
ABSTRACT
GBM is one of the most malignant tumor in central nervous system. The resistance to temozolomide (TMZ) is inevitable in GBM and the characterization of TMZ resistance seriously hinders clinical treatment. It is worthwhile exploring the underlying mechanism of aggressive invasion and TMZ resistance in GBM treatment. Bioinformatic analysis was used to analyze the association between RND1 and a series of EMT-related genes. Colony formation assay and cell viability assay were used to assess the growth of U87 and U251 cells. The cell invasion status was evaluated based on transwell and wound-healing assays. Western blot was used to detect the protein expression in GBM cells. Treatment targeted RND1 combined with TMZ therapy was conducted in nude mice to evaluate the potential application of RND1 as a clinical target for GBM. The overexpression of RND1 suppressed the progression and migration of U87 and U251 cells. RND1 knockdown facilitated the growth and invasion of GBM cells. RND1 regulated the EMT of GBM cells via inhibiting the phosphorylation of AKT and GSK3-β. The promoted effects of RND1 on TMZ sensitivity was identified both in vitro and in vivo. This research demonstrated that the overexpression of RND1 suppressed the migration and EMT status by downregulating AKT/GSK3-β pathway in GBM. RND1 enhanced the TMZ sensitivity of GBM cells both in vitro and in vivo. Our findings may contribute to the targeted therapy for GBM and the understanding of mechanisms of TMZ resistance in GBM.
Introduction
Glioblastoma (GBM) is the most malignant brain tumor in adults that exhibits aggressive cell proliferation and invasion.Citation1 Although maximum safe surgical resection can prolong survival of patients, nearly all GBM patient relapse.Citation2 Temozolomide (TMZ) is currently one of the standard treatment in GBM which also promoted the survival of patients.Citation3 However, the resistance to TMZ is inevitable in GBM and the characterization of TMZ resistance seriously hinders clinical treatment.Citation4 Hence, it is worthwhile exploring the underlying mechanism of aggressive invasion and TMZ resistance in GBM treatment.
The Rho family GTPase 1 (RND1), also known as RHOS and RHO6, is a member of the RND family.Citation5 The RND family is an atypical class of Rho-GTPases consisting of RND1, RND2 and RND3 that lacks GTPase hydrolytic activity.Citation6 RND family proteins are highly expressed in brain tissue and play an important role in the generation and migration of neurons.Citation7 Recent research has demonstrated the function of RND family in regulating GBM progression. RND2 attenuates apoptosis and autophagy in glioblastoma cells by targeting the p38 MAPK signaling pathway.Citation8 RND3 induces GBM cell apoptosis via NF‐κB signaling.Citation9 Meanwhile, a research identified that RND1 regulated migration of human glioblastoma stem-like cells according to their anatomical localization and defines a prognostic signature in glioblastoma.Citation10 In other malignancies, RND1 also acts as a tumor suppressor gene. For instance, RND1 inhibits tumor progression and invasion in triple-negative breast cancer and hepatocellular carcinoma.Citation11,Citation12 Even though the function of RND1 to inhibit glioma stem-like cell invasion has been revealed, the specific regulatory mechanism in GBM cells has not been elucidated.
Although temozolomide is the first‐line pharmaceutical in glioma with excellent capability in crossing the blood‐brain barrier, acquired TMZ resistance is unavoidable in most GBM patients.Citation13 Methylguanine DNA methyltransferase (MGMT) can repair the mutagenic effect of TMZ on DNA and GBM patients expressing high levels of MGMT exhibit significant resistance to TMZ treatment.Citation14 In addition, hypoxia and epithelial-mesenchymal transition (EMT) status of glioma cells caused by chemotherapy was closely related to drug resistance.Citation15,Citation16 Emerging studies have shown that phosphatidylinositol 3-kinase/Akt (PI3K/Akt) signaling participated in the regulation of chemoresistance in glioma. The application of TMZ significantly activated the cascade of PI3K/Akt/GSK-3, and the use of LY294002 and Tubeimoside-I to inhibit AKT activation can effectively suppress EMT and enhance the efficacy of TMZ in glioma.Citation17,Citation18 Whether RND1 affects the sensitivity of glioma cells to TMZ remain unknown and is worthy of further investigation.
In this research, we revealed an anti-tumor activity and EMT-suppression of RND1 in GBM via AKT signaling. Overexpression of RND1 inhibited the phosphorylation of AKT protein and suppressed the invasion of GBM cells. Considering the relationship between EMT status and tumor drug resistance is recognized,Citation19 we further researched the role of RND1 in regulating GBM resistance to TMZ. On top of that, we found that the inhibitory regulation of AKT phosphorylation by RND1 enhanced the sensitivity of GBM cells to TMZ. Our research further explored the tumor suppressor role and regulatory mechanism of RND1 in GBM, which is expected to provide new ideas for targeted therapy in GBM.
Materials and methods
Cell culture
Human GBM cell lines U87 and U251 were obtained from Shanghai Institute of Cell Biology, Chinese Academy of Sciences (Shanghai, China). Authentication of GBM cells was conducted by short tandem repeat (STR) in Procell Life Science & Technology (Wuhan, China). GBM cells were cultured in high-glucose DMEM supplemented with 10% fetal bovine serum (10099141C, Gibco, USA) at 37°C with 5% CO2. The TMZ-resistant U87 cell line (U87-TR) was established by exposing normal U87 cell lines to TMZ at gradient ascending doses (final dose at 400 μM) for 6 months. The establishment of TMZ-resistant U87 cell line was confirmed via cell viability assay (Supplemental ).
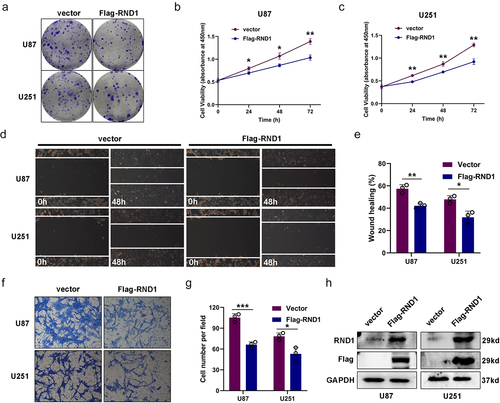
Figure 1. Overexpression of RND1 suppressed the progression and migration of GBM cells. (a) The colony formation assay was performed when RND1 was overexpressed in U87 and U251 cells. (b-c) the cell viability of U87 and U251 was detected when RND1 was upregulated. (d-e) wound-healing assay was performed in U87 and U251 cells transfected with flag-RND1. (f-g) Transwell assay was performed to assess the migration ability of U87 and U251 cells. (H) RND1 overexpression was confirmed by western blot analysis. Results were obtained in three independent experiments and quantified. *, p < .05; **, p < .01; ***, p < .001.
Reagents and antibodies
DMSO was purchased from Servicebio (GC203005). The AKT inhibitor SC66 was obtained from MedChemExpress (HY-19832). TMZ was purchased from Selleck (S1237).
The following antibodies were used: anti-RND1 (GTX83709, GeneTex), anti-GAPDH (#5174, Cell Signalling Technology, CST), anti-Flag (M185, Medical Biological Laboratories), anti-E-cadherin (20874–1-AP, Proteintech), anti-N-cadherin (22018–1-AP, Proteintech), anti-Vimentin (10366–1-AP, Proteintech), anti-Snail1 (sc -28,199, Santa Cruz), anti-mmp2 (10373–2-AP, Proteintech), anti-AKT (GTX121937, GeneTex), anti-Phospho-AKT (#4060, CST), anti-GSK-3β (#12456, CST), anti-Phospho-GSK-3β (#9323, CST), anti-β-catenin (51067–2-AP, Proteintech), anti-MAPK (#4511, CST), anti-Phospho-MAPK (#9212, CST) and anti-Smad3 (66516–1-Ig, Proteintech).
Bioinformatics
The association between RND1 and a series of EMT-related genes (MMP2, MMP3, MMP9, SNAI1, SNAI2, TJP1, CDH1, CDH2, and VIM) was analyzed based on RNAseq data of 703 glioma tissues from TCGA database. P-values were obtained from Pearson correlation.
Western blot analysis
Cell samples were lysed in Radio Immunoprecipitation Assay (RIPA) buffer (P0013B, Beyotime) containing the protease inhibitor Cocktail (4693116001, Roche). Protein concentration was measured by the BCA Kit (P0012S, Beyotime) and 5×loading buffer (P0015, Beyotime) was added. Protein samples (20 μg/lane) were electrophoretic in 10–12% SDS-PAGE gels and transferred to poly vinylidene fluoride (PVDF) membranes. The membranes were incubated with primary antibodies and HRP-conjugated secondary antibodies. Finally, the membranes were prepared with ECL Western blotting substrate Kit (BL520A, Biosharp) and captured using a ChemiDoc Imaging System (Bio-Rad, USA). The analysis was conducted using Image J.
Plasmid construction of RND1
The construction of the overexpression plasmid pCDNA3.1-Flag-RND1 and specific shRNA targeting of human RND1 (shRND1) was helped with Miaoling Biology (Wuhan, China) and described previously.Citation20 The sequences of pCDNA3.1-Flag-RND1 and shRND1 were provided in Supplemental Table S1.
Cell viability assay
Cell viability of U87 and U251 was measured by a Cell Counting Kit-8 (CCK-8) Kit (CK04, Dojindo Molecular Technologies). Cells were cultured in 96-well plates with plasmid transfection or drug action and detected at specified time points (0 h, 24 h, 48 h and 72 h) with the help of CCK8 Kits. After CCK8 reagent was added, cells were placed in an incubator at 37°C for 2 h. The OD value was measured at 450 nm using a Multimode Plate Reader (PerkinElmer, Germany).
Colony formation assay
U87 and U251 cells were seeded into six-well plates (500 cells per well) and incubated at 37°C with 5% CO2 for 2 weeks. The colonies were fixed with 4% formaldehyde for and stained with .2% crystal violet at room temperature. The representative colonies were captured and quantified.
Wound healing assay
U87 and U251 cells in 6-well plates were scraped with a 200 μL pipette tip to create the wounds. To exclude the effect of cell proliferation, only 1% FBS was added in DMEM. Cells that migrated to the wound area were photographed at 0 h and 48 h. Representive boundaries of the wound were drawn with straight lines. Wound healing ratio was calculated and analyzed.
Transwell assay
U87 and U251 cells were seeded into the upper chamber of polycarbonate transwell filters in serum-free DMEM. DMEM containing 10% FBS was added in lower chambers as chemoattractant. Cells were incubated for 24 h and fixed with 4% paraformaldehyde. The cells on the lower chamber were dyed with crystal violet (Beyotime). Images were captured by an inverted microscope (Olympus BX51, Japan) and the number of cells was counted.
Generation of stable cell line and subcutaneous xenograft mouse model
To establish the stable U87 cell line with RND1 overexpression and carrying luciferase, pLVX-CMV-Luciferase-RND1-puro was transfected with pMD2.G and psPAX2 into 293T cells to produce lentiviruse. Then, U87 cell line was cultured in lentiviruse for 48 h and selected by puromycin.
Four-week-old male Balb/c nude mice were purchased from Shulaibao (Wuhan, China) Biotechnology Co., Ltd. In the subcutaneous xenograft model, 1 × 106 stable cells (pLVX-luciferase and pLVX-RND1-luciferase) were subcutaneously injected into armpits of nude mice. TMZ treatment started 7 days after injection, the mice were injected intraperitoneally with two cycles of TMZ (50 mg/kg) for 5 days. All nude mice were captured by IVIS (In Vivo Imaging System) at the 21st day after tumor injection and sacrificed at were sacrificed at 28th day. Animal experiment of this research was approved by the institutional animal care and use committee of Renmin Hospital of Wuhan University.
In vivo imaging of mouse xenografts
Tumor-bearing mice were injected with 10 mg D-Luciferin potassium (ST196, Beyotime, China) via caudal vein. Luminescence was captured and calculated by an In Vivo Imaging System (IVIS) (Bruker Xtreme BI, Bruker, USA).
Statistical analysis
Experiments were repeated three times and data are presented as the mean ± standard deviation. Data were analyzed using SPSS (Version 19.0) and GraphPad Prism (Version 8.0). Student’s t-test was used for data comparisons between two groups. Comparisons among multiple groups were analyzed using one-way ANOVA followed by Tukey’s post hoc test. p < .05 was considered as statistical significance.
Results
Overexpression of RND1 suppressed the progression and migration of U87 and U251 cells
We first overexpressed RND1 via the transfection of Flag-RND1 plasmid in U87 and U251 cells (). Colony formation assay and cell viability assay were used to assess the growth of U87 and U251 cells when RND1 was overexpressed. Results indicated that the overexpression of RND1 obviously suppressed the growth of U87 and U251 cells (). We evaluated the EMT status of U87 and U251 cells based on transwell and wound-healing assays. From the results, we observed that the cell invasion ability of U87 and U251 was significantly inhibited when RND1 was up-regulated (). Hence, RND1 suppressed the growth and invasion status of U87 and U251 cells.
RND1 knockdown facilitated the growth and invasion of U87 and U251 cells
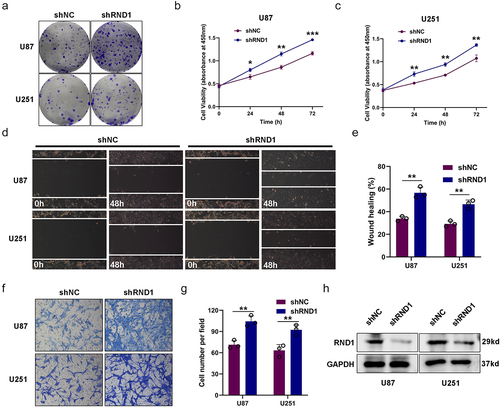
We further knockdown RND1 via the transfection of shRND1 plasmid in U87 and U251 cells and the efficiency of knockdown was confirmed by western blot analysis (). Colony formation assay and cell viability assay were used to assess the growth of U87 and U251 cells when RND1 was knocked down. Results indicated that the knockdown of RND1 obviously facilitated the growth of U87 and U251 cells (). Meanwhile, we evaluated the EMT status of U87 and U251 cells based on transwell and wound-healing assays. We found that the cell invasion of U87 and U251 was significantly promoted when RND1 was down-regulated (). Thus, knockdown of RND1 promoted GBM invasion and progression.
Figure 2. RND1 knockdown facilitated the growth and invasion of GBM cells. (a) The colony formation assay was performed when RND1 was knocked down in U87 and U251 cells. (b-c) the cell viability of U87 and U251 was detected when RND1 was downregulated. (d-e) wound-healing assay was performed in U87 and U251 cells transfected with shRND1. (f-g) Transwell assay was performed to assess the migration ability of U87 and U251 cells. (h) RND1 knockdown efficiency was confirmed by western blot analysis. Results were obtained in three independent experiments and quantified. *, p < .05; **, p < .01; ***, p < .001.
RND1 inhibited the EMT in GBM
To further confirm the effect of RND1 on EMT state of GBM cells, we first analyzed the association between RND1 and a series of EMT-related genes (MMP2, MMP3, MMP9, SNAI1, SNAI2, TJP1, CDH1, CDH2 and VIM) based on RNAseq data of 703 glioma tissues from TCGA database. The results significantly indicated that RND1 was positively correlated with anti-EMT genes (TJP1 and CDH1) and negatively correlated with pro-EMT genes (MMP2, MMP3, MMP9, SNAI1, SNAI2, CDH2 and VIM) ().
Figure 3. RND1 inhibited the EMT in GBM. (a-i) the correlation between RND1 and a series of EMT-related genes was analyzed based on RNAseq data of 703 glioma tissues from TCGA database. P-values were obtained from Pearson correlation. RND1 was positively correlated with anti-EMT genes (TJP1 and CDH1) and negatively correlated with pro-EMT genes (MMP2, MMP3, MMP9, SNAI1, SNAI2, CDH2 and VIM). SNAI1, also known as Snail1; SNAI2, also known as Slug; TJP1, also known as ZO-1; CDH1, also known as E-Cadherin; CDH-2, also known as N-cadherin; VIM, also known as Vimentin. (j-l) the protein (E-cadherin, N-cadherin, Vimentin, Snail1 and mmp2) expression was detected via western blot assay when RND1 was overexpressed or knocked down in U87.
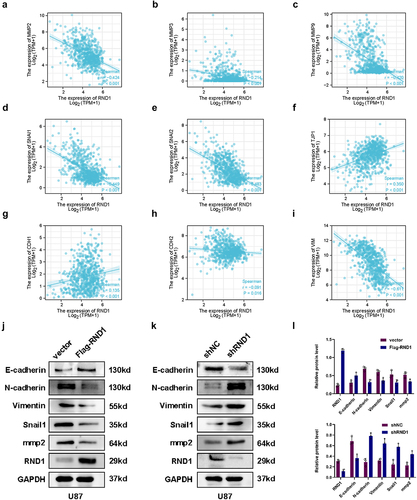
In vitro experiments, we analyzed the expression level of EMT-related genes using western blot analysis when RND1 was up-regulated or down-regulated in U87. Results showed that E-cadherin was upregulated when RND1 was overexpressed in U87 (). Meanwhile, N-cadherin, Vimentin, Snail1 and mmp2 was downregulated (). Consistently, when RND1 was knocked down, E-cadherin was downregulated and N-cadherin, Vimentin, Snail1, and mmp2 was upregulated (). Thus, through the detection and analysis of the expression of EMT-related genes, we discovered that RND1 inhibited the EMT status in GBM
RND1 regulated the EMT of GBM cells via AKT signal
To investigate the potential mode of RND1 on GBM invasion, we detected the several signals regulating tumor EMT. Considering TGF-β-Smad, PI3K-AKT, MAPK-p38, and Wnt-β-catenin signals were core pathways regulating tumor EMT,Citation21,Citation22 we researched the protein levels of these signals via western blot analysis when the expression of RND1 was changed. The results showed that the overexpression of RND1 did not affect the protein level of p-MAPK, Smad3, and β-catenin but inhibit the phosphorylation of AKT (p-AKT) (). Meanwhile, to confirm the effect of RND1 on AKT signal, we also detected the protein level of total and phosphorylated GSK3-β, an important signal downstream of AKT.Citation23 We discovered a significant decrease of phosphorylated GSK3-β (p-GSK3-β) protein when RND1 was overexpressed in U87 cells (), indicating a convincing negative regulation of RND1 on AKT signal.
Figure 4. RND1 regulated the EMT of GBM cells via AKT signal. (a) Protein (AKT, p-AKT, GSK3-β, p-GSK3-β, MAPK, p-MAPK, Smad3, and β-catenin) levels were detected via western blot assay when RND1 was upregulated in U87 cells. Protein levels of western blots were quantified (d). (b) Protein (AKT, p-AKT, GSK3-β, p-GSK3-β, E-cadherin, Vimentin, mmp2, and Snail1) levels were detected via western blot assay when RND1 was knocked down in U87 cells with the treatment of SC66 (an inhibitor of AKT phosphorylation). (c) Transwell assay was performed to assess the migration ability of U87 and representative images were shown. Knockdown of RND1 facilitated the invasion of U87 cells, which could be reversed by SC66. (e) Invasive cells in transwell assay were quantified. *, p < .05; **, p < .01; ***, p < .001; ns, no significance.
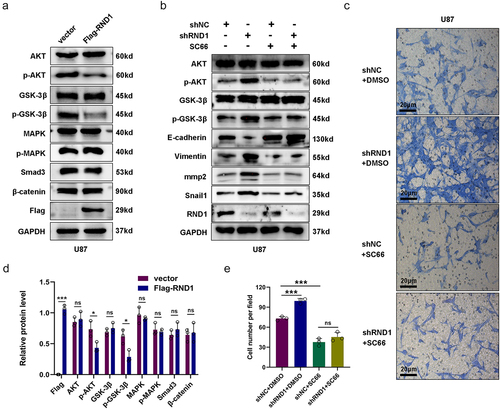
Moreover, we also detected the AKT signal activity when RND1 was knocked down in U87 cells. Results from western blot analysis indicated that downregulation of RND1 obviously increased the phosphorylation of AKT and GSK3-β and regulated EMT-related genes including E-cadherin, Vimentin, Snail1, and mmp2 (). Meanwhile, we discovered that the use of SC66 (an inhibitor of AKT phosphorylationCitation24) significantly decreased phosphorylated AKT and GSK3-β and reversed the effect of RND1 on EMT-related proteins (). Furthermore, transwell assay was performed in U87 cells with the treatment of SC66. Results showed that knockdown of RND1 facilitated the invasion of U87 cells, which could be reversed by SC66 ().
RND1 enhanced the TMZ sensitivity of normal and TMZ-resistant GBM cells
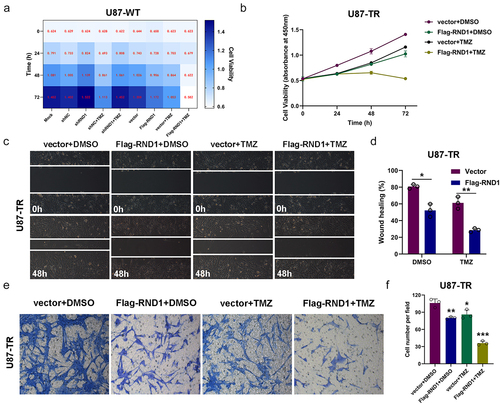
Next, to determine the impact of RND1 on the TMZ sensitivity of GBM cells, we overexpressed or knocked down RND1 in U87 cells and treated U87 cells with 400 μM TMZ (). The results of cell viability assay showed that RND1 enhanced the TMZ sensitivity of normal U87 cells. While in clinical practice, resistance to TMZ in GBM is a common phenomenon. To continue the research on the role of RND1 in GBM resistance, we established the TMZ-resistant U87 cell line (U87-TR) by exposing normal U87 cell lines to TMZ at a high dose of 400 μM for 6 months. The establishment of U87-TR was confirmed via cell viability assay (Supplemental ). The cell viability assay in U87-TR indicated that RND1 overexpression significantly enhanced the anti-tumor activity of TMZ in drug-resistant U87-TR (). Consistently, in the transwell and wound-healing assays, upregulation of RND1 combined with the use of TMZ showed the strongest inhibition of cell invasion in U87-TR ().
Figure 5. RND1 enhanced the TMZ sensitivity of normal and TMZ-resistant GBM cells. (a) The cell viability assay was performed in normal U87 cells (U87-WT) treated with TMZ when RND1 was overexpressed or knocked down. Results indicated that RND1 enhanced the TMZ sensitivity in U87-WT. (b) The cell viability assay was performed in TMZ-resistant U87 cells (U87-TR) treated with TMZ when RND1 was overexpressed. Results indicated that RND1 enhanced the TMZ sensitivity in U87-TR. (c) Wound-healing assay was conducted in U87-TR treated with TMZ when RND1 was overexpressed. The percentage of wound-healing was quantified (d). (e) Transwell assay was performed to assess the migration ability of U87-TR cells treated with TMZ and flag-RND1. (f) Invasive cells in transwell assay were quantified. *, p < .05; **, p < .01; ***, p < .001.
RND1 enhanced the TMZ sensitivity of GBM cells via inhibiting AKT signal
As a next step, the specific mechanism of RND1’s regulation on the TMZ sensitivity of GBM cells should be identified. We first detected the activity of AKT signal and EMT status in U87-TR cells. The results of western blot analysis showed that U87-TR cells obviously had a stronger AKT signaling pathway activity and more malignant EMT status compared to U87-WT cells (). Moreover, in U87-TR cells, RND1 also showed a function of inhibiting the activity of AKT signaling pathway and improving the EMT status ().
Figure 6. RND1 enhanced the TMZ sensitivity of GBM cells via inhibiting AKT signal. (a) Protein (AKT, p-AKT, GSK3-β, p-GSK3-β, E-cadherin, Vimentin, mmp2 and Snail1) levels were detected via western blot assay in U87-WT and U87-TR cells. Protein levels of western blots were quantified (c). (b) Protein (AKT, p-AKT, GSK3-β, p-GSK3-β, N-cadherin, Vimentin and Snail1) levels were detected via western blot assay in U87-TR cells transfected with flag-RND1. Protein levels of western blots were quantified (d). (e) The cell viability assay was performed when RND1 was overexpressed in U87-TR cells with the treatment of SC79 (an activator of AKT phosphorylation). Overexpression of RND1 enhanced the TMZ sensitivity of U87-TR cells, which could be reversed by SC79. (f) Protein (AKT and p-AKT) levels were detected via western blot assay in U87-TR cells transfected with flag-RND1 when SC79 was used. *, p < .05; **, p < .01; ***, p < .001; ns, no significance.
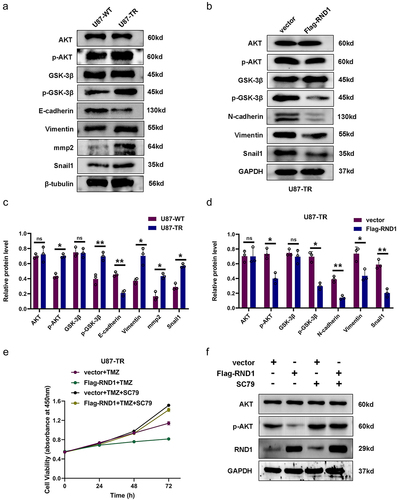
Furthermore, we overexpressed RND1 and treated U87-TR cells with SC79 (an activator of AKT phosphorylationCitation25). The results of cell viability assay showed that upregulation of RND1 suppressed the growth of U87-TR cells, which could be reversed by SC79 (). Meanwhile, from western blot analysis, we discovered that the use of SC79 significantly reversed the effect of RND1 on AKT signaling pathway ().
RND1 enhanced the TMZ sensitivity of GBM in xenograft mouse model
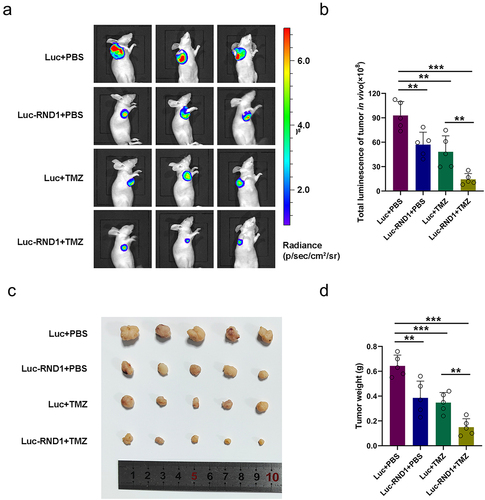
To better evaluate the potential application of RND1 as a clinical target for GBM, treatment targeted RND1 combined with TMZ therapy was conducted in vivo. Stable U87 cell line (U87-Luc-RND1) overexpressing RND1 was established using lentivirus and injected into armpits for the generation of subcutaneous xenograft model. 7 days after tumor injection, two groups (Luc+TMZ and Luc-RND1+TMZ) of mice received two cycles of intraperitoneal treatment of TMZ (50 mg/kg/day, 5 day/cycle). Subsequently, tumor growth was evaluated by IVIS and the fluorescence showed that progression of subcutaneous tumor was suppressed when RND1 was overexpressed in U87 (). Meanwhile, it was observed that RND1 overexpression enhanced the TMZ sensitivity of treatment in U87-tumor-bearing mice (). By comparing the relative size and weight of tumors from each group, it was observed that tumor growth was significantly inhibited in Luc-RND1 group and RND1 enhanced the TMZ sensitivity in GBM ().
Figure 7. RND1 enhanced the TMZ sensitivity of GBM in xenograft mouse model. (a-b) RND1 suppressed the growth and enhanced the TMZ sensitivity of GBM in xenograft mouse model. Luciferase fluorescence of mouse xenografts was detected and quantified using IVIS at the 21st day. Luc groups, n = 5; Luc-RND1 groups, n = 5; Luc+TMZ groups, n = 5; Luc-RND1+TMZ groups, n = 5. (c-d) subcutaneous tumors were excised and weighed after mice sacrifice. The weight of tumors was analyzed. *, p < .05; **, p < .01; ***, p < .001.
Discussion
GBM is one of the most malignant tumor in central nervous system (CNS). The poor prognosis of GBM is associated with its resistance to radiotherapy and chemotherapy.Citation4,Citation26 In this research, we discussed RND1-induced EMT inhibition which enhanced the GBM sensitivity to TMZ. This study may improve the understanding of the glioma EMT regulation and bring hope to the treatment of GBM.
An important physiological role of RND1 is to participate in neuronal development and growth. RND1 stimulates neurites in PC12 cells and promotes neuronal axon formation and stimulates dendrite growth in hippocampal neurons.Citation27–29 As a key protein regulating the cytoskeleton and intercellular connections, RND1 also plays an important role in the tumor EMT regulatory network. In breast cancer, RND1 induces epithelial-mesenchymal transition (EMT) in breast cancer epithelial cells by activating the Ras and MAPK pathways.Citation11 In hepatocellular carcinoma, the reduction of RND1 expression is related to cancer progression, and RND1 may inhibit tumor invasion and migration by regulating the Ras signaling pathway.Citation30 In glioma, it was reported that RND1 induced ferroptosis via regulating p53-SLC7A11 signal and inhibited the migration of glioma stem-like cells in the periventricular zone (PVZ).Citation10,Citation20 However, previous study did not elucidate the function and specific regulatory mechanism of RND1 in GBM EMT.
To begin with, we overexpressed or knocked down RND1 in GBM cell lines U87 and U251 and performed the cell viability assay and colony formation assay to evaluate the growth of GBM cells. It was observed that RND1 significantly suppressed the growth of U87 and U251 cells. Meanwhile, from the phenotype of EMT based on transwell and wound-healing assays, we identified that RND1 obviously inhibited the invasion of GBM cells. As a next step, we found that RND1 affected GBM EMT on protein levels. Consistently, based on RNAseq data of 703 glioma tissues from TCGA database, we found that RND1 was positively correlated with anti-EMT genes (TJP1 and CDH1) and negatively correlated with pro-EMT genes (MMP2, MMP3, MMP9, SNAI1, SNAI2, CDH2 and VIM). Hence, we demonstrated that RND1 inhibited EMT and suppressed the progression of GBM.
Notably, we found that RND1-mediated EMT was critically regulated by AKT signaling in glioma. Previous studies have revealed that TGF-β-Smad, PI3K-AKT, MAPK-p38 and Wnt-β-catenin signals were core pathways regulating tumor EMT.Citation15,Citation16 Results from western blot analysis revealed that the overexpression of RND1 did not affect the protein level of p-MAPK, Smad3 and β-catenin but inhibit the phosphorylation of AKT and GSK3-β. SC66, an inhibitor of AKT phosphorylation,Citation31 significantly decreased phosphorylated AKT and GSK3-β and reversed the effect of RND1 on EMT-related proteins and cell invasion. It has confirmed that PI3K/Akt signal pathway after activation of EGFR plays an important role in the genesis and development of glioma.Citation32 Meanwhile, the PI3K-AKT inhibitors as a single agent or combined with other therapies are being tested in a number of clinical trials.Citation33 In our research, we revealed that RND1 is a new target for regulating AKT/GSK3-β signal, which is expected to provide new ideas for targeted therapy of PI3K-AKT.
TMZ is the core treatment in GBM, but TMZ resistance is a major problem in at least 50% GBM patients.Citation4 The promoter methylation status of O6-methylguanine methyltransferase (MGMT) and DNA repair function are main controlling factors of GBM resistance to TMZ.Citation34 In glioma, the nuclear factor-Kappa B (NF-kB), STAT3 and JNK signals were involved in the regulation of TMZ resistance.Citation35–37 In our research, we identified a function of RND1 to enhance TMZ sensitivity of U87 wild type (U87-WT) cells. U87 was reported to be a TMZ-sensitive cell line and adapted TMZ resistant U87 cell line could be generated from stepwise exposure of the cells to TMZ.Citation38,Citation39 We built a TMZ resistant U87 (U87-TR) cell line and also found a TMZ sensitization induced by RND1 in U87-TR. Previous study has pointed out the activation of the AKT signaling pathway in TMZ resistant GBM cells.Citation40 Consistently, we detected stronger phosphorylation of AKT and GSK3-β in U87-TR cells, which exhibited greater invasiveness. Interestingly, RND1 also showed a function of inhibiting the activity of AKT signaling pathway and improving the EMT status in U87-TR cells. Additionally, SC79, an activator of AKT signal,Citation41 reversed the effect of RND1 on AKT signaling pathway and the growth of U87-TR cells. These results indicated that RND1 could enhance the TMZ sensitivity of GBM cells by downregulating AKT/GSK3-β pathway.
Furthermore, to better evaluate the potential application of RND1 as a target for GBM, treatment targeted RND1 combined with TMZ therapy was conducted in vivo. As a result, the growth of subcutaneous tumor was significantly suppressed when RND1 was overexpressed and RND1 also enhanced the TMZ sensitivity of U87 cells in xenograft model. Our work revealed a potential chemotherapeutic role of RND1 and provided a new combination with TMZ for GBM patients. However, there still exists insufficient considerations in our research design. GBM is highly inter-tumorally and inter-tumorally heterogeneous at both the molecular and histological levels.Citation42 On the one hand, the discrepancy of RND1 gene status and TMZ sensitivity between GBM patients need to be taken into account. On the other hand, intra-tumoral heterogeneity is an important factor in GBM’s resistance to TMZ or other therapies.Citation43 It is probably attributed to the presence of multiple subclone tumor cells and non-neoplastic cells, including infiltrating and resident immune cells, vascular cells, and other glial cells, which have the ability to evade chemotherapy and regenerate tumors in GBM.Citation44 Our in vitro and in vivo experiments were conducted in U87 and U251 cell lines, which cannot reflect the complex and heterogeneous status of GBM. Hence, the function of RND1 should be evaluated in more cell lines including patient-derived primary cell lines and patient-derived tumor xenografts ought to be used to simulate the primary and heterogeneous status of GBM. Moreover, right combination of targeting different signaling pathways or biological processes could enhance efficacy in a synergistic or additive manner in GBM.Citation45,Citation46 In the future, combining RND1 with other targets or immunotherapy may also be a promising direction for GBM treatment.
Conclusion
In summary, our study demonstrated that overexpression of RND1 suppressed the migration and EMT status in GBM. We verified that RND1 inhibited EMT status of GBM cells via regulating AKT/GSK3-β pathway. The effects of RND1 on TMZ sensitivity was identified both in vitro and in vivo. Furthermore, RND1 enhanced the TMZ sensitivity of GBM cells by downregulating AKT/GSK3-β pathway. Our findings may contribute to the targeted therapy for GBM and the understanding of mechanisms of TMZ resistance in GBM.
Supplemental Material
Download Zip (355.3 KB)Acknowledgments
We appreciate all the staff in central laboratory at the Renmin Hospital of Wuhan University.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data used to support the findings of this study are available from the corresponding author upon request.
Supplemental material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15384047.2024.2321770
Additional information
Funding
Notes on contributors
Qian Sun
Qian Sun, is studying for an M.D. at Wuhan University, specializing in targeted treatment of glioma.
Junjie Xu
Junjie Xu, is currently working in Office of Director, Renmin Hospital of Wuhan University.
Fan’en Yuan
Fan’en Yuan, is graduated from Wuhan University with a major in neurosurgery, and is currently a postdoctoral fellow in University of Pittsburgh.
Yan Liu
Yan Liu, is currently working in Department of Anesthesiology, Renmin Hospital of Wuhan University.
Qianxue Chen
Qianxue Chen, MD, Professor, Supervisor, is currently the Director of Department of Neurosurgery, Renmin Hospital of Wuhan University.
Lirui Guo
Lirui Guo, is currently the head nurse of Department of Neurosurgery, Renmin Hospital of Wuhan University.
Huimin Dong
Huimin Dong, MD, is currently the associate senior doctor of Department of Neurology, Renmin Hospital of Wuhan University.
Baohui Liu
Baohui Liu, MD, Associate Professor, is currently the senior doctor of Department of Neurosurgery, Renmin Hospital of Wuhan University.
References
- Paolillo M, Boselli C, Schinelli S. Glioblastoma under Siege: an Overview of Current Therapeutic Strategies. Brain Sci. 2018;8(1):15. doi:10.3390/brainsci8010015.
- Li YM, Suki D, Hess K, Sawaya R. The influence of maximum safe resection of glioblastoma on survival in 1229 patients: can we do better than gross-total resection? J Neurosurg. 2016;124(4):977–13. doi:10.3171/2015.5.JNS142087.
- Tan AC, Ashley DM, López GY, Malinzak M, Friedman HS, Khasraw M. Management of glioblastoma: state of the art and future directions. CA Cancer J Clin. 2020;70(4):299–312. doi:10.3322/caac.21613.
- Lee SY. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016;3(3):198–210. doi:10.1016/j.gendis.2016.04.007.
- Mouly L, Gilhodes J, Lemarié A, Cohen-Jonathan Moyal E, Toulas C, Favre G, Sordet O, Monferran S. The RND1 Small GTPase: main functions and emerging role in Oncogenesis. Int J Mol Sci. 2019;20(15):3612. doi:10.3390/ijms20153612.
- Chardin P. Function and regulation of Rnd proteins. Nature reviews Molecular cell biology. Nat Rev Mol Cell Biol. 2006;7(1):54–62. doi:10.1038/nrm1788.
- Basbous S, Azzarelli R, Pacary E, Moreau V. Pathophysiological functions of rnd proteins. Small GTPases. 2021;12(5–6):336–357. doi:10.1080/21541248.2020.1829914.
- Xu Y, Sun Q, Yuan F, Dong H, Zhang H, Geng R, Qi Y, Xiong X, Chen Q, Liu B. RND2 attenuates apoptosis and autophagy in glioblastoma cells by targeting the p38 MAPK signalling pathway. J Exp Clin Cancer Res. 2020;39(1):174. doi:10.1186/s13046-020-01671-2.
- Sun Q, Dong H, Li Y, Yuan F, Xu Y, Mao S, Xiong X, Chen Q, Liu B. Small GTPase RHOE/RND3, a new critical regulator of NF-κB signalling in glioblastoma multiforme? Cell Prolif. 2019;52(5):e12665. doi:10.1111/cpr.12665.
- Boyrie S, Delmas C, Lemarié A, Lubrano V, Dahan P, Malric L, Luis J, Gilhodes J, Tosolini M, Mouly L, et al. RND1 regulates migration of human glioblastoma stem-like cells according to their anatomical localization and defines a prognostic signature in glioblastoma. Oncotarget. 2018;9(73):33788–33803. doi:10.18632/oncotarget.26082.
- Okada T, Sinha S, Esposito I, Schiavon G, López-Lago MA, Su W, Pratilas CA, Abele C, Hernandez JM, Ohara M, et al. The Rho GTPase Rnd1 suppresses mammary tumorigenesis and EMT by restraining ras-MAPK signalling. Nat Cell Biol. 2015;17(1):81–94. doi:10.1038/ncb3082.
- Qin CD, Ma DN, Zhang SZ, Zhang N, Ren ZG, Zhu XD, Jia QA, Chai ZT, Wang CH, Sun HC, et al. The Rho GTPase Rnd1 inhibits epithelial-mesenchymal transition in hepatocellular carcinoma and is a favorable anti-metastasis target. Cell Death Disease. 2018;9(5):486. doi:10.1038/s41419-018-0517-x.
- Zhao J, Yang S, Cui X, Wang Q, Yang E, Tong F, Hong B, Xiao M, Xin L, Xu C, et al. A novel compound EPIC-0412 reverses temozolomide resistance via inhibiting DNA repair/MGMT in glioblastoma. Neuro Oncol. 2023;25(5):857–870. doi:10.1093/neuonc/noac242.
- Kinslow CJ, Rae AI, Taparra K, Kumar P, Siegelin MD, Grinband J, Gill BJA, McKhann GM, Sisti MB, Bruce JN, et al. MGMT promoter methylation predicts overall survival after chemotherapy for 1p/19q-codeleted gliomas. Clin Cancer Res. 2023;29(21):4399–4407. doi:10.1158/1078-0432.CCR-23-1295.
- Maugeri G, D’Amico AG, Saccone S, Federico C, Rasà DM, Caltabiano R, Broggi G, Giunta S, Musumeci G, D’Agata V. Effect of PACAP on hypoxia-induced angiogenesis and epithelial–mesenchymal transition in Glioblastoma. Biomedicines. 2021;9(8):965. doi:10.3390/biomedicines9080965.
- D’Amico AG, Maugeri G, Vanella L, Pittalà V, Reglodi D, D’Agata V. Multimodal Role of PACAP in Glioblastoma. Brain Sci. 2021;11(8):994. doi:10.3390/brainsci11080994.
- Tomar VS, Patil V, Somasundaram K. Temozolomide induces activation of Wnt/β-catenin signaling in glioma cells via PI3K/Akt pathway: implications in glioma therapy. Cell Biol Toxicol. 2020;36(3):273–278. doi:10.1007/s10565-019-09502-7.
- Tang Q, Cao H, Tong N, Liu Y, Wang W, Zou Y, Xu L, Zeng Z, Xu W, Yin Z, et al. Tubeimoside-I sensitizes temozolomide-resistant glioblastoma cells to chemotherapy by reducing MGMT expression and suppressing EGFR induced PI3K/Akt/mTOR/NF-κB-mediated signaling pathway. Phytomedicine. 2022;99:154016. doi:10.1016/j.phymed.2022.154016.
- Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. nature reviews clinical oncology. Nat Rev Clin Oncol. 2017;14(10):611–629. doi:10.1038/nrclinonc.2017.44.
- Sun Q, Xu Y, Yuan F, Qi Y, Wang Y, Chen Q, Liu B. Rho family GTPase 1 (RND1), a novel regulator of p53, enhances ferroptosis in glioblastoma. Cell & Bioscience. 2022;12(1):53. doi:10.1186/s13578-022-00791-w.
- De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13(2):97–110. doi:10.1038/nrc3447.
- Brabletz S, Schuhwerk H, Brabletz T, Stemmler MP. Dynamic EMT: a multi-tool for tumor progression. EMBO J. 2021;40(18):e108647. doi:10.15252/embj.2021108647.
- Song M, Bode AM, Dong Z, Lee MH. AKT as a therapeutic target for cancer. Cancer Res. 2019;79(6):1019–1031. doi:10.1158/0008-5472.CAN-18-2738.
- Chen W, Zhao S, Yu W, Rao T, Ruan Y, Zhu S, Xia Y, Song H, Cheng F. SC66 inhibits the proliferation and induces apoptosis of human bladder cancer cells by targeting the AKT/β-catenin pathway. J Cell Mol Med. 2021;25(22):10684–10697. doi:10.1111/jcmm.17005.
- Wen L, Wang K, Zhang F, Tan Y, Shang X, Zhu Y, Zhou X, Yuan H, Hu F. AKT activation by SC79 to transiently re-open pathological blood brain barrier for improved functionalized nanoparticles therapy of glioblastoma. Biomaterials. 2020;237:119793. doi:10.1016/j.biomaterials.2020.119793.
- Anjum K, Shagufta BI, Abbas SQ, Patel S, Khan I, Shah SAA, Akhter N, Hassan SSU. Current status and future therapeutic perspectives of glioblastoma multiforme (GBM) therapy: a review. Biomed Pharmacother Biomed Pharmacotherapie. 2017;92:681–689. doi:10.1016/j.biopha.2017.05.125.
- de Souza LE, Moura Costa MD, Bilek ES, Lopes MH, Martins VR, Püschel AW, Mercadante AF, Nakao LS, Zanata SM, de Souza LER. STI1 antagonizes cytoskeleton collapse mediated by small GTPase Rnd1 and regulates neurite growth. Exp Cell Res. 2014;324(1):84–91. doi:10.1016/j.yexcr.2014.03.017.
- Li YH, Ghavampur S, Bondallaz P, Will L, Grenningloh G, Pu Schel AW. Rnd1 regulates axon extension by enhancing the microtubule destabilizing activity of SCG10. J Biol Chem. 2009;284(1):363–371. doi:10.1074/jbc.M808126200.
- Ishikawa Y, Katoh H, Negishi M. Small GTPase Rnd1 is involved in neuronal activity-dependent dendritic development in hippocampal neurons. Neurosci Lett. 2006;400(3):218–223. doi:10.1016/j.neulet.2006.02.064.
- Komatsu H, Iguchi T, Masuda T, Hirata H, Ueda M, Kidogami S, Ogawa Y, Sato K, Hu Q, Nambara S, et al. Attenuated RND1 expression confers malignant phenotype and predicts poor prognosis in hepatocellular carcinoma. annals of surgical oncology. Ann Surg Oncol. 2017;24(3):850–859. doi:10.1245/s10434-016-5573-9.
- Gao L, Liu J, Xu P, Deng G, Liu B, Yuan F, Tan Y, Sun Q, Xu Y, Zhang H, et al. AKT inhibitor SC66 inhibits proliferation and induces apoptosis in human glioblastoma through down-regulating AKT/β-catenin pathway. Front Pharmacol. 2020;11:1102. doi:10.3389/fphar.2020.01102.
- Li X, Wu C, Chen N, Gu H, Yen A, Cao L, Wang E, Wang L. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget. 2016;7(22):33440–33450. doi:10.18632/oncotarget.7961.
- Westhoff MA, Karpel-Massler G, Brühl O, Enzenmüller S, La Ferla-Brühl K, Siegelin MD, Nonnenmacher L, Debatin KM. A critical evaluation of PI3K inhibition in Glioblastoma and neuroblastoma therapy. Mol Cell Ther. 2014;2(1):32. doi:10.1186/2052-8426-2-32.
- Butler M, Pongor L, Su YT, Xi L, Raffeld M, Quezado M, Trepel J, Aldape K, Pommier Y, Wu J. MGMT Status as a Clinical Biomarker in Glioblastoma. Trends Cancer. 2020;6(5):380–391. doi:10.1016/j.trecan.2020.02.010.
- Wang X, Jia L, Jin X, Liu Q, Cao W, Gao X, Yang M, Sun B. NF-κB inhibitor reverses temozolomide resistance in human glioma TR/U251 cells. Oncol Lett. 2015;9(6):2586–2590. doi:10.3892/ol.2015.3130.
- Kohsaka S, Wang L, Yachi K, Mahabir R, Narita T, Itoh T, Tanino M, Kimura T, Nishihara H, Tanaka S. STAT3 inhibition overcomes temozolomide resistance in glioblastoma by downregulating MGMT expression. Mol Cancer Ther. 2012;11(6):1289–1299. doi:10.1158/1535-7163.MCT-11-0801.
- Ueno H, Tomiyama A, Yamaguchi H, Uekita T, Shirakihara T, Nakashima K, Otani N, Wada K, Sakai R, Arai H, et al. Augmentation of invadopodia formation in temozolomide-resistant or adopted glioma is regulated by c-jun terminal kinase-paxillin axis. Biochem Bioph Res Co. 2015;468(1–2):240–247. doi:10.1016/j.bbrc.2015.10.122.
- Montaldi AP, Godoy PR, Sakamoto-Hojo ET. APE1/REF-1 down-regulation enhances the cytotoxic effects of temozolomide in a resistant glioblastoma cell line. Mutat Res Genet Toxicol Environ Mutagen. 2015;793:19–29. doi:10.1016/j.mrgentox.2015.06.001.
- Ujifuku K, Mitsutake N, Takakura S, Matsuse M, Saenko V, Suzuki K, Hayashi K, Matsuo T, Kamada K, Nagata I, et al. miR-195, miR-455-3p and miR-10a(*) are implicated in acquired temozolomide resistance in glioblastoma multiforme cells. Cancer Lett. 2010;296(2):241–248. doi:10.1016/j.canlet.2010.04.013.
- Dai S, Yan Y, Xu Z, Zeng S, Qian L, Huo L, Li X, Sun L, Gong Z. SCD1 confers temozolomide resistance to human glioma cells via the Akt/GSK3β/β-catenin signaling axis. Front Pharmacol. 2017;8:960. doi:10.3389/fphar.2017.00960.
- Liu W, Jing ZT, Wu SX, He Y, Lin YT, Chen WN, Lin XJ, Lin X. A novel AKT activator, SC79, prevents acute hepatic failure induced by fas-mediated apoptosis of hepatocytes. Am J Pathol. 2018;188(5):1171–1182. doi:10.1016/j.ajpath.2018.01.013.
- Bikfalvi A, da Costa CA, Avril T, Barnier JV, Bauchet L, Brisson L, Cartron PF, Castel H, Chevet E, Chneiweiss H, et al. Challenges in glioblastoma research: focus on the tumor microenvironment. Trends Cancer. 2023;9(1):9–27. doi:10.1016/j.trecan.2022.09.005.
- Nam Y, Koo H, Yang Y, Shin S, Zhu Z, Kim D, Cho HJ, Mu Q, Choi SW, Sa JK, et al. Pharmacogenomic profiling reveals molecular features of chemotherapy resistance in IDH wild-type primary glioblastoma. Genome Med. 2023;15(1):16. doi:10.1186/s13073-023-01165-8.
- Tatari N, Khan S, Livingstone J, Zhai K, McKenna D, Ignatchenko V, Chokshi C, Gwynne WD, Singh M, Revill S, et al. The proteomic landscape of glioblastoma recurrence reveals novel and targetable immunoregulatory drivers. Acta Neuropathol. 2022;144(6):1127–1142. doi:10.1007/s00401-022-02506-4.
- Romeo G, Ciaffaglione V, Amata E, Dichiara M, Calabrese L, Vanella L, Sorrenti V, Grosso S, D’Amico AG, D’Agata V, et al. Combination of Heme Oxygenase-1 Inhibition and Sigma Receptor Modulation for Anticancer Activity. Molecules. 2021;26(13):3860. doi:10.3390/molecules26133860.
- Bausart M, Préat V, Malfanti A. Immunotherapy for glioblastoma: the promise of combination strategies. J Exp Clin Cancer Res. 2022;41(1):35. doi:10.1186/s13046-022-02251-2.