
ABSTRACT
Thioredoxin Reductase (TrxR) functions to recycle thioredoxin (Trx) during hydroperoxide metabolism mediated by peroxiredoxins and is currently being targeted using the FDA-approved anti-rheumatic drug, auranofin (AF), to selectively sensitize cancer cells to therapy. AF treatment decreased TrxR activity and clonogenic survival in small cell lung cancer (SCLC) cell lines (DMS273 and DMS53) as well as the H727 atypical lung carcinoid cell line. AF treatment also significantly sensitized DMS273 and H727 cell lines in vitro to sorafenib, an FDA-approved multi-kinase inhibitor that depleted intracellular glutathione (GSH). The pharmacokinetic, pharmacodynamic, and safety profile of AF was examined in nude mice with DMS273 xenografts administered AF intraperitoneally at 2 mg/kg or 4 mg/kg (IP) once (QD) or twice daily (BID) for 1−5 d. Plasma levels of AF were 10–20 μM (determined by mass spectrometry of gold), and the optimal inhibition of TrxR activity was obtained at 4 mg/kg once daily, with no effect on glutathione peroxidase 1 activity. This AF treatment extended for 14 d, inhibited TrxR (>75%), and resulted in a significant prolongation of median overall survival from 19 to 23 d (p = .04, N = 30 controls, 28 AF). In this experiment, there were no observed changes in animal bodyweight, complete blood counts (CBCs), bone marrow toxicity, blood urea nitrogen, or creatinine. These results support the hypothesis that AF effectively inhibits TrxR both in vitro and in vivo in SCLC, sensitizes NETs and SCLC to sorafenib, and could be repurposed as an adjuvant therapy with targeted agents that induce disruptions in thiol metabolism.
Introduction
Pulmonary neuroendocrine tumors (NET) and carcinomas (NEC) stem from neuroendocrine cells, which produce hormones, and are defined by four histological variants, namely typical carcinoid (TC), atypical carcinoid (AC), large cell neuroendocrine carcinoma (LCNEC), and small cell lung carcinoma (SCLC). These tumors represent a group of heterogeneous neoplasms that are classified according to their degree of differentiation, mitosis, and degree of necrosis.Citation1 Clinically, TC are low-grade malignant tumors, AC are intermediate-grade malignant tumors, and SCLC/LCNEC are high-grade malignant full-blown carcinomas with no significant differences in survival between them.Citation1 For lung NETs, the 5-y survival is 35% for patients presenting with distant metastases, which is the initial presentation in 35% of patients. The survival rate drops to 4% in poorly differentiated NETs.Citation2 The average survival time for patients with lung NEC (including SCLC) is 10 months, with a 2-y survival of only 6%.Citation3,Citation4 Therefore, improved treatment options are of utmost importance for pulmonary NETs and NECs.
SCLC is characterized by loss of RB1 and TP53 accompanied by neuroendocrine differentiation; however; recently, distinct subtypes of SCLC, defined by patterns of transcription factor activation, have been defined and shown to have unique therapeutic vulnerabilities.Citation5,Citation6 It has been demonstrated that the non-neuroendocrine subtype (defined by the expression of RE-1 silencing transcription factor and vimentin) is highly susceptible to glutathione-dependent lipid oxidative cell death while the more classic neuroendocrine (defined by the expression of ASCL1 and/or NEUROD1) is reliant on the thioredoxin pathway for cellular health.Citation6 However, SCLCs have extensive intertumoral heterogeneity as well as subtype plasticityCitation7 leading to difficulty in treatment and development of therapy resistance. Logically, using a combination treatment that targets both classic neuroendocrine transcription subsets will presumably result in a more complete therapeutic response.
A hallmark of most cancers is the increased steady-state levels of reactive oxygen species (ROS) often stemming from one electron reductions of O2 in mitochondrial electron transport chains to form superoxide radical (O2·-) which is rapidly converted to H2O2.Citation8 Cancer cells adapt to increased levels of ROS by upregulating endogenous antioxidant systems, including the scavenging enzymes in the glutathione-dependent and thioredoxin-dependent hydroperoxide metabolic pathways. The glutathione and thioredoxin systems play key roles in the overall cellular oxidation state because of their intracellular dithiol recycling reactions in hydroperoxide metabolism and redox signaling.Citation9 In particular, the thioredoxin system consists of the redox-active proteins thioredoxin (Trx) and the NADPH electron acceptor thioredoxin reductase (TrxR) which primarily function to reduce peroxiredoxin (Prx) during the catalytic reduction of hydroperoxides. TrxR has three known mammalian isoforms that can be found in the cytosol, mitochondria, and one specific to spermatozoa.Citation10 According to the Project Score database, knock-out of TrxR has shown an anti-tumoral effect in 21% of 324 cell lines subjected to CRISPR-Cas9 screensCitation11 clearly indicating TrxR as a potential target for cancer treatment. In addition, Bebber et al. found that the overall survival of SCLC patients with low TrxR expression was significantly better than patients with tumors with high TrxR expression.Citation6 These data provide the rationale for targeting both Trx and glutathione pathways in SCLC.
Sorafenib is a kinase inhibitor that has activity against many protein kinases including vascular endothelial growth factor, platelet-derived growth factor receptor, and Raf kinase as well as being currently approved for the treatment of advanced renal cell carcinoma, hepatocellular carcinoma, and thyroid cancer.Citation12 Sorafenib is known to inhibit the cystine/glutamate antiporter SLC7A11 (commonly known as xCT) which functions to import cystine for glutathione biosynthesis.Citation13 It has been previously demonstrated that sorafenib treatment results in a decrease in glutathione, iron-dependent lipid oxidative cell death, and cisplatin sensitivity of cancer cells.Citation14,Citation15
Auranofin (AF), a gold phosphine complex, was developed as a rheumatic drug as it was originally thought to exert its effects by inhibiting humoral immunity. The most common acute adverse reactions associated with AF treatment were gastrointestinal with loose stools or diarrhea occurring in 39% of patients. Rashes and pruritis were also common, occurring in >10% of patients.Citation16–18 In addition, chronic use of gold complexes caused thrombocytopenia in 0.7% of patients.Citation19 Although diarrhea and stomach upset occurred in the initial month of treatment, other adverse events typically occurred only after prolonged therapy.Citation20 Clinical AF use has declined due to the availability of more effective and well-tolerated biological treatments for rheumatoid arthritis.
However, AF was also discovered to irreversibly inhibit both cytosolic and mitochondrial TrxR.Citation21,Citation22 This discovery has led to the active investigation of repurposing AF as an anticancer agent. Currently, four clinical trials using AF are underway for chronic lymphocytic leukemia (NCT01419691, NCT01747798), ovarian cancer (NCT03456700), and recurrent lung cancer (NCT01737502). We have previously shown simultaneous disruption of glutathione and thioredoxin metabolism enhanced non-small cell lung cancer cell killing and sensitivity to chemotherapy.Citation23 In addition, we demonstrated that treatment of breast cancer cells with AF before external beam radiation decreased the number of invading metastatic breast cancer cells.Citation24 We also demonstrated that suppressing TrxR with AF can sensitize breast cancer stem cells to ROS-induced stem cell transitions associated with epithelial-to-mesenchymal transition and cytotoxicity associated with 2-deoxyglucose treatment.Citation25
The current study examined the effects of AF in lung NETs and SCLCs in vitro as well as in responses to sorafenib. We demonstrated the effects of AF and sorafenib treatment on antioxidant defense and clonogenic survival. In addition, because little is known about the pharmacokinetics of AF in mice, we developed a SCLC xenograft model and determined a safe and efficacious dose of AF that significantly inhibited TrxR in tumors without affecting glutathione peroxidase 1. After a 2-week dosing schedule of AF at 4 mg/kg IP once daily, complete blood counts as well as hematopoietic stem cell and progenitor cell populations were measured to show that AF was well-tolerated by the hematopoietic system of tumor-bearing mice. Renal and liver function, evaluated by serum chemistry analyses, as well as weight loss and animal body conditioning also indicated that AF was well-tolerated in this model system. Finally, the median overall survival of the mice with SCLC xenografts was significantly prolonged by treatment with AF. These results provide a rigorous characterization of the effects of AF on a SCLC xenograft mouse model and continue to support the hypothesis that repurposing of AF as an adjuvant to cancer therapy in combination with agents that disrupt thiol metabolism is well-justified.
Results
AF inhibits TrxR and decreases clonogenic survival in lung NET and NECs via a thiol-mediated mechanism
We and others have demonstrated that treatment with physiologically relevant doses of AF decreases TrxR activity and sensitizes a variety of cancer cells to chemotherapy and ionizing radiation both in vitro and in vivo.Citation23,Citation24,Citation26,Citation27 Recently, it was demonstrated that an SCLC subpopulation with a neuroendocrine-like phenotype was resistant to cell death from lipid oxidation and also demonstrated a clear dependence on the Trx-driven anti-oxidant pathway for survival.Citation6 We also have demonstrated that a subpopulation of breast cancer stem cells was more dependent on the Trx system.Citation25 To extend these observations, we treated two SCLC cell lines (DMS53, DMS273) (NECs) and one lung NET cell line (H727) with AF followed by the TrxR activity assay and the clonogenic survival assay. The NEC cell lines DMS273 expresses NEUROD1 but not ASCL1 transcription factorCitation28 while DMS53 expresses both subtype transcription factors.Citation29 The bronchial carcinoid cell line H727 expresses ASCL1.Citation30 demonstrates that 1 μM AF for 24 h significantly decreases TrxR activity and clonogenic survival in lung NET and NECs. We have previously demonstrated in lung, prostate, and breast cancer that simultaneous inhibition of Trx- and glutathione-dependent pathways of hydroperoxide metabolism resulted in enhanced cell death via a mechanism of thiol-mediated oxidative stress.Citation23–25,Citation31 It has also been demonstrated in SCLC cells that targeting both the glutathione and Trx-dependent pathways simultaneously prevents cancer stem cell plasticity and improves overall survival.Citation6 Others have found that the clinically approved anti-cancer drug sorafenib inhibits xCT function resulting in decreased intracellular glutathione and an increase in lipid oxidation stress.Citation13,Citation14
Figure 1. AF decreases TrxR activity and clonogenic survival of lung NET and NECs. AF enhances clonogenic cell death with sorafenib treatment. (a) Small cell lung cancer DMS273 and DMS53 and bronchial carcinoid H727 cell line were treated with 1 µM AF for 24 h (gray bars) and then collected for TxrR enzyme activity. All activity was normalized to H727 control cells. (b) Same cell lines and treatment as in (a) with the clonogenic assay. Normalized to each cell line’s respective control. * p < .05 compared to untreated cells two-way ANOVA with Fishers LSD n = 3. (c) Exponentially growing DMS273 and H727 cells were treated for 48 h with sorafenib at 2.5 or 5 µM followed by harvest and glutathione assay. (d,e) DMS273 and H727 cells were treated with 48 h sorafenib and the last 24 h with 1 µM AF followed by clonogenic assay. Clonogenic survival colony counts were normalized to killing with the treatment of AF alone. n ≥ 3 independent experiments two-way ANOVA Fishers LSD * significantly different than control (p < .05), # significantly different than either drug alone (p < .05).
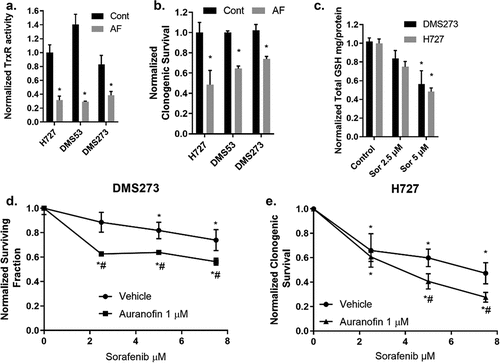
Therefore, we hypothesized that combining AF and sorafenib would decrease NEC and NET cell survival. Initially, we treated H727 and DMS273 cells (cell lines expressing different neuroendocrine transcription subtypes) with pharmacologically relevant doses of sorafenib followed by quantification of intercellular glutathione verifying a dose-dependent reduction of total intracellular glutathione in each cell line (). To test a combination of AF + sorafenib, all cell lines were grown in the exponential phase and then given escalating doses of sorafenib for 48 h with or without AF for the final 24 h. AF significantly enhanced clonogenic cell death of sorafenib in both DMS273 and H727 cells (). These results underline the importance of the hydroperoxide detoxification pathways in cancer cells as well as the simultaneous targeting of both major thiol-mediated detoxification pathways for maximal anticancer effects.
AF dosing schedule to determine maximal TrxR Inhibitory activity inhibition in SCLC xenografts
Although AF has been used historically, little is known about its pharmacokinetic and pharmacodynamic capacity in preclinical mouse models of SCLC. We sought to determine a safe and effective dose of AF in mice using a DMS273 SCLC xenograft mouse model. AF (2 or 4 mg/kg) was administered IP once (QD) or twice (BID) daily, three nude mice per group. Mice were euthanized at 24 h and 5 d after the start of injections, and the TrxR activity was measured in the xenograft tumors, kidneys, and livers. 24 h of AF administration resulted in no significant effects on TrxR activity in the tumors, but by 5 consecutive days of dosing, TrxR activity was inhibited by 50% at 2 mg/kg BID, 4 mg/kg QD, and 4 mg/kg BID (). To investigate the effects of AF treatment on organs that are frequent sights of damage from cancer therapies, kidneys and livers were harvested and TrxR activity was determined. TrxR activity in kidneys was significantly reduced by 2 mg/kg QD and 4 mg/kg BID at 24 h. TrxR activity in kidneys was significantly inhibited by 5 consecutive days of dosing in 2 mg/kg BID, 4 mg/kg QD, and 4 mg/kg BID (). TrxR inhibition in the liver was only observed at an AF dose of 4 mg/kg BID at 5 d ().
Figure 2. Dose escalation of AF selectively decreases TrxR activity in SCLC xenografts, kidneys, and liver but does not affect Gpx1 activity. (a, b, c) Female athymic nude mice (6 per group) were xenografted with 5 × 106 DMS273 cells (2 tumors/mouse one on each flank) and given AF IP injections for 24 h or 5 d either QD or BID. Kidney, liver, and tumor samples were collected for TrxR activity. (d) Plasma samples were collected 9 h after the final BID dose (5 d of therapy), and gold content was determined. (e) Gpx1 activity was determined in xenograft tumors after 5 d of AF therapy. * = p-value <.05, ** = p-value <.01, *** = p-value <.001. Analyzed by mix-effects ANOVA with Fishers LSD comparisons.
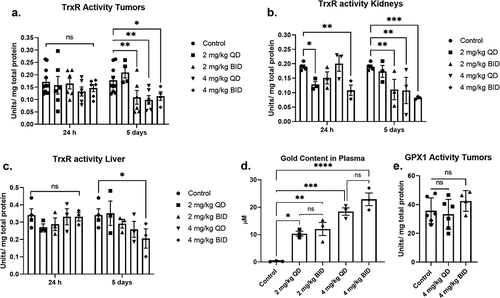
To demonstrate the bioavailability of AF and to afford comparisons with human blood levels, plasma gold levels were measured 9 h after the last BID dosing or 21 h after the last QD dosing. There was a significant increase in plasma gold content of all mice treated with AF (). As expected, the 4 mg/kg dosing resulted in significantly more gold in the plasma than the 2 mg/kg dosing. There was no significant difference between QD and BID dosing indicating that steady-state levels of gold in the plasma can be maintained with 5 d of QD dosing as expected with the long half-life of AF reported in humans and rodents.Citation16,Citation32 Similar to TrxR, Gpx1 is also a selenium-containing enzyme with an active thiol moiety, and it has been reported that high doses of AF can inhibit its activity in vitro.Citation33 GPx1 activity was measured in the tumors after 5 d of treatment with 4 mg/kg QD and BID. Treatment with AF resulted in no significant change in GPx1 activity in the xenograft tumors () indicating that the dose used in this study effectively inhibits TrxR activity while GPx1 is not changed in vivo. Together, these results suggest that 4 mg/kg QD is the optimal AF dose to achieve inhibition of TrxR activity in DMS273 tumors. However, this dose also inhibited TrxR activity in the kidneys indicating the potential for nephrotoxicity when combined with other agents.
Efficacy of AF as a single agent in an SCLC Model
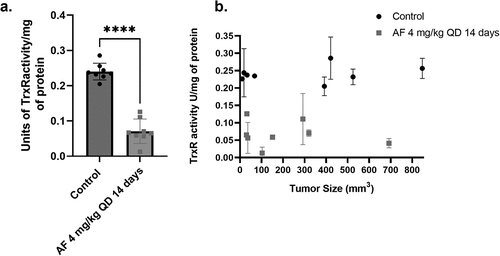
To determine the single-agent activity of AF in vivo, DMS273 SCLC xenografted mice were injected IP with 4 mg/kg QD AF for 14 consecutive days. Two mice in the AF treatment group and the control group (each bearing two bilateral flank tumors) and four mice from the control and AF treatment groups (each with one tumor) were euthanized 24 h after their final dose of AF (day 15) to determine the ability of AF to inhibit TrxR activity (eight tumors total from each group were analyzed). AF treatment for 14 d significantly inhibited TrxR activity in the tumors by ~75% (). Importantly, AF was equally effectively independent of tumor size, confirming that AF can penetrate large tumors and effectively inhibit TrxR activity ().
Figure 3. AF decreases TrxR activity in DMS273 tumors in vivo independent of tumor size. Two mice each with two flank DMS273 xenografts in the first cohort and four mice with one flank DMS273 xenografts in the third cohort were treated with AF 4 mg/kg IP QD for 14 d and then euthanized and evaluated for TrxR enzyme activity assays, and results are plotted in aggregate (a) or as a function of tumor size at the end of treatment (b). * p < .05 compared to untreated cells two-way ANOVA with Fishers LSD comparisons.
To examine the safety and antitumor activity of AF as a single agent, three separate cohorts of mice with DMS273 xenograft tumors were treated with either vehicle control or 4 mg/kg AF IP every day for 14 d. Mice were considered to have met euthanasia criteria when tumors reached 1000 mm3 for 2 consecutive days. The three separate cohorts had 9–10 animals/group for a total of 28 AF-treated mice and 30 vehicle control-treated mice, and tumor size was monitored as a function of time (Supplemental Figure S1). shows the mice from both AF-treated and vehicle control gained weight equally, demonstrating no signs of distress as assessed by all the general activity endpoints specified in our IACUC-approved protocol. Common acute side effects of AF therapy in humans include diarrhea, skin rash, and pruritus.Citation20 None of these side effects were noted in any animals after the 14 d of continuous AF treatment. These results support the hypothesis that this AF dosing scheme was well-tolerated in tumor-bearing mice for 14 d.
Figure 4. 4 mg/kg QD AF IP for 14 d is nontoxic to bone marrow, liver, and kidneys of mice. Female nude mice-bearing DMS273 xenografts were treated with either vehicle or 4 mg/kg AF IP every day for 14 d. Mice were euthanized when tumors were ≥1000 mm3. This experiment was done on three separate cohorts (9–10 animals/cohort) of animals for a total of 28 AF-treated mice and 30 vehicle control-treated mice. (a) Mice were monitored daily, and the % of the initial weight was plotted as a function of time from the beginning of AF treatment. (b, c) Bone marrow from 9 AF-treated mice (5 from the first cohort, 4 from the second cohort) and 13 control mice (7 from the first cohort, 6 from the second cohort) was harvested from the femurs at euthanasia and subjected to flow cytometry for HSCs (lineage (Lin)− sca-1+ c-Kit+, LSK) shown in (b) and HSPCs (lin- cKit+ Sca1+ CD135-) populations shown in (c). (d-I) Blood was drawn from seven control mice and five AF-treated mice from the third cohort) via cardiac puncture at euthanasia and evaluated for clinical chemistry endpoints by Antech Diagnostics.
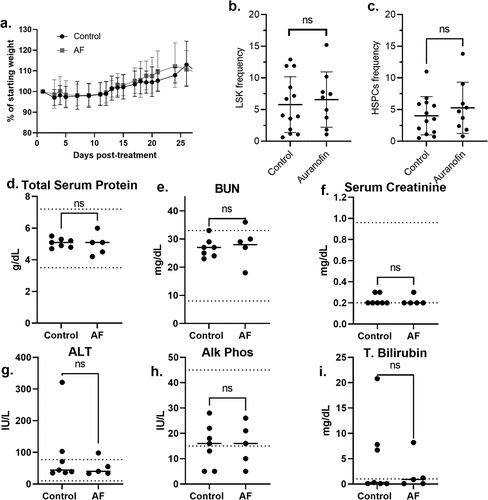
In humans, thrombocytopenia or leukopenia have been reported in patients with AF.Citation20 When complete blood counts (CBCs) were analyzed in tumor-bearing mice being treated with AF using a Siemens ADVIA 120 Hematology Analyzer (), there were no significant changes in WBCs, neutrophils, lymphocytes, RBCs, hemoglobin, hematocrit, mean corpuscular volume (mcv), or platelets. In order to determine any possible effects of AF on hematopoietic stem cells (HSCs and HSPCs), bone marrow from 9 AF-treated mice (5 from the first cohort, 4 from the second cohort) and 13 control mice (7 from the first cohort, 6 from the second cohort) were harvested from the femurs at euthanasia and subjected to flow cytometry for HSCs (Lineage (Lin)− Sca-1+ c-Kit+, LSK) and HSPCs (Lin- cKit+ Sca1+ CD135-). shows no significant differences in HSCs or HSPCs with AF treatment. In addition, no significant changes in LT-HSC or ST-HSC populations were noted (data not shown). Overall, the analysis of hematological data indicated that this AF dosing scheme had no adverse toxic effects on CBCs or bone marrow proliferation during the 14-d exposure. Blood drawn from seven control mice and five AF-treated mice from the third cohort via cardiac puncture at euthanasia were evaluated for clinical chemistry endpoints by Antech Diagnostics () and demonstrated no significant differences in total serum protein, BUN, serum creatinine, ALT, Alk Phos, or total bilirubin.
Table 1. CBC analysis of peripheral blood during AF treatment. Number of mice in each group in parentheses.
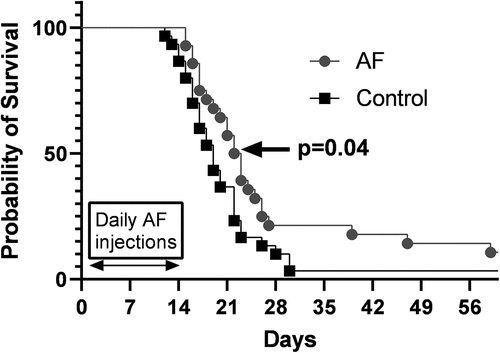
When tumor growth (Supplemental Figure S1A-F) and Kaplan−Meier survival curves () combining all three cohorts of mice exposed to AF for 14 d were analyzed, the median overall survival of the AF-treated animals was found to be significantly increased (median overall survival of 19 d in control versus 23 d in AF-treated animals (p = .04) (). These results support the hypothesis that 14-d AF treatment can significantly inhibit TrxR activity in SCLC xenografts, demonstrating modest but significant single-agent anti-tumor efficacy with no measurable systemic toxicity to the liver, kidney, or hematopoietic system.
Figure 5. 4 mg/kg QD AF IP for 14 d prolongs survival in nude mice with DMS273 xenografts. Tumors from all mice shown in were measured via calipers, and tumor volumes were calculated. When tumor volumes measured 1000 mm3, mice were considered to have reached euthanasia criteria and Kaplan−Meier curves (three cohorts combined) were used to estimate the overall survival. Log-rank (Mantel Cox) test was used to determine significance (p < .04).
Discussion
AF has been FDA-approved and used for rheumatoid arthritis for decades and the kinetics of the orally administered drug in humans was thoroughly studied in the 1980s,Citation32,Citation34 by following the gold content in the patient’s serum. It was found that maximum gold levels were not reached until 16 weeks of treatment.Citation35 An interesting question not addressed by these early studies was whether AF’s gold phosphine complex acted as a prodrug (inactive precursor) that administers the active gold molecule, or if the gold atom remains attached to the phosphine ligand during treatment. Although AF’s antiproliferative properties have been known for some time, the ability of AF to inhibit TrxR was not discovered until 1998.Citation22 Based on those results, different gold (I) complexes have been synthesized and tested as prospective anticancer drugs. Many of these gold (I) complexes show sub-micromolar anticancer activities against several cancer cell lines. The mechanism of action of many gold (I) complexes has been investigated and is thought to cause mitochondrial disruption via inhibition of selenium- and sulfur-containing enzymes including TrxR, glutathione peroxidase, cysteine protease, or glutathione-S-transferase.Citation36,Citation37
Targeting TrxR with AF has recently been suggested as having the potential to selectively enhance cancer therapy responses using conventional radio-chemotherapies based on the tumor cells’ upregulation of hydroperoxide metabolic pathways that are dependent on TrxR.Citation6,Citation8,Citation23,Citation24,Citation27,Citation31 Furthermore, we have shown combining inhibition of TrxR using AF and depletion of GSH metabolism using buthionine sulfoximine, which dramatically enhances the cancer cell-specific cytotoxicity and hydroperoxide-mediated metabolic oxidative stress.Citation23 In the current study, we now show that AF significantly enhances the toxicity of the FDA-approved broad-spectrum kinase inhibitor, sorafenib, via GSH inhibition.Citation15,Citation38,Citation39 Interestingly, this enhancement happened in both NEC and NET settings under varying contexts of transcriptional regulation, demonstrating the universality of the treatment combination. Treatment with sorafenib in humans has a low incidence of serious adverse effects including myelosuppression, electrolyte disorders, and damaged heart and kidneys,Citation40 and further experiments conducted in vivo combining AF with sorafenib will be designed to monitor for added toxicity.
Although AF has been used therapeutically for arthritis in humans for 40 y, preclinical mouse models have not been extensively studied for injury markers in the context of dosing schemes for preclinical cancer therapy studies. Consequently, we have established a once-daily, nontoxic dosing regimen of AF that effectively inhibits TrxR in a mouse SCLC xenograft model. These studies demonstrated that AF treatment resulted in dose-dependent increases in plasma drug concentration that led to significant inhibition in TrxR (but not GPx1) in DMS273 xenograft tumors without any indication of weight loss, bone marrow, kidney, or liver toxicities. To compare the bioavailability of AF in mice to that of humans we measured the gold in the plasma of mice. We found the average level of the gold in the plasma with 4 mg/kg IP dosing in mice (18.4 µM) was an 11-fold higher concentration than the average concentration found in humans (1.6 µM) taking the recommended dose of 6 mg/day orally for 7 d for rheumatoid arthritis.Citation41 The long half-life (35 d in humans) of AF suggests that higher serum and tumor drug levels can be achieved with continued dosing.Citation41 In fact, the primary objective of an ongoing clinical trial (NCT01737502) is to establish the maximum tolerated dose of AF in combination with sirolimus in lung cancer, including SCLC.
In normal cells, redox metabolism is tightly coupled via non-equilibrium steady-state fluxes of reactive metabolic byproducts and electron carriers through redox-sensitive signal transduction pathways. It is increasingly recognized that dysregulation of mitochondrial redox metabolism in cancer cells leads to increased steady-state levels of reactive species including H2O2 and organic hydroperoxides that are compensated for by increased hydroperoxide metabolic pathways dependent upon NADPH and thiol-containing antioxidant enzyme pathways using glutathione and thioredoxin as co-factors.Citation8,Citation42–44 Thioredoxin (Trx) is a small disulfide-containing protein that is maintained in a reduced state by the NADPH-dependent thioredoxin reductases (TrxR) and acts as an antioxidant both by facilitating the reduction of other proteins by cysteine thiol-disulfide exchange as well as acting as a cofactor in the peroxiredoxin-mediated detoxification of hydroperoxides.Citation31 Since Trx/TrxR-dependent antioxidant pathways have been shown to be critical for cancer cells to maintain a viable steady-state in the face of increased fluxes of hydroperoxides by mitochondrial metabolism, TrxR has been proposed to be a selective metabolic target for enhancing tumor cell responses to cancer therapy.Citation45
To understand normal versus tumor tissue responses to AF, previous studies in isolated rat heart mitochondria and tumor cells were accomplished.Citation21,Citation46–49 AF was found to be an inhibitor of isolated rat heart mitochondrial TrxR activity. In isolated mitochondria, AF had little effect on oxygen consumption and did not alter the redox properties of the isolated mitochondrial purine nucleotides. AF treatment did increase total thiol groups indicating a possible adaptive response to disruption in thiol metabolism. Also, AF (1–5 µM) was found to increase H2O2 levels and oxidized peroxiredoxin 3 in isolated mitochondria as well as cells in the presence of electron transport chain inhibitors that increase ROS levels.Citation21 This effect was suggested to depend on the decrease in the removal of H2O2 in the AF-treated isolated mitochondria.Citation48 Despite these previous studies of AF in rat livers and heart mitochondria, the current study did not find any in vivo evidence of AF toxicity in animals treated with 4 mg/kg daily for 2 weeks including weight gain, liver enzymes, kidney enzymes, CBCs, or bone marrow. These results support the hypothesis that during short-term (2-week) exposures to AF at doses associated with cancer therapy, the drug is well-tolerated by normal tissues.
Conclusion
In vitro AF treatment on high-grade lung NETs and NECs decreases TrxR activity, clonogenic cell survival, and enhances the toxicity of an FDA-approved inhibitor sorafenib, which decreases total GSH. These data support the hypothesis that repurposing of AF as an adjuvant to cancer therapy in combination with targeted agents that disrupt thiol metabolism is well-justified. In vivo work showed that AF as a single agent at 4 mg/kg daily for 2 weeks increased the overall survival with no significant systemic toxicities. Overall, these results support the hypothesis that during short-term (2-week) exposures to AF at cancer therapy-relevant doses, the drug is well-tolerated and could be repurposed as an adjuvant to enhance cancer therapeutic responses in future preclinical and clinical studies.
Methods and materials
Cell culture
DMS53 human small cell lung cancer cells (Neuroendocrine Carcinoma) (NECs) and H727 human (atypical lung neuroendocrine tumor cells) (NETs) were obtained from American Type Culture Collection (ATCC-CRL-2062; CRL-5815). DMS273 human small cell lung cancer cells (NECs) were obtained from the European Collection of Authenticated Cell Cultures (UACAC-95062830). Cells were maintained in RPMI 1640 media (gibco-11,875–093) with 10% fetal bovine serum (R&D sytems-S11150). Cultures were maintained in 5% CO2 and humidified in a 37°C incubator. Cells were used within 20 passages from ATCC and tested as mycoplasma negative.
Clonogenic cell survival assay
Exponentially growing DMS273, DMS53, and H727 cells on 60 mm dishes were treated with increasing doses of sorafenib (Cayman Chemicals -10,009,644) for 48 h with or without 1 μM AF for the last 3 h. Floating cells in a medium were collected and combined with the attached cells from the same dish that were trypsinized with 1 ml trypsin-EDTA (gibco-252,000). Samples were centrifuged and cells were counted using a Beckman Coulter Counter. Cells were plated at low density (150–20,000 cells per dish), and clones were allowed to grow for 10–14 d in maintenance media with 0.1% gentamycin added. Cells were then fixed with 70% ethanol and stained with Coomassie blue for the analysis of clonogenic survival. Individual assay colony counts were normalized to that of control with at least three cloning dishes per condition, repeated in at least three separate experiments.
Thioredoxin reductase assay
TrxR activity was determined spectrophotometrically using the reduction of 5,5’-dithiobis(2-nitrobenzoic) acid (DTNB) with NADPH to 5-thio-2-nitrobenzoic acid (TNB), which generates a yellow color at 412 nm (Millipore-Sigma CS0170). Enzymatic activity was determined by subtracting the time-dependent increase in absorbance at 412 nm in the presence of the TrxR activity inhibitor, aurothioglucose from total activity (Sigma-CS0170). One unit of activity was defined as 1 μM TNB formed/(min·mg protein). Protein concentrations were determined by the Lowry assay.Citation50
Glutathione assay
Exponentially growing DMS273 and H727 cells on 100 mm dishes were treated for 48 h with sorafenib. Immediately following treatment, the cultures were washed in cold PBS, and then, the cells were scrape harvested in 300 μL 5% 5-sulfosalicylic acid (SSA; Sigma-S2130) in water and stored at −20°C for a maximum of 72 h. Total glutathione (GSH; Sigma-G4251) content was determined by the method of GriffithCitation51 using NADPH (Sigma-N6505). The rates of the reaction were compared to similarly prepared GSH standard curves. Glutathione determinations were normalized to the protein content of the insoluble pellet from the SSA extracts 2.5% SDS in 0.1 N bicarbonate using the BCA Protein Assay Kit (Thermo Scientific, Rockford, IL. REF-23225).
Glutathione peroxidase 1 (GPx1) activity assay
Glutathione peroxidase was determined spectrophotometrically using the method of Lawrence and Burk as described previously.Citation52,Citation53 Enzymatic activity was determined by measuring the disappearance of NADPH at 340 nm in the presence of GPx1 from cell pellets or standards. One unit of activity was defined as 1 µmole NAPDH oxidized/min at a specified glutathione (GSH) concentration. Protein concentrations were determined by the Lowry assay.Citation50
Animal experiments
Female 6–8-week-old athymic-nu/nu nude mice were purchased from Envigo (Athymic Nude-Foxn1nu). Female mice were used for these studies as they have been shown to be more sensitive to the toxic effects of AF.Citation54 Mice were housed in a pathogen-free barrier room in the Animal Care Facility at the University of Iowa and handled using aseptic procedures. All procedures were approved by the IACUC committee of the University of Iowa and conformed to the guidelines established by NIH. Mice were allowed at least one week to acclimate prior to beginning experimentation; food and water were made freely available. Tumor cells were inoculated into nude mice by subcutaneous injection of 0.1 mL aliquots of saline containing 1 × 106 DMS273 cells into the right and/or left flank using 25-gauge needles. Mouse health and activity levels were monitored daily during drug treatment as per our IACUC-approved protocol. Mice were terminally anesthetized using 200 µl IP injection of ketamine/xylazine 17.5/2.5 µg/µl mix. When analgesia was achieved, as determined by lack of toe-pinch response, blood was withdrawn via cardiac puncture followed by cervical dislocation as per our IACUC-approved protocol.
In vivo AF dose escalation
In the pilot dose escalation study to determine what AF dosing scheme gave the most rapid and durable inhibition of TrxR activity based on our previous work,Citation24,Citation25 five groups of six mice/group (DMS273 tumors growing on both flanks; two tumors/mouse) were given AF IP once (QD) or twice (BID) a day for 1 or 5 d. We sacrificed half of the mice (3 mice/group) after 24 h to determine whether the effects of AF on TrxR activity could be seen quickly (). The other three mice per group were sacrificed after 5 d as it has been demonstrated in humans that gold levels in plasma reach a 1 µM steady-state after several doses (a level of AF shown to inhibit TrxR in vitro).Citation41 Group 1 was given vehicle injection; group 2 was given 2 mg/kg AF QD; group 3 was given 2 mg/kg AF BID; group 4 was given 4 mg/kg AF QD; and group 5 was given 4 mg/kg AF BID. AF was made by dissolving 5.0 mg AF in 250 µl of absolute ethanol in a 10 ml glass beaker. When completely dissolved, 250 µl Kolliphor EL (Sigma C5135) and 4 ml of 0.9% normal saline for injection (Hospira-PAA128035) were added and pH was adjusted to 7–8 range using approximately 25 µl 8.4% sodium bicarbonate for injection (Hospira-RL-0059) followed by the addition of normal saline for injection to adjust final volume to 8.33 ml and filtered using 200-micron filter into a sterile vial. The final AF concentration was 0.6 mg/ml, and it was made fresh daily. Mice in the control group were administered vehicle without drugs. Mice were euthanized 21 h after the last QD dose and 9 h after the last BID dose; tumors and organs were frozen in liquid nitrogen for later evaluation for TrxR activity. Terminal blood was collected via cardiac puncture under anesthesia, and plasma was separated via centrifugation at 10,000 rcf for 10 min.
Determining gold content in plasma
Plasma samples collected 9 h after the final BID dose (5 d of therapy) and gold content determined were prepared by treatment with aqua regia followed by dilution and analysis using inductively coupled plasma mass spectrometry (ICPMS). Aqua regia was prepared from optima grade hydrochloric and nitric acids (Fisher-Cas 7647-01-0 and Cas 7697-37-5) (3:1, respectively). A 100-µL aliquot of plasma was transferred to a 15-mL centrifuge tube, 500 μL aqua regia was added, and the mixture was heated on a hot block at 90 ± 4°C for 30 minutes. After cooling, the volume was brought to 2.0 mL using deionized water. A 500-μL aliquot of the digestate was mixed with an equal volume of internal standard (10 μg/L Iridium) and analyzed using ICPMS (Agilent 7700) with an external calibration curve.
Tumor growth and survival
In the survival experiments, three cohorts of mice (randomly assigned groups of 9–10 mice/group bearing DMS273 flank tumors) were administered AF at 4 mg/kg or vehicle intraperitoneally (IP) once a day for 14 consecutive days ( and Supplemental Figure S1). When xenograft tumor volumes were measured, approximately 100 mm3 treatments were started. Additionally, in the first cohort, two mice with bilateral flank tumors in the AF group and in the control group were euthanized 24 h after the last dose of 4 mg/kg AF (day 15), and tumors were frozen at −80°C to determine TrxR activity assay. Similarly, in the third experiment, four mice in the control group and four mice in the AF group with one tumor/mouse were likewise sacrificed on day 15 to determine TrxR activity (). On day 15, blood was obtained via mandibular vein and diluted 1:10 in PBS and a complete blood cell count (CBC) was processed in a Siemens ADVIA 120 Hematology Analyzer. The remaining mice were monitored for signs of toxicity by examining body weight and general activity level, and tumors were measured daily by a blinded investigator using Vernier calipers (Vol = (Length x Width2/2)). Mice were euthanized on the day when tumor size exceeded 1000 mm3 for 2 consecutive days (a size determined to not cause discomfort or mobility issues in the mice). No mice died before reaching this endpoint. Blood was collected from a select group of seven control and five AF mice from the third cohort via cardiac puncture. Biochemical assays for AST, ALT, BUN, and creatinine were standard systemic toxicity biomarker assays performed commercially by Antech Diagnostics using protocols described on their website https://www.antechdiagnostics.com/test-guide/ ().
Bone marrow harvest and flow cytometry analysis
Immediately after euthanasia, whole bone marrow cells were obtained from 9 AF-treated and 13 control mice per group, by flushing one femur with PBS 2% FBS followed by erythrocyte elimination with ACK Lysing Buffer (10-548E, BioWhittaker, Lonza) (). The remaining cells were filtered through a 40 μm pore size cell strainer and counted. 1 × 106 cells were stained with Zombie Aqua (423102, BioLegend) for 20 min at room temperature for live/dead cells discrimination, followed by a 30-min surface staining on ice using PBS 1% BSA as staining buffer with allophycocyanin (APC)-conjugated, lineage negative (Lin-) cocktail (BD Bioscience), PerCP/Cy5.5-conjugated anti-Sca1 (Clone D7, BioLegend), APC/Cy7-conjugated anti-cKit (Clone 2B8, BioLegend), PE-conjugated anti-CD135 (Clone A2F10, BioLegend), FITC-conjugated anti-CD48 (Clone HM48–1, BioLegend), PE/Cy7-conjugated anti-CD150 (Clone TC15-12F12.2, BioLegend), and BV421-conjugated anti-CXCR4 (Clone L276F12, BioLegend). Cells were immediately analyzed using an LSR II flow cytometer (Becton Dickinson). The biomarkers were applied to identify multipotent hematopoietic stem cells (HSC) (Lin- cKit+ Sca1+, LSK), ST-HSC (short-term hematopoietic stem cells, Lin- cKit+ Sca1+ CD135- CD48- CD150-), LT-HSC (long-term hematopoietic stem cells, Lin- cKit+ Sca1+ CD135- CD48+CD150-), and ST-HSC/LT-HSC and committed progenitors (hematopoietic stem and progenitor cells, HSPC, Lin- cKit+ Sca1+ CD135-) using FlowJo Software (Treestar, Ashland, OR).
Statistical analysis
The biochemical data analysis was performed using ANOVA multiple comparisons with uncorrected Fishers LSD on GraphPad Prism software. The in vivo regression analysis was used to model tumor growth as a nonlinear function of follow-up time and to make treatment group comparisons of estimated tumor means. Survival graphs were obtained with the methods of Kaplan–Meier and compared with log-rank tests.
Glutathione peroxidase was determined spectrophotometrically using the method of Lawrence and Burk as described previously.Citation52 Enzymatic activity was determined by measuring the disappearance of NADPH at 340 nm in the presence of GPx1 from cell pellets or standards. One unit of activity was defined as 1 µmole NAPDH oxidized/min at a specified glutathione (GSH) concentration. Protein concentrations were determined by the Lowry assay.
Suplim Fig 1.tif
Download TIFF Image (863.8 KB)Acknowledgments
We would like to thank the Holden Comprehensive Cancer Center, the University of Iowa College of Medicine Flow Cytometry Facility, the University of Iowa Office of Animal Resource, the University of Iowa Radiation and Free Radical Research Core, and the Holden Comprehensive Cancer Center Biostatistical Core.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The authors confirm that the data supporting the findings of this study are available within the article or its supplementary materials. Any data that support the findings of this study that are not found within the article are available from the corresponding author, [MAF], upon reasonable request.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15384047.2024.2382524
Additional information
Funding
References
- Pelosi G, Sonzogni A, Harari S, Albini A, Bresaola E, Marchiò C, Massa F, Righi L, Gatti G, Papanikolaou N, et al. Classification of pulmonary neuroendocrine tumors: new insights. Transl Lung Cancer Res. 2017 Oct. 6(5):513–11. doi:10.21037/tlcr.2017.09.04.
- Torniai M, Scortichini L, Tronconi F, Rubini C, Morgese F, Rinaldi S, Mazzanti P, Berardi R. Systemic treatment for lung carcinoids: from bench to bedside. Clin Transl Med. [2019 Jul 4]. 8(1):22. doi:10.1186/s40169-019-0238-5.
- Rudin CM, Brambilla E, Faivre-Finn C, Sage J. Small-cell lung cancer. Nat Rev Dis Primers. [2021 Jan 14]. 7(1):3. doi:10.1038/s41572-020-00235-0.
- Lu M, Zhang R, Qi LS, Wang YL, Sun XX, You J. Pathologic responses to neoadjuvant chemoimmunotherapy in primary limited-stage small-cell lung cancer. Thorac Cancer. 2022 Nov. 13(22):3208–16. doi:10.1111/1759-7714.14679.
- Gay CM, Stewart CA, Park EM, Diao L, Groves SM, Heeke S, Nabet BY, Fujimoto J, Solis LM, Lu W, et al. Patterns of transcription factor programs and immune pathway activation define four major subtypes of SCLC with distinct therapeutic vulnerabilities. Cancer Cell. [2021 Mar 8]. 39(3):346–60 e7. doi:10.1016/j.ccell.2020.12.014.
- Bebber CM, Thomas ES, Stroh J, Chen Z, Androulidaki A, Schmitt A, Höhne MN, Stüker L, de Pádua Alves C, Khonsari A, et al. Ferroptosis response segregates small cell lung cancer (SCLC) neuroendocrine subtypes. Nat Commun. [2021 Apr 6]. 12(1):2048. doi:10.1038/s41467-021-22336-4.
- Ireland AS, Micinski AM, Kastner DW, Guo B, Wait SJ, Spainhower KB, Conley CC, Chen OS, Guthrie MR, Soltero D, et al. MYC drives temporal evolution of small cell lung cancer subtypes by reprogramming neuroendocrine fate. Cancer Cell. [2020 Jul 13]. 38(1):60–78 e12. doi:10.1016/j.ccell.2020.05.001.
- Zhou D, Shao L, Spitz DR. Reactive oxygen species in normal and tumor stem cells. Adv Cancer Res. 2014;122:1–67. doi:10.1016/B978-0-12-420117-0.00001-3.
- Holmgren A, Johansson C, Berndt C, Lonn ME, Hudemann C, Lillig CH. Thiol redox control via thioredoxin and glutaredoxin systems. Biochem Soc Trans. 2005 Dec. 33(Pt 6):1375–77. doi:10.1042/BST0331375.
- Lu J, Holmgren A. The thioredoxin antioxidant system. Free radical biology & medicine. Free Radical Biol And Med. 2014 Jan. 66:75–87. doi:10.1016/j.freeradbiomed.2013.07.036.
- Behan FM, Iorio F, Picco G, Gonçalves E, Beaver CM, Migliardi G, Santos R, Rao Y, Sassi F, Pinnelli M, et al. Prioritization of cancer therapeutic targets using CRISPR–Cas9 screens. Nature. 2019 Apr. 568(7753):511–16. doi:10.1038/s41586-019-1103-9.
- Hasskarl J. Sorafenib: targeting multiple tyrosine kinases in cancer. Recent Results Cancer Res. 2014;201:145–64. doi:10.1007/978-3-642-54490-3_8.
- Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS, et al. Pharmacological inhibition of cystine–glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. [2014 May 20]. 3:e02523. doi:10.7554/eLife.02523.
- Li Y, Yan H, Xu X, Liu H, Wu C, Zhao L. Erastin/sorafenib induces cisplatin-resistant non-small cell lung cancer cell ferroptosis through inhibition of the Nrf2/xCT pathway. Oncol Lett. 2020 Jan. 19(1):323–33. doi:10.3892/ol.2019.11066.
- Roh JL, Kim EH, Jang H, Shin D. Aspirin plus sorafenib potentiates cisplatin cytotoxicity in resistant head and neck cancer cells through xCT inhibition. Free Radical Biol & Med. 2017 Mar. 104:1–9. doi:10.1016/j.freeradbiomed.2017.01.002.
- Furst DE. Mechanism of action, pharmacology, clinical efficacy and side effects of auranofin. An orally administered organic gold compound for the treatment of rheumatoid arthritis. Pharmacotherapy. 1983 Sep. 3(5):284–98. doi:10.1002/j.1875-9114.1983.tb03277.x.
- Horton RJ. Comparative safety and efficacy of auranofin and parenteral gold compounds: a review. Scand J Rheumatol Supplement. 1983;51(suppl51):100–10. doi:10.3109/03009748309095361.
- Liu J, Akahoshi T, Namai R, Matsui T, Kondo H. Effect of auranofin, an antirheumatic drug, on neutrophil apoptosis. Inflamm Res. 2000 Sep. 49(9):445–51. doi:10.1007/s000110050615.
- Bakke E, Myklebust G, Gran JT. Thrombocytopenia induced by auranofin treatment. Tidsskr Nor Laegeforen. [1997 Nov 20]. 117(28):4081–82. Trombocytopeni utlost ved auranofinbehandling.
- Bandilla KK, Missler B. Longterm results of auranofin therapy. Clin Rheumatol. 1987 Sep. 6(2):35–42.10.1007/BF02203383.
- Rigobello MP, Scutari G, Folda A, Bindoli A. Mitochondrial thioredoxin reductase inhibition by gold(I) compounds and concurrent stimulation of permeability transition and release of cytochrome c. Biochemical Pharmacol. [2004 Feb 15]. 67(4):689–96. doi:10.1016/j.bcp.2003.09.038.
- Gromer S, Arscott LD, Williams CH Jr., Schirmer RH, Becker K. Human placenta thioredoxin reductase. Isolation of the selenoenzyme, steady state kinetics, and inhibition by therapeutic gold compounds. The J Biol Chem. [1998 Aug 7]. 273(32):20096–101. doi:10.1074/jbc.273.32.20096.
- Fath MA, Ahmad IM, Smith CJ, Spence J, Spitz DR. Enhancement of carboplatin-mediated lung cancer cell killing by simultaneous disruption of glutathione and thioredoxin metabolism. Res Support, N.I.H. Extramural. Clin Cancer Res: an Off J Am Assoc For Cancer Res [2011 Oct 1]. 17(19):6206–17. doi:10.1158/1078-0432.CCR-11-0736.
- Rodman SN, Spence JM, Ronnfeldt TJ, Zhu Y, Solst SR, O’Neill RA, Allen BG, Guan X, Spitz DR, Fath MA. Enhancement of radiation response in breast cancer stem cells by inhibition of thioredoxin- and glutathione-dependent metabolism. Radiat Res. 2016 Oct. 186(4):385–95. doi:10.1667/RR14463.1.
- Luo M, Shang L, Brooks MD, Jiagge E, Zhu Y, Buschhaus JM, Conley S, Fath MA, Davis A, Gheordunescu E, et al. Targeting breast cancer stem cell state equilibrium through modulation of redox signaling. Cell Metab. [2018 Jul 3]. 28(1):69–86 e6. doi:10.1016/j.cmet.2018.06.006.
- Nag D, Bhanja P, Riha R, Sanchez-Guerrero G, Kimler BF, Tsue TT, Lominska C, Saha S. Auranofin protects intestine against radiation injury by modulating p53/p21 pathway and radiosensitizes human colon tumor. Clin Cancer Res: an Off J Am Assoc For Cancer Res. [2019 Aug 1]. 25(15):4791–807. doi:10.1158/1078-0432.CCR-18-2751.
- Wang H, Bouzakoura S, de Mey S, Jiang H, Law K, Dufait I, Corbet C, Verovski V, Gevaert T, Feron O, et al. Auranofin radiosensitizes tumor cells through targeting thioredoxin reductase and resulting overproduction of reactive oxygen species. Oncotarget. [2017 May 30]. 8(22):35728–42. doi:10.18632/oncotarget.16113.
- Chen H, Gesumaria L, Park YK, Oliver TG, Singer DS, Ge K, Schrump DS. BET inhibitors target the SCLC-N subtype of small-cell lung cancer by blocking NEUROD1 transactivation. Mol Cancer Res. [2023 Feb 1]. 21(2):91–101. doi:10.1158/1541-7786.MCR-22-0594.
- Manzo A, Sforza V, Carillio G, Palumbo G, Montanino A, Sandomenico C, Costanzo R, Esposito G, Laudato F, Mercadante E, et al. Lurbinectedin in small cell lung cancer. Front Oncol. 2022;12:932105. doi:10.3389/fonc.2022.932105.
- Augustyn A, Borromeo M, Wang T, Fujimoto J, Shao C, Dospoy PD, Lee V, Tan C, Sullivan JP, Larsen JE, et al. ASCL1 is a lineage oncogene providing therapeutic targets for high-grade neuroendocrine lung cancers. Proc Natl Acad Sci USA. [2014 Oct 14]. 111(41):14788–93. doi:10.1073/pnas.1410419111.
- Li L, Fath MA, Scarbrough PM, Watson WH, Spitz DR. Combined inhibition of glycolysis, the pentose cycle, and thioredoxin metabolism selectively increases cytotoxicity and oxidative stress in human breast and prostate cancer. Redox Biol. 2015;4:127–35. doi:10.1016/j.redox.2014.12.001.
- Van Riel PL, Gribnau FW, de Putte Lb V, Arts CW, Van Aernsbergen A. Serum gold concentrations during treatment with auranofin. Res Support, Non-U.S. Gov’t. Clin Rheumatol. 1987 Mar. 6(1):50–54. doi:10.1007/BF02201000.
- Chaudiere J, Wilhelmsen EC, Tappel AL. Mechanism of selenium-glutathione peroxidase and its inhibition by mercaptocarboxylic acids and other mercaptans. Research support, U.S. Gov’t, P.H.S. The J Biol Chem. [1984 Jan 25]. 259(2):1043–50. doi:10.1016/S0021-9258(17)43563-0.
- Blocka K, Furst DE, Landaw E, Dromgoole S, Blomberg A, Paulus HE. Single dose pharmacokinetics of auranofin in rheumatoid arthritis. J Rheumatol Suppl. 1982 Jul. 8:110–19.
- Van Riel PL, Gribnau FW, de Putte Lb V, Arts CW, Van Aernsbergen A. Serum gold concentrations during treatment with auranofin. Clin Rheumatol. 1987 Mar. 6(1):50–54. doi:10.1007/BF02201000.
- Lu Y, Ma X, Chang X, Liang Z, Lv L, Shan M, Lu Q, Wen Z, Gust R, Liu W, et al. Recent development of gold(I) and gold(III) complexes as therapeutic agents for cancer diseases. Chem Soc Rev. [2022 Jul 4]. 51(13):5518–56. doi:10.1039/d1cs00933h.
- Stenger-Smith JR, Mascharak PK. Gold drugs with Au(PPh(3))(+) moiety: advantages and medicinal applications. ChemMedchem. [2020 Nov 18]. 15(22):2136–45. doi:10.1002/cmdc.202000608.
- Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, Negrier S, Chevreau C, Solska E, Desai AA, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. [2007 Jan 11]. 356(2):125–34. doi:10.1056/NEJMoa060655.
- Chen D, Fan Z, Rauh M, Buchfelder M, Eyupoglu IY, Savaskan N. ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner. Oncogene. [2017 Oct 5]. 36(40):5593–608. doi:10.1038/onc.2017.146.
- Li Y, Gao ZH, Qu XJ. The adverse effects of sorafenib in patients with advanced cancers. Basic Clin Pharma Tox. 2015 Mar. 116(3):216–21. doi:10.1111/bcpt.12365.
- Capparelli EV, Bricker-Ford R, Rogers MJ, Jh M, Reed SL. Phase I clinical trial results of auranofin, a novel antiparasitic agent. Antimicrob Agents Chemother. 2017 Jan. 61(1). doi:10.1128/AAC.01947-16.
- Spitz DR. Manipulations of redox metabolism for enhancing radiation therapy responses: a historical perspective and novel hypothesis. Semin Radiat Oncol. 2019 Jan. 29(1):1–5. doi:10.1016/j.semradonc.2018.10.010.
- Aykin-Burns N, Ahmad IM, Zhu Y, Oberley LW, Spitz DR. Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. Biochem J. [2009 Feb 15]. 418(1):29–37. doi:10.1042/BJ20081258.
- Scarbrough PM, Mapuskar KA, Mattson DM, Gius D, Watson WH, Spitz DR. Simultaneous inhibition of glutathione- and thioredoxin-dependent metabolism is necessary to potentiate 17AAG-induced cancer cell killing via oxidative stress. Res Support, N.I.H. Extramural Research support. Non-U.S. Gov’t. Free radical biology & medicine. Jan 15 2012;52(2):436–43. doi:10.1016/j.freeradbiomed.2011.10.493.
- Nguyen P, Awwad RT, Smart DD, Spitz DR, Gius D. Thioredoxin reductase as a novel molecular target for cancer therapy. Cancer Lett. [2006 May 18]. 236(2):164–74. doi:10.1016/j.canlet.2005.04.028.
- Folda A, Citta A, Scalcon V, Calì T, Zonta F, Scutari G, Bindoli A, Rigobello MP. Mitochondrial thioredoxin system as a modulator of cyclophilin D redox state. Sci Rep. [2016 Mar 15]. 6(1):23071. doi:10.1038/srep23071.
- Marzano C, Gandin V, Folda A, Scutari G, Bindoli A, Rigobello MP. Inhibition of thioredoxin reductase by auranofin induces apoptosis in cisplatin-resistant human ovarian cancer cells. Free Radical Biol & Med. [2007 Mar 15]. 42(6):872–81. S0891-5849(06)00814-8 [pii] doi:10.1016/j.freeradbiomed.2006.12.021.
- Rigobello MP, Folda A, Baldoin MC, Scutari G, Bindoli A. Effect of auranofin on the mitochondrial generation of hydrogen peroxide. Role of thioredoxin reductase. Free Radical Res. 2005 Jul. 39(7):687–95. Q100X5571110W745 [pii] doi:10.1080/88010715760500135391.
- Rigobello MP, Folda A, Scutari G, Bindoli A. The modulation of thiol redox state affects the production and metabolism of hydrogen peroxide by heart mitochondria. Arch Biochem Biophys. [2005 Sep 15]. 441(2):112–22. doi:10.1016/j.abb.2005.07.007.
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. The J Biol Chem. 1951 Nov. 193(1):265–75. doi:10.1016/S0021-9258(19)52451-6.
- Griffith OW. Determination of glutathione and glutathione disulfide using glutathione reductase and 2-vinylpyridine. Anal Biochem. [1980 Jul 15]. 106(1):207–12. doi:10.1016/0003-2697(80)90139-6.
- Sullivan SJ, Oberley TD, Roberts RJ, Spitz DR. A stable O2-resistant cell line: role of lipid peroxidation byproducts in O2-mediated injury. Am J Physiol. 1992 June. 262(6 Pt 1):L748–56. doi:10.1152/ajplung.1992.262.6.L748.
- Lawrence RA, Burk RF. Glutathione peroxidase activity in selenium-deficient rat liver. Biochem Biophys Res Commun. [1976 Aug 23]. 71(4):952–58. doi:10.1016/0006-291x(76)90747-6.
- Yang L, Wang H, Yang X, Wu Q, An P, Jin X, Liu W, Huang X, Li Y, Yan S, et al. Auranofin mitigates systemic iron overload and induces ferroptosis via distinct mechanisms. Signal Transduct Target Ther. [2020 Jul 31]. 5(1):138. doi:10.1038/s41392-020-00253-0.