
ABSTRACT
Background
Chronic stress can induce stress-related hormones; norepinephrine (NE) is considered to have the highest potential in cancer. NE can stimulate the expression of hypoxia-inducible factor-1α (HIF-1α), which is associated with vascular endothelial growth factor (VEGF) secretion and tumor angiogenesis. However, the underlying mechanisms are poorly understood.
Methods
Tumor-bearing mice were subjected to chronic restraint stress and treated with normal saline, human monoclonal VEGF-A neutralizing antibody bevacizumab, or β-adrenergic receptor (β-AR) antagonist (propranolol). Tumor growth and vessel density were also evaluated. Human colorectal adenocarcinoma cells were treated with NE, propranolol, or the inhibitor of transforming growth factor-β (TGF-β) receptor Type I kinase (Ly2157299) in vitro. TGF-β1 in mouse serum and cell culture supernatants was quantified using ELISA. The expression of HIF-1α was measured using Real time-PCR and western blotting. Cell migration and invasion were tested.
Results
Chronic restraint stress attenuated the efficacy of bevacizumab and promoted tumor growth and angiogenesis in a colorectal tumor model. Propranolol blocked this effect and inhibited TGF-β1 elevation caused by chronic restraint stress or NE. NE upregulated HIF-1α expression, which was reversed by propranolol or Ly2157299. Propranolol and Ly2157199 blocked NE-stimulated cancer cell migration and invasion.
Conclusions
Our results demonstrate the effect of NE on tumor angiogenesis and the critical role of TGF-β1 signaling during this process. In addition, β-AR/TGF-β1 signaling/HIF-1α/VEGF is a potential signaling pathway. This study also indicates that psychosocial stress might be a risk factor which weakens the efficacy of anti-angiogenic therapy.
1. Introduction
Angiogenesis is one of the hallmarks of the occurrence and development of solid tumors, during which vascular endothelial growth factor A (VEGF-A) plays a critical role.Citation1 Bevacizumab, a human monoclonal VEGF-A neutralizing antibody, was the first anti-angiogenesis drug approved by the Food and Drug Administration (FDA) for metastatic colon cancer. Although bevacizumab provides a clinical benefit to a subset of patients, others do not respond or gradually develop resistance.Citation2 No clinical benefit has been shown in monotherapy with bevacizumab but has been demonstrated in combination with chemotherapy or immunotherapy.Citation3,Citation4 However, bevacizumab alone has shown promising antitumor activity in animal models.Citation5 The possible mechanisms underlying the differences between patients and animals warrant further exploration.
The majority of cancer patients suffer from enduring negative emotions, including anxiety and depression, and are in a state of chronic stress.Citation6,Citation7 Numerous epidemiological studies have identified chronic stress as a pivotal element in oncogenesis and progression.Citation8,Citation9 Stress-related hormones (SRHs), including epinephrine, norepinephrine, and glucocorticoids etc., are released in response to chronic stress, which mediates the hypothalamic-pituitary-adrenal axis and sympathetic nervous system.Citation10,Citation11 Notably, norepinephrine (NE) is regarded as the most influential tumor-related SRH and is widely used to simulate chronic stress.Citation12
Previous studies have reported that NE can act on various cancer cells, such as breast cancer, ovarian cancer, colon cancer, lung cancer, and prostatic cancer, upregulate VEGF, IL-6, IL-8, and matrix metalloproteinase, induce epithelial – mesenchymal transition (EMT), and further increase migration or invasion through β-adrenergic receptor (β-AR) activation. Propranolol, an antagonist of β-AR, can block the NE effect.Citation13–18 Chronic stress is known to facilitate the growth, progression, and metastasis of human tumor xenografts. Propranolol was also found to counteract the impacts of chronic stress.Citation14–16,Citation19 In our previous studies, we demonstrated that NE induced cancer cells to secrete VEGF, which in turn promoted tumor angiogenesis and further attenuated the anti-angiogenic efficacy of sunitinib, a small-molecule targeted drug.Citation15,Citation17 Theoretically, norepinephrine is stimulated in chronic stress, and VEGF is secreted, impairing the efficacy of bevacizumab. Chronic stress seems to be a potential promoter of tumor angiogenesis and may influence the effect of bevacizumab. However, this phenomenon has not been proven in monoclonal antibodies and the underlying mechanisms remain to be elucidated.
Glucocorticoids and catecholamines can stimulate the secretion of TGF-β1,Citation20,Citation21 which is an independent prognostic risk factor for cancer patients.Citation22,Citation23 HIF-1α, a downstream signaling molecule of TGF-β1, can be activated upon the interaction of TGF-β1 with its receptor I (TGF-βRI) and further mediates VEGF secretion.Citation24,Citation25 Inhibition of TGF-βRI has been shown to be associated with lower VEGF levels and less angiogenesis, invasion, and metastasis in hepatocellular carcinoma.Citation26,Citation27 The attenuated activity of TGF-β1 contributes to reduced tumor hypoxia.Citation28 Moreover, at the protein level, decreased expression of HIF-1α and downregulated VEGF levels occurred in parallel in a time-dependent manner.Citation29 Therefore, TGF-β signaling and HIF-1α may be two significant factors involved in chronic stress-related tumor angiogenesis.
This study aimed to verify the efficacy of bevacizumab in the treatment of chronic stress. Another purpose of this study was to explore the role of TGF-β1 and HIF-1α and investigate its potential mechanism.
2. Results
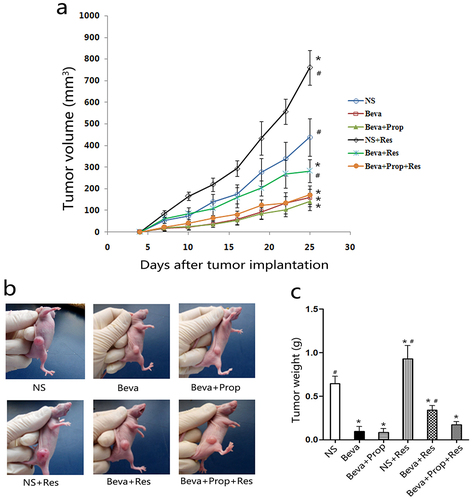
2.1. Chronic restraint stress promoted tumor growth and weakened the efficacy of bevacizumab
To confirm chronic stress could impair the efficacy of bevacizumab in vivo, we established tumor model and CRS model in mice to observe the changes of tumor, according to tumor volume and tumor weight. When tumors in the right flank were palpable, the mice were administered normal saline, bevacizumab, propranolol, or CRS for 21 days. As shown in , bevacizumab treatment (Beva) led to a flatter tumor volume curve (), smaller tumor volume (), and lighter tumor weight (), while chronic restraint (NS+Res) resulted in a larger volume and heavier weight compared to the control group (NS). Moreover, CRS accelerated the tumor growth rate in mice treated with Beva (Beva+Res) compared to the Beva group. This indicated that bevacizumab was effective in HT-29 cells in vivo and that the efficacy of bevacizumab was impaired by CRS. However, the tumor volume and tumor weight in the Beva+Prop+Res group were lower than those in the NS and Beva+Res groups. There were no significant differences between Beva, Beva+Prop, and Beva+Prop+Res. This means that bevacizumab was not affected by propranolol, and that propranolol could block the effect of CRS. This means that β-AR is involved in this process as well.
Figure 1. Chronic restraint stress promoted tumor growth and weakened the efficacy of bevacizumab.
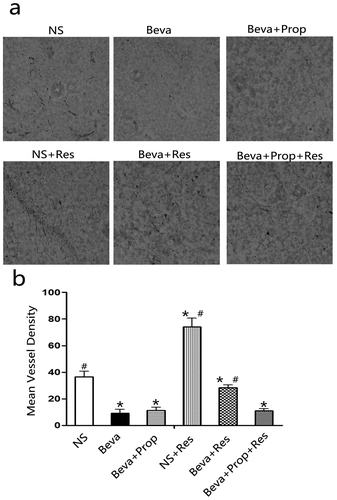
2.2. Chronic stress stimulated angiogenesis in HT-29 model and was blocked by propranolol
Bevacizumab is a humanized monoclonal antibody against VEGF-A, a key factor inducing the formation of blood vessels (angiogenesis) in tumors, and the number of vessels in tumor tissues could reflect the efficacy of bevacizumab to some degree. To test the hypothesis that CRS could stimulate angiogenesis in vivo and further impair the efficacy of bevacizumab, tumor tissues from the tumor-bearing mice were sliced into cryosections and stained with a murine endothelial cell marker (CD31) to quantify the mean vessel density (MVD) counts, which represent the number of blood vessels. shows the representative photographs of the cryosections in each group. As shown in , MVD counts significantly increased in the NS + Res group. Bevacizumab inhibited tumor angiogenesis because of the minimum MVD, but higher MVD counts were observed in the Beva+Res group. However, the MVD counts were lower in the Beva+Prop+Res group than in the Beve+Res group. No statistical significance was observed when comparing Beva, Beva+Prop, and Beva+Prop+Res. The results not only verified the efficacy of bevacizumab but also confirmed the role of CRS in angiogenesis. This also indicated that the effect of CRS is dependent on β-AR.
Figure 2. Propranolol inhibited chronic restraint stress-stimulated angiogenesis in vivo.
2.3. The effects of chronic restraint stress and norepinephrine on TGF-β1 secretion and TGF-βRI expression
As described in the Introduction section, TGF-β1 can be induced by catecholamines. To verify whether CRS or NE could stimulate TGF-β1 secretion, TGF-β1 levels in mouse serum and culture supernatant were measured by ELISA (). CRS significantly increased TGF-β1 levels in mouse serum, regardless of whether the mice were treated with bevacizumab (Beva+Res group) or not (NS+Res group). However, in the Beva+Prop+Res group, the TGF-β1 level remained at the baseline level. There was no statistical significance among the NS, Beva, Beva+Prop, and Beva+Prop+Res groups. This indicated that CRS increased TGF-β1 in vivo in a β-AR-dependent manner.
Figure 3. Chronic restraint stress and norepinephrine induced TGF-β1 secretion.
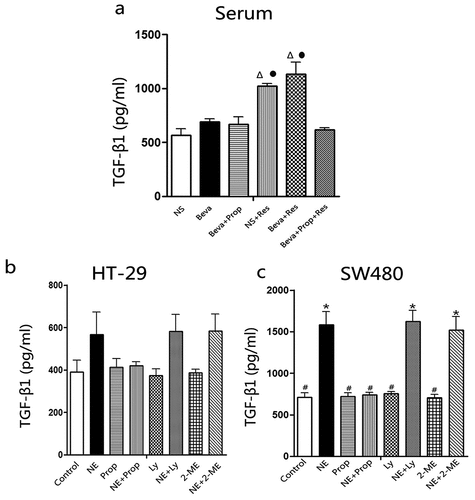
For the SW480 culture supernatant, TGF-β1 levels increased after NE-treatment for 48 h. Prop inhibited the NE effect, but Ly2157299 and 2-ME did not. TGF-β1 remained at the baseline level when the cells were only treated with Prop, Ly2157299, or 2-ME (). The TGF-β1 level in HT-29 cells did not significantly increase after NE-treatment for 48 h, but a trend was observed (). Interestingly, in another study, we found that TGF-β1 was significantly induced in HT-29 cells after NE-treatment for 72 h and Prop also played an inhibitive role.Citation30
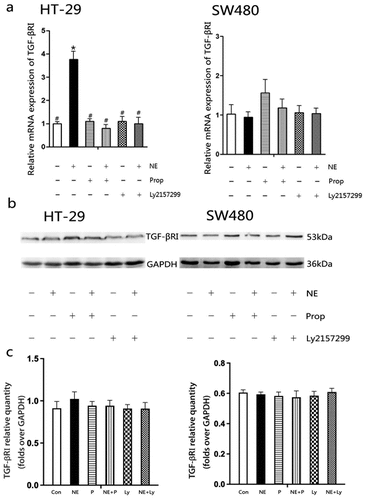
The mRNA and protein expression of TGF-βRI were further analyzed because of its potential positive correlation with angiogenesis, in order to observe the effect of NE on TGF-βRI (). After exposure to NE for 48 h, the mRNA expression of TGF-βRI was upregulated in HT-29 cells, while it remained at the baseline level in SW480 cells (). However, no significant difference was observed in HT-29 and SW480 cells at the protein level (). The quantification of proteins showed no statistically significant differences ().
Figure 4. The mRNA and protein expression of TGF-βRI in HT-29 and SW480 cells after norepinephrine treatment.
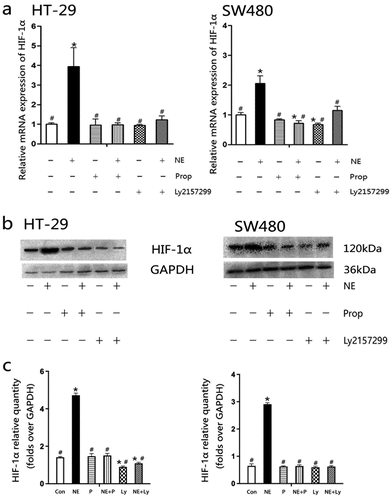
2.4. Norepinephrine promoted HIF-1α expression
HIF-1α is a downstream signal of TGF-β1 and plays a key role in VEGF secretion and tumor angiogenesis.Citation24,Citation25,Citation29 Therefore, measurement of HIF-1α after NE treatment is critical. NE stimulated HIF-1α transcription in HT-29 and SW480 cells (). Both the β-AR antagonist Prop and TGF-βRI inhibitor Ly2157299 were able to block the effect of NE. shows that NE increased HIF-1α protein expression, and protein quantification () showed similar results to mRNA. These results demonstrate that HIF-1α is a downstream signal of β-AR and TGF-β1. According to the results above, it also indicated that the β-AR/TGF-β1 signaling/HIF-1α might be a possible signaling pathway in this study.
Figure 5. Propranolol or Ly2157299 blocked norepinephrine-induced mRNA and protein expression of HIF-1α.
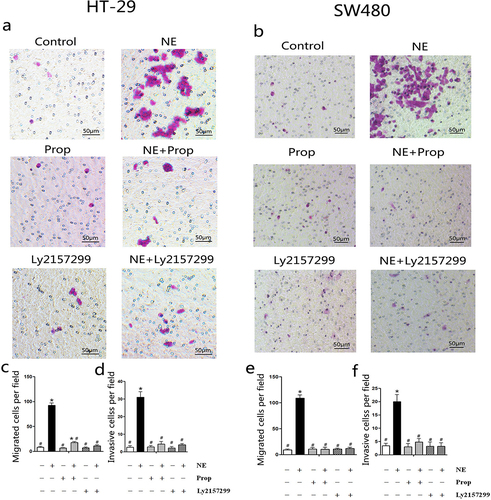
2.5. Prop and Ly2157299 blocked norepinephrine-induced cancer cell migration and invasion
NE and VEGF are associated with enhanced cancer cell migration and invasion.Citation15,Citation19,Citation31 Transwell migration and Matrigel invasion assays were used to observe the migration and invasion abilities of the cells after NE treatment, respectively. As shown in the representative photographs (), the number of migratory () and invasive cells () in the NE-treated group was greater than that in the other groups. The results showed that NE stimulated the migration and invasion of HT-29 and SW480 cells. We also observed that Prop and Ly2157299 inhibited this process. This indicated that NE induced cell migration and invasion through β-AR and TGF-β signaling.
Figure 6. Norepinephrine stimulated HT-29 and SW480 cells migration and invasion.
3. Discussion
Chronic stress is a well-known risk factor for immune and cardiovascular diseases.Citation32,Citation33 It also increases the risk of tumorigenesis and progression via a variety of mechanisms, such as decreasing splenocytes, interferon-γ, tumor-infiltrating lymphocytes, and cytotoxic T-lymphocytes,Citation34 attenuating P53 functionCitation35 and augmenting regulatory T cell (Treg) infiltration,Citation36 leading to impaired anti-tumor immunity. On the other hand, promoting tumor angiogenesis is another mechanism by which chronic stress is involved.Citation14,Citation37 Retrospective clinical analyses have revealed that beta-blockers, including propranolol, decrease mortality and recurrence of breast cancer, ovarian cancer, and prostatic cancer and prolong disease-free survival and overall survival.Citation38–41 We have reported that chronic stress-induced colorectal adenocarcinoma HT-29 cell EMT stimulated HT-29 cell migration and invasion, and weakened the anti-tumor effect of sunitinib in vivo.Citation15,Citation30 Unlike sunitinib, bevacizumab, a monoclonal antibody, was used in the present study. CRS exacerbated tumor load () and angiogenesis () in the HT-29 tumor model. Furthermore, propranolol blocks the effects of CRS. This indicated that CRS could impair the effect of bevacizumab via β-AR activation. Meanwhile, it might account for the unsatisfactory clinical results of single-agent bevacizumab in one aspect.
The CRS model is a classic and well-established framework for examining the effect of chronic stress on cancer in animal-based studies.Citation14,Citation15,Citation42 It has been demonstrated to elicit depression-like or anxiety-like behaviors in mice,Citation43,Citation44 and to increase serum NE and VEGF in mice.Citation15,Citation42 Adenylyl cyclase is activated after NE binding to β-AR, leading to enhanced synthesis of cAMP. This activates the cAMP-dependent dependent protein kinase A (PKA), thereby initiating series of downstream molecules.Citation13,Citation30,Citation45 NE is believed to promote TGF-β1 secretion.Citation20 Our results also showed that CRS and NE elevated TGF-β1 levels in the serum and cell supernatant, respectively. This augmentation of TGF-β1 levels was inhibited by propranolol but not by Ly2157299 or 2-ME (). TGF-β1 appears to be the downstream signaling pathway of β-AR, which can be induced through β-AR activation. Although there was no statistical difference in TGF-β1 levels between the NE-treated cells and the control at the time of 48 h in HT-29 cells, an increasing trend was discernible (). In another study, TGF-β1 levels were significantly increased by NE at 72 hCitation30. In addition, at the 48-hour time point, TGF-βRI transcription was upregulated in HT-29 cells treated with NE, but no difference was observed at the protein level (). Until the 96 h, TGF-βRI protein expression in HT-29 cells was significantly increased by NE.Citation30 In contrast to the observations in HT-29 cells, no similar phenomenon was detected in the SW480 cells. We considered that cell specificity contributed to the different cellular responses to NE treatment.
TGF-β1 is believed to be associated with poor prognosis in colorectal cancer and is a predictor of hepatic metastasis.Citation23 Evidences have shown that TGF-β1 weakens cancer immunotherapy,Citation46,Citation47 induces cancer cell EMTCitation30,Citation48,Citation49 and promotes VEGF secretion.Citation50–52 A clinical study reported that serum VEGF and TGF-β1 levels were positively correlated in cancer patients.Citation53 As mentioned previously, HIF-1α is the downstream signaling pathway of TGF-β1 and plays a critical role in the stimulation of VEGF secretion.Citation24,Citation25,Citation29 However, another study indicated that HIF-1α is an upstream signaling factor of TGF-β1.Citation54 According to the results, propranolol inhibited NE-induced TGF-β1 secretion, but Ly2157299 and 2-ME did not (). Even in the HT-29 culture supernatant, an increase in TGF-β1 level was observed in the NE, NE+Prop, and NE + 2-ME groups (). It is possible that HIF-1α is a downstream signaling pathway of TGF-β1 in the present study. As shown in , the mRNA and protein expression of HIF-1α was upregulated by NE treatment and was reversed by additional propranolol or Ly2157299 treatment. In another study, the β2-AR/HIF-1α/VEGF axis was found to regulate stress-induced tumor angiogenesis.Citation55 Therefore, we hypothesized that β-AR/TGF-β signaling/HIF-1α/VEGF might be a signaling pathway involved in angiogenesis, which is stimulated by chronic stress. This may also influence the effect of bevacizumab.
EMT is a key factor in tumor invasion and metastasis.Citation56,Citation57 However, VEGF is an important determinant of the increased oncogenesis of cells undergoing EMT.Citation58,Citation59 Inhibition of the VEGF signaling pathway leads to the suppression of EMT. This is attributed to the role of VEGF in enhancing the migratory and invasive properties of MCF-7 cells.Citation60 We found that NE could induce EMT in HT-29 cells and that β-AR/TGF-β1 signaling/p-Smad3/Snail and β-AR/TGF-β1 signaling/HIF-1α/Snail were two signaling pathways involved in this process.Citation30 As shown in , the migration and invasion of cells were enhanced after NE treatment. Therefore, we speculated that chronic stress could induce cancer cell EMT through several signaling pathways,Citation30,Citation48,Citation49 including the promotion of angiogenesis and the further stimulation of EMT. However, the detailed mechanism requires further investigation.
4. Conclusions
Our study demonstrated that chronic stress stimulated tumor growth and angiogenesis and weakened the efficacy of bevacizumab, but was reversed by the β-AR antagonist propranolol. To our knowledge, this is the first study to investigate the effects of chronic stress on monoclonal antibodies. Norepinephrine, the most significant tumor-related SRHs, acts on β-AR and induces TGF-β1 and HIF-1α expression, leading to VEGF secretion. Taken together, β-AR/TGF-β1 signaling/HIF-1α/VEGF is a potential signaling pathway involved.
5. Material and methods
5.1. Reagents
Norepinephrine, propranolol, and 2-methoxyestradiol (2-ME, an HIF-1α inhibitor) were procured from Sigma-Aldrich (USA) and Enzo (Germany), respectively. Ly2157299, a TGF-β receptor Type I (TGF-βRI) kinase inhibitor, was supplied by Selleck (USA). Bevacizumab was obtained from Roche (Basel, Switzerland). Rabbit monoclonal antibodies against human HIF-1α and TGF-βRI were obtained from Cell Signaling (USA), and CD31 (rat anti-mouse monoclonal antibody) was purchased from BD Pharmingen (USA). Boster (China) provided the rabbit anti-human GAPDH (Glyceraldehyde-3-phosphate dehydrogenase), along with HRP-conjugated secondary rabbit anti-rat and goat anti-rabbit. TRIzol and the One Step SYBR® PrimeScript™ RT-PCR Kit were acquired from Thermo Fisher (USA) and TaKaRa (Japan), respectively. Human and mouse TGF-β1 ELISA kits (Neobioscience Technology Co., Ltd., China), Matrigel (BD Biosciences, USA), and 24-well Transwell (8-μm pore size, 6.5-mm diameter, Corning Costar Corporation, USA) were used.
5.2. Cell culture and treatment
The human colorectal adenocarcinoma cell lines HT-29 and SW480, kindly provided by the State Key Laboratory of Biotherapy (Sichuan University, Chengdu, China), were cultivated in RPMI1640 complete medium (Hyclone) supplemented with 10% fetal bovine serum (Hyclone), penicillin (100 U/mL), and streptomycin (10 mg/L) at 37°C in a humidified atmosphere of 5% CO2.
For the experiments in vitro, cells were allowed to reach 80% confluence, underwent serum starvation overnight, and were treated for 48 h according to the following six groups:1) serum-free culture medium (control); 2) 10 μM norepinephrine (NE); 3) 10 μM propranolol (Prop); 4) 10 μM NE +10 μM Prop (NE+P); 5) 10 μM Ly2157299 (Ly); 6) 10 μM NE +10 μM Ly2157299 (NE+Ly). All reagents were prepared in serum-free culture media to prevent interference from serum components. Propranolol and Ly2157299 were added to the culture media one hour prior to the addition of norepinephrine. Each group contained three replicates.
5.3. Establishment of tumor model and chronic restraint stress model
Five- to 7-week-old female athymic BALB/c nude mice were obtained from Beijing HFK Bioscience Co., Ltd. (Beijing, China) and were granted approval by the Sichuan University Animal Care and Use Committee. All mice were housed in a germ-free environment with food and water ad libitum, a regular 12-hour day/night cycle, and stable temperature ranging from 21–25°C. A tumorigenic challenge was initiated by inoculating the mice with 2.5 × 106 HT-29 cells into the right flank. Upon the formation of palpable tumors, the mice were randomly distributed into six groups (five mice per group) and were administered the following treatments:1) NS, received normal saline 100 μL via intraperitoneal injection (i.p.) daily; 2) Beva, received normal saline 100 μL i.p. daily and bevacizumab i.p. twice a week; 3) Beva+Prop, received propranolol i.p. daily and bevacizumab i.p. twice a week; 4) NS+Res, received normal saline 100 μL i.p. daily and restraint stress daily; 5) Beva+Res, received normal saline 100 μL i.p. daily, restraint stress daily, and bevacizumab i.p. twice a week; and 6) Beva+Prop+Res, received propranolol i.p. daily, restraint stress daily, and bevacizumab i.p. twice a week. The dosages of bevacizumab and propranolol were 5 mg/kg/100 μL and 1 μmol/100 g/100 μL, respectively. The chronic restraint stress (CRS) model has been delineated in prior studies.Citation14,Citation15,Citation42 Briefly, mice were constrained horizontally in well ventilated 50 mL conical centrifuge tubes (Corning, USA) for 2 h daily for 21 consecutive days. Mice in the tubes were able to breathe freely but were unable to move.
A caliper was utilized to monitor the longest dimension (length) and shortest dimension (width) of each tumor every 3 days. Tumor volume was estimated using the formula: volume (mm3) = length × widthCitation2 ×0.52. Following 21 consecutive days of restraint, all mice were anesthetized using chloral hydrate and subsequently sacrificed. Serum samples were collected and stored at −80°C for ELISA. Tumors in the right flank were carefully separated, weighed, immediately embedded in OCT compound (Sakura Finetek USA, USA), and conserved at −80°C for immunohistochemistry.
5.4. Immunohistochemistry for CD31
To evaluate vessel densities in tumor tissues, frozen tumors were sliced into 5 μm at −20°C and then stained with monoclonal rat anti-mouse CD31, a marker for murine endothelial cell. Briefly, frozen sections were fixed in cold acetone for 20 min, followed by three washed in phosphate-buffered saline (PBS). After incubation with 3% H2O2 for 10 min and washing with PBS, sections were incubated with CD31 (1:400) at 4°C overnight. The next day, the sections underwent a final PBS wash, followed by a 2-hour incubation with a rabbit anti-rat antibody at a temperature of 37°C. The sections were examined using a microscope. The MVD of each sample was calculated by determining the average count of immunoreactive endothelial cells in five random fields (200×).
5.5. Assessment of TGF-β1
HT-29 and SW480 cells were treated for 48 h according to the following eight groups:1) control, 2) 10 μM NE; 3) 10 μM Prop; 4) 10 μM NE +10 μM Prop, 5) 10 μM Ly; 6) 10 μM NE +10 μM Ly, 7) 10 μM 2-ME; 8) 10 μM NE +10 μM 2-ME. Following the treatment, the supernatant from the cell cultures was harvested for ELISA analysis. The mouse serum described above was stored at −80°C for ELISA. Adhering to the guidelines provided by the manufacturer, the levels of TGF-β1 in the mouse serum and culture supernatant were detected using mouse and human TGF-β1 ELISA kits, respectively. The luminescence plate reader (PerkinElmer, USA) was used to read the plates with a wavelength setting of 450 nm. The concentration of each sample was calculated by using a standard curve. Each sample was tested in triplicates.
5.6. Cell migration and invasion assay
To explore the impact of NE on the migration and invasive capabilities of cancer cells, the Transwell migration assay and the Matrigel invasion assay were performed as described in previous studies.Citation15 HT-29 and SW480 cells, which had been deprived of serum, were suspended in serum-free medium at a density of 2.5 × 106/mL. To serve as a chemoattractant, 500 μL of medium containing 10% fetal bovine serum was added to each well of lower chamber. A volume of 100 μL cell suspension (HT-29 or SW480, total number of each well was 2.5 × 105) was transferred to the upper wells. Subsequently, the cells were subjected to treatment with NE, Prop, or Ly2157299, in accordance with the six groups outlined for cell culture and treatment protocols, and were subsequently incubated for 12 hours at 37°C
Cotton swabs were used to remove cells from the upper surface of the separating membranes. After immersion in 4% paraformaldehyde for 30 min, the separated membranes were stained with 0.1% crystal violet (Beyotime, China) for 20 min. The migratory cells remained on the lower surface of the separating membranes and were stained purple. Cell migration was evaluated by averaging the total number of migratory cells from five randomly selected views under a microscope with a 200× magnification.
The Matrigel invasion assay followed a similar procedure to the Transwell migration assay. The upper chambers of the transwell were coated with matrigel (100 μL of 1.0 mg/mL) beforehand. Medium (500 μL) supplemented with 10% fetal bovine serum was added to each lower chamber. Serum-starved 5 × 105/100 μL HT-29 or SW480 cells were seeded in the upper chambers, exposed to treatments with NE, Prop, or Ly2157299 according to the groups, and incubated for 24 h at 37°C. The remaining procedure was the same as that used in the Transwell migration assay.
5.7. Real-time quantitative PCR (RT-PCR)
Following the RNA extraction protocols, total RNA from HT-29 and SW480 cells in the six groups described in Cell culture and treatment was isolated using TRIzol reagent (15596018, Thermo Fisher). RT-PCR was conducted using the One Step SYBR® PrimeScript™ RT-PCR Kit and the CFX 96™ Real-Time System in a C1000™ Thermal Cycler (Bio-Rad, USA). The sequences of the primers used in this study were as follows:
TGF-βRI sense 5′-GCTGTGAAGCCTTGAGAGTAATGG-3′,
anti-sense 5′-TTCCTGTTGACTGAGTTGCGATAA-3′;
Accession:NM_001130916.
HIF-1α sense 5′-ACTGCACAGGCCACATTCACG-3′,
anti-sense 5′-GGTTCACAAATCAGCACCAAGC-3′;
Accession:NM_001243084.
GAPDH sense 5-CTTTGGTATCGTGGAAGGACTC-3′,
anti-sense 5′-GTAGAGGCAGGGATGATGTTCT-3′.
Accession:NM_001256799.
GAPDH was used as an internal positive control. RT-PCR was executed according to the manufacturer’s guidelines. Each sample was processed at least in triplicate, and the relative mRNA expression levels of TGF-β1 and HIF-1α were calculated using the following formula: relative expression = 2−(ΔCt), ΔCt= (Ct gene of interest – Ct GAPDH).
5.8. Western blot analysis
Proteins of the cells in the six groups described in the “Cell culture and treatment” section were extracted for western blot analysis. Briefly, cells were washed and collected by gentle scraping with cold PBS. The suspension was centrifuged and the precipitate was resuspended in RIPA buffer (150 mM NaCl, 0.1% SDS, 1 mM sodium orthovanadate, 1% TritonX-100, 0.5% sodium deoxycholate, 50 mM Tris-base, 10 mM sodium fluoride, 1% phosphatase inhibitor cocktail, and 1% protease inhibitor cocktail). The mixture was incubated on ice for 30 min, followed by a centrifugation step to remove the debris. Approximate 10–30 μg of proteins were resuspended in SDS-PAGE loading buffer, separated on polyacrylamide gel and electro-transferred into nitrocellulose membranes (Millipore, USA). The membranes were blocked with 5% nonfat milk for 1 h at room temperature and incubated with rabbit monoclonal antibodies against human HIF-1α (1:1000) or TGF-βRI (1:1000) at 4°C overnight. After extensive washing with TBST (Tris Buffer saline containing 0.1% Tween-20 (TBST) to remove unbound primary antibodies, the membranes were incubated with HRP-conjugated secondary goat anti-rabbit antibody, diluted to 1:3000, for a period of 2 hours at room temperature. The membranes were washed again with TBST, and the immunoreactive bands were visualized using Chemiluminescent HRP Substrate (Millipore, USA) and analyzed using Image-Pro Plus 6.0.
5.9. Statistical analysis
The data are presented as mean ± standard deviation (SD). All statistical analyses were performed using SPSS version 23.0. One-way analysis of variance (ANOVA) and repeated-measures ANOVA were used to analyze the differences between groups. A two-sided p < .05 was considered statistically significant.
Author contributions
Professor Yu Jiang and Jie Zhang designed this study; Jie Zhang and Yao-tiao Deng carried out the experiments and drafted the manuscript; Jie Zhang and Jie Liu collected and analyzed the data; Lu Gan and Yu Jiang revised the manuscript. All authors have reviewed and approved the final manuscript.
Ethical approval
This article does not include any studies with human participants performed by any of the authors.
This study has been approved by the Sichuan University Animal Care and Use Committee.
All applicable international, national, and Sichuan University guidelines for the care and use of animals were followed in this study.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data supporting the findings of this study are available from the corresponding author, JY, upon reasonable request.
The primer sequences used in this study were obtained from the National Center for Biotechnology Information (NCBI): https://www.ncbi.nlm.nih.gov/guide/dna-rna/.
Additional information
Funding
References
- Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473(7347):298–12. doi:10.1038/nature10144.
- Lambrechts D, Lenz HJ, de Haas S, Carmeliet P, Scherer SJ. Markers of response for the antiangiogenic agent bevacizumab. J Clin Oncol. 2013;31(9):1219–30. doi:10.1200/JCO.2012.46.2762.
- Recondo G Jr., Diaz-Canton E, de la Vega M, Greco M, Recondo G Sr., Valsecchi ME. Advances and new perspectives in the treatment of metastatic colon cancer. World J Gastrointest Oncol. 2014;6(7):211–24. doi:10.4251/wjgo.v6.i7.211.
- Garcia J, Hurwitz HI, Sandler AB, Miles D, Coleman RL, Deurloo R, Chinot OL. Bevacizumab (Avastin(r)) in cancer treatment: A review of 15 years of clinical experience and future outlook. Cancer Treat Rev. 2020;86:102017. doi:10.1016/j.ctrv.2020.102017.
- Chen N, Ren M, Li R, Deng X, Li Y, Yan K, Xiao L, Yang Y, Wang L, Luo M. et al. Bevacizumab promotes venous thromboembolism through the induction of PAI-1 in a mouse xenograft model of human lung carcinoma. Mol Cancer. 2015;14(1):140. doi:10.1186/s12943-015-0418-x.
- Linden W, Vodermaier A, Mackenzie R, Greig D. Anxiety and depression after cancer diagnosis: prevalence rates by cancer type, gender, and age. J Affect Disord. 2012;141(2–3):343–51. doi:10.1016/j.jad.2012.03.025.
- Deng YT, Zhong WN, Jiang Y. Measurement of distress and its alteration during treatment in patients with nasopharyngeal carcinoma. Head Neck. 2014;36(8):1077–86. doi:10.1002/hed.23412.
- Pinquart M, Duberstein PR. Depression and cancer mortality: a meta-analysis. Psychol Med. 2010;40(11):1797–810. doi:10.1017/S0033291709992285.
- Antoni MH. Psychosocial intervention effects on adaptation, disease course and biobehavioral processes in cancer. Brain Behav Immun. 2013;30:S88–98. doi:10.1016/j.bbi.2012.05.009.
- Spiegel D. Mind matters in cancer survival. Psychooncology. 2012;21(6):588–93. doi:10.1002/pon.3067.
- Thornton LM, Andersen BL, Blakely WP. The pain, depression, and fatigue symptom cluster in advanced breast cancer: Covariation with the hypothalamic–pituitary–adrenal axis and the sympathetic nervous system. Health Psychol. 2010;29(3):333–37. doi:10.1037/a0018836.
- Yang EV, Kim SJ, Donovan EL, Chen M, Gross AC, Webster Marketon JI, Barsky SH, Glaser R. Norepinephrine upregulates VEGF, IL-8, and IL-6 expression in human melanoma tumor cell lines: implications for stress-related enhancement of tumor progression. Brain Behav Immun. 2009;23(2):267–75. doi:10.1016/j.bbi.2008.10.005.
- Sloan EK, Priceman SJ, Cox BF, Yu S, Pimentel MA, Tangkanangnukul V, Arevalo JM, Morizono K, Karanikolas BD, Wu L. et al. The sympathetic nervous system induces a metastatic switch in primary breast cancer. Cancer Res. 2010;70(18):7042–52. doi:10.1158/0008-5472.CAN-10-0522.
- Thaker PH, Han LY, Kamat AA, Arevalo JM, Takahashi R, Lu C, Jennings NB, Armaiz-Pena G, Bankson JA, Ravoori M. et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat Med. 2006;12(8):939–44. doi:10.1038/nm1447.
- Liu J, Deng GH, Zhang J, Wang Y, Xia XY, Luo XM, Deng YT, He SS, Mao YY, Peng XC. et al. The effect of chronic stress on anti-angiogenesis of sunitinib in colorectal cancer models. Psychoneuroendocrinology. 2015;52:130–42. doi:10.1016/j.psyneuen.2014.11.008.
- Cheng Y, Gao XH, Li XJ, Cao QH, Zhao DD, Zhou JR, Wu HX, Wang Y, You LJ, Yang HB. et al. Depression promotes prostate cancer invasion and metastasis via a sympathetic-cAMP-FAK signaling pathway. Oncogene. 2018;37(22):2953–66. doi:10.1038/s41388-018-0177-4.
- Deng GH, Liu J, Zhang J, Wang Y, Peng XC, Wei YQ, Jiang Y. Exogenous norepinephrine attenuates the efficacy of sunitinib in a mouse cancer model. J Exp Clin Cancer Res. 2014;33(1):21. doi:10.1186/1756-9966-33-21.
- Yang EV, Sood AK, Chen M, Li Y, Eubank TD, Marsh CB, Jewell S, Flavahan NA, Morrison C, Yeh PE. et al. Norepinephrine up-regulates the expression of vascular endothelial growth factor, matrix metalloproteinase (MMP)-2, and MMP-9 in nasopharyngeal carcinoma tumor cells. Cancer Res. 2006;66(21):10357–64. doi:10.1158/0008-5472.CAN-06-2496.
- Krizanova O, Babula P, Pacak K. Stress, catecholaminergic system and cancer. Stress. 2016;19(4):419–28. doi:10.1080/10253890.2016.1203415.
- Elenkov IJ, Chrousos GP. Stress hormones, proinflammatory and antiinflammatory cytokines, and autoimmunity. Ann N Y Acad Sci. 2002;966(1):290–303. doi:10.1111/j.1749-6632.2002.tb04229.x.
- Sigala B, McKee C, Soeda J, Pazienza V, Morgan M, Lin CI, Selden C, Vander Borght S, Mazzoccoli G, Roskams T. et al. Sympathetic nervous system catecholamines and neuropeptide Y neurotransmitters are upregulated in human NAFLD and modulate the fibrogenic function of hepatic stellate cells. PLOS ONE. 2013;8(9):e72928. doi:10.1371/journal.pone.0072928.
- Calon A, Espinet E, Palomo-Ponce S, Tauriello DV, Iglesias M, Cespedes MV, Sevillano M, Nadal C, Jung P, Zhang XH. et al. Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis Initiation. Cancer Cell. 2012;22(5):571–84. doi:10.1016/j.ccr.2012.08.013.
- Tsushima H, Ito N, Tamura S, Matsuda Y, Inada M, Yabuuchi I, Imai Y, Nagashima R, Misawa H, Takeda H. et al. Circulating transforming growth factor beta 1 as a predictor of liver metastasis after resection in colorectal cancer. Clin Cancer Res. 2001;7(5):1258–62.
- Shi X, Guo LW, Seedial SM, Si Y, Wang B, Takayama T, Suwanabol PA, Ghosh S, DiRenzo D, Liu B. et al. TGF-β/Smad3 inhibit vascular smooth muscle cell apoptosis through an autocrine signaling mechanism involving VEGF-A. Cell Death Disease. 2014;5(7):e1317. doi:10.1038/cddis.2014.282.
- Wu Y, Lucia K, Lange M, Kuhlen D, Stalla GK, Renner U. Hypoxia inducible factor-1 is involved in growth factor, glucocorticoid and hypoxia mediated regulation of vascular endothelial growth factor-A in human meningiomas. J Neurooncol. 2014;119(2):263–73. doi:10.1007/s11060-014-1503-5.
- Mazzocca A, Fransvea E, Lavezzari G, Antonaci S, Giannelli G. Inhibition of transforming growth factor β receptor I kinase blocks hepatocellular carcinoma growth through neo-angiogenesis regulation. Hepatology. 2009;50(4):1140–51. doi:10.1002/hep.23118.
- Fransvea E, Angelotti U, Antonaci S, Giannelli G. Blocking transforming growth factor-beta up-regulates E-cadherin and reduces migration and invasion of hepatocellular carcinoma cells. Hepatology. 2008;47(5):1557–66. doi:10.1002/hep.22201.
- Varadan V, Kamalakaran S, Gilmore H, Banerjee N, Janevski A, Miskimen KL, Williams N, Basavanhalli A, Madabhushi A, Lezon-Geyda K. et al. Brief-exposure to preoperative bevacizumab reveals a TGF-β signature predictive of response in HER2-negative breast cancers. Int J Cancer. 2016;138(3):747–57. doi:10.1002/ijc.29808.
- Wang Z, Zhou J, Fan J, Tan CJ, Qiu SJ, Yu Y, Huang XW, Tang ZY. Sirolimus inhibits the growth and metastatic progression of hepatocellular carcinoma. J Cancer Res Clin Oncol. 2009;135(5):715–22. doi:10.1007/s00432-008-0506-z.
- Zhang J, Deng YT, Liu J, Wang YQ, Yi TW, Huang BY, He SS, Zheng B, Jiang Y. Norepinephrine induced epithelial–mesenchymal transition in HT-29 and A549 cells in vitro. J Cancer Res Clin Oncol. 2016;142(2):423–35. doi:10.1007/s00432-015-2044-9.
- Barrett CS, Millena AC, Khan SA. TGF-β effects on prostate cancer cell migration and invasion require FosB. Prostate. 2017;77(1):72–81. doi:10.1002/pros.23250.
- Gu HF, Tang CK, Yang YZ. Psychological stress, immune response, and atherosclerosis. Atherosclerosis. 2012;223(1):69–77. doi:10.1016/j.atherosclerosis.2012.01.021.
- Jeon SW, Kim YK. Neuroinflammation and cytokine abnormality in major depression: Cause or consequence in that illness? World J Psychiatry. 2016;6(3):283–93. doi:10.5498/wjp.v6.i3.283.
- Dhabhar FS, Saul AN, Holmes TH, Daugherty C, Neri E, Tillie JM, Kusewitt D, Oberyszyn TM, Rameshwar P. High-anxious individuals show increased chronic stress burden, decreased protective immunity, and increased cancer progression in a mouse model of squamous cell carcinoma. PLOS ONE. 2012;7(4):e33069. doi:10.1371/journal.pone.0033069.
- Feng Z, Liu L, Zhang C, Zheng T, Wang J, Lin M, Zhao Y, Wang X, Levine AJ, Hu W. Chronic restraint stress attenuates p53 function and promotes tumorigenesis. Proc Natl Acad Sci USA. 2012;109(18):7013–18. doi:10.1073/pnas.1203930109.
- Kim HR, Moon S, Lee HK, Kang JL, Oh S, Seoh JY. Immune dysregulation in chronic stress: a quantitative and functional assessment of regulatory T cells. Neuroimmunomodulation. 2012;19(3):187–94. doi:10.1159/000331586.
- Wu X, Liu BJ, Ji S, Wu JF, Xu CQ, Du YJ, You XF, Li B, Le JJ, Xu HL. et al. Social defeat stress promotes tumor growth and angiogenesis by upregulating vascular endothelial growth factor/extracellular signal-regulated kinase/matrix metalloproteinase signaling in a mouse model of lung carcinoma. Mol Med Rep. 2015;12(1):1405–12. doi:10.3892/mmr.2015.3559.
- Barron TI, Connolly RM, Sharp L, Bennett K, Visvanathan K. Beta blockers and breast cancer mortality: a population- based study. J Clin Oncol. 2011;29(19):2635–44. doi:10.1200/JCO.2010.33.5422.
- Powe DG, Voss MJ, Zanker KS, Habashy HO, Green AR, Ellis IO, Entschladen F. Beta-blocker drug therapy reduces secondary cancer formation in breast cancer and improves cancer specific survival. Oncotarget. 2010;1(7):628–38. doi:10.18632/oncotarget.197.
- Watkins JL, Thaker PH, Nick AM, Ramondetta LM, Kumar S, Urbauer DL, Matsuo K, Squires KC, Coleman RL, Lutgendorf SK. et al. Clinical impact of selective and nonselective beta-blockers on survival in patients with ovarian cancer. Cancer. 2015;121(19):3444–51. doi:10.1002/cncr.29392.
- Lu H, Liu X, Guo F, Tan S, Wang G, Liu H, Wang J, He X, Mo Y, Shi B. Impact of beta-blockers on prostate cancer mortality: a meta-analysis of 16,825 patients. Onco Targets Ther. 2015;8:985–90. doi:10.2147/OTT.S78836.
- Lin Q, Wang F, Yang R, Zheng X, Gao H, Zhang P, Zhou Y. Effect of chronic restraint stress on human colorectal carcinoma growth in mice. PLOS ONE. 2013;8(4):e61435. doi:10.1371/journal.pone.0061435.
- Crema L, Schlabitz M, Tagliari B, Cunha A, Simao F, Krolow R, Pettenuzzo L, Salbego C, Vendite D, Wyse AT. et al. Na+, K+ ATPase activity is reduced in amygdala of rats with chronic stress-induced anxiety-like behavior. Neurochem Res. 2010;35(11):1787–95. doi:10.1007/s11064-010-0245-9.
- Christiansen SH, Olesen MV, Wortwein G, Woldbye DP. Fluoxetine reverts chronic restraint stress-induced depression-like behaviour and increases neuropeptide Y and galanin expression in mice. Behav Brain Res. 2011;216(2):585–91. doi:10.1016/j.bbr.2010.08.044.
- Yang R, Lin Q, Gao HB, Zhang P. Stress-related hormone norepinephrine induces interleukin-6 expression in GES-1 cells. Braz J Med Biol Res. 2014;47(2):101–09. doi:10.1590/1414-431X20133346.
- Wu M, Chen X, Lou J, Zhang S, Zhang X, Huang L, Sun R, Huang P, Wang F, Pan S. TGF-β1 contributes to CD8+ Treg induction through p38 MAPK signaling in ovarian cancer microenvironment. Oncotarget. 2016;7(28):44534–44. doi:10.18632/oncotarget.10003.
- Rouce RH, Shaim H, Sekine T, Weber G, Ballard B, Ku S, Barese C, Murali V, Wu MF, Liu H. et al. The TGF-β/SMAD pathway is an important mechanism for NK cell immune evasion in childhood B-acute lymphoblastic leukemia. Leukemia. 2016;30(4):800–11. doi:10.1038/leu.2015.327.
- Choi MJ, Cho KH, Lee S, Bae YJ, Jeong KJ, Rha SY, Choi EJ, Park JH, Kim JM, Lee JS. et al. hTERT mediates norepinephrine-induced Slug expression and ovarian cancer aggressiveness. Oncogene. 2015;34(26):3402–12. doi:10.1038/onc.2014.270.
- Shan T, Cui X, Li W, Lin W, Li Y, Chen X, Wu T. Novel regulatory program for norepinephrine-induced epithelial–mesenchymal transition in gastric adenocarcinoma cell lines. Cancer Sci. 2014;105(7):847–56. doi:10.1111/cas.12438.
- Gupta DK, Singh N, Sahu DK. TGF-beta Mediated Crosstalk Between Malignant Hepatocyte and Tumor Microenvironment in Hepatocellular Carcinoma. Cancer Growth Metastasis. 2014;7:1–8. doi:10.4137/CGM.S14205.
- Xie Q, Xie J, Zhong J, Cun X, Lin S, Lin Y, Cai X. Hypoxia enhances angiogenesis in an adipose-derived stromal cell/endothelial cell co-culture 3D gel model. Cell Prolif. 2016;49(2):236–45. doi:10.1111/cpr.12244.
- Li GC, Zhang HW, Zhao QC, Sun LI, Yang JJ, Hong L, Feng F, Cai L. Mesenchymal stem cells promote tumor angiogenesis via the action of transforming growth factor β1. Oncol Lett. 2016;11(2):1089–94. doi:10.3892/ol.2015.3997.
- Liu SG, Yuan SH, Wu HY, Liu J, Huang CS. The clinical research of serum VEGF, TGF-β1, and endostatin in non-small cell lung cancer. Cell Biochem Biophys. 2015;72(1):165–69. doi:10.1007/s12013-014-0431-5.
- Tang L, Pei H, Yang Y, Wang X, Wang T, Gao E, Li D, Yang Y, Yang D. The inhibition of calpains ameliorates vascular restenosis through MMP2/TGF-β1 pathway. Sci Rep. 2016;6(1):29975. doi:10.1038/srep29975.
- Shan T, Ma J, Ma Q, Guo K, Guo J, Li X, Li W, Liu J, Huang C, Wang F. et al. β2-AR-HIF-1α: A novel regulatory axis for stress-induced pancreatic tumor growth and angiogenesis. Curr Mol Med. 2013;13(6):1023–34. doi:10.2174/15665240113139990055.
- Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–90. doi:10.1016/j.cell.2009.11.007.
- Shenoy AK, Jin Y, Luo H, Tang M, Pampo C, Shao R, Siemann DW, Wu L, Heldermon CD, Law BK. et al. Epithelial-to-mesenchymal transition confers pericyte properties on cancer cells. J Clin Invest. 2016;126(11):4174–86. doi:10.1172/JCI86623.
- Fantozzi A, Gruber DC, Pisarsky L, Heck C, Kunita A, Yilmaz M, Meyer-Schaller N, Cornille K, Hopfer U, Bentires-Alj M. et al. VEGF-mediated angiogenesis links EMT-induced cancer stemness to tumor initiation. Cancer Res. 2014;74(5):1566–75. doi:10.1158/0008-5472.CAN-13-1641.
- Kong D, Zhou H, Neelakantan D, Hughes CJ, Hsu JY, Srinivasan RR, Lewis MT, Ford HL. VEGF-C mediates tumor growth and metastasis through promoting EMT-epithelial breast cancer cell crosstalk. Oncogene. 2021;40(5):964–79. doi:10.1038/s41388-020-01539-x.
- Shilpa P, Kaveri K, Salimath BP. Anti-metastatic action of anacardic acid targets VEGF-induced signalling pathways in epithelial to mesenchymal transition. Drug Discov Ther. 2015;9(1):53–65. doi:10.5582/ddt.2014.01042.