line indicates for unlabeled cells, green for cells treated with DMSO and Orange (50 μM) and magenta (100 μM) for cells treated with nimesulide. (c) Cancer cells were treated with or without nimesulide (50 μM) and incubated for 24 hours. After incubation cells were washed with PBS and lysed with native lysis buffer. Equal amounts of total protein were loaded and resolved by 4–20% Tris-glycine SDS−PAGE gels and immunoblotted with antibodies against DR5 and β-Actin.
Effect of nimesulide on surface expression of DR4
To further verify the specificity of nimesulide, we tested its effect on expression of death receptor 4 (DR4), which is a homologous member of the TNF receptor superfamily (51% identical and 68% like DR5) that binds to TRAIL and triggers apoptosis.Citation51,Citation52 We tested the expression of DR4 in all three cancer cell lines in the presence and absence of nimesulide using a western blot. It has been reported that the neuroblastoma cell-line SHSY5Y expresses both DR5 and DR4,Citation53 so we have used the lysate from SHSY5Y cells as a positive control. No significant expression of DR4 was observed in the presence or absence of nimesulide (Supplementary Figure 3B-C) in all three cancer cell lines. Some studies have previously shown that these cancer cell lines have very low or no DR4 expression.Citation54,Citation55 Furthermore, it has been reported that TRAIL-induced apoptosis is preferentially mediated via DR5 in AsPC-1 and Panc1.Citation54,Citation55 These results suggest that DR4 is not contributing to TRAIL-induced apoptosis, and nimesulide does not affect the DR4 expression levels in all these cell lines.
Effect of nimesulide FADD overexpression-induced apoptosis
To rule out nonspecific downstream signaling effects of nimesulide, we tested their effects on FADD overexpression-induced apoptosis. DR5-independent apoptosis pathways can be interrogated in HEK293 cells due to their negligible expression of DR5.Citation56,Citation57 It has been previously shown that overexpressing FADD in HEK293 cells induces apoptosis-independent of the TRAIL-DR5 pathway.Citation58–60 Thus, any effect on FADD-induced apoptosis in the presence of the small molecules suggests that they are acting through a mechanism that is not DR5-dependent. To test the effects of DR5 hit compounds on FADD overexpression-induced apoptosis, we first confirmed that the FADD plasmid could induce overexpression of FADD in HEK293 cells, which was determined using western blot ( and Supplementary Figure 4). Transfected cells showed a significant band at 23 kDa corresponding to FADD compared to untransfected cells (Supplementary Figure 4). Next, we tested the effect of FADD overexpression on caspase-8 activity. Caspase-8 activity was significantly increased (2.5-fold) in cells transfected with FADD when compared with untrasfected HEK293 cells ()
Figure 9. Effect of nimesulide on FADD overexpression-induced apoptosis in HEK293 cells. (a) Western blot analysis of HEK293 cell lysates of untransfected and FADD transfected cells (b) Overexpression of FADD increases caspase-8 activity in HEK293 cells. (c) Effect of nimesulide on endogenous caspase-8 activity in HEK293 cells. (d) Effect of nimesulide on FADD-induced caspase-8 activity in HEK293 cells overexpressing FADD. Data are presented as mean ± standard deviation (N = 3). ****P < .0001 compared to control by two-tailed unpaired t test.
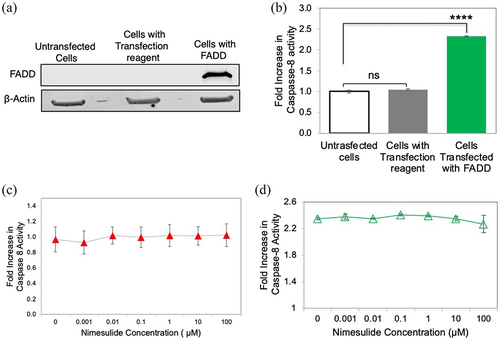
In order to establish that the mode of action of nimesulide occurs through DR5 and not an independent caspase-8 pathway, we tested the effects of the nimesulide on basal caspase-8 activity in HEK293 cells, which have very low levels of endogenous DR5.Citation56,Citation57 Nimesulide had no effect on basal caspase-8 activity in HEK293s (), which suggests that nimesulide is not increasing caspase-8 activity in a DR5-independent manner. We then tested the effects of nimesulide on FADD-induced apoptosis, to determine whether the changes in caspase-8 activity are due to a nonspecific interaction with FADD. Nimesulide had little to no effect on FADD-induced apoptosis, even at relatively high concentrations (). These results confirm that the mode of action of nimesulide is not due to interference with FADD-induced apoptosis.
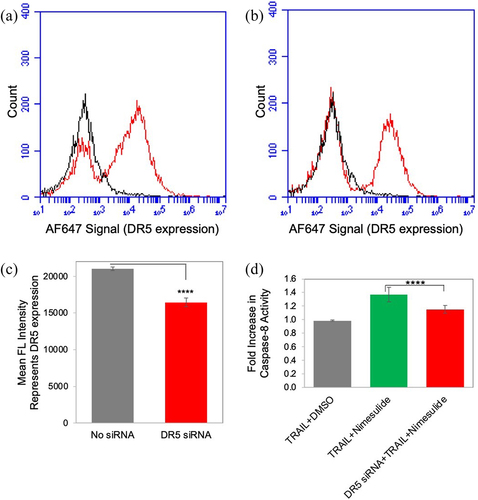
Knockdown of DR5 decreased TRAIL-and nimesulide-induced caspase-8 activity
To determine the specificity of nimesulide in the enhancement of TRAIL-induced apoptosis, we downregulated DR5 expression in pancreatic cancer cells using DR5 siRNA and tested caspase-8 activity in these cells. However, unfortunately, DR5 siRNA knockdown was only partially successful in Panc1 cells (). DR5 expression was reduced by 22% after DR5 siRNA transfection of Panc1 cells. We showed that nimesulide-TRAIL-induced caspase-8 activity was decreased in DR5 siRNA transfected cells compared to untransfected cells (). These results suggest that increased caspase-8 activity in the presence of nimesulide originates from DR5 rather than a nonspecific interaction.
Figure 10. Effect of nimesulide on DR5 siRNA transfected cells. FACS data demonstrate surface expression of DR5 on Panc1 cells (a) and with DR5 siRNA transfected Panc1 (b) cells. (c) Mean FL intensity of transfected and untransfected panc1 cells. Cells were transfected with DR5 siRNA oligonucleotides (25 nM) using lipofectamine 3000. Transfected cells were incubated for 48 hours before further analysis. DR5 expression was determined using anti-DR5 antibody and analyzed by flow cytometry. (d) Caspase-8 activity was measured in siRNA transfected and untransfected panc1 cell. Cells were treated with TRAIL (0.1 μg/ml) +DMSO or TRAIL+nimesulide (50 μM). Data are presented as mean ± standard deviation (N = 3) ****P < .0001 compared to TRAIL nimesulide control by two-tailed unpaired t test.
Discussion
Taken together, our results and other published studies suggest that nimesulide has the ability to act on different targets at the same time (e.g., PTEN and DR5).Citation29–31 In general, multi-target drugs have attracted attention as potential therapeutic solutions to fight against complex health conditions linked to drug-resistance issues.Citation61–64 However, despite their therapeutic promise in cancer, nimesulide and other FDA approved COX-2 inhibitors (e.g., valdecoxib and rofecoxib) have had limited reach (including being banned in several countries) because of their hepatotoxicity and associated cardiovascular problems.Citation65 Despite these issues, the European Medicines Agency has confirmed that the hepatic risks associated with nimesulide can be limited by controlling the dose and duration of treatment,Citation66,Citation67 and a review of safety standards concluded that nimesulide’s overall benefits outweigh the risks.Citation22,Citation68 That said, in part based on these concerns, multiple studies have been done to modify the nimesulide scaffold so as to develop more potent and less hepatoxic anticancer agents.Citation23,Citation69,Citation70
As we have now shown, a common feature between nimesulide and DuP-697 is that both compounds promote DR5 clustering and induce apoptosis in TRAIL resistant cancer cells. Exactly how these small molecules achieve this clustering effect remains an important, unanswered question. Despite the intact expression of DR5, pancreatic cancer cells are not capable of initiating DR5 clustering () and caspase-8 activity in the presence of ligand alone ( and supplementary Figure 5). It is interesting to note that nimesulide treatment alone, while promoting DR5 clustering (), wasn’t sufficient to induce caspase-8 activity (). Similar results were reported in TRAIL resistant colon cancer cells treated with DuP-697.Citation20 It’s likely that while these small molecules can enhance clustering, concomitant TRAIL binding is necessary to force DR5 into a signaling-competent conformational state. Indeed, we have previously suggested that TRAIL binding causes a conformational change in DR5 that occurs in tandem with clustering, and that specific conformational states of the receptor within clusters are necessary for activation; that is, at the molecular scale, not all clusters are the same.Citation48,Citation71
While drugs like nimesulide and DuP-697 show promise, there is nonetheless an ongoing need to develop small molecules that enhance TRAIL sensitivity in cancer cells. There are no existing high-throughput screening (HTS) platforms that enable discovery of small molecules that induce DR5 clustering. For example, neither DuP-697 nor bioymifi were identified through screens based on their direct effect on DR5. In the current and previous studiesCitation35,Citation72 we have shown that it is possible to monitor a combination of DR5 clustering and conformational changes using a cellular TR-FRET-based DR5 biosensor (). We recently showed that this biosensor is HTS-compatible and used it to identify DR5 inhibitors from the Selleck library of small molecules.Citation73 The screening platform utilizes a high-precision fluorescence lifetime plate readerCitation74 (see Methods) that we have also used recently for small molecule discovery targeting TNFR1.Citation33,Citation75 Here, we first identified nimesulide as a promising DR5-targeted compound via a pilot HTS screen of the 1280 compounds in the Sigma Aldrich Library of Pharmacologically Active Compounds (LOPAC) library. In this very limited proof-of-principle screen, we identified nine compounds that increased FRET (Supplementary Figure 6A). These hit compounds have interesting known biological actions. Rutaecarpine is a COX-2 inhibitor, and our study provides the first evidence of its relevance in the DR5 pathway.Citation76 Etodolac, gossypol, and bropirimine exhibit an antitumor effect on several different cancer cells. SC-514 and etodolac are anti-inflammatory compounds.Citation77,Citation78 A3 hydrochloride is a nonselective casein kinase inhibitor,Citation79 Isoguvacine hydrochloride is a γ-aminobutyric acid type A receptor agonist and GR 55562 dihydrobromide is a serotonin receptor antagonist.Citation80 Functionality of a random subset of the hit compounds were evaluated using caspase-8 assay. Of five compounds tested, only nimesulide increased caspase-8 activity, and the other compounds either decreased or had no effect on caspase activity (Supplementary Figure 6B) and hence did not warrant additional investigation here.
Our cell-based TR-FRET platform was designed around HEK293 cells (a nonmalignant line) because of their ease of transfection and history of success in drug discovery campaigns using a high-sensitivity fluorescence plate reader technology.Citation33,Citation81 Future FRET studies will be done in TRAIL-resistant pancreatic cancer cell lines in order to account for at least two important differences in these cells. First, receptor expression levels impact its functionCitation82–84 and clustering,Citation85 and lines expressing the fluorescently labeled receptors should be engineered to match native levels of DR5 in the plasma membrane of resistant pancreatic cancer cells – though this is a non-trivial undertaking. Second, cancer cells are known to have altered phospholipid and cholesterol content in the plasma membraneCitation86–88 and we have shown this can alter DR5 clustering and activity.Citation32 Even without these potential improvements, the success we have shown here with nimesulide suggests that the existing platform holds promise for ongoing efforts to study small molecule activators of DR5. One limitation of this new technology is that it does not allow a deconvolution of two related phenomena: an increase in FRET could reflect clustering of DR5 or structural rearrangements in the DR5 backbone. Thus, despite the likelihood of false-positives (e.g., small molecules that increase FRET due to non-activating molecular reorganizations), we are hopeful that given larger libraries this platform will be successful in identifying lead compounds that are more potent and more specific – and thus safer – than nimesulide.
Statistical Analysis
Statistical analyses were performed using Prism 8.0 software (GraphPad Software, Inc., CA, USA) and the P-values were calculated using a two-tailed unpaired t test or one-way ANOVA.
*p-value<0.05; **p-value<0.01; ***p-value<0.001 and ****p-value<0.0001.
IRB or Ethical Review Board approval
This research does not require IRB or ethics review board approval.
Supplemental Material
Download Zip (900.7 KB)Acknowledgments
We thank Samantha Yuen from the David Thomas group for technical discussions.
Disclosure statement
D.D.T. holds equity in and serves as executive officers for Photonic Pharma LLC, a company that owns intellectual property related to technology used in part of this project. These relationships have been reviewed and managed by the University of Minnesota in accordance with its conflict of interest policies. The remaining authors declare no competing interests.
Data availability statement
The datasets used or analyzed during the current study are available from the corresponding author on reasonable request.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15384047.2023.2176692
Additional information
Funding
Notes on contributors
Nagamani Vunnam
Nagamani Vunnam is a research scientist at University of Minnesota.
Malaney C. Young
Malaney C. Young received her B.S. in Chemical Engineering from Rensselaer Polytechnic Institute in 2015, and her PhD in Biomedical Engineering from University of Minnesota in 2021. Her PhD research focused on the development and implementation of FRET-based high-throughput screening assays to identify small-molecule drug candidates which target pathological protein interactions involved in neurodegenerative disorders and cancer. She joined the Drug Discovery Lab at UCLA as a postdoctoral researcher in 2021 where she continued to further her drug discovery experience by developing AlphaLISA-based HTS assays, implementing stem cell research, and leading the mouse behavioral team. She is currently a Staff Scientist at City of Hope Medical Center in Duarte, CA, where she is working on translational cancer research.
Elly E. Liao
Elly E. Liao is a research scientist at University of Minnesota.
Chih Hung Lo
Chih Hung Lo obtained his PhD from University of Minnesota, and he is currently a Dean's Postdoctoral Fellow in Lee Kong Chian School of Medicine, Nanyang Technological University, Singapore. His research interests focus on understanding TNF receptor structure-function relationship and role of TNF receptor in neuroinflammation and neurodegeneration.
Evan Huber
Evan Huber received his master’s degree in biomedical engineering from University of Minnesota, and he is currently an advanced research associate at Bio-techne.
MaryJane Been
MaryJane Been received her master’s degree in biomedical engineering from University of Minnesota, and she is currently a R&D Scientist - Immunoassays at Qorvo Biotechnologies.
David D. Thomas
David D. Thomas is a professor at University of Minnesota.
Jonathan N. Sachs
Jonathan N. Sachs is a professor at University of Minnesota.
References
- Dai X, Zhang J, Arfuso F, Chinnathambi A, Zayed ME, Alharbi SA, Kumar AP, Ahn KS, Sethi G. Targeting TNF-related apoptosis-inducing ligand (TRAIL) receptor by natural products as a potential therapeutic approach for cancer therapy. Exp Biol Med (Maywood). 2015;240(6):760–14. doi:10.1177/1535370215579167.
- Trivedi R, Mishra DP. Trailing TRAIL resistance: novel targets for TRAIL sensitization in cancer cells. Front Oncol. 2015;5:69. doi:10.3389/fonc.2015.00069.
- Snajdauf M, Havlova K, Vachtenheim J Jr., Ozaniak A, Lischke R, Bartunkova J, Smrz D, Strizova Z. The TRAIL in the treatment of human cancer: an update on clinical trials. Front Mol Biosci. 2021;8:628332. doi:10.3389/fmolb.2021.628332.
- Dimberg LY, Anderson CK, Camidge R, Behbakht K, Thorburn A, Ford HL. On the TRAIL to successful cancer therapy? Predicting and counteracting resistance against TRAIL-based therapeutics. Oncogene. 2013;32(11):1341–1350. doi:10.1038/onc.2012.164.
- Twomey JD, Kim SR, Zhao L, Bozza WP, Zhang B. Spatial dynamics of TRAIL death receptors in cancer cells. Drug Resist Updat. 2015;19:13–21. doi:10.1016/j.drup.2015.02.001.
- Wang S, El-Deiry WS. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene. 2003;22(53):8628–8633. doi:10.1038/sj.onc.1207232.
- Lee SH, Shin MS, Kim HS, Lee HK, Park WS, Kim SY, Lee JH, Han SY, Park JY, Oh RR, et al. Alterations of the DR5/TRAIL receptor 2 gene in non-small cell lung cancers. Cancer Res. 1999;59(22):5683–5686.
- Eggert A, Grotzer MA, Zuzak TJ, Wiewrodt BR, Ho R, Ikegaki N, Brodeur GM. Resistance to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in neuroblastoma cells correlates with a loss of caspase-8 expression. Cancer Res. 2001;61(4):1314–1319.
- Zhang L, Fang B. Mechanisms of resistance to TRAIL-induced apoptosis in cancer. Cancer Gene Ther. 2005;12(3):228–237. doi:10.1038/sj.cgt.7700792.
- Slomp A, Peperzak V. Role and regulation of pro-survival BCL-2 proteins in multiple myeloma. Front Oncol. 2018;8:533. doi:10.3389/fonc.2018.00533.
- Owens TW, Gilmore AP, Streuli CH, Foster FM. Inhibitor of apoptosis proteins: promising targets for cancer therapy. J Carcinog Mutagen. 2013;(Suppl 14): S14–004.
- Daniel PT, Wieder T, Sturm I, Schulze-Osthoff K. The kiss of death: promises and failures of death receptors and ligands in cancer therapy. Leukemia. 2001;15(7):1022–1032. doi:10.1038/sj.leu.2402169.
- Dang CT, Shapiro CL, Hudis CA. Potential role of selective COX-2 inhibitors in cancer management. Oncology (Williston Park). 2002;16(5 Suppl 4):30–36.
- Dannenberg AJ, Altorki NK, Boyle JO, Dang C, Howe LR, Weksler BB, Subbaramaiah K. Cyclo-oxygenase 2: a pharmacological target for the prevention of cancer. Lancet Oncol. 2001;2(9):544–551. doi:10.1016/S1470-2045(01)00488-0.
- Romano M, Claria J. Cyclooxygenase-2 and 5-lipoxygenase converging functions on cell proliferation and tumor angiogenesis: implications for cancer therapy. FASEB J. 2003;17(14):1986–1995. doi:10.1096/fj.03-0053rev.
- Dannenberg AJ, Lippman SM, Mann JR, Subbaramaiah K, DuBois RN. Cyclooxygenase-2 and epidermal growth factor receptor: pharmacologic targets for chemoprevention. J Clin Oncol. 2005;23(2):254–266. doi:10.1200/JCO.2005.09.112.
- Dannenberg AJ, Zakim D. Chemoprevention of colorectal cancer through inhibition of cyclooxygenase-2. Semin Oncol. 1999;26(5):499–504.
- Subbaramaiah K, Dannenberg AJ. Cyclooxygenase 2: a molecular target for cancer prevention and treatment. Trends Pharmacol Sci. 2003;24(2):96–102. doi:10.1016/S0165-6147(02)00043-3.
- Xie C, Xu X, Wang X, Wei S, Shao L, Chen J, Cai J, Jia L. Cyclooxygenase-2 induces angiogenesis in pancreatic cancer mediated by prostaglandin E2. Oncol Lett. 2018;16(1):940–948. doi:10.3892/ol.2018.8786.
- Martin S, Phillips DC, Szekely-Szucs K, Elghazi L, Desmots F, Houghton JA. Cyclooxygenase-2 inhibition sensitizes human colon carcinoma cells to TRAIL-induced apoptosis through clustering of DR5 and concentrating death-inducing signaling complex components into ceramide-enriched caveolae. Cancer Res. 2005;65(24):11447–11458. doi:10.1158/0008-5472.CAN-05-1494.
- Totzke G, Schulze-Osthoff K, Janicke RU. Cyclooxygenase-2 (COX-2) inhibitors sensitize tumor cells specifically to death receptor-induced apoptosis independently of COX-2 inhibition. Oncogene. 2003;22(39):8021–8030. doi:10.1038/sj.onc.1206837.
- Caiazzo E, Ialenti A, Cicala C. The relatively selective cyclooxygenase-2 inhibitor nimesulide: what’s going on? Eur J Pharmacol. 2019;848:105–111. doi:10.1016/j.ejphar.2019.01.044.
- Catarro M, Serrano JL, Ramos SS, Silvestre S, Almeida P. Nimesulide analogues: from anti-inflammatory to antitumor agents. Bioorg Chem. 2019;88:102966. doi:10.1016/j.bioorg.2019.102966.
- Sengel-Turk CT, Hascicek C, Bakar F, Simsek E. Comparative evaluation of nimesulide-loaded nanoparticles for anticancer activity against breast cancer cells. AAPS PharmSciTech. 2017;18(2):393–403. doi:10.1208/s12249-016-0514-2.
- Barac A, Mitulovic G, Hallstrom S, Zehetmayer S, Grasl MC, Erovic BM. Impact of combined treatment with nimesulide and cisplatin on oral carcinoma cells. Onco Targets Ther. 2017;10:3607–3616. doi:10.2147/OTT.S131106.
- Liu JR, Wu WJ, Liu SX, Zuo LF, Wang Y, Yang JZ, Nan YM. Nimesulide inhibits the growth of human esophageal carcinoma cells by inactivating the JAK2/STAT3 pathway. Pathol Res Pract. 2015;211(6):426–434. doi:10.1016/j.prp.2015.01.007.
- Nam KT, Hahm KB, Oh SY, Yeo M, Han SU, Ahn B, Kim YB, Kang JS, Jang DD, Yang KH, et al. The selective cyclooxygenase-2 inhibitor nimesulide prevents Helicobacter pylori-associated gastric cancer development in a mouse model. Clin Cancer Res. 2004;10(23):8105–8113. doi:10.1158/1078-0432.CCR-04-0896.
- Suleyman H, Cadirci E, Albayrak A, Halici Z. Nimesulide is a selective COX-2 inhibitory, atypical non-steroidal anti-inflammatory drug. Curr Med Chem. 2008;15(3):278–283. doi:10.2174/092986708783497247.
- Chu M, Wang T, Sun A, Chen Y. Nimesulide inhibits proliferation and induces apoptosis of pancreatic cancer cells by enhancing expression of PTEN. Exp Ther Med. 2018;16(1):370–376. doi:10.3892/etm.2018.6191.
- Eibl G, Reber HA, Wente MN, Hines OJ. The selective cyclooxygenase-2 inhibitor nimesulide induces apoptosis in pancreatic cancer cells independent of COX-2. Pancreas. 2003;26(1):33–41. doi:10.1097/00006676-200301000-00007.
- Ferreira RG, Narvaez LEM, Espindola KMM, Rosario A, Lima WGN, Monteiro MC. Can nimesulide nanoparticles be a therapeutic strategy for the inhibition of the KRAS/PTEN signaling pathway in pancreatic cancer? Front Oncol. 2021;11:594917. doi:10.3389/fonc.2021.594917.
- Lewis AK, Valley CC, Peery SL, Brummel B, Braun AR, Karim CB, Sachs JN. Death receptor 5 networks require membrane cholesterol for proper structure and function. J Mol Biol. 2016;428(24 Pt A):4843–4855. doi:10.1016/j.jmb.2016.10.001.
- Lo CH, Vunnam N, Lewis AK, Chiu TL, Brummel BE, Schaaf TM, Grant BD, Bawaskar P, Thomas DD, Sachs JN. An innovative high-throughput screening approach for discovery of small molecules that inhibit TNF receptors. SLAS Discov. 2017;22(8):950–961. doi:10.1177/2472555217706478.
- Stroik DR, Yuen SL, Janicek KA, Schaaf TM, Li J, Ceholski DK, Hajjar RJ, Cornea RL, Thomas DD. Targeting protein-protein interactions for therapeutic discovery via FRET-based high-throughput screening in living cells. Sci Rep. 2018;8(1):12560. doi:10.1038/s41598-018-29685-z.
- Vunnam N, Lo CH, Grant BD, Thomas DD, Sachs JN. Soluble extracellular domain of death receptor 5 inhibits TRAIL-induced apoptosis by disrupting receptor-receptor interactions. J Mol Biol. 2017;429(19):2943–2953. doi:10.1016/j.jmb.2017.08.009.
- von Stetten D, Noirclerc-Savoye M, Goedhart J, Gadella TW Jr., Royant A. Structure of a fluorescent protein from aequorea victoria bearing the obligate-monomer mutation A206K. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2012;68(Pt 8):878–882. doi:10.1107/S1744309112028667.
- Zacharias DA, Violin JD, Newton AC, Tsien RY. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science. 2002;296(5569):913–916. doi:10.1126/science.1068539.
- Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281(5381):1305–1308. doi:10.1126/science.281.5381.1305.
- Kischkel FC, Lawrence DA, Chuntharapai A, Schow P, Kim KJ, Ashkenazi A. Apo2L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptors 4 and 5. Immunity. 2000;12(6):611–620. doi:10.1016/S1074-7613(00)80212-5.
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1–2):55–63. doi:10.1016/0022-1759(83)90303-4.
- Ashkenazi A. Targeting the extrinsic apoptotic pathway in cancer: lessons learned and future directions. J Clin Invest. 2015;125(2):487–489. doi:10.1172/JCI80420.
- Dumitru CA, Gulbins E. TRAIL activates acid sphingomyelinase via a redox mechanism and releases ceramide to trigger apoptosis. Oncogene. 2006;25(41):5612–5625. doi:10.1038/sj.onc.1209568.
- Pan L, Fu TM, Zhao W, Zhao L, Chen W, Qiu C, Liu W, Liu Z, Piai A, Fu Q, et al. Higher-order clustering of the transmembrane anchor of DR5 drives signaling. Cell. 2019;176(6):1477–1489 e14. doi:10.1016/j.cell.2019.02.001.
- Song JH, Tse MC, Bellail A, Phuphanich S, Khuri F, Kneteman NM, Hao C. Lipid rafts and nonrafts mediate tumor necrosis factor related apoptosis-inducing ligand induced apoptotic and nonapoptotic signals in non small cell lung carcinoma cells. Cancer Res. 2007;67(14):6946–6955. doi:10.1158/0008-5472.CAN-06-3896.
- Wang G, Wang X, Yu H, Wei S, Williams N, Holmes DL, Halfmann R, Naidoo J, Wang L, Li L, et al. Small-molecule activation of the TRAIL receptor DR5 in human cancer cells. Nat Chem Biol. 2013;9(2):84–89. doi:10.1038/nchembio.1153.
- Naismith JH, Devine TQ, Brandhuber BJ, Sprang SR. Crystallographic evidence for dimerization of unliganded tumor necrosis factor receptor. J Biol Chem. 1995;270(22):13303–13307. doi:10.1074/jbc.270.22.13303.
- Lee HW, Lee SH, Lee HW, Ryu YW, Kwon MH, Kim YS. Homomeric and heteromeric interactions of the extracellular domains of death receptors and death decoy receptors. Biochem Biophys Res Commun. 2005;330(4):1205–1212. doi:10.1016/j.bbrc.2005.03.101.
- Valley CC, Lewis AK, Mudaliar DJ, Perlmutter JD, Braun AR, Karim CB, Thomas DD, Brody JR, Sachs JN. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) induces death receptor 5 networks that are highly organized. J Biol Chem. 2012;287(25):21265–21278. doi:10.1074/jbc.M111.306480.
- Abdelrahim M, Newman K, Vanderlaag K, Samudio I, Safe S. 3,3’-diindolylmethane (DIM) and its derivatives induce apoptosis in pancreatic cancer cells through endoplasmic reticulum stress-dependent upregulation of DR5. Carcinogenesis. 2006;27(4):717–728. doi:10.1093/carcin/bgi270.
- Park EJ, Min KJ, Choi KS, Kubatka P, Kruzliak P, Kim DE, Kwon TK. Chloroquine enhances TRAIL-mediated apoptosis through up-regulation of DR5 by stabilization of mRNA and protein in cancer cells. Sci Rep. 2016;6(1):22921. doi:10.1038/srep22921.
- Pan G, Ni J, Wei YF, Yu G, Gentz R, Dixit VM. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science. 1997;277(5327):815–818. doi:10.1126/science.277.5327.815.
- Pan G, O’Rourke K, Chinnaiyan AM, Gentz R, Ebner R, Ni J, Dixit VM. The receptor for the cytotoxic ligand TRAIL. Science. 1997;276(5309):111–113. doi:10.1126/science.276.5309.111.
- Ruggeri P, Cappabianca L, Farina AR, Gneo L, Mackay AR. NGF sensitizes TrkA SH-SY5Y neuroblastoma cells to TRAIL-induced apoptosis. Cell Death Discov. 2016;2(1):16004. doi:10.1038/cddiscovery.2016.4.
- Artykov AA, Yagolovich AV, Dolgikh DA, Kirpichnikov MP, Trushina DB, Gasparian ME. Death receptors DR4 and DR5 undergo spontaneous and ligand-mediated endocytosis and recycling regardless of the sensitivity of cancer cells to TRAIL. Front Cell Dev Biol. 2021;9:733688. doi:10.3389/fcell.2021.733688.
- Mohr A, Yu R, Zwacka RM. TRAIL-receptor preferences in pancreatic cancer cells revisited: both TRAIL-R1 and TRAIL-R2 have a licence to kill. BMC Cancer. 2015;15(1):494. doi:10.1186/s12885-015-1508-2.
- Gibson SB, Oyer R, Spalding AC, Anderson SM, Johnson GL. Increased expression of death receptors 4 and 5 synergizes the apoptosis response to combined treatment with etoposide and TRAIL. Mol Cell Biol. 2000;20(1):205–212. doi:10.1128/MCB.20.1.205-212.2000.
- Vunnam N, Szymonski S, Hirsova P, Gores GJ, Sachs JN, Hackel BJ. Noncompetitive allosteric antagonism of death receptor 5 by a synthetic affibody ligand. Biochemistry. 2020;59(40):3856–3868. doi:10.1021/acs.biochem.0c00529.
- Kim PK, Park SY, Koty PP, Hua Y, Luketich JD, Billiar TR. Fas-associating death domain protein overexpression induces apoptosis in lung cancer cells. J Thorac Cardiovasc Surg. 2003;125(6):1336–1342. doi:10.1016/S0022-5223(02)73227-3.
- Ranjan K, Surolia A, Pathak C. Apoptotic potential of Fas-associated death domain on regulation of cell death regulatory protein cFLIP and death receptor mediated apoptosis in HEK 293T cells. J Cell Commun Signal. 2012;6(3):155–168. doi:10.1007/s12079-012-0166-2.
- Ranjan K, Pathak C. FADD regulates NF-kappaB activation and promotes ubiquitination of cFLIPL to induce apoptosis. Sci Rep. 2016;6(1):22787. doi:10.1038/srep22787.
- Koeberle A, Werz O. Multi-target approach for natural products in inflammation. Drug Discov Today. 2014;19(12):1871–1882. doi:10.1016/j.drudis.2014.08.006.
- Lee JW, Dworetzky B. Rational polytherapy with antiepileptic drugs. Pharmaceuticals (Basel). 2010;3(8):2362–2379. doi:10.3390/ph3082362.
- Makhoba XH, Viegas C Jr., Mosa RA, Viegas FPD, Pooe OJ. Potential impact of the multi-target drug approach in the treatment of some complex diseases. Drug Des Devel Ther. 2020;14:3235–3249. doi:10.2147/DDDT.S257494.
- Zheng H, Fridkin M, Youdim M. From single target to multitarget/network therapeutics in Alzheimer’s therapy. Pharmaceuticals (Basel). 2014;7(2):113–135. doi:10.3390/ph7020113.
- Kwon J, Kim S, Yoo H, Lee E, Zheng Y-W. Nimesulide-induced hepatotoxicity: a systematic review and meta-analysis. PLoS One. 2019;14(1):e0209264. doi:10.1371/journal.pone.0209264.
- Donati M, Conforti A, Lenti MC, Capuano A, Bortolami O, Motola D, Moretti U, Vannacci A, Rafaniello C, Vaccheri A, et al. Risk of acute and serious liver injury associated to nimesulide and other NSAIDs: data from drug-induced liver injury case-control study in Italy. Br J Clin Pharmacol. 2016;82(1):238–248. doi:10.1111/bcp.12938.
- Nimesulide. In LiverTox: clinical and research information on drug-induced liver injury. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases. 2012.
- Rainsford KD. Members of the consensus report group on, N., Nimesulide – a multifactorial approach to inflammation and pain: scientific and clinical consensus. Curr Med Res Opin. 2006;22(6):1161–1170. doi:10.1185/030079906X104849.
- Renard JF, Julemont F, de Leval X, Pirotte B. The use of nimesulide and its analogues in cancer chemoprevention. Anticancer Agents Med Chem. 2006;6(3):233–237. doi:10.2174/187152006776930855.
- Zhong B, Cai X, Chennamaneni S, Yi X, Liu L, Pink JJ, Dowlati A, Xu Y, Zhou A, Su B. From COX-2 inhibitor nimesulide to potent anti-cancer agent: synthesis, in vitro, in vivo and pharmacokinetic evaluation. Eur J Med Chem. 2012;47(1):432–444. doi:10.1016/j.ejmech.2011.11.012.
- Lewis AK, James ZM, McCaffrey JE, Braun AR, Karim CB, Thomas DD, Sachs JN. Open and closed conformations of the isolated transmembrane domain of death receptor 5 support a new model of activation. Biophys J. 2014;106(6):L21–4. doi:10.1016/j.bpj.2014.01.044.
- Vunnam N, Campbell-Bezat CK, Lewis AK, Sachs JN, Receptor D. 5 activation is energetically coupled to opening of the transmembrane domain dimer. Biophys J. 2017;113(2):381–392. doi:10.1016/j.bpj.2017.05.038.
- Young M, Vunnam N, Reddeck R, Yuen S, Thomas DD, Sachs JN. Naturally occurring compounds that inhibit TRAIL-induced DR5 activation. J Nat Prod Submitted. 2022;41(3): 818–28.
- Schaaf TM, Peterson KC, Grant BD, Thomas DD, Gillispie GD. Spectral unmixing plate reader: high-throughput, high-precision FRET assays in living cells. SLAS Discov. 2017;22(3):250–261. doi:10.1177/1087057116679637.
- Lo CH, Schaaf TM, Grant BD, Lim CK, Bawaskar P, Aldrich CC, Thomas DD, Sachs JN. Noncompetitive inhibitors of TNFR1 probe conformational activation states. Sci Signal. 2019;12(592):592. doi:10.1126/scisignal.aav5637.
- Jia S, Hu C. Pharmacological effects of rutaecarpine as a cardiovascular protective agent. Molecules. 2010;15(3):1873–1881. doi:10.3390/molecules15031873.
- Chen YF, Jobanputra P, Barton P, Bryan S, Fry-Smith A, Harris G, Taylor RS. Cyclooxygenase-2 selective non-steroidal anti-inflammatory drugs (etodolac, meloxicam, celecoxib, rofecoxib, etoricoxib, valdecoxib and lumiracoxib) for osteoarthritis and rheumatoid arthritis: a systematic review and economic evaluation. Health Technol Assess. 2008;12(11):1–278. doi:10.3310/hta12110. iii.
- Kishore N, Sommers C, Mathialagan S, Guzova J, Yao M, Hauser S, Huynh K, Bonar S, Mielke C, Albee L, et al. A selective IKK-2 inhibitor blocks NF-kappa B-dependent gene expression in interleukin-1 beta-stimulated synovial fibroblasts. J Biol Chem. 2003;278(35):32861–32871. doi:10.1074/jbc.M211439200.
- Hu G, Jang GF, Cowan CW, Wensel TG, Palczewski K. Phosphorylation of RGS9-1 by an endogenous protein kinase in rod outer segments. J Biol Chem. 2001;276(25):22287–22295. doi:10.1074/jbc.M011539200.
- Krogsgaard-Larsen P, Schultz B, Mikkelsen H, Aaes-Jorgensen T, Bogeso KP. THIP, isoguvacine, isoguvacine oxide, and related GABA agonists. Adv Biochem Psychopharmacol. 1981;29:69–76.
- Lo CH, Schaaf TM, Thomas DD, Sachs JN. Fluorescence-based TNFR1 biosensor for monitoring receptor structural and conformational dynamics and discovery of small molecule modulators. Methods Mol Biol. 2021;2248:121–137.
- Cheng J, Hylander BL, Baer MR, Chen X, Repasky EA. Multiple mechanisms underlie resistance of leukemia cells to Apo2 Ligand/TRAIL. Mol Cancer Ther. 2006;5(7):1844–1853. doi:10.1158/1535-7163.MCT-06-0050.
- Jin Z, McDonald ER 3rd, Dicker DT, El-Deiry WS. Deficient tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptor transport to the cell surface in human colon cancer cells selected for resistance to TRAIL-induced apoptosis. J Biol Chem. 2004;279(34):35829–35839. doi:10.1074/jbc.M405538200.
- Tang X, Sun YJ, Half E, Kuo MT, Sinicrope F. Cyclooxygenase-2 overexpression inhibits death receptor 5 expression and confers resistance to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human colon cancer cells. Cancer Res. 2002;62(17):4903–4908.
- Cazanave SC, Mott JL, Bronk SF, Werneburg NW, Fingas CD, Meng XW, Finnberg N, El-Deiry WS, Kaufmann SH, Gores GJ. Death receptor 5 signaling promotes hepatocyte lipoapoptosis. J Biol Chem. 2011;286(45):39336–39348. doi:10.1074/jbc.M111.280420.
- Alves AC, Ribeiro D, Nunes C, Reis S. Biophysics in cancer: the relevance of drug-membrane interaction studies. Biochim Biophys Acta. 2016;1858(9):2231–2244. doi:10.1016/j.bbamem.2016.06.025.
- Koundouros N, Poulogiannis G. Reprogramming of fatty acid metabolism in cancer. Br J Cancer. 2020;122(1):4–22. doi:10.1038/s41416-019-0650-z.
- Szlasa W, Zendran I, Zalesinska A, Tarek M, Kulbacka J. Lipid composition of the cancer cell membrane. J Bioenerg Biomembr. 2020;52(5):321–342. doi:10.1007/s10863-020-09846-4.