
ABSTRACT
Poly ADP-ribose polymerase (PARP) inhibitors are effective therapies for cancer patients with homologous recombination (HR) deficient tumors. The imipridone ONC206 is an orally bioavailable dopamine receptor D2 antagonist and mitochondrial protease ClpP agonist that has anti-tumorigenic effects in endometrial cancer via induction of apoptosis, activation of the integrated stress response and modulation of PI3K/AKT signaling. Both PARP inhibitors and imipridones are being evaluated in endometrial cancer clinical trials but have yet to be explored in combination. In this manuscript, we evaluated the effects of the PARP inhibitor olaparib in combination with ONC206 in human endometrioid endometrial cancer cell lines and in a genetically engineered mouse model of endometrial cancer. Our results showed that simultaneous exposure of endometrial cancer cells to olaparib and ONC206 resulted in synergistic anti-proliferative effects and increased cellular stress and apoptosis in both cell lines, compared to either drug alone. The combination treatment also decreased expression of the anti-apoptotic protein Bcl-2 and reduced phosphorylation of AKT and S6, with greater effects compared to either drug alone. In the transgenic model of endometrial cancer, the combination of olaparib and ONC206 resulted in a more significant reduction in tumor weight in obese and lean mice compared to ONC206 alone or olaparib alone, together with a considerably decreased Ki-67 and enhanced H2AX expression in obese and lean mice. These results suggest that this novel dual therapy may be worthy of further exploration in clinical trials.
Inroduction
Endometrial carcinoma (EC) originates from the epithelial cells of the endometrium and accounts for 90% of uterine cancersCitation1. In 2022, there will be an estimated 65,950 new cases of endometrial cancer and 12,550 deaths in the USCitation2. EC is one of the few cancers with both increasing incidence and mortality. This increase has been attributed to rising rates of obesity as obesity is a well-known driver of EC pathogenesisCitation3. Obesity is associated with both higher risk of developing and dying from ECCitation3–6. Based on the differences between molecular, clinical, and histopathological features, EC has been divided into two histopathological groups – type I and type II. Type I EC is estrogen-dependent, moderately differentiated to well-differentiated, surface-invasive endometrioid neoplasms that account for more than 70% of endometrial cancers. Type II EC is a non-estrogen-dependent, non-endometrioid, more aggressive growth with a high risk of recurrence and occurs in older women who are not obeseCitation1,Citation7. While the prognosis is favorable for women who present with early-stage EC, survival rates for women with metastatic EC are less than 20%Citation8. Thus, novel therapies are desperately needed for patients with advanced and recurrent cancer.
Poly(ADP-ribose) polymerase (PARP) has been implicated in DNA single-strand breaks, gene transcription and apoptosisCitation9,Citation10. PARP inhibitors are a class of drugs that inhibit PARP enzymes and, thus, prevent single-strand DNA break repair which leads to accumulation of double-stranded DNA breaksCitation10. In tumor cells with BRCA mutations and/or deficient homologous recombination repair, PARP inhibition has greater impact on DNA repair, resulting in inhibition of tumor cell proliferation and induction of tumor regressionCitation11,Citation12. While PARP inhibitors are FDA-approved as monotherapy in ovarian cancer, PARP monotherapy has not shown promising efficacy in clinical trials for ECCitation13. Combining PARP inhibitors with agents that inhibit homologous recombination or with platinum-based agents as chemosensitizers may be effective strategies to increase the efficacy of PARP inhibitors in the treatment of ECCitation13,Citation14. This strategy has been successful in pre-clinical studies in multiple other tumor typesCitation15–20.
Imipridones are a novel class of cancer therapy that target dopamine receptor D2 signaling, induce TRAIL, and activate the integrated stress response via mitochondrial caseinolytic protease P (ClpP)Citation21–24. ONC201, the first member of the imipridone class, is orally bioavailable and well toleratedCitation25. ONC201 is currently being evaluated in numerous phase I and II clinical trials, including in EC. ONC206 is a derivative and chemical analogue of ONC201 that exhibits enhanced potencyCitation26,Citation27 and is currently being evaluated in Phase I trials. Our recent study shows that ONC201 or ONC206 effectively inhibits tumor cell proliferation and tumor growth through inhibition of the Akt/mTOR pathway in EC cell lines and a transgenic mouse model of ECCitation28–31.
PTEN and PIK3CA, which are frequently mutated in both type I (endometrioid) and type II (serous) ECsCitation32,Citation33, have been shown to play an indirect role in homologous recombinationCitation15,Citation16,Citation34. PI3K inhibition leads to downregulation of BRCA, and loss of homologous recombination results in increased DNA damage and sensitivity to PARP inhibitorsCitation15,Citation16. The inhibitory effects of ONC201 and ONC206 on tumor growth are thought to be via their effects on the PI3K/AKT/mTOR pathway as well as DNA repairCitation21,Citation29,Citation35,Citation36, suggesting that ONC201 and ONC206 may be logical therapeutic partners for PARP inhibition in EC. Since PARP inhibitors and imipridones are both being evaluated in EC clinical trials but have yet to be explored in combination in pre-clinical models, the objectives of the present study were to determine the anti-proliferative effects of olaparib in combination with ONC206 in EC cell lines and a transgenic mouse model of endometrioid EC.
Results
Olaparib and ONC206 effectively inhibited cell proliferation and had synergistic anti-proliferative effects in EC cells
To investigate whether olaparib and ONC206 exhibits anti-proliferative activity against EC cells, we treated two EC cell lines, ECC-1 and HEC-1A, with different concentrations of olaparib and ONC206 for 72 h. Confirming our prior work, the MTT assay showed ONC206 potently inhibited cell proliferation in a dose-dependent manner in both cell lines with an IC50 of 0.19 µM for ECC-1 and 0.31 µM for HEC-1A (). A similar inhibitory effect was observed in olaparib treated ECC-1 and HEC-1A cells. Olaparib significantly suppressed growth of both cell lines in a dose-dependent manner with an IC50 of 8.64 µM for ECC-1 and 42.06 µM for HEC-1A ().
Figure 1. Synergistic effects of olaparib and ONC206 in EC cells. ECC-1 and HEC-1A cell lines were treated with varying doses of olaparib and ONC206 as single agents for 72 h. Cell proliferation was assessed by MTT assay. Olaparib and ONC206 significantly inhibited cell proliferation in a dose dependent manner (a and b). Simultaneous exposure to both drugs at low doses resulted in synergistic inhibition of cell proliferation in both cell lines (c). The combination index (CI) was calculated by the Bliss Independence model (d). Western blotting results indicated that ONC206, olaparib and the combination treatment reduced phosphorylation of AKT and S6 in both cell lines after 24 h of treatment (e). OLA is an abbreviation for Olaparib. (* = p < .05, ** = p < .01).
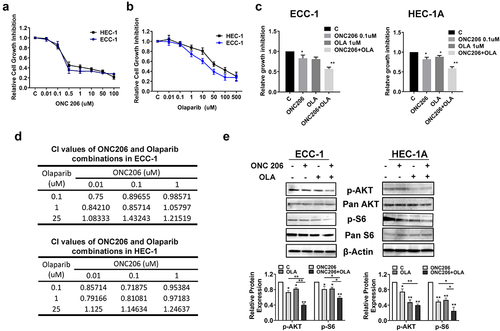
We next explored the efficacy of the combination of olaparib and ONC206 in EC cells. Co-treatment with different concentrations of olaparib and ONC206 resulted in greater inhibition of cell proliferation than with either drug alone after 72 h of treatment (). To determine whether the effects of simultaneous exposure were synergistic, the combination index (CI) was calculated using the Bliss Independence model. Synergistic anti-proliferative effects (CI < 1) were found with olaparib at doses of 0.1–1 µM and ONC206 at doses of 0.01–0.1 µM. In contrast, the combination of high doses (25 µM) of olaparib and varying doses of ONC206 did not induce synergistic reduction in cell viability ().
Given that imipridones inhibit cell proliferation and tumor growth through effects on the AKT/mTOR pathway in ECCitation36, we next investigated whether the AKT/mTOR pathway was involved in the synergistic anti-proliferative effects of olaparib and ONC206 in the EC cell lines. Both cell lines were treated with ONC206, olaparib, and the combination for 24 h. Western immunoblotting results show that olaparib and ONC206 significantly inhibited phosphorylation of AKT and S6. Dual therapy resulted in even greater effects on reducing phosphorylation of AKT and S6 in both cell lines (). Together, these results suggest that the combination of olaparib and ONC206 synergistically induced growth inhibition through modulation of the AKT/mTOR pathway in EC cell lines.
The combination of olaparib and ONC206 effectively induced apoptosis in EC cells
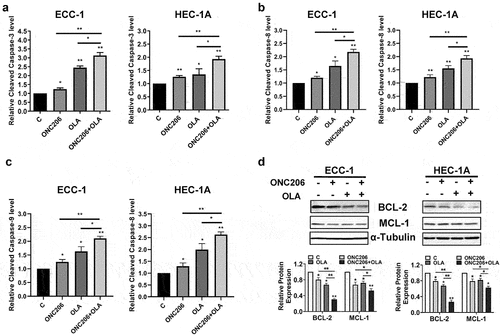
Olaparib and ONC206 have been known to both induce apoptosis in EC cellsCitation27,Citation37. To determine the effect of co-treatment with olaparib and ONC206 on apoptosis in EC cells, an ELISA cleaved caspase-3 assay was performed after treatment with ONC206, olaparib, and the combination to evaluate the effect of treatment on cleaved caspase-3 activity. Treatment with ONC206 (0.1 µM) effectively increased cleaved caspase-3 activity by 1.23 times in the ECC-1 cells and 1.26 times in the HEC-1A cells compared to controls, respectively. Olaparib at 1 µM increased cleaved caspase-3 activity by 2.45 times in the ECC-1 cells and by 1.35 times in the HEC-1A cells. The combination of both drugs led to a greater effect compared to either drug alone in both cell lines (). Consistent with the results of cleaved caspase-3, ONC206, olaparib, and the combination exhibited similar effects on cleaved caspase-8 and −9 activities in both cell lines, with the combination treatment producing the greatest effects (). Western immunoblotting demonstrated decreased expression of the anti-apoptotic proteins BCL-2 and MCL-1 in both cell lines that was further enhanced by combination treatment, mirroring the caspase-3 assay in the ECC-1 and HEC-1A cells (). These results suggest that induction of apoptosis by olaparib, ONC206, or the combination may be dependent on caspase pathways in EC cells.
Figure 2. Olaparib and ONC206 induced apoptosis in EC cells. ECC-1 and HEC-1A cells were treated with 0.1 µM ONC206, 1 µM olaparib and the combination of olaparib and ONC206 for 12 h. Olaparib and ONC206 increased cleaved caspase-3 activity in both cell lines with greater effects for the combination compared to either drug alone (a). Western immunoblotting demonstrated decreased expression of the anti-apoptotic proteins BCL-2 and MCL-1, with olaparib and ONC206 treatment (b). OLA is an abbreviation for Olaparib. (* = p < .05, ** = p < .01).
The combination of olaparib and ONC206 effectively increased cellular stress in EC cells
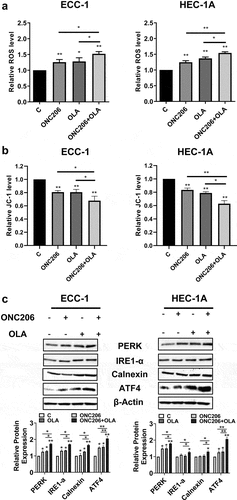
Given evidence that imipridones activate the integrated stress responseCitation25,Citation27, we examined the effects of olaparib and ONC206 on production of reactive oxygen species (ROS) using the DCFH-DA assay. ONC206 (0.1 µM) treatment resulted in a 1.25-fold increase in ROS in the ECC-1 cells and a 1.31-fold increase in the HEC-1A cells. Olaparib (1 µM) treatment resulted in a 1.28-fold increase in the ECC-1 cells and a 1.37-fold increase in the HEC-1A cells. Combination olaparib and ONC206 therapy led to a 1.5-fold increase in ROS over controls in both cell lines, which was also significantly increased compared to either drug alone (). Similar results were observed via the JC-1 assay. ONC206 or olaparib reduced mitochondrial membrane potential with the greatest effects seen after combination treatment in both cell lines (). Confirmatory western blots demonstrated increased expression of the cellular stress proteins PERK, ATF4, IRE, and calnexin with ONC206, olaparib, or combination treatment in both cell lines after 16 h of treatment ().
Figure 3. Olaparib and ONC206 induced cellular stress in EC cells. ECC-1 and HEC-1A cells were treated with 0.1 µM ONC206, 1 µM olaparib and the combination. ROS was assessed by DCFDA assay. Combination therapy with olaparib and ONC206 resulted in increased reactive oxygen species and mitochondrial membrane potential as compared to either drug alone (a and b). Confirmatory western blot assays demonstrated increased expression of the cellular stress proteins PERK, IRE, ATF4 and calnexin with combination treatment (c). OLA is an abbreviation for Olaparib. (* = p < .05, ** = p < .01).
The combination of olaparib and ONC206 reduced tumor weight in vivo
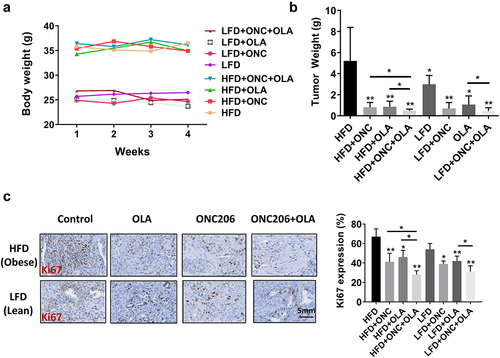
In order to further verify the inhibitory effect of ONC206, olaparib, and the combination in vitro, we treated the Lkb1fl/flp53fl/fl genetically engineered mouse model of EC with ONC206, olaparib, and the combination of both drugs for 4 weeks under obese and lean conditions (15 mice/group). Obese mice weighed significantly more at the start of treatment compared to lean mice (35.7 g in obese vs 25.6 g in lean, p < .01). During the varying treatments, all mice exhibited normal activities with no changes in body weight, and both drugs were well tolerated individually and in combination in the mice (). As seen in our prior studies using the same mouse model, obese mice had significantly greater tumor weights compared to lean mice across all treatment groups (5.29 g vs 2.98 g, p < .04) Treatment with ONC206 decreased tumor weight by 84% in obese vs 77% in lean mice. Olaparib decreased tumor weight by 83% in obese vs 65% in lean mice. The combination decreased tumor weight by 91% in obese vs 85% in lean mice. The reduction in combination treatment compared to olaparib monotherapy was significant for both obese and lean mice (p < .05). The reduction in combination treatment compared to ONC206 monotherapy was significant for obese mice only (p < .05, ). Immunohistochemistry showed that olaparib and ONC206 significantly reduced expression of Ki-67, a marker of tumor cell proliferation, in both obese and lean mice, with more potent effects with combination treatment ().
Figure 4. Synergistic effect of treatment with olaparib and ONC206 in vivo. Lkb1fl/flp53 fl/fl mice were fed HFD (obese) or LFD (lean) starting at 3 weeks of age. After injection of AdCre into one uterine horn at 8 weeks, the mice were treated with ONC206 (100 mg/kg weekly), olaparib (25 mg/kg daily) and the combination for 4 weeks. Diet-induced obesity significantly increased the body weights of Lkb1fl/flp53 fl/fl mice (a). Olaparib and ONC206 effectively reduced tumor weights, and combination treatment led to a significantly greater reduction in tumor weight compared to either drug alone under both obese and lean conditions (b). Immunohistochemistry showed that olaparib and ONC206 significantly reduced expression of Ki-67, a marker of tumor cell proliferation, in both obese and lean mice, with more potent effects seen with combination treatment (c). OLA is an abbreviation for Olaparib. (* = p < .05, ** = p < .01).
The combination of olaparib and ONC206 resulted in a greater inhibitory effect on DNA repair
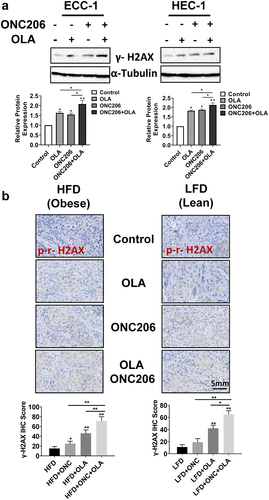
In order to better understand how ONC206, olaparib and the combination treatment affect DNA repair, we determined the protein expression of phosphorylated γ-H2A× following treatment with ONC206, olaparib, and the combination in vitro and in vivo. Western blotting results showed that after treatment with olaparib and ONC206 for 24 h, levels of phosphorylated γ-H2A× (a marker of DNA damage) were increased in both the ECC-1 and HEC-1A cells compared with vehicles. Furthermore, phosphorylated γ-H2A× levels were most pronounced with the combination of olaparib and ONC206 treatment in both cell lines (). To confirm the immunoblotting results in EC cells, the expression of phosphorylated γ-H2A× was examined with immunohistochemistry in endometrial tumors under obese and lean conditions. Single agent olaparib and ONC206 treatments increased phosphorylated γ-H2A× in the tumors of obese and lean mice. Combined olaparib and ONC206 treatment significantly enhanced phosphorylated γ-H2A× expression over that of either drug alone ().
Figure 5. Olaparib and ONC206 induced the expression of phosphorylated γ-H2A× in vitro and in vivo. ECC-1 and HEC-1A cells were treated with 0.1 µM ONC206, 1 µM olaparib and the combination for 24 h. Western blotting showed that olaparib and ONC206 increased the expression of phosphorylated γ-H2A× in both cell lines with greater expression in the combination treatment (a). The expression of phosphorylated γ-H2A× in tumor tissues was detected by immunohistochemical staining after Lkb1fl/flp53 fl/fl mice were treated with ONC206, olaparib and the combination for 4 weeks. Dual therapy with olaparib and ONC206 increased gamma-H2A× as compared to treatment with either drug alone in obese and lean mice (b). OLA is an abbreviation for Olaparib. (* = p < .05, ** = p < .01).
Discussion
Given the poor prognosis associated with advanced and recurrent EC and the steady rise in mortality from EC, there is a critical need to develop novel therapies for patients with EC. Olaparib as a single agent or as a chemosensitizer has shown promising clinical efficacy in preclinical models and clinical trials in multiple types of cancer with BRCA mutations and/or a homologous recombination deficiency phenotypeCitation13,Citation38. In addition, some studies have found that the inhibitory effects of olaparib do not always depend on BRCA1/2 status in ovarian and breast cancers. The combination of AKT inhibitors, Topo I inhibitors, and regulators of homologous recombination has clear therapeutic potential in pre-clinical modelsCitation39,Citation40. The anti-tumorigenic activities of ONC201 have been extensively studied in pre-clinical models as well as clinical trialsCitation25. In understanding its unique mechanism on inhibition of tumor growth, ONC201 has exhibited effective synergy in combination with chemotherapeutic drugs and other small-molecule inhibitors that work by complementary mechanismsCitation41–43. ONC206, a derivative and chemical analogue of ONC201, demonstrates enhanced noncompetitive DRD2 antagonism and bioavailability compared to ONC201 and is currently being assessed in a first-in-human trialCitation26,Citation30,Citation44. Our previous studies demonstrated that ONC206 caused apoptosis and inhibited tumor growth in a transgenic mouse model of EC through inhibition of the AKT/mTOR pathway. Since inhibition of the AKT/mTOR pathway leads to downregulation of BRCA1/2 and homologous recombination and olaparib activates AKT/mTOR signalingCitation15,Citation19,Citation45, the combination of olaparib and ONC206 may have a synergistic effect in inhibiting tumor growth in EC via dual inhibition of the AKT/mTOR pathway. In addition, the unique pharmacological properties and robust safety profile of ONC206 also provide opportunities to combine with other chemotherapeutics and targeted therapies, including olaparib, without additional toxicities for patients. In this study, we have demonstrated a synergistic relationship between olaparib and ONC206 with combination treatment condition in vitro and in vivo, resulting in inhibition of cell proliferation, induction of apoptosis and cellular stress, induction of DNA damage, and reduction in tumor growth in both obese and lean mice. These results suggest that the olaparib could improve clinical benefit in combination with ONC206 in EC patients.
Somatic mutation or deletion of the PTEN tumor suppressor is the most frequent genetic alteration in endometrioid ECCitation33. PTEN is a major negative controller for the down regulation of the PI3K/Akt pathway that controls cell proliferation, growth, survival, and genomic instabilityCitation33,Citation46. Whether PI3K/Akt activity affects the sensitivity of PARP inhibitors to inhibit tumor growth is still controversial, with response in some but not all cancer cell typesCitation13. It has been reported that loss of PTEN function results in higher response of PARP inhibitors through regulation of homologous recombination repair in EC cellsCitation20,Citation47. However, recent studies found that mutated PTEN or PTEN-deficiency in EC cell lines did not predict the sensitivity to olaparib in regard to inhibition of cell proliferationCitation19,Citation37. Other studies have found that PTEN status may influence the response of EC cells to combined PARP inhibitors and PI3K inhibitors but not PARP inhibitors aloneCitation19. In this study, we used PTEN mutated ECC-1 cells and PTEN wild-type HEC-1A cells to evaluate the efficacy of olaparib alone and in combination with ONC206Citation19,Citation48. The results showed that olaparib and ONC206 inhibited cell proliferation in both cell lines, whereas ECC-1 cells showed higher sensitivity to olaparib, indicating that PTEN status and activity of the PI3K/AKT pathway are relevant to the response to olaparib but not to ONC206. The combination of low doses of olaparib and ONC206 synergized to achieve anti-proliferative effects in both cell lines, regardless of PTEN status. Moreover, combination treatment of olaparib and ONC206 for 4 weeks synergistically inhibited tumor growth in our Lkb1fl/fl p53fl/fl transgenic mouse model of endometrioid EC without obvious toxicity, implying that this combination treatment may have inhibitory effects in EC patients with wild type PTEN or PTEN loss tumors.
To better understand the functional mechanisms of olaparib and ONC206 in inhibiting EC cell proliferation and tumor growth, we explored the effects of both drugs and the combination on apoptosis and cellular stress. Induction of apoptosis via TRAIL and activation of the integrated stress response via ClpP are known mechanisms of imipridones that have been previously demonstrated in ECCitation24,Citation27,Citation29,Citation36,Citation49. Similarly, PARP inhibitors also lead to induction of apoptosis by inducing ROS and increasing non-reparable DNA damage and expression of FAS and TRAIL-related death receptor 5Citation50. Monotherapy with both olaparib and ONC206 resulted in increased cellular stress and cleaved caspase-3, −8 and −9 activities compared to controls. The combination of both drugs led to significant increases in both caspase activities and cellular stress as well as in anti-apoptotic proteins and cellular stress-related proteins including BCL-2, MCL-1, PERK, BIP, and calnexin. Although slightly increased gamma-H2A× protein levels were observed in both cell lines following olaparib and ONC206 treatment, the combination of olaparib and ONC206 significantly increased phosphorylated γ-H2A× expression compared to monotherapy or control in EC cells and in endometrial tumors of the Lkb1fl/fl p53fl/fl mouse model. These results imply that decreased DNA repair and increased activity of apoptosis may contribute to the anti-proliferative effects of the olaparib/ONC206 combination treatment in EC cells and endometrial tumors.
Targeting the PI3K/AKT pathway may be another underlying mechanism of combination treatment with olaparib and ONC206 to improve the synergistic inhibition of cell proliferation and tumor growth in EC. This pathway exerts a profound impact on downregulation of BRCA1 and BRCA2, the process of DNA damage repair, and the maintenance of genome stability. Activation of the PI3K/AKT pathway is associated with acquired resistance to PARP inhibitor treatment and may compromise the efficacy of PARP inhibitorsCitation19,Citation51,Citation52. PTEN-deficient tumors present an enhanced sensitivity to the combination of PI3K and PARP inhibition in EC cells and in a PTEN-deficient mouse model of ECCitation19,Citation53. The effect of PARP inhibitors on the PI3K/AKT pathway remains elusive. Olaparib has been shown to reduce phosphorylation of PI3K and AKT in gastric adenocarcinoma cells via the ClC-3/SGK1 regulatory axisCitation54. In BRCA-proficient triple negative breast cancer cells, treatment with olaparib did not show significant regulation in the expression and phosphorylation of AKT, but targeting of PI3K activity by BKM120 synergistically increased sensitivity to olaparib in inhibition of cell proliferationCitation55. Interestingly, recent studies have also found that olaparib can increase phosphorylation of AKT and activate PI3K/AKT pathway in tumor cells and mouse tumor models, resulting in compromised efficacy of olaparibCitation19,Citation45,Citation56. Thus, targeting both PI3K/AKT and PARP may lead to improved anti-tumor activity of PARP inhibitors. Our previous studies have confirmed that ONC201 and ONC206 effectively inhibited the PI3K/AKT/mTOR pathway in EC cellsCitation27,Citation29,Citation36. In this study, we found that the combination of olaparib and ONC206 showed significant effects on the AKT/mTOR/S6 pathway compared to ONC206 or olaparib alone. The finding of increased inhibition of the PI3K/AKT/mTOR pathway with the combination therapy is consistent with prior studies evaluating this treatment strategyCitation15,Citation45,Citation51,Citation57.
Given the role of obesity in the pathogenesis of EC and effects of estrogen and hyperinsulinemia on the PI3K/AKT/mTOR pathwayCitation3, we explored the effects of olaparib and ONC206 treatment on obese versus lean mice using our Lkb1fl/flp53fl/fl model. Tumor weights were significantly increased in the obese mice, consistent with our previous work showing that a high fat diet promotes tumor growth in this modelCitation36,Citation58,Citation59. ONC201 and ONC206 have been shown to decrease endometrial tumor growth in both obese and lean mice, with suggestion of possible greater effects in obese miceCitation36. Correspondingly, olaparib effectively reduced tumor weight in obese and lean mice, and exhibited a more effective inhibitory effect in obese mice in this study. Combination treatment with olaparib and ONC206 was effective in both obese and lean mice with a 91% reduction in tumor weight in obese mice and an 85% reduction in tumor weight in lean mice without any observed side effects. ONC206 has exhibited an excellent safety profile in the clinical trials, and our results suggest that the combination of olaparib and ONC206 should be well tolerated per this pre-clinical mouse study.
In conclusion, treatment with olaparib in combination with ONC206 resulted in synergistic inhibition of EC cell proliferation and reduction in EC tumor weight under obese and lean conditions. Combination treatment increased apoptosis and cellular stress compared to treatment with either drug alone. Dual therapy also led to inhibition of the PI3K/AKT/mTOR pathway. A first-in-human trial of ONC206 is currently underway in central nervous system malignancies. These preclinical studies provide rationale for further exploration of this novel dual therapy of olaparib and ONC206 in clinical trials in EC.
Methods
Cell culture and reagents
Two human EC cell lines were used for all experiments: ECC-1 and HEC-1A. ECC-1 cells were maintained in RPMI supplemented with 5% FBS and penicillin-streptomycin. HEC1 cells were maintained in McCoy’s 5A medium with 10% FBS and penicillin-streptomycin. The cells were cultured in a humidified incubator with 5% CO2 at 37°C. ONC206 was obtained from Oncoceutics/Chimerix (Philadelphia, PA). Olaparib was obtained from MedChemExpress LLC (Monmouth Junction, NJ). Reagents were purchased from Sigma-Aldrich (St. Louis, MO). Antibodies for western blot analysis were purchased from Cell Signaling technology (Danvers, MA), Santa Cruz Biotechnology (Santa Cruz, CA), and Abcam (Cambridge, MA).
MTT assay
The effects of ONC206, olaparib, and combination treatment were assessed using the MTT assay. The cell lines were grown in 96-well plates at a concentration of 4000–6000 cells/well for 24 h. The cells were then treated with varying doses of olaparib and ONC206 individually and in combination for a period of 72 h. Five-microliter MTT dissolved in PBS (5 mg/ml) was added to each well. After an additional hour of incubation, the supernatant was discarded, and the reaction was terminated with the addition of 100 µl DMSO. MTT was quantified by measuring the light absorbance at 570 nm using the Tecan microplate reader (Morrisville, NC). The Quest Graph IC50 Calculator (AAT Bioquest, Sunnyvale, CA) was used to calculate the IC50s for each drug. The combination index (CI) was calculated by the Bliss Independence model to determine whether the drug effects were additive (CI = 1), synergistic (CI < 1), or antagonistic (CI > 1)Citation60. Each experiment was performed in triplicate and repeated for consistency of results.
Cleaved caspase-3, -8, and -9 assays
Apoptosis was assessed following treatment with ONC206, olaparib, the combination, or vehicle using the colorimetric cleaved caspase-3, -8, and −9 assays (AAT Bioquest, Sunnyvale, CA). The ECC-1 and HEC1A cells were plated in 6-well plates at a concentration of 2.5 × 10Citation5. After incubation for 24 h, both cell lines were treated with different concentrations of olaparib, ONC206, the combination, or vehicle. After 12 h of treatment, 180 µl of caspase lysis buffer was added to each well. Protein concentration was determined using the BCA assay. Lysates were then seeded on black 96-well plates and incubated with caspase buffer and DEVD substrate. A Tecan microplate reader was then used to determine the fluorescence of each well: Ex/Em = 400/505 nm for cleaved caspase-3, Ex/Em = 376/482 for cleaved caspase-8, and Ex/Em = 341/441 for cleaved caspase-9. Each experiment was performed in triplicate and repeated for consistency of results.
Reactive oxygen species (ROS) assay
ROS was evaluated using the DCFH-DA assay to measure reactive oxygen species. Cells were plated on 96-well plates and incubated for 24 h. The cells were then treated with olaparib, ONC206, the combination, or vehicle for 16 h. The oxidation-sensitive probe DCFH-DA was then added (10 µM) to each well for 30 min. Fluorescence was measured at an excitation wavelength of 485 nm and an emission wavelength of 530 nm using a Tecan plate reader. Each experiment was performed in triplicate and repeated for consistency of results.
JC-1 assay
Mitochondrial membrane potential was analyzed with the specific fluorescent probe JC-1 (AAT Bioquest, Sunnyvale, CA). The ECC-1 and HEC-1A cells were plated in 96 well plates overnight and treated with olaparib, ONC206, the combination, or vehicle for 16 h. The cells were then treated with 2 µM JC-1 for 30 min at 37°C. The microplate reader measured the fluorescence intensity at the Ex/Em = 480/590 nm for the JC-1 assay. Each experiment was repeated three times to assess for consistency of results.
Western immunoblotting
The ECC-1 and HEC-1A cells were seeded in 6-well plates at a concentration of 2.5 × 105 and incubated for 24 h. The cells were then treated with olaparib, ONC206, the combination, or vehicle for 18–36 h. After treatment, 180 µl of RIPA buffer was added to each well, and cell lysates were collected. Protein concentration was determined using the BCA assay. Equal amounts of protein were denatured at 95°C, separated by gel electrophoresis, and transferred to a PVDF membrane. Membranes were blocked with 5% nonfat dry milk and then incubated with primary antibodies overnight at 4°C. The membranes were then washed and incubated with the appropriate peroxidase-conjugated secondary antibody for 1 h. Antibody-bound proteins were then detected using the ChemiDoc Image System (Bio-Rad, Hercules, CA). The membranes were then stripped and re-probed using B-actin or a-tubulin antibodies to confirm equal protein loading. Primary antibodies used in this study included BCL-2, MCL-1, PERK, IRE, calnexin, phosphorylated (phos)-S6, pan-S6, phos-AKT, pan-AKT, a-tubulin, and B-actin. Experiments were repeated three times to confirm consistent results.
Lkb1fl/flp53fl/fl mouse model of endometrioid EC
The Lkb1fl/flp53fl/fl genetically engineered mouse model of endometrioid EC was used in this studyCitation58. Given that obesity is a driver of EC and results in more aggressive tumor behavior in our mouse model, we decided to look at the effects of olaparib and ONC206 in both lean and obese mice as we have in prior studiesCitation36,Citation59. To model diet-induced obesity, mice were treated with either a low fat diet (consisting of 10% of calories from fat, “lean”) or high fat diet (consisting of 60% of calories from fat, “obese”) at 3 weeks of age. At 6–8 weeks of age, mice were injected with adenovirus Ad5-CMV-Cre (AdCre, Transfer Vector Core, University of Iowa) into one uterine horn. Mice were divided into eight groups—four lean and four obese with ten mice in each group. The mice were treated by oral gavage with either placebo, ONC206 at a dose of 100 mg/kg weekly, olaparib 25 mg/kg daily, or the combination. All mice were euthanized after 4 weeks of treatment. Tumor tissues and blood samples were collected and stored at −70°C. The protocol for the animal experiments was reviewed and approved by the Institutional Animal Care and Use Committee at the University of North Carolina at Chapel Hill.
Immunohistochemistry
Tumors from the Lkb1fl/flp53fl/fl mice were formalin-fixed and paraffin embedded using standard methodologies at the Animal Histopathology Core Facility at the University of North Carolina at Chapel Hill. Slides (5 µm) were stained with anti-Ki-67 and anti-H2A×. Slides were then scanned using Motic (Houston, TX) and scored by ImagePro software (Vista, CA).
Statistical analysis
Descriptive statistics were used to summarize data. Student’s t-test and ANOVA were used for parametric data, and Wilcoxon and Kruskal-Wallis were used for non-parametric data to evaluate statistical significance between treatment groups. Differences were considered significant if the p-value was less than 0.05. GraphPad Prism 6 (La Jolla, CA) was used for all graphs and significance tests.
Disclosure statement
VVP and JEA are employees and stockholders of Oncoceutics/Chimerix. Oncoceutics/Chimerix provided ONC201 and ONC206 for these studies. No potential conflicts of interest were disclosed by the other authors.
Data availability statement
All data generated or analyzed during this study are included in this article. The datasets used and/or analyzed during the current study are available from the corresponding authors on reasonable requests.
Additional information
Funding
Notes on contributors
Sarah E. Paraghamian
Sarah E. Paraghamian, Gyencologic Oncology fellow, Division of Gyencologic Oncology, University of North Carolina at Chapel Hill, USA
Jianqing Qiu
Dr. Jianqing Qiu, Associate professor, Department of Obstetrics and Gyencology, the Second Hospital of Shandong University, Jinan, China
Gabrielle M. Hawkins
Dr. Gabrielle M. Hawkins, Gyencologic Oncology fellow, Division of Gynecologic Oncology, University of North Carolina at Chapel Hill, USA
Ziyi Zhao
Dr. Ziyi Zhao, Graduate student, Department of Gyencologic Oncology, Beijing Obstetrics and Gynecology Hospital, Capital Medical University, Beijing, China
Wenchuan Sun
Dr. Wenchuan Sun, Technician, Division of Gynecologic Oncology, University of North Carolina at Chapel Hill, USA
Yali Fan
Dr. Yali Fan, Graduate student, Department of Gynecologic Oncology, Beijing Obstetrics and Gynecology Hospital, capital Medical University, Beijing, China
Xin Zhang
Dr. Xin Zhang, Graduate student, Department of Gynecologic Oncology, Beijing Obstetrics and Gynecology Hospital, Capital Medical University, Beijing, China
Hongyan Suo
Dr. Hongyan Suo, Graduate student, Department of Gynecologic Oncology, Beijing Obstetrics and Gyencology Hospital, Capital Medical University, Beijing, China
Tianran Hao
Miss. Tianran Hao, Technician, Division of Gynecologic Oncology, University of North Carolina at Chapel Hill, USA
Varun Vijay Prabhu
Dr. Varun Vijay Prabhu, Scientist, Chimerix, Durham, NC, USA
Joshua E. Allen
Dr. Joshua E. Allen, Scientist, Chimerix Durham, NC, USA
Chunxiao Zhou
Dr. Chunxiao Zhou, Associate professor, Division of Gyencologic Oncology, University of North Carolina at Chapel Hill, USA
Victoria Bae-Jump
Dr. Victoria Bae-Jump, Professor, Division of Gynecologic Oncology, University of North Carolina at Chapel Hill, USA
References
- Fontenot VE, Tewari K. 2023. The current status of immunotherapy in the treatment of primary advanced and recurrent endometrial cancer. Curr Opin Obstet Gynecol. 35(1):34–10. doi:10.1097/GCO.0000000000000839.
- Siegel RL, Miller KD, Fuchs HE, Jemal A. 2022. Cancer statistics, 2022. CA Cancer J Clin. 72(1):7–33. doi:10.3322/caac.21708.
- Onstad MA, Schmandt RE, Lu KH. 2016. Addressing the role of obesity in endometrial cancer risk, prevention, and treatment. J Clin Oncol. 34(35):4225–4230. doi:10.1200/JCO.2016.69.4638.
- Renehan AG, Tyson M, Egger M, Heller RF, Zwahlen M. 2008. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet. 371(9612):569–578. doi:10.1016/S0140-6736(08)60269-X.
- Bhaskaran K, Douglas I, Forbes H, dos-Santos-Silva I, Leon DA, Smeeth L. 2014. Body-mass index and risk of 22 specific cancers: a population-based cohort study of 5·24 million UK adults. Lancet. 384(9945):755–765. doi:10.1016/S0140-6736(14)60892-8.
- Calle EE, Kaaks R. 2004. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer. 4(8):579–591. doi:10.1038/nrc1408.
- Brooks RA, Fleming GF, Lastra RR, Lee NK, Moroney JW, Son CH, Tatebe K, Veneris JL. Current recommendations and recent progress in endometrial cancer. CA Cancer J Clin. 2019;69:258–279. doi:10.3322/caac.21561.
- Siegel RL, Miller KD, Fuchs HE, Jemal A. 2021. Cancer Statistics, 2021. CA Cancer J Clin. 71(1):7–33. doi:10.3322/caac.21654.
- Sousa FG, Matuo R, Soares DG, Escargueil AE, Henriques JA, Larsen AK, Saffi J. 2012. PARPs and the DNA damage response. Carcinogenesis. 33(8):1433–1440. doi:10.1093/carcin/bgs132.
- McNevin CS, Cadoo K, Baird AM, Finn SP, McDermott R. 2022. PARP inhibitors in advanced prostate cancer in tumors with DNA damage signatures. Cancers. 14(19):4751. doi:10.3390/cancers14194751.
- Miller RE, Elyashiv O, El-Shakankery KH, Ledermann JA. Ovarian cancer therapy: homologous recombination deficiency as a predictive biomarker of response to PARP inhibitors. Onco Targets Ther. 2022;15:1105–1117. doi:10.2147/OTT.S272199.
- Bound NT, Vandenberg CJ, Kartikasari AER, Plebanski M, Scott CL. 2022. Improving PARP inhibitor efficacy in high-grade serous ovarian carcinoma: a focus on the immune system. Front Genet. 13. doi:10.3389/fgene.2022.886170.
- Musacchio L, Caruso G, Pisano C, Cecere SC, Di Napoli M, Attademo L, Tambaro R, Russo D, Califano D, Palaia I, et al. parp inhibitors in endometrial cancer: current status and perspectives. Cancer Manag Res. 2020;12:6123–6135. doi:10.2147/CMAR.S221001.
- Evers B, Drost R, Schut E, de Bruin M, van der Burg E, Derksen PW, Holstege H, Liu X, van Drunen E, Beverloo HB, et al. Selective inhibition of BRCA2-deficient mammary tumor cell growth by AZD2281 and cisplatin. Clin Cancer Res. 2008;14(12):3916–3925. doi:10.1158/1078-0432.CCR-07-4953.
- Juvekar A, Burga LN, Hu H, Lunsford EP, Ibrahim YH, Balmañà J, Rajendran A, Papa A, Spencer K, Lyssiotis CA, et al. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov. 2012;2(11):1048–1063. doi:10.1158/2159-8290.CD-11-0336.
- Ibrahim YH, García-García C, Serra V, He L, Torres-Lockhart K, Prat A, Anton P, Cozar P, Guzmán M, Grueso J, et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2012;2(11):1036–1047. doi:10.1158/2159-8290.CD-11-0348.
- Konstantinopoulos PA, Barry WT, Birrer M, Westin SN, Cadoo KA, Shapiro GI, Mayer EL, O’Cearbhaill RE, Coleman RL, Kochupurakkal B, et al. Olaparib and α-specific PI3K inhibitor alpelisib for patients with epithelial ovarian cancer: a dose-escalation and dose-expansion phase 1b trial. Lancet Oncol. 2019;20(4):570–580. doi:10.1016/S1470-2045(18)30905-7.
- Hinchcliff E, Chelariu-Raicu A, Westin SN. 2021. Current and future landscape of poly (ADP-ribose) polymerase inhibition resistance. Curr Opin Obstet Gynecol. 33(1):19–25. doi:10.1097/GCO.0000000000000678.
- Bian X, Gao J, Luo F, Rui C, Zheng T, Wang D, Wang Y, Roberts TM, Liu P, Zhao JJ, et al. PTEN deficiency sensitizes endometrioid endometrial cancer to compound PARP-PI3K inhibition but not PARP inhibition as monotherapy. Oncogene. 2018;37(3):341–351. doi:10.1038/onc.2017.326.
- Philip CA, Laskov I, Beauchamp MC, Marques M, Amin O, Bitharas J, Kessous R, Kogan L, Baloch T, Gotlieb WH, et al. Inhibition of PI3K-AKT-Mtor pathway sensitizes endometrial cancer cell lines to PARP inhibitors. BMC Cancer. 2017;17(1):638. doi:10.1186/s12885-017-3639-0.
- Allen JE, Kline CL, VV P, Wagner J, Ishizawa J, Madhukar N, Lev A, Baumeister M, Zhou L, Lulla A, et al. Discovery and clinical introduction of first-in-class imipridone ONC201. Oncotarget. 2016;7(45):74380–74392. doi:10.18632/oncotarget.11814.
- Ishizawa J, Kojima K, Chachad D, Ruvolo P, Ruvolo V, Jacamo RO, Borthakur G, Mu H, Zeng Z, Tabe Y, et al. ATF4 induction through an atypical integrated stress response to ONC201 triggers p53-independent apoptosis in hematological malignancies. Sci Signal. 2016;9(415):ra17. doi:10.1126/scisignal.aac4380.
- Graves PR, Aponte-Collazo LJ, Fennell EMJ, Graves AC, Hale AE, Dicheva N, Herring LE, Gilbert TSK, East MP, McDonald IM, et al. Mitochondrial protease ClpP is a target for the anticancer compounds ONC201 and related analogues. ACS Chem Biol. 2019;14(5):1020–1029. doi:10.1021/acschembio.9b00222.
- Wang S, Dougan DA. 2019. The direct molecular target for imipridone ONC201 is finally established. Cancer Cell. 35(5):707–708. doi:10.1016/j.ccell.2019.04.010.
- Prabhu VV, Morrow S, Rahman KA, Zhou L, Ralff M, Ray J, Jhaveri A, Ferrarini I, Lee Y, Parker C, et al. ONC201 and imipridones: anti-cancer compounds with clinical efficacy. Neoplasia. 2020;22(12):725–744. doi:10.1016/j.neo.2020.09.005.
- Wagner J, Kline CL, Ralff MD, Lev A, Lulla A, Zhou L, Olson GL, Nallaganchu BR, Benes CH, Allen JE, et al. Preclinical evaluation of the imipridone family, analogs of clinical stage anti-cancer small molecule ONC201, reveals potent anti-cancer effects of ONC212. Cell Cycle. 2017;16(19):1790–1799. doi:10.1080/15384101.2017.1325046.
- Zhang Y, Huang Y, Yin Y, Fan Y, Sun W, Zhao X, Tucker K, Staley A, Paraghamian S, Hawkins G, et al. ONC206, an imipridone derivative. Induces Cell Death Through Activation of the Integrated Stress Response in Serous Endometrial Cancer in VitroFrontiers in Oncology. 2020;10:577141. doi:10.3389/fonc.2020.577141.
- Pierce SR, Fang Z, Yin Y, West L, Asher M, Hao T, Zhang X, Tucker K, Staley A, Fan Y, et al. 2021. Targeting dopamine receptor D2 as a novel therapeutic strategy in endometrial cancer. J Experimental Clinical Cancer Res CR. 40:021–01842.
- Fang Z, Wang J, Clark LH, Sun W, Yin Y, Kong W, Pierce SR, West L, Sullivan SA, Tran AQ, Prabhu VV et al. 2018. ONC201 demonstrates anti-tumorigenic and anti-metastatic activity in uterine serous carcinoma in vitro. Am J Cancer Res. 8:1551–1563.
- Zhang Y, Huang Y, Yin Y, Fan Y, Sun W, Zhao X, Zhao X, Tucker K, Staley A, Paraghamian S, Hawkins G, et al. 2020. ONC206, an imipridone derivative induces cell death through activation of the integrated stress response in serous endometrial cancer in vitro. Frontiers in Oncol. 10:577214.
- Staley A, Tucker K, Yin Y, Zhang X, Fan Y, Zhang Y, Fang Z, Sun W, Suo H, Zhao X et al. 2021. Highly potent dopamine receptor D2 antagonist ONC206 demonstrates anti-tumorigenic activity in endometrial cancer. Am J Cancer Res. 11:5374–5387.
- O’Hara AJ, Bell DW. 2012. The genomics and genetics of endometrial cancer. Adv Genomics Genet. 2012:33–47.
- Yeramian A, Moreno-Bueno G, Dolcet X, Catasus L, Abal M, Colas E, Reventos J, Palacios J, Prat J, Matias-Guiu X. et al. 2013. Endometrial carcinoma: molecular alterations involved in tumor development and progression. Oncogene. 32:403–413.
- Kumar A, Fernandez-Capetillo O, Fernadez-Capetillo O, Carrera AC. 2010. Nuclear phosphoinositide 3-kinase beta controls double-strand break DNA repair. Proc Natl Acad Sci U S A. 107:7491–7496.
- Amoroso F, Glass K, Singh R, Liberal F, Steele RE, Maguire S, Tarapore R, Allen JE, Van Schaeybroeck S, Butterworth KTet al. 2021. Modulating the unfolded protein response with ONC201 to impact on radiation response in prostate cancer cells. Sci Rep. 11:4252.
- Pierce SR, Fang Z, Yin Y, West L, Asher M, Hao T, Zhang X, Tucker K, Staley A, Fan Y, et al. 2021. Targeting dopamine receptor D2 as a novel therapeutic strategy in endometrial cancer. J Experimental Clinical Cancer Res CR. 40:61.
- Miyasaka A, Oda K, Ikeda Y, Wada-Hiraike O, Kashiyama T, Enomoto A, Hosoya N, Koso T, Fukuda T, Inaba K et al. 2014. Anti-tumor activity of olaparib, a poly (ADP-ribose) polymerase (PARP) inhibitor, in cultured endometrial carcinoma cells. BMC Cancer. 14:179.
- Bartoletti M, Musacchio L, Giannone G, Tuninetti V, Bergamini A, Scambia G, et al. 2021. Lorusso D, Valabrega G, Mangili G, Puglisi F Emerging molecular alterations leading to histology-specific targeted therapies in ovarian cancer beyond PARP inhibitors. Cancer Treat Rev. 101:102298.
- Cardillo TM, Sharkey RM, Rossi DL, Arrojo R, Mostafa AA, Goldenberg DM. 2017. Synthetic lethality exploitation by an anti-trop-2-SN-38 antibody-drug conjugate, IMMU-132, plus PARP inhibitors in BRCA1/2-wild-type triple-negative breast cancer. Clin Cancer Res. 23:3405–3415.
- McCann KE, Hurvitz SA. 2018. Advances in the use of PARP inhibitor therapy for breast cancer. Drugs Context. 7:212540.
- Lim B, Peterson CB, Davis A, Cho E, Pearson T, Liu H, Hwang M, Ueno NT, Lee J. et al. 2021 ONC201 and an MEK inhibitor trametinib synergistically inhibit the growth of triple-negative breast cancer cells. Biomedicines. 9:1410.
- Borsuk R, Zhou L, Chang WI, Zhang Y, Sharma A, Prabhu VV, Tapinos N, Lulla RR, El-Deiry WS, et al. 2021. Potent preclinical sensitivity to imipridone-based combination therapies in oncohistone H3K27M-mutant diffuse intrinsic pontine glioma is associated with induction of the integrated stress response, TRAIL death receptor DR5, reduced ClpX and apoptosis. Am J Cancer Res. 11:4607–4623.
- Talekar MK, Allen JE, Dicker DT, El-Deiry WS. 2015. ONC201 induces cell death in pediatric non-Hodgkin’s lymphoma cells. Cell Cycle. 14:2422–2428.
- Ishida CT, Zhang Y, Bianchetti E, Shu C, Nguyen TTT, Kleiner G, Sanchez-Quintero MJ, Quinzii CM, Westhoff MA, Karpel-Massler G et al. 2018. Metabolic reprogramming by dual AKT/ERK Inhibition through imipridones elicits unique vulnerabilities in glioblastoma. Clin Cancer Res. 24:5392–5406.
- González-Billalabeitia E, Seitzer N, Song SJ, Song MS, Patnaik A, Liu XS, Papa A, Hobbs RM, Chen M, Lunardi A, et al. 2014. Vulnerabilities of PTEN-TP53-deficient prostate cancers to compound PARP-PI3K inhibition. Cancer Discov. 4:896–904.
- Slomovitz BM, Coleman RL. 2012. The PI3K/AKT/mTOR pathway as a therapeutic target in endometrial cancer. Clin Cancer Res. 18:5856–5864.
- Dedes KJ, Wetterskog D, Mendes-Pereira AM, Natrajan R, Lambros MB, Geyer FC, Vatcheva R, Savage K, Mackay A, Lord CJ, et al. 2010. PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Sci Transl Med. 2:53ra75.
- Devor EJ, Gonzalez-Bosquet J, Thiel KW, Leslie KK. 2020. Genomic characterization of five commonly used endometrial cancer cell lines. Int J Oncol. 57:1348–1357.
- Ray JE, Ralff MD, Jhaveri A, Zhou L, Dicker DT, Ross EA, El-Deiry WS. 2021. Antitumorigenic effect of combination treatment with ONC201 and TRAIL in endometrial cancer in vitro and in vivo. Cancer Biol Ther. 22:554–563.
- Macciò A, Madeddu C. The mechanism of cancer cell death by PARP inhibitors goes beyond DNA damage alone. Int J Cancer. 2019 Nov 1Epub 2019 May 16;145(9):2594–2596. doi:10.1002/ijc.32392.
- Ma S, Pradeep S, Villar-Prados A, Wen Y, Bayraktar E, Mangala LS, Kim MS, Wu SY, Hu W, Rodriguez-Aguayo C, et al. 2019. GnRH-R-targeted lytic peptide sensitizes BRCA wild-type ovarian cancer to PARP inhibition. Mol Cancer Ther. 18:969–979.
- Cardnell RJ, Feng Y, Diao L, Fan YH, Masrorpour F, Wang J, Shen Y, Mills GB, Minna JD, Heymach JV, et al. 2013. Proteomic markers of DNA repair and PI3K pathway activation predict response to the PARP inhibitor BMN 673 in small cell lung cancer. Clin Cancer Res. 19:6322–6328.
- Philip CA, Laskov I, Beauchamp MC, Marques M, Amin O, Bitharas J, Kessous R, Kogan L, Baloch T, Gotlieb WH, et al. 2017. Inhibition of PI3K-AKT-Mtor pathway sensitizes endometrial cancer cell lines to PARP inhibitors. BMC Cancer. 17:017–3639.
- Gu Z, Wang L, Yao X, Long Q, Lee K, Li J, Yue D, Yang S, Liu Y, Li N et al. 2020. ClC-3/SGK1 regulatory axis enhances the olaparib-induced antitumor effect in human stomach adenocarcinoma. Cell Death & Disease. 11:020–03107.
- Li Y, Wang Y, Zhang W, Wang X, Chen L, Wang S. 2021. BKM120 sensitizes BRCA-proficient triple negative breast cancer cells to olaparib through regulating FOXM1 and Exo1 expression. Sci Rep. 11:4774.
- Santiago-O’Farrill JM, Weroha SJ, Hou X, Oberg AL, Heinzen EP, Maurer MJ, Pang L, Rask P, Amaravadi RK, Becker SE et al. 2020. Poly(adenosine diphosphate ribose) polymerase inhibitors induce autophagy-mediated drug resistance in ovarian cancer cells, xenografts, and patient-derived xenograft models. Cancer. 126:894–907.
- Wang D, Li C, Zhang Y, Wang M, Jiang N, Xiang L, Li T, Roberts TM, Zhao JJ, Cheng H, et al. 2016. Combined inhibition of PI3K and PARP is effective in the treatment of ovarian cancer cells with wild-type PIK3CA genes. Gynecol Oncol. 142:548–556.
- Guo H, Kong W, Zhang L, Han J, Clark LH, Yin Y, Fang Z, Sun W, Wang J, Gilliam TP et al. 2019. Reversal of obesity-driven aggressiveness of endometrial cancer by metformin. Am J Cancer Res. 9:2170–2193.
- Roque DR, Zhang L, Wysham WZ, Han J, Sun W, Yin Y, Livingston JN, Batchelor KW, Zhou C, Bae-Jump VL et al. 2021. The effects of NT-1044, a novel AMPK activator, on endometrial cancer cell proliferation, apoptosis, cell stress and in vivo tumor growth. Front Oncol. 11:690435.
- Foucquier J, Guedj M. 2015. Analysis of drug combinations: current methodological landscape. Pharmacol Res Perspect. 3:e00149.