
ABSTRACT
Acute myeloid leukemia (AML) is a heterogeneous hematologic malignancy characterized by abnormal cell proliferation, apoptosis repression and myeloid differentiation blockade of hematopoietic stem/progenitor cells. Developing and identifying novel therapeutic agents to reverse the pathological processes of AML are of great significance. Here in this study, we found that a fungus-derived histone deacetylase inhibitor, Apicidin, presents promising therapeutic effect on AML by inhibiting cell proliferation, facilitating apoptosis and inducing myeloid differentiation of AML cells. Mechanistic investigation revealed that QPCT is identified as a potential downstream target of Apicidin, which exhibits significantly decreased expression in AML samples compared with the normal controls and is remarkably up-regulated in AML cells upon Apicidin management. Functional study and rescue assay demonstrated that QPCT depletion further promotes cell proliferation, inhibits apoptosis and impairs myeloid differentiation of AML cells, alleviating the anti-leukemic effect of Apicidin on AML. Our findings not only provide novel therapeutic target for AML, but also lay theoretical and experimental foundation for the clinical application of Apicidin in AML patients.
Introduction
Acute myeloid leukemia (AML) is a malignant hematopoietic neoplasm characterized by arrest of myeloid differentiation, rapid growth and apoptotic repression of leukemic blasts arisen from the hematopoietic stem/progenitor cells (HSPCs) population within the bone marrowCitation1. The incidence rate of AML is 3–5 per 100,000Citation2. Older people are more susceptible, with a median onset age of 68 years. AML is the most common type of acute leukemia in adults with a low survival probability, particularly in older patientsCitation3. The chemotherapeutic regimens and allogeneic hematopoietic stem cell transplantation (allo-HCT) remain the first-line treatment choices for AML patients, which could achieve 5-year survivals of 30% to 35% in younger patients (age <60 years) and 10%-15% in older patients (age ≥60 years)Citation4. In the past decades, great progress has been made in understanding the genomic landscape and pathogenesis of AML, which has accelerated translational discoveries and is contributing to a steady improvement in the outlook and prognosis of AMLCitation5. In recent years, several small molecule-targeted therapies such as the fms-like tyrosine kinase 3 (FLT3) and isocitrate dehydrogenase (IDH) inhibitors and the BCL2 inhibitor venetoclax have been approved, and brought more choices and great benefits for AML patients unable to receive intensive chemotherapy and allo-HCTCitation6,Citation7. However, the outcome in older patients remains dismal, with a median survival of only 5 to 10 monthsCitation8. It is still in urgent need to develop novel therapies with less intensive and more tolerable therapeutic effects for the most older AML patients.
Accumulating evidences indicate that epigenetic aberration contributes to the oncogenic transforming including AMLCitation9,Citation10. Histone deacetylases (HDAC), as one of the major epigenetic regulators, are key enzymes involved in epigenetic modulation and catalyze the removal of acetyl from lysine residues in histones and non-histone proteins, causing gene transcriptional repression and subsequent changes in signaling eventsCitation11. HDACs have been intensively studied in the past few decades and increasing evidence indicates that HDACs play diverse functions in a wide range of physiological and pathological processesCitation12. Presently, HDAC family members are appealing targets for drug exploration and a number of HDAC inhibitors have been developed, which are emerging as a new class of targeting agents especially in various cancers including AMLCitation13,Citation14. Chidamide, a novel and orally available benzamide-type HDAC inhibitor, selectively inhibiting the activity of HDAC1, 2, 3, and 10, has been demonstrated to confer promising therapeutic effect in AML by use alone or in combination with other chemotherapeutic or targeting agentsCitation15–17. Apicidin is a fungal metabolite with potent HDAC inhibitor activity and has been reported to inhibit cell proliferation in several human cancer cell lines, including leukemiaCitation18. However, it remains to be further investigated about whether Apicidin could be used as a therapeutic agent in AML.
Here in this study, we found that Apicidin presents promising therapeutic effect on AML by inhibiting cell proliferation, promoting apoptosis and inducing myeloid differentiation of AML cells. Mechanistic investigation revealed that glutaminyl-peptide cyclotransferase (QPCT) is up-regulated by Apicidin treatment, presents aberrantly decreased expression in AML patients compared with normal controls and functions as an important regulator in myeloid differentiaiton and AML pathogenesis, partly underlying the anti-leukemic effect of Apicidin on AML. Our findings not only provide novel therapeutic target for AML, but also lay theoretical and experimental foundation for the clinical application of Apicidin in AML patients.
Materials and methods
Human samples
Human bone marrow samples of AML patients and iron deficiency anemia (IDA) normal controls were collected from the hematology department of the affiliated hospital of Jining Medical University. Informed consent to perform the biological studies was obtained from the individuals examined, and the related study was approved by the medical scientific research ethics committee of Affiliated Hospital of Jining Medical University and has been conducted in accordance with the Declaration of Helsinki. Mononuclear cells (MNCs) of the samples were isolated by Ficoll density gradient [d = 1.077 g/ml] for subsequent RNA extraction or culture with Apicidin treatment. The characteristics of AML patients used in this study were presented in Table S1.
Cell culture and treatment
THP-1 and HL-60 were purchased from cell resource center of Shanghai Institutes for Biological Science, Chinese Academy of Sciences; 293T was purchased from cell resource center of Institutes of Basic Medical Sciences, Chinese Academy of Medical Sciences; NB4 was purchased from cell resource center of Procell company. THP-1 and NB4 were cultured in PRMI 1640 medium (Hyclone); HL-60 and primary MNCs were cultured in Iscove’s modified Dubecco’s Medium (IMDM) (Hyclone); 293T was cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Hyclone). All cultures were supplemented with 10% fetal bovine serum (FBS) (Gibco), 100 U/ml penicillin and 100 μg/ml streptomycin (Sigma-Aldrich) at 37°C in 5% CO2. The macrophage-like differentiation of THP-1 and HL-60 was induced with PMA (Sigma-Aldrich) at final concentration of 10 nM. The granulocytic differentiation of NB4 and HL-60 was induced with ATRA (Sigma-Aldrich) at final concentration of 2 μM. Apicidin (Cat. No.: HY-N6735) was purchased from MedChemExpress (MCE) and used to treat AML cells at final concentration of 1 μM.
RNA extraction and Qrt-PCR analysis
Total RNA was extracted from cell samples using TRIzol Reagent (Invitrogen) and quantified using the NanoDrop 2000 spectrophotometer (Thermo Scientific). The first strand of cDNA was synthesized using Moloney murine leukemia virus (M-MLV) reverse transcriptase (Invitrogen) according to the manufacturer’s instructions. Oligo (dT) was used as the primer for reverse transcription of mRNA. GAPDH was used as the loading control. qRT-PCR was performed in a Bio-Rad CFX-96 System (Bio-Rad) using the SYBR Premix (ABclonal). The primers used for reverse transcription and qRT-PCR are listed in Table S2.
Plasmid construction
The shRNA sequences for QPCT and corresponding control were synthesized, annealed and inserted into pSIH1-H1-copGFP (System Biosciences). The oligonucleotides used for plasmid construction are listed in Table S3.
Lentivirus production and cell infection
The recombination lentiviruses for knockdown were produced in 293T cells using pSIH1-H1-copGFP-based constructs. Lentivirus packaging was performed using the pPACKH1 HIV Lentivector Packaging Kit (LV500A–1, System Biosciences) according to the manufacturer’s instructions. The virus particles in the culture supernatant were condensed by ultracentrifugation and subsequently used to infect the AML cells in 6-well plates with 5 μg/mL polybrene (SigmaAldrich).
Cell proliferation assay
AML cells, infected with lentivirus and/or treated with Apicidin, were plated in 96-well plate at a density of 10,000 cells/well and incubated in 10% CCK-8 (Dojindo) diluted in normal culture medium at 37°C for 1.5 hours. Then proliferation rates were determined at 0, 24, 48, and 72 h by measuring the absorbance (A) with a microplate reader set at 450 and 630 nm. All experiments were performed in triplicate. The relative cell viability in each time point was calculated as follows: Atest group/Acontrol group, in which the relative cell viability in control group was normalized as “1” in each time point.
Cell apoptosis assay
AML cells, infected with lentivirus and/or treated with Apicidin, were starved in low FBS medium for 48 h and then collected, washed once with PBS and re-suspended in the 1× binding buffer. Apoptotic cells were stained with APC-Annexin V and 7-AAD (Biolegend) at room temperature for 20 min and immediately analyzed by flow cytometry.
Flow cytometry analysis
AML cells were treated with Apicidin or infected with lentivirus followed by induction toward macrophage-like and granulocytic differentiation, respectively, and harvested at 48 h of differentiation. The cells were rinsed twice with PBS, resuspended in 100 μl PBS and incubated with APC-conjugated anti-CD14 or anti-CD11b (eBioscience) at 4°C for 30 min. Then the cells were washed with 1 ml PBS, re-suspended in 300 μl PBS and analyzed immediately using a DxFLEX flow cytometer (Beckman).
Chromatin Immunoprecipitation (ChIP)
HL-60 cells were seeded onto 10-cm plates and grown to approximately 70% confluence. Then the cells were treated with dimethyl sulfoxide (DMSO) or Apicidin for 48 h and ChIP was performed using Chromatin Immunoprecipitation Kit (WLA106a, Wanleibio) according to the manufacturer’s instructions. The precipitated DNA was amplified by PCR. All the primers used for ChIP-PCR are listed in Table S4. Primary antibodies against the following proteins were used: H3K27ac (ab177178; Abcam), H3K9ac (ab177177; Abcam), and IgG provided in the ChIP Kit (WLA106a, Wanleibio).
Western blot
Total proteins were extracted from AML cells and/or corresponding controls using RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1% sodium deoxycholate, 0.1%SDS) supplemented with 1 mM PMSF and Protease inhibitor mixture (PI0015, Leagene Bio). The protein concentration was determined using BCA Protein Assay Kit (P0010, Beyotime). Protein samples were separated on 10% SDS-PAGE gels and then transferred to PVDF membranes. Membranes were blocked with 5% skimmed milk in TBS-T and incubated with primary antibodies at 4°C overnight. Primary antibodies against the following proteins were used: QPCT (A6711; ABclonal), and actin (AC004; ABclonal). Horseradish peroxidase-conjugated secondary antibodies were used (ZSGB-BIO). Signals were detected using an ECL (enhanced chemiluminescence) kit (Millipore).
Statistical analysis
Student’s t-test (two-tailed) was performed to analyze the data. The statistically significant difference was considered only when P value was less than 0.05. All data were shown as mean ± SD. *P < .05; **P < .01, ***P < .001.
Results
Apicidin presents significant anti-leukemic effect on AML cells
Apicidin is a cyclic tetrapeptide isolated from Fusariumsp with HDAC inhibitor activity, whose chemical structure is shown in . To investigate whether Apicidin confers anti-leukemic effect on AML cells, we used Apicidin to treat AML cell lines (THP-1, NB4 and HL-60) and primary AML cells derived from the bone marrows of three different AML patients. Then cell proliferation, apoptosis and myeloid differentiation of AML cells were evaluated. Apicidin presented striking inhibitory effect on the proliferation of AML cell lines (, top panel) and remarkably facilitated cell apoptosis (2A-C, middle panel). The expression of myeloid differentiation markers CD14 or/and CD11b also exhibited significantly increased expression in the three AML cell lines after Apicidin management according to the flow cytometric analyses (, bottom panel). The same results were also observed in the primary AML cells (). Collectively, the results indicate that Apicidin may be used as a promising and alternative therapeutic agent for AML by inhibiting proliferation, faciliating apoptosis and inducing myeloid differentiation of AML cells.
Figure 1. Chemical structure of Apicidin.
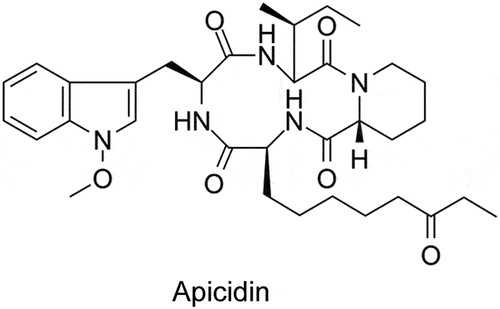
Figure 2. Apicidin inhibits proliferation, promotes apoptosis and induces myeloid differentiation of AML cells. (a-c) THP-1 (a), NB4 (b), and HL-60 (c) cells were treated with Apicidin for 48-72h. Then cell proliferation was evaluated by CCK-8 assay, cell apoptosis analyses were performed using double staining of Annexin V and 7-AAD followed by flow cytometry assay, and myeloid differentiation markers CD14 and/or CD11b were characterized by flow cytometric analyses. (d-f) The bone marrow MNCs were isolated from three independent AML patients and treated with Apicidin for indicated times. Then cell proliferation was evaluated by CCK-8 assay, cell apoptosis analyses were performed using double staining of Annexin V and 7-AAD followed by flow cytometry assay, and myeloid differentiation markers CD14 and CD11b were characterized by flow cytometric analyses. *P< .05, **P< .01, ***P< .001, Student’s t-test.
Apicidin increases QPCT expression in AML cells
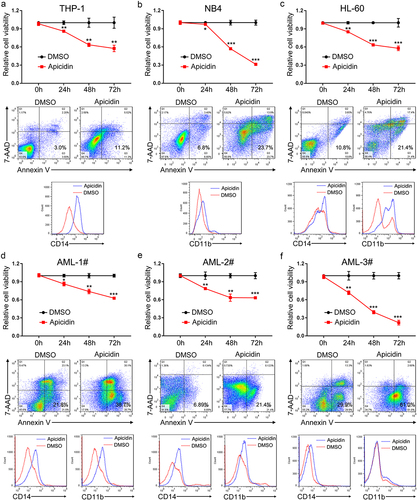
Different HDAC inhibitors may act on different HDAC family members, generally leading to increased histone acetylation level and subsequent gene expression alterationsCitation19. To further reveal the underlying molecular mechanism of Apicidin in AML treatment, we consulted the expression profiling data of AML (GSE79605) and Apicidin treatmentCitation20, and also the profiling data of chidamide management (GSE199466), another HDAC inhibitor like Apicidin. Many candidate genes were selected for experimental validation in Apicidin-handled AML cells (data not shown). QPCT, encoding a pituitary glutaminyl cyclotransferase and responsible for the presence of pyroglutamyl residues in many neuroendocrine peptidesCitation21, is one of the identified differential genes which presented remarkably increased expression in AML cell lines () and primary AML cells () upon Apicidin treatment. To demonstrate whether increased QPCT expression in Apicidin-treated AML cells is associated with alteration of H3K27ac and H3K9ac levels in QPCT promoter region, we conducted H3K27ac and H3K9ac ChIP experiment using HL-60 cells treated with DMSO and Apicidin. Ultrasonication effiency of the genomic DNA was detected by agarose gel electrophoresis and presented in . Four pairs of PCR primers (primer pairs 1–4) were designed covering four different possible regions binded by H3K27ac and H3K9ac in QPCT promoter (). The H3K27ac and H3K9ac ChIP-PCR results indicated that Apicidin management significantly enhanced H3K27ac and H3K9ac binding to the primer pair 1, primer pair 2 and primer pair 3 expanded promoter regions of QPCT, compared with DMSO group (). Collectively, the results demonstrated that Apicidin may facilitate QPCT expression in AML cells by increasing H3K27ac and H3K9ac levels in QPCT promoter region.
Figure 3. QPCT is identified as a potential downstream target of Apicidin in AML cells. (a) QPCT protein expression was detected by western blot in THP-1, NB4 and HL-60 cells treated with Apicidin or DMSO for 48h. (b-c) Qrt-PCR (b) and western blot (c) analyses of QPCT expression in Apicidin-treated AML MNCs. (d) Ultrasonication effiency of the genomic DNA detected by agarose gel electrophoresis. (e) Sketch map showing the distribution of primers used to expand the putative H3K27ac and H3K9ac binding sites in QPCT promoter region. (f) ChIP-Qrt-PCR analysis of H3K27ac and H3K9ac binding level in the QPCT promoter region after Apicidin treatment in HL-60 cells. *P< .05, **P< .01, ns: not significant, Student’s t-test.
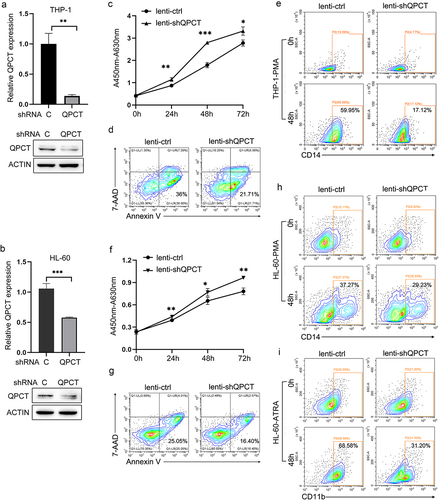
QPCT exhibits aberrantly decreased expression in AML patients and increases upon myeloid induction of AML cell lines
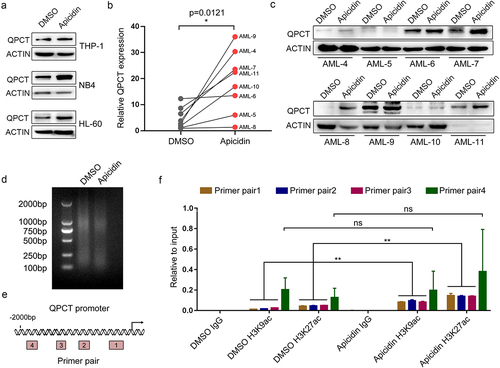
Presently, the role of QPCT in AML pathogenesis has never been reported. To evaluate its function in AML, we first detected QPCT expression in the bone marrow-derived mononuclear cells (MNCs) of AML patients and normal controls, which indicates that QPCT presented significantly decreased expression in AML samples at both the mRNA and protein levels (). No significant correlation between QPCT expression and different AML karyotypes was observed in our samples. AML development is often accompanied with myeloid differentiation blockadeCitation22–24. To explore whether aberrant expression of QPCT influences the myeloid differentiation process, we also analyzed the expression of QPCT using qRT-PCR and western blot during the PMA induced macrophage-like differentiation of THP-1 and HL-60 cells, and ATRA induced granulocytic differentiation of NB4 and HL-60 cells. The results showed that QPCT presents gradually increased expression during the PMA/ATRA-induced myeloid differentiation of AML cell lines at both mRNA and protein levels (). These results sugggest that aberrant expression of QPCT in AML may correlate with myeloid differentiation blockade and mediate AML pathogenesis.
Figure 4. QPCT presents decreased expression in AML patients and increases upon myeloid induction. (a) Qrt-PCR analysis of QPCT mRNA expression in bone marrow MNCs derived from 29 AML patients (FAB M1 to M5 subtypes) and 28 normal controls. (b) Western blot analysis of QPCT protein level in 12 AML patients and 12 normal controls. (c-f) Qrt-PCR and western blot analyses of QPCT expression during PMA-induced macrophage-like differentiation of THP-1 (c) and HL-60 (d) cells and ATRA-induced granulocytic differentiation of NB4 (e) and HL-60 (f) cells. ***P< .001, Student’s t-test.
QPCT acts as an important regulator in AML leukemogenesis
To further elucidate the role of QPCT in AML, we make use of the recombined lentiviruses that express specific shRNA for QPCT (lenti-shQPCT) to infect THP-1 and HL-60 cells, respectively, which resulted in significant down-regulation of QPCT expression at both mRNA and protein levels in THP-1 () and HL-60 () cells. Decreased QPCT expression in THP-1 cells contributed to remarkable cell proliferation () and apoptosis repression (), and also strikingly impaired PMA induced macrophage-like differentiation as revealed by decreased CD14 expression according to the flow cytometric analysis (). Likewise, lentivirus-mediated knock-down of QPCT expression in HL-60 cells further exacerbated the malignant phenotype of leukemia, characterized by increased cell proliferation (), reduced apoptosis () and myeloid differentiation blockade with decreased CD14 () and CD11b () expression. Taken together, these results demonstrated that low expression of QPCT in AML cells may contribute to the pathogenesis and progression of AML by simultaneously increasing cell proliferation, reducing apoptosis and arresting myeloid differentiation.
Figure 5. QPCT regulates the proliferation, apoptosis and myeloid differentiation of AML cells. (a-b) THP-1 and HL-60 cells were infected with lenti-ctrl and lenti-shQPCT, replaced with fresh medium 24h later and cultured for another 48h. Then QPCT expression was detected by Qrt-PCR and western blot in THP-1 (a) and HL-60 cells (b). (c-d) Cell proliferation was evaluated by CCK-8 assay (c) and cell apoptosis analyses were performed using double staining of Annexin V and 7-AAD followed by flow cytometry assay (d) in infected THP-1 cells. (e) CD14 expression was evaluated by cytometric analyses in THP-1 cells that were infected with lenti-shQPCT (or lenti-ctrl) followed by PMA induction. (f-g) Cell proliferation was evaluated by CCK-8 assay (f) and cell apoptosis analyses were performed using double staining of Annexin V and 7-AAD followed by flow cytometry assay (g) in infected HL-60 cells. (h) CD14 expression was evaluated by cytometric analyses in HL-60 cells that were infected with lenti-shQPCT (or lenti-ctrl) followed by PMA induction. (i) CD11b expression was evaluated by cytometric analyses in infected and ATRA-induced HL-60 cells. *P< .05, **P< .01 and ***P< .001, Student’s t-test.
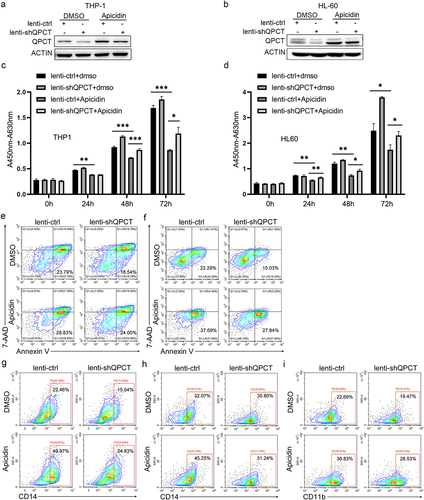
QPCT partly underlies the anti-leukemic effect of Apicidin on AML
To further demonstrate that whether Apicidin exerts its anti-leukemic role in AML by increasing QPCT expression, we performed rescue assay. THP-1 and HL-60 cells were infected with lenti-shQPCT or lenti-ctrl twenty-four hours later, the cells were changed into fresh complete medium with Apicidin or DMSO. As expected, lenti-shQPCT infection resulted in significant down-regulation of QPCT in both DMSO and Apicidin treated groups, and Apicidin up-regulated QPCT expression even in the presence of shQPCT in THP-1 () and HL-60 () cells. Lenti-shQPCT-mediated depletion of QPCT partly alleviated the anti-leukemic effect of Apicidin as revealed by increased cell proliferation of THP-1 and HL-60 () cells, partial apoptosis repression () and decreased CD14 () or/and CD11b () expression as comparedto lenti-shQPCT+Apicidin with lenti-ctrl+Apicidin. These results demonstrated that QPCT may act as a potential downstream target and partly mediates the therapeutic effect of Apicidin on AML.
Figure 6. QPCT partly underlies the therapeutic effect of Apicidin on AML. THP-1 and HL-60 cells were infected with lenti-shQPCT or lenti-ctrl, and twenty-four hours later, the cells were changed into fresh complete medium with Apicidin or DMSO. (a-b) QPCT protein expression was evaluated by western blot in THP-1 (a) and HL-60 (b) cells. (c-d) Cell proliferation was analyzed by CCK-8 in THP-1 (c) and HL-60 (d) cells. (e-f) Cell apoptosis analyses were performed using double staining of Annexin V and 7-AAD followed by flow cytometry assay in THP-1 (e) and HL-60 cells (f). (g-h) Myeloid differentiation marker CD14 was characterized by flow cytometric in THP-1 (g) and HL-60 cells (h). (i) Myeloid differentiation marker CD11b was characterized by flow cytometric analysis in HL-60 cells. *P< .05, **P< .01 and ***P< .001, Student’s t-test.
Discussion
AML is a heterogeneous and aggressive hematopoietic disorder characterized by malignant proliferation and apoptotic repression of clonal neoplastic cells and also differentiation blockade of myeloid blastsCitation25,Citation26. Mechanistic insights reveal that increasing numbers of gene mutations, chromosome translocations, deletions and other genetic abnormalities contribute to the onset and progression of AMLCitation27,Citation28. Accumulating evidence also demonstrates that epigenetic dysregulations including DNA methylation and histone deacetylation also play essential roles in the pathogenesis of AML, which further aggravates the biological complexity of AMLCitation29,Citation30. On the other hand, the great progress in epigenetic regulators contributing to AML development supports the rationale for the use of epigenetic drugs in patients with AMLCitation31. In recent years, small molecule-mediated targeting of key regulators in epigenetic aberrancy represents a novel and alternative therapeutic strategy for AML, especially for the refractory/relapsed patientsCitation32. Azacitidine and decitabine belong to the cytidine analogues and could be used as hypomethylating agents by inhibiting the activity of DNA methyltransferaseCitation33, which in combination with venetoclax presented promising therapeutic effect and well-tolerable results in elderly patients with AMLCitation34. Besides, different kinds of HDAC inhibitors such as Panobinostat, Vorinostat and Trichostatin A have also been developed and found to promote cell death, autophagy, apoptosis, or growth arrest in preclinical AML modelsCitation35. Apicidin, a cyclic tetrapeptide, is a natural fungal metabolite and inhibits HDACs at nanomolar concentrationsCitation36, which has been reported to induce apoptosis via a mitochondrial/cytochrome-c-dependent pathway in AML cell linesCitation18. Here in this study, we further demonstrated that Apicidin exhibits encouraging anti-leukemic activity in AML cell lines and primary AML cells by not only promoting cell apoptosis, but also inhibiting cell proliferation and inducing myeloid differentiation.
Subsequently, mechanistic investigation revealed that QPCT may act as a potential downstream target of Apicidin, which presented significantly increased expression in Apicidin-handled AML cells. Here, QPCT was chosen mainly because of our preivous work in which QPCT was identified as one of the top fifty up-regulated genes elicted by Chidamide treatment in bone marrow derived mesenchymal stem cells according to the profiling data. And it has been reported that both Apicidin and Chidamide could inhibit the activity of HDAC2 and HDAC3, resulting in elevation of histone acetylation level and subsequent increased gene expressionCitation37,Citation38. Increasing evidence indicates that histone acetylation is an important determinant of gene expression by regulating transcription, chromatin remodeling and nuclear architecture, whose abnormal alterations mediated by the dysfunction of histone acetyltransferase (HAT) and HDAC have been implicated in various pathophysiological processes, including AML pathogenesisCitation39,Citation40. QPCT was further demonstrated to present aberrantly decreased expression in AML patient samples compared with the normal controls and exerts tumor suppressive role in AML development by regulating cell proliferation, apoptosis and myeloid differentiation. We also demonstrated that increased binding of H3K27ac and H3K9ac in QPCT specific promoter regions after Apicidin management may contribute to enhanced QPCT expression in Apicidin-treated AML cells. However, as to whether H3K27ac and H3K9ac alterations in QPCT promoter regions mediate the low expression of QPCT in AML samples, it is not invloved in this work and will be further investigated in the future.
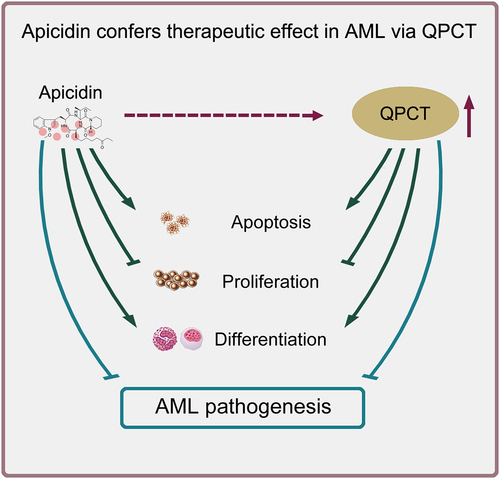
Taken together, we summarized a model of Apicidin- and QPCT-mediated involvement in AML pathogenesis (). Apicidin presents outstanding therapeutic effect on AML by inhibiting cell proliferation, facilitating apoptosis and inducing myeloid differentiation of AML cells. QPCT is identified as a potential downstream target of Apicidin, which exhibits remarkably increased expression in AML cells upon Apicidin management. Functional study and rescue assay reveal that QPCT depletion further promotes cell proliferation, inhibits apoptosis and impairs myeloid differentiation of AML cells, alleviating the anti-leukemic effect of Apicidin on AML. Our findings not only provide novel therapeutic target for AML, but also lay theoretical and experimental foundation for the clinical application of Apicidin in AML patients.
Figure 7. Schematic representation of Apicidin-mediated therapeutic effect on AML. Apicidin increases QPCT expression and confers therapeutic effect on AML by simultaneously inhibiting cell proliferation, promoting apoptosis and inducing myeloid differentiation of AML cells.
Availability of data and materials
All the data and material in the manuscript are available from the corresponding author on reasonable request.
Authorship
L.L.L., H.H.L. and L.L. performed most of the experiments, and L.L.L. interpreted the data. Q.H., C.Y.Y., P.P.C., J.J.Z., S.S.R., M.X.Y., X.Y.M., W.J.S., L.L.C. and H.Z. helped collect the samples and performed partial experiments. M.T.C. conceived the study and wrote the manuscript., X.N.Z. directed the experiments, analyzed the data and provided critical advice during the preparation of the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Written informed consent to perform the biological studies was obtained from the individuals examined. This study was approved by the Ethics Committee of Affiliated Hospital of Jining Medical University (2017-FY-001).
Supplemental Material
Download MS Word (108 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15384047.2023.2228497
Additional information
Funding
Notes on contributors
Lulu Liu
Lulu Liu, received her BS and Master degree from Qufu Normal University in 2010 and 2013. Now, she is an assistant researcher in the Affiliated Hospital of Jining Medical University. Her research focuses on hematopoietic regulation in acute myeloid leukemia and aplastic anemia.
Haihui Liu
Haihui Liu, received her BS degree from Medical College of Shandong First Medical University in 2015 and her Master degree in Hematology from Jining Medical University in 2018. Now, she is a physician in the hematology department of Affiliated Hospital of Jining Medical University. Her research focuses on the diagnosis and treatment of hematological diseases.
Lei Liu
Lei Liu, received his BS degree from Jining Medical University in 2013 and his Master degree from Soochow University in 2016. Now, he is a physician in the hematology department of Affiliated Hospital of Jining Medical University. His research focuses on the pathogenesis and treatment of acute myeloid leukemia.
Qian Huang
Qian Huang, received her BS degree from Shandong First Medical University in 2007. Now, she is a physician in the hematology department of Affiliated Hospital of Jining Medical University. Her research focuses on the diagnosis and treatment of acute leukemia.
Chunyan Yang
Chunyan Yang, received her BS degree from Jining Medical University in 2006 and her Master degree in Hematology from Dalian Medical University in 2009. Now, she is a physician in Hematology department, Affiliated Hospital of Jining Medical University. Her research focuses on the pathogenesis and treatment of aplastic anemia.
Panpan Cheng
Panpan Cheng, received her BS degree from Qingdao University Qingdao Medical College in 2007 and her Master degree in Clinical Laboratory Diagnostics from Shantou University Medical College in 2010. Now, she is a clinical pathologist in the Affiliated Hospital of Jining Medical University. Her research focuses on hematopathology including morphology and immunology.
Saisai Ren
Saisai Ren, received her BS degree and Master degree from Jining Medical University in 2014 and 2017. Now, she is a physician in the hematology department of Affiliated Hospital of Jining Medical University. Her research focuses on the diagnosis and treatment of hematological diseases.
Jingjing Zhang
Jingjing Zhang, received her BS degree from Zunyi Medical College in 2007. Now, she is an associate chief physician in hematology department, Affiliated Hospital of Jining Medical University. Her research focuses on the pathogenesis and treatment of acute myeloid leukemia.
Mingxiao Yu
Mingxiao Yu, received her BS degree from Shandong First Medical University in 2019 and her Master degree in Clinical Medicine from Jining Medical University in 2023. Her research focuses on NK cell mediated cytotoxicity in acute myeloid leukemia.
Xinying Ma
Xinying Ma, received her BS degree from Inner Mongolia Medical University in 2018 and her Master degree in Clinical Medicine from Jining Medical University in 2023. Her research focuses on hematopoietic regulation of acute myeloid leukemia and multiple myeloma.
Wenjun Song
Wenjun Song, received her BS degree and Master degree from Jining Medical University in 2019 and 2023. Her research focuses on mesenchymal stem cell mediated marrow microenvironment changes in acute myeloid leukemia.
Lulu Chen
Lulu Chen, received her BS degree and Master degree from Jining Medical University in 2018 and 2021. Now, she is a physician in the hematology department of Affiliated Hospital of Jining Medical University. Her research focuses on the pathogenesis and treatment of acute myeloid leukemia.
Hao Zhang
Hao Zhang, received his BS degree in Clinical Medicine Science from Xinxiang Medical University in 1999 and his Master degree in internal medicine from Wuhan University in 2002. Later, he obtained his PhD in hematology from Fujian Medical University in 2008. Now, Prof. Zhang is a Chief physician in hematology department, Affiliated Hospital of Jining Medical University. His research focuses on the basic and translational study of hematopoietic stem cell transplantation on the treatment of various hematological disorders.
Mingtai Chen
Mingtai Chen, received his BS degree in Biological science from Shandong University in 2011. Later, he obtained his PhD in Biochemistry and molecular biology from Peking Union Medical College / Chinese Academy of Medical Science in 2016. Now, Prof. Chen is a full-time principle investigator at the medical research center, Affiliated Hospital of Jining Medical University. His research focuses on hematopoietic regulation in acute myeloid leukemia and aplastic anemia.
Xianning Zhang
Xianning Zhang, received his BS degree in Biological Science from Shandong Agricultural University in 2015 and his Master degree in Immunology from University of Chinese Academy of Sciences in 2018. Now, he is an assistant researcher in the Affiliated Hospital of Jining Medical University. His research focuses on the pathogenesis and treatment of aplastic Anemia.
References
- Newell LF, Cook RJ. Advances in acute myeloid leukemia. BMJ. 2021;375n2026:n2026. doi:10.1136/bmj.n2026.
- Thomas X, Elhamri M, Heiblig M. Emerging pharmacotherapies for elderly acute myeloid leukemia patients. Expert Rev Hematol. 2020;13(6):619–11. doi:10.1080/17474086.2020.1758058.
- Rowe JM. Changing trends in the therapy of acute myeloid leukemia. Best Pract Res Clin Haematol. 2021;34(4):101333. doi:10.1016/j.beha.2021.101333.
- Kantarjian HM, Kadia TM, DiNardo CD, Welch MA, Ravandi F. Acute myeloid leukemia: treatment and research outlook for 2021 and the MD Anderson approach. Cancer. 2021;127(8):1186–1207. doi:10.1002/cncr.33477.
- Kishtagari A, Levine RL, Viny AD. Driver mutations in acute myeloid leukemia. Curr Opin Hematol. 2020;27(2):49–57. doi:10.1097/MOH.0000000000000567.
- Cerrano M, Itzykson R. New treatment options for acute Myeloid Leukemia in 2019. Curr Oncol Rep. 2019;21(2):16. doi:10.1007/s11912-019-0764-8.
- Ball B, Mei M, Otoukesh S, Stein A. 2021. Current and emerging therapies for acute Myeloid Leukemia. Cancer Treat Res. 181:57–73.
- Dohner H, Weisdorf DJ, Bloomfield CD, Longo DL. Acute Myeloid Leukemia. N Engl J Med. 2015;373(12):1136–1152. doi:10.1056/NEJMra1406184.
- Goldman SL, Hassan C, Khunte M, Soldatenko A, Jong Y, Afshinnekoo E, Mason CE. Epigenetic modifications in acute Myeloid Leukemia: prognosis, treatment, and heterogeneity. Front Genet. 2019;10:133. doi:10.3389/fgene.2019.00133.
- Zhang J, Gao X, Yu L. Roles of histone deacetylases in acute Myeloid Leukemia with fusion proteins. Front Oncol. 2021;11:741746. doi:10.3389/fonc.2021.741746.
- Milazzo G, Mercatelli D, Di Muzio G, Triboli L, De Rosa P, Perini G, Giorgi FM. Histone Deacetylases (HDACs): evolution, specificity, role in transcriptional complexes, and pharmacological actionability. Genes (Basel). 2020;11(5):11. doi:10.3390/genes11050556.
- Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10(1):32–42. doi:10.1038/nrg2485.
- Li Y, Seto E. Hdacs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb Perspect Med. 2016;6(10):6. doi:10.1101/cshperspect.a026831.
- Mehrpouri M, Pourbagheri-Sigaroodi A, Bashash D. The contributory roles of histone deacetylases (HDACs) in hematopoiesis regulation and possibilities for pharmacologic interventions in hematologic malignancies. Int Immunopharmacol. 2021;100:108114. doi:10.1016/j.intimp.2021.108114.
- Liu L, Zhang J, Zhang X, Cheng P, Liu L, Huang Q, Liu H, Ren S, Wei P, Wang C, et al. HMGB1: an important regulator of myeloid differentiation and acute myeloid leukemia as well as a promising therapeutic target. J Mol Med (Berl). 2021;99(1):107–118. doi:10.1007/s00109-020-01998-5.
- Li Y, Wang Y, Zhou Y, Li J, Chen K, Zhang L, Deng M, Deng S, Li P, Xu B, et al. Cooperative effect of chidamide and chemotherapeutic drugs induce apoptosis by DNA damage accumulation and repair defects in acute myeloid leukemia stem and progenitor cells. Clin Epigenetics. 2017;9(1):83. doi:10.1186/s13148-017-0377-8.
- Li G, Li D, Yuan F, Cheng C, Chen L, Wei X. Synergistic effect of chidamide and venetoclax on apoptosis in acute myeloid leukemia cells and its mechanism. Ann Transl Med. 2021;9(20):1575. doi:10.21037/atm-21-5066.
- Kwon SH, Ahn SH, Kim YK, Bae GU, Yoon JW, Hong S, Lee HY, Lee Y-W, Lee H-W, Han J-W, et al. Apicidin, a histone deacetylase inhibitor, induces apoptosis and Fas/Fas ligand expression in human acute promyelocytic leukemia cells. J Biol Chem. 2002;277(3):2073–2080. doi:10.1074/jbc.M106699200.
- Ma X, Ezzeldin HH, Diasio RB. Histone deacetylase inhibitors: current status and overview of recent clinical trials. Drugs. 2009;69(14):1911–1934. doi:10.2165/11315680-000000000-00000.
- You JS, Kang JK, Seo DW, Park JH, Park JW, Lee JC, Jeon YJ, Cho EJ, Han J-W. Depletion of embryonic stem cell signature by histone deacetylase inhibitor in NCCIT cells: involvement of Nanog suppression. Cancer Research. 2009;69(14):5716–5725. doi:10.1158/0008-5472.CAN-08-4953.
- Ezura Y, Kajita M, Ishida R, Yoshida S, Yoshida H, Suzuki T, Hosoi T, Inoue S, Shiraki M, Orimo H, et al. Association of multiple nucleotide variations in the pituitary glutaminyl cyclase gene (QPCT) with low radial BMD in adult women. J Bone Miner Res. 2004;19(8):1296–1301. doi:10.1359/JBMR.040324.
- Cheng H, Cheng T. ‘Waterloo’: when normal blood cells meet leukemia. Curr Opin Hematol. 2016;23(4):304–310. doi:10.1097/MOH.0000000000000253.
- Chen MT, Dong L, Zhang XH, Yin XL, Ning HM, Shen C, Su R, Li F, Song L, Ma Y-N, et al. ZFP36L1 promotes monocyte/macrophage differentiation by repressing CDK6. Sci Rep. 2015;5(1):16229. doi:10.1038/srep16229.
- Chen MT, Lin HS, Shen C, Ma YN, Wang F, Zhao HL, Yu J, Zhang J-W. PU.1-regulated long noncoding RNA lnc-MC controls human monocyte/macrophage differentiation through interaction with MicroRNA 199a-5p. Molecular And Cellular Biology. 2015;35(18):3212–3224. doi:10.1128/MCB.00429-15.
- De Kouchkovsky I, Abdul-Hay M. ‘Acute myeloid leukemia: a comprehensive review and 2016 update’. Blood Cancer Journal. 2016;6(7):e441. doi:10.1038/bcj.2016.50.
- Garg M, Nagata Y, Kanojia D, Mayakonda A, Yoshida K, Haridas Keloth S, Zang ZJ, Okuno Y, Shiraishi Y, Chiba K, et al. Profiling of somatic mutations in acute myeloid leukemia with FLT3-ITD at diagnosis and relapse. Blood. 2015;126(22):2491–2501. doi:10.1182/blood-2015-05-646240.
- Yang JJ, Park TS, Wan TS. 2017. Recurrent cytogenetic abnormalities in acute Myeloid Leukemia. Methods Mol Biol. 1541:223–245.
- Foucar K, Anastasi J. Acute Myeloid Leukemia with recurrent cytogenetic abnormalities. Am J Clin Pathol. 2015;144(1):6–18. doi:10.1309/AJCPI9C8UILYQTNS.
- Huang HT, Figueroa ME. Epigenetic deregulation in myeloid malignancies. Blood. 2021;138(8):613–624. doi:10.1182/blood.2019004262.
- Liu XL, Liu HQ, Li J, Mao CY, He JT, Zhao X. Role of epigenetic in leukemia: from mechanism to therapy. Chem Biol Interact. 2020;317:108963. doi:10.1016/j.cbi.2020.108963.
- Wang P, Wang Z, Liu J. Role of HDACs in normal and malignant hematopoiesis. Mol Cancer. 2020;19(1):5. doi:10.1186/s12943-019-1127-7.
- Thol F, Heuser M. Treatment for relapsed/refractory acute myeloid Leukemia. Hemasphere. 2021;5(6):e572. doi:10.1097/HS9.0000000000000572.
- Bohl SR, Bullinger L, Rucker FG. Epigenetic therapy: azacytidine and decitabine in acute myeloid leukemia. Expert Rev Hematol. 2018;11(5):361–371. doi:10.1080/17474086.2018.1453802.
- DiNardo CD, Pratz K, Pullarkat V, Jonas BA, Arellano M, Becker PS, Frankfurt O, Konopleva M, Wei AH, Kantarjian HM, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2019;133(1):7–17. doi:10.1182/blood-2018-08-868752.
- San Jose-Eneriz E, Gimenez-Camino N, Agirre X, Prosper F. HDAC inhibitors in acute Myeloid Leukemia. Cancers Basel. 2019;11(11):11. doi:10.3390/cancers11111794.
- Kouraklis G, Theocharis S. Histone deacetylase inhibitors: a novel target of anticancer therapy (review). Oncol Rep. 2006;15:489–494. doi:10.3892/or.15.2.489.
- Khan N, Jeffers M, Kumar S, Hackett C, Boldog F, Khramtsov N, Qian X, Mills E, Berghs S, Carey N, et al. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem J. 2008;409(2):581–589. doi:10.1042/BJ20070779.
- Ning ZQ, Li ZB, Newman MJ, Shan S, Wang XH, Pan DS, Zhang J, Dong M, Du X, Lu X-P, et al. Chidamide (CS055/HBI-8000): a new histone deacetylase inhibitor of the benzamide class with antitumor activity and the ability to enhance immune cell-mediated tumor cell cytotoxicity. Cancer Chemother Pharmacol. 2012;69(4):901–909. doi:10.1007/s00280-011-1766-x.
- Neganova ME, Klochkov SG, Aleksandrova YR, Aliev G. Histone modifications in epigenetic regulation of cancer: perspectives and achieved progress. Semin Cancer Biol. 2020;83:452–471. doi:10.1016/j.semcancer.2020.07.015.
- Spreafico M, Gruszka AM, Valli D, Mazzola M, Deflorian G, Quinte A, Totaro MG, Battaglia C, Alcalay M, Marozzi A, et al. HDAC8: a promising therapeutic target for acute Myeloid Leukemia. Front Cell Dev Biol. 2020;8:844. doi:10.3389/fcell.2020.00844.