
Abstract
Expression of oncogenes or short telomeres can trigger an anticancer response known as cellular senescence activating the p53 and RB tumor suppressor pathways. This mechanism is switched off in most tumor cells by mutations in p53 and RB signaling pathways. Surprisingly, p53 disabled tumor cells could be forced into senescence by expression of a mutant allele of the nuclear envelope protein lamin A. The pro-senescence lamin A mutant contains a deletion in the sequence required for processing by the protease ZMPSTE24 leading to accumulation of farnesylated lamin A in the nuclear envelope. In addition, the serine at position 22, a target for CDK1-dependent phosphorylation, was mutated to alanine, preventing CDK1-catalyzed nuclear envelope disassembly. The accumulation of this mutant lamin A compromised prophase to prometaphase transition leading to invaginations of the nuclear lamina, nuclear fragmentation and impaired chromosome condensation. Cells exited this impaired mitosis without cytokinesis and re-replicated their DNA ultimately arresting in interphase as polyploid cells with features of cellular senescence including increased expression of inflammatory gene products and a significant reduction of tumorigenicity in vivo.
Introduction
Cellular senescence is a permanent cell cycle arrest with a dual role in biology. First, senescence is a barrier for tumorigenesis since it prevents the expansion of cells with the potential to form malignant tumors. On the other hand, senescent cells accumulate in old tissues and could be responsible for age-associated diseases.Citation1 The mechanisms promoting senescence during aging are currently unknown. Senescent cells accumulate in mouse models of premature aging where the underlying defect affects mitosis.Citation2,3 In humans, the Hutchinson-Gilford Progeria Syndrome (HGPS) mimics some aspects of human aging.Citation4-6 HGPS cells accumulate a variant of the nuclear envelope protein lamin A, known as progerin, which is responsible for the disease. Progerin can induce cellular senescence although the direct mechanism remains to be identified. Cells expressing progerin have genomic instability,Citation7 telomere dysfunction,Citation8 altered epigenetic modifications of histones,Citation9,10 abnormal chromosome segregation, binucleationCitation11 and changes in nuclear architecture.Citation12 The exact molecular mechanism explaining these alterations and their connection to cellular senescence is unknown. It is plausible that a defect in some key function of the nuclear lamina underlies cellular senescence and aging.
The nuclear lamina is mostly composed of several types of lamins and their associated proteins. Lamins of type A and B undergo C-terminal farnesylation, a process that contributes to their localization at the nuclear membrane. However, the C-terminus of lamin A is subsequently cleaved-off by the protease ZMPSTE24/FACE1.Citation13 Progerin is a lamin A mutant with 50-aa deletion near the C-terminus.Citation4,5 The deleted fragment contains the cleavage site for the ZMPSTE24 protease, so progerin cannot go through the last endoproteolytic step that cleaves the C-terminal extension of pre-lamin A and stays permanently farnesylated in the nuclear lamina.Citation14 Inhibitors of farnesyl transferases can rescue some of the defects of cells expressing progerinCitation12 suggesting that permanently farnesylated lamin A is responsible for the disease. Hutchinson-Gilford Progeria Syndrome is not accompanied by cancer developmentCitation15 despite the accelerated aging caused by progerin expression. Although, it is plausible that children with this disease do not live long enough to show an increase in cancer incidence, it is also likely that the ability of progerin to impair cell cycle progression and regulate senescenceCitation8 can halt cancer progression in these patients perhaps pointing to a novel mechanism to treat human cancers.
During every cell cycle the nuclear lamina is disassembled at prophase and then reassembled during cytokinesis.Citation16 The mitotic CDK/cyclin complexes phosphorylate the N-terminus conserved domain of lamin A at serine 22 (S22) and the C-terminus, at serine 392 (S392), inducing lamina depolymerization.Citation17,18 Mutations in these phosphorylation sites in lamin A inhibit nuclear lamina disassembly.Citation19 Here, we report that mutating the serine 22 phosphorylation site of progerin (S22A-progerin) enhances its ability to induce cellular senescence characterized by DNA damage, expression of inflammatory cytokines and inhibition of tumor progression in mice. The effects requires farnesylation and the formation of a thick nuclear envelope, is associated with nuclear membrane invaginations, nuclear fragmentation and polyploidy and is independent of p53.
Results
Accumulation of farnesylated prelamin A inhibits tumor cell proliferation
Progerin and L647Rprelamin A are mutant alleles of prelamin A with a disabled ZMPSTE24 cleavage site.Citation20 Stable expression of these mutant prelamin A genes from retroviral vectors in the human osteosarcoma cell line U-2 OS induced a strong growth inhibition over 6 d of culture while overexpression of wild type prelamin A had a lesser effect (). We also expressed wild type prelamin A and its mutants in HeLa cervical cancer cells (containing the papillomavirus proteins E6 and E7 that inactivate p53 and RB), H 1299 lung cancer cells (with mutant p53) and PC-3 prostate cancer cells (with mutant p53). Again, mutant prelamin A alleles inhibited the proliferation of these tumor cells over a 6 day period of culture although the effect was not as striking as in U-2 OS cells (Fig. S1A-C).
Figure 1. Accumulation of lamin (A) in the nuclear envelope inhibits cancer cell growth. (A) Immunoblot for lamin A/C of U-2 OS cells expressing vector (V), wild type lamin A (wt), progerin (Pr) or L647R lamin A (LR). (B) Growth curves of U-2 OS cells expressing vector, progerin, L647R lamin A or wild type lamin A (wtLaminA). Cells were plated in triplicate 4 d after infection with a retroviral vector expressing wtLamin A, progerin, or L647R lamin A mutant. Relative cell numbers were estimated from a crystal violet staining assay. (C) qPCR showing ZMPSTE24 mRNA levels of U-2 OS cells with a control shRNA (NTC) or shZMPSTE24. (D) Growth curves of U-2 OS cells expressing shZMPSTE24 or NTC. (E) Immunoblots for the indicated proteins of U-2 OS cells and (F) H 1299 with a control shRNA (NTC) or shZMPSTE24. (G) Growth curves of H 1299 cells expressing wild type lamin A and NTC (wtLaminA/NTC), shZMPSTE24 and vector (shZMPSTE24/V), or wild type lamin A and shZMPSTE24 (wtLaminA/shZMPSTE24). (H) Immunofluorescence images of H 1299 cells expressing the vectors indicated in (G). Magnification = 10 μm.
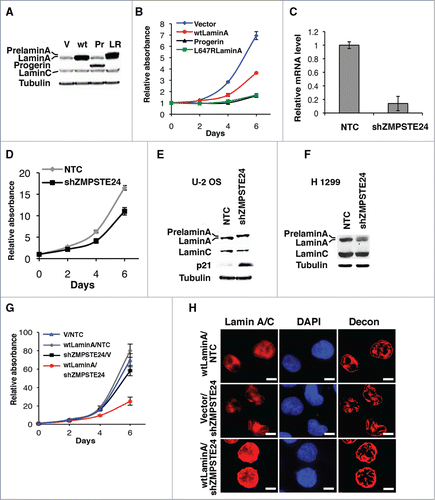
Since progerin and L647R prelamin A remain both farnesylated we mutated the cysteine in the farnesylation site (the CaaX-motif) into serine to prevent farnesylation.Citation14 Non-farnesylated progerin (ProgCS) and L647R mutant (L647RCS) had the same proliferation rates as control cells (Fig. S1D and E). Farnesylation of prelamin A contributes to targeting prelamin A to the nuclear membrane.Citation20 In agreement, farnesylated mutant lamins strongly accumulated in the nuclear lamina of interphase cells, while elimination of farnesylation led to different patterns for progerin and L647R staining (Fig. S1F). Non-farnesylated progerin (ProgCS) formed intranuclear foci in around 50% of cells and the rest of the cells had low lamin A/C signal localized both at the nuclear envelope and the nucleoplasm. Non-farnesylated L647R did not form intranuclear foci as ProgCS but its accumulation at the nuclear envelope was reduced in comparison with farnesylated L647R. These results are consistent with previous work reporting that non-farnesylated progerin was unable to induce nuclear alterations in cell cultureCitation21 or the progeria phenotype in mice.Citation22 We conclude that farnesylation is required for the growth inhibition induced by progerin and the L647R mutant prelamin A.
The farnesylated C-terminus of prelamin A is cleaved off by the zinc metallopeptidase ZMPSTE24/FACE-1Citation23 in the inner nuclear membrane.Citation24 It is known that siRNAs against ZMPSTE24/FACE-1 lead to accumulation of farnesylated prelamin A and growth inhibition in HeLa cells.Citation25 Using an shRNA against ZMPSTE24/FACE-1 (shZMPSTE24) () we confirmed this growth inhibition phenotype in osteosarcoma U-2 OS cells (). Western blot analysis revealed accumulation of uncleaved prelamin A in U-2 OS cells expressing shZMPSTE24 and induction of the p53 target p21 (). In p53 null H 1299 cells, shZMPSTE24 was not as efficient as in U-2 OS cells in leading to accumulation of prelamin A and growth inhibition (). Combining expression of wild type lamin A with shZMPSTE24 induced a stronger growth arrest in H 1299 cells () suggesting that growth inhibition depends on the amount of prelamin A. This accumulation of prelamin A in the nuclear lamina was observed by immunofluorescence using an antibody against lamin A/C in H 1299 cells ().
Inhibition of phosphorylation at serine 22 in progerin increases its ability to inhibit tumor cell proliferation
Phosphorylation of the N-terminus of lamin A is required for disassembly of the nuclear lamina during mitosis.Citation18,19 Synchronization of U-2 OS cells expressing progerin, or non-farnesylated progerin by nocodazole treatment revealed that progerin could be serine 22-phosphorylated at G2/M (Supplemental Fig. S2A). Nevertheless, accumulation of progerin compromised proper nuclear lamina disassembly (Fig. S2B) resulting in moderate cell growth inhibition (). We thus reasoned that a progerin mutant unable to undergo phosphorylation at serine 22 should accumulate in the nuclear lamina at higher levels and inhibit further tumor cell proliferation. We expressed S22A-progerin in several tumor cell lines and compared the effects with the same cells expressing progerin or an empty vector control. In all cases, S22A-progerin led to stronger growth arrest as measured in growth curves over 6 d or a colony formation assay over 13 d ().
Figure 2. S22A-progerin triggers a cell proliferation arrest and a multinucleated phenotype in tumor cells. (A-C) Growth curves of different cancer cell lines expressing the indicated vectors. (D) Colony formation assay of cell lines as in (A-C). (E) Quantitation of cell growth in the colony formation assay using crystal violet staining. (F) Immunofluorescence for lamin A/C of U-2 OS cells expressing a vector control, progerin (Pr) or S22A-progerin (S22A). Decon means deconvolution option of Metamorph software. (G) Immunofluorescence showing the cell nucleus (blue-DAPI staining) and the cytoskeleton (red-Tubulin) in the indicated cell lines expressing vector, progerin or S22A-progerin. The percent and S.D of multinuclear cells are indicated at the bottom right of each panel. Magnification = 10 μm.
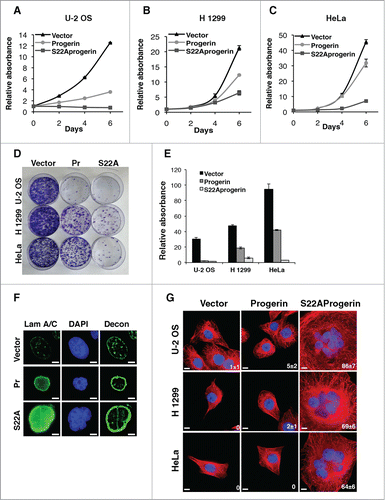
For visualization of S22A-progerin and progerin we performed immunofluorescence and found that S22A-progerin formed a thicker nuclear lamina than progerin ( see also ). Intriguingly, the majority of the S22A-progerin expressing cells displayed multiple nuclei and a very large nuclear and cell size (). To investigate whether farnesylation was also required for growth inhibition by S22A-progerin we created a mutant in which cysteine in the CaaX-motif was replaced by serine preventing farnesylation.Citation14 S22A-progerinCS, like progerinCS did not affect cell growth in U-2 OS cells (Fig. S3A and B) and did not accumulate in the nuclear envelope (Fig. S3C). Importantly, the level of expression of S22A-progerinCS was higher than S22A-progerin indicating that interfering with nuclear envelope functions rather than an overexpression artifact is the cause of the cell cycle arrest. Together, these results suggest that a stable lamin A cage around the nucleus impairs cell proliferation.
S22A-progerin induces nuclear envelope invaginations and polyploidy
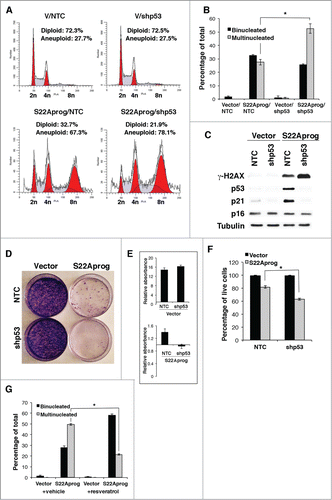
We next characterized in detail the phenotype of large multinucleated cells induced by S22A-progerin. DNA content analysis revealed that a large portion of S22A-progerin-expressing cells had abnormally high levels of DNA peaking at 4 times the normal diploid genome (). The low expression of cyclin B1 and cyclin A () suggests that the S22A-progerin cell population contained a significant fraction of cells arrested at G1, since these 2 proteins mainly peak at G2/M. However, the reduction of levels of cyclin A and B is moderate and we can not discard that some cells are also arrested in G2. Simultaneous expression of S22A-wild type-lamin A (S22ALam) with shZMPSTE24 also induced polyploidy in U-2 OS cells, although in smaller fraction of cells compared with S22A-progerin (). Careful observations of nuclei in U-2 OS or HeLa cells shortly after expressing S22A-progerin indicates first an accumulation of binucleated cells that later become multinucleated without overall increase in cell numbers ().
Figure 3. DNA content analysis reveals the polyploidy of the S22A-progerin-expressing cells. (A) DNA content analysis was performed by fluorescence activated cell sorting (FACS) of U-2 OS cells expressing vector, progerin, or S22A-progerin. Quantification of the DNA content analysis (%) is shown in each panel. (B) Immunoblots for the indicated proteins of U-2 OS cells expressing the vectors as in (A). (C) FACS of U-2 OS cells expressing vector and a control shRNA (NTC), vector (V) and shZMPSTE24, S22A-wild type lamin A (S22ALam) and NTC, S22ALam and shZMPSTE24, or S22A-progerin and NTC. Quantification of DNA content analysis (%) of FACS is shown in each panel. (D) Quantification of cells with 2 or more nuclei in U-2 OS or HeLa cells after infections with a retroviral vector expressing S22A-progerin or an empty vector control.
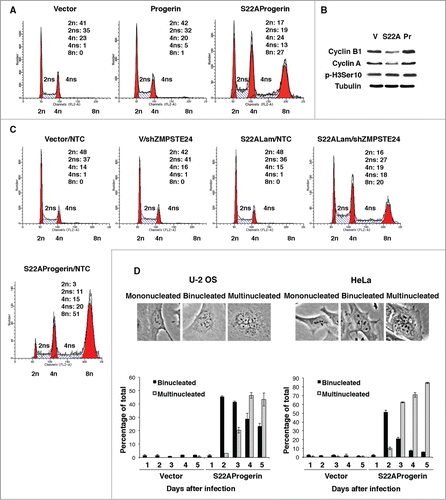
Figure 4. S22A-progerin expressing cells display invaginations of the nuclear envelope, nuclear fragmentation and a failure in chromosome condensation. (A) Immunofluorescence with a progerin specific antibody in U-2 OS cells expressing vector, progerin, or S22A-progerin. Magnification = 10 μm. (B) Electron microscopy showing a cell where a nuclear envelope invagination divides the nuclei in 2 fragments. The black arrows indicate the outer nuclear membrane (ONM) and the white arrows the inner nuclear membrane (INM). (C) Dapi staining of U-2 OS cells expressing vector, progerin, or S22A-progerin. The percent of cells with condensed chromosomes is indicated at the bottom right of each panel. Magnification = 10 μm.
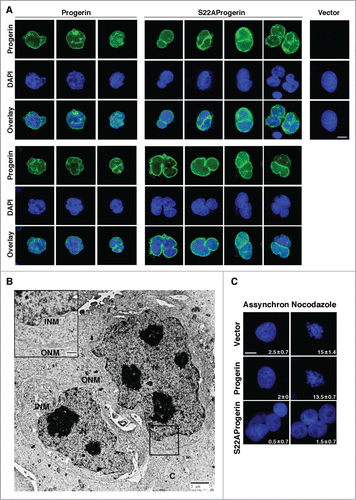
Multinucleation could arise by normal mitotic progression with a failure in cytokinesisCitation26 or by nuclear fragmentation as seen in cancer cells treated with chemotherapeutic drugs.Citation27 Visualization of progerin at early times after their expression, when cells are not yet multinucleated revealed an invagination of progerin and presumably its attached nuclear membrane in cells that express S22A-progerin consistent with a nuclear fragmentation model for multinucleation (). Electron microscopy clearly revealed invaginations of the nuclear envelope that occasionally fragmented the nucleus ( and Fig. S4). Treatment of S22A- progerin expressing cells with nocodozale did not allow visualizing condensed chromosomes as seen in progerin expressing cells or in control cells with an empty vector (). This result suggests that S22A-progerin-expressing cells are compromised at the transition between prophase and prometaphase, the stage at which chromosomes condense and the nuclear envelope dismantles.
We noticed that the proportion of multinucleated cells was higher in the p53 disabled cell line HeLa when compared with the p53 wild type U 2-OS (). To investigate whether p53 limits multinucleation in cells expressing S22A-progerin we inactivated p53 expression in U 2-OS using a validated shRNA. We found that expression of S22A progerin in U 2-OS cells expressing a shRNA against p53 expression increased the number of cells with a polyploid DNA content () and multiple nuclei (). Immunoblots confirmed that the shRNA decreased p53 expression and its target p21 that were both induced by S22A progerin. In contrast, p16INK4a expression was not affected by expression of S22A progerin (). S22A progerin increased DNA damage signals as measured with anti-γH2AX antibody and inactivation of p53 further amplified this signal (). Likewise, S22A progerin decreased colony formation and inactivation of p53 also magnified this effect (). Although most cells expressing S22A progerin remained viable, p53 inhibition increased the proportion of cell death as well (). These results indicate that p53 is required for a checkpoint response that limits DNA replication, DNA damage and multinucleation in cells with altered nuclear envelope but is not required for growth arrest in response to S22A-progerin expression. Further proof that checkpoint functions of p53 limit multinucleation was obtained by using resveratrol, a drug that can prevent cell cycle progression at S phase.Citation28 Treatment of S22A progerin expressing cells with resveratrol induced accumulation of cells in S phase (Fig. S5) and reduced the number of multinucleated cells ().
Figure 5. Role of p53 in S22A-progerin expressing cells. (A) DNA content analysis of U-2 OS cells expressing vector or S22A-progerin with shRNA control (NTC) or shRNA against p53. Quantification of the DNA content analysis (%) is shown in each panel. (B) Quantification of cells with 2 or more nuclei in U-2 OS as in (A). (C) Immunoblots for the indicated proteins using extracts from cells as in (A). (D) Colony formation assay of cell lines as in (A). (E) Quantitation of cell growth in the colony formation assay using crystal violet staining. (F) Cell viability in the indicated cell lines using a trypan blue exclusion assay. (G) Quantitation of cells with 2 or more nuclei in U-2 OS expressing the indicated vectors and treated with 40 μM resveratrol or vehicle for 48 hours.
S22A-progerin induces tumor cell senescence
Drugs that interfere with mitosis usually kill cells by apoptosis in a process that requires the spindle checkpoint.Citation29 However, most cells expressing S22A-progerin remained viable. Since normal fibroblasts expressing progerin prematurely senesceCitation8 we next investigated the presence of senescence markers in tumor cells after introduction of progerin or S22A-progerin. We first used the senescence associated β-galactosidase marker and found that H 1299 and PC-3 cells expressing S22A-progerin but not progerin were positive for this marker (). We also found DNA damage foci labeled with anti-γH2AX antibody in H1299 and PC3 cells expressing S22A progerin (). We could not stain U-2 OS for senescence associated β-galactosidase because this cell line may have defects in the expression of this enzyme. However, many other senescence markers were positive in U-2 OS cells expressing S22A-progerin but not progerin. These markers included PML bodies () and DNA damage foci (). Intriguingly, DNA damage foci in S22A progerin expressing were in association to the nuclear envelope decorating the nuclear periphery or the lamin A invaginations described above ( and E). S22A progerin induced senescence was also characterized by low expression of the proliferation antigen Ki-67 () and the E2F target MCM6 () and high expression of the CDK inhibitor p21 (). We also compared the gene expression pattern of U-2 OS cells expressing S22A-progerin with the cells expressing wild type lamin A. Data from 3 independent experiments were analyzed with Flexarray 1.6.1.1 using robust multiarray average for normalization. Data is available at the GEO repository (GSE51349). Gene expression changes showing a fold of 2 or more were used for pathway analysis using the platform Babelomics.Citation30 The main functional category of upregulated genes involved cytokine and extracellular factors (Fig. S6A, B and ) consistent with the concept that senescent cells exhibit a senescence associated secretory phenotype or SASP.Citation31 The expression of some of these extracellular factors was then confirmed by qPCR (Fig. S6C). Gene Set Enrichment Analysis (GSEA) revealed an overlap with gene sets related to senescence and the NF-κB transcription factor (Supplemental tables I and II). Immunoblots show phosphorylation of RelA at serine 536 and the p38 MAPK at the activation loop Thr180/Tyr182 both in progerin and S22A-progerin expressing cells indicating activation of these pathways by both mutant lamins (). This result suggests that additional mechanisms may explain the increased expression of cytokines genes in S22A-progerin expressing cells. Both progerin and S22A progerin induced accumulation of the DNA damage marker γ-H2AX but the effect was much higher in S22A progerin expressing cells. Taken together, the results indicate that S22A-progerin, unlike progerin can force tumor cells to senesce. Progerin can induce senescence in normal human cells and this event is associated with telomere dysfunction.Citation32 The failure of progerin to induce senescence in a significant number of tumor cells could be due to better telomere maintenance in tumor cells or higher CDK activity due to defects in p16INK4a and other CDK inhibitors. In contrast, S22A-progerin creates a thicker lamin A cage around the nucleus () activating a senescence program in tumor cells with different genetic backgrounds and telomere maintenance.
Figure 6. S22A-progerin induces tumor cell senescence. (A) Senescence associated β-galactosidase staining in the indicated cell lines expressing wild type lamin A (wtlamin A), progerin or S22A-progerin. (B) Immunofluorescence for γ-H2AX in the indicated cell lines. (C-D) Immunofluorescence for PML bodies (C) or γ-H2AX (D) of U-2 OS cells expressing wild type lamin A (wt), progerin (prog) or S22A-progerin (S22Aprog). The percent and S.D labeled cells are indicated at the bottom right of each panel. Magnification = 10 μm. (E) Immunoflourescence of cells co-stained for lamin A and γ-H2AX (F-H) qPCR for genes whose expression change with senescence in U-2 OS cells. (I) Immunoblots for the indicated proteins of U-2 OS cells with empty vector (V), S22A-progerin (S22A) or progerin (Pr). All markers in A-I were measured at day 5 post-infection.
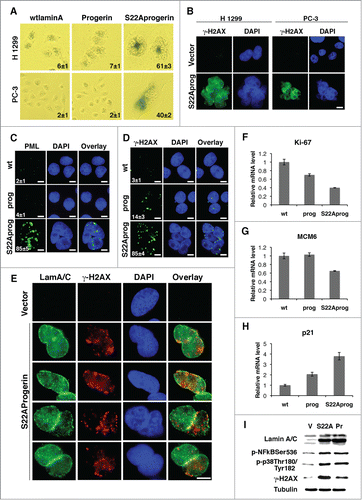
Table 1. The secretome of S22 A-progerin induced senescence in U-2 OS cells
To investigate whether the growth inhibition and senescence phenotype induced by S22A-progerin in tumor cells is sufficient to inhibit tumor formation in mice we inoculated 106 H 1299 cells expressing S22A-progerin or a vector control into nude mice. Most injections with H 1299 cells (9 out of 10) developed tumors that were detected 10 d after inoculation and grew aggressively 2 weeks after. In contrast, only 4 out of 10 injections with S22A-progerin formed tumors that appeared much later (25 d after inoculation) and were considerably smaller ().
Figure 7. S22A-progerin blocks tumor progression in mice. (A) Tumor formation in BALB/c nude mice injected with 106 H 1299 cells expressing vector or S22A-progerin (S22Aprog). (B) Relative tumor growth in BALB/c nude mice injected with H 1299 cells expressing a vector or S22A-progerin. * = p value <0.05. Error bars indicate SEM. (C) Summary of number of tumors developed in BALB/c nude mice. (D) Immunofluorescence for lamin A in tumor sections from mice inoculated with H 1299 cells expressing vector or S22A-progerin. Right panel shows the same staining in H 1299 cells before inoculation in mice. Magnification = 10 μm. (E) qPCR for LMNA (isoform A-delta50) in H 1299 cells expressing vector (V) of S22A-progerin (S22A) or from tumors as in (A). (F) Immunoblots for lamin A/C in tumors and H 1299 cells as in (A). Numbers indicate different tumors.
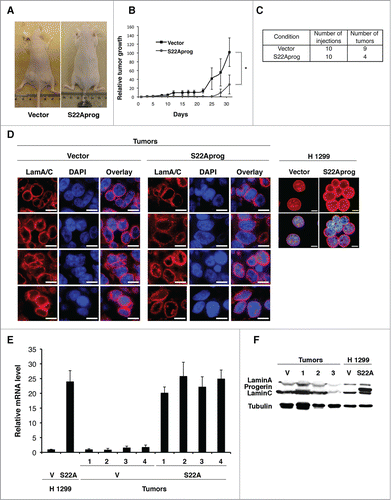
It is likely that the few and late growing tumors observed in mice injected with S22A-progerin expressing cells are the result of cells that express lower levels of lamin A. To investigate this issue we isolated tumors from mice inoculated with H 1299 cells expressing a vector control of S22A-progerin. Then we fixed the tumors and stained sections with an anti-lamin A antibody. We found a similar level of lamin A staining around the nucleus when compared tumors with empty vectors or S22A-progerin. These levels were not comparable to the high levels of lamin A around the nucleus in H 1299 cells expressing S22A-progerin before inoculation (), suggesting that tumorigenesis selected against cells with a S22A-progerin nuclear cage. Intriguingly, qPCR analysis of S22A-progerin gene expression showed that tumors expressing this gene had high levels of the mRNA comparable to the cells before inoculation () but the protein was not detectable (). The events leading to loss of S22A-progerin protein remain to be investigated. It has been reported that senescent cells can generate proliferating cells by a special kind of asymmetric cell division involving budding.Citation33 It is plausible that this mechanism may generate the tumor cells expressing low levels of S22A-progerin.
Discussion
We show here that tumor cells can be forced into a senescent cell cycle arrest by expression of a mutated form of lamin A with 2 molcular defects. First, our lamin A mutant cannot be processed by the protease ZMPSTE24/FACE1 and stays farnesylated in the nuclear lamina. Second, we mutated the phosphorylation site at serine 22, implicated in nuclear envelope disassembly during mitosis.Citation19 As a result, cells expressing S22A-progerin arrest as multinucleated senescent cells, expressing senescence biomarkers including the senescence associated secretory phenotype.
Senescence induction by S22A-progerin involves the ability of this protein to form a thick stable nuclear lamina that interferes with mitotic progression and promotes nuclear fragmentation. Consistent with this model, overexpression of S22A progerin with a mutation in its farnesylation signal prevented its localization to the nuclear membrane and totally abolished its ability to cause growth arrest or senescence. We propose that a lamin A cage physically interferes with nuclear mechanics during prophase inhibiting the nuclear envelope breakdown required for karyokinesis.Citation34 A failure to disassemble the nuclear envelope can inhibit the transition from prophase to prometaphase and eventually chromosome disjunction leading to aberrant mitosis, also called mitotic catastrophe.Citation35 Morphologically, mitotic catastrophe is associated with multinucleated cells, which ultimately undergo cell death or senescence.Citation35 Aberrant mitosis of nontransformed cells commonly leads to accumulation of G1-arrested tetraploid binucleated cellsCitation36 exhibiting markers of senescence.Citation37 In normal human cells, this G1 arrest is dependent on p53 and the p38 MAPK.Citation38 In contrast, transformed cells after tetraploidization continue DNA synthesis leading to polyploidy,Citation36,39 multinucleationCitation11 and death,Citation39 the latter associated to the spindle checkpoint.Citation29 The fact that most S22A progerin-expressing cells remain alive but senescent suggests that they do not activate the spindle checkpoint-dependent apoptosis pathway but a novel pathway linking altered prophase to senescence.
S22A-progerin induced senescence is p53-independent since it occurs in cell lines that do not express p53 (PC3 and H1299 cells) or in U 2-OS cells where p53 was knocked down by RNA interference. In fact, p53 restricted polyploidy in S22A-progerin-expressing cells. Since hyperploid can trigger senescence,Citation40 p53 null cells could be more sensitive to treatments capable of activating the S22A-progerin senescence pathway. Consistent with this interpretation, p53 can prevent cellular senescence by decreasing TOR activity and forcing cells into quiescence.Citation41 S22A-progerin induced senescence was associated to inhibition of chromosome condensation, invaginations of the nuclear membrane and DNA damage. Intriguingly, DNA damage foci in S22A-progerin expressing cells were associated to the nuclear envelope suggesting a direct effect of progerin on DNA damage. Lamins are known to bind DNA, mainly at sequences that bind the nuclear matrix called matrix attachment regions or MARs.Citation42 It is plausible that tension promoted by S22A-progerin and to a lesser extent progerin can damage DNA at these sites. Chromosome condensation requires the CDK1/cyclin B complexes and is inhibited by calcium.Citation43 It is intriguing that nuclear membrane invaginations which involve the nuclear membraneCitation44 can lead to nuclear fragmentation but also connect the calcium rich endoplasmic reticulum to the nucleusCitation45 providing a potential for calcium mediated signals to regulate mitotic progression.Citation43 These invaginations of the nuclear envelope were previously noticed in cells expressing progerinCitation46 or overexpressing wild type lamin A Citation44 and we report here that mutation of serine 22 in progerin magnifies this phenotype. The mechanism leading to nuclear invaginations remain to be investigated but as proposed above to explain DNA damage they could result from increased tension created at the nuclear envelope but higher levels of lamin A. Progerin is highly expressed in HGPS cells but is also present in old cells at lower concentrations.Citation47 CDK inhibitors such as p16INK4a increase their expression with ageCitation48 suggesting an endogenous pathway that inhibits CDKs and could prevent phosphorylation of progerin at serine 22. p16INK4a or any other factor that inhibits CDK-dependent phosphorylation of progerin can in principle potentiate its ability to induce senescence and aging.
Our results thus provide evidence for a new pathway to induce senescence effective in p53 disabled tumor cells, which is based on mechanical obstacles to karyokinesis. This pathway involves an alternative route in cell cycle progression where cells experiencing a block in prophase sidestep the rest of mitosis entering G1 and S, increasing their DNA content. A similar cell cycle variation termed mitotic slippage was described in cells where the mitotic spindle was compromised with drugs. In this case some cells manage to gradually exit mitosis entering G1 and progressing to the rest of the cell cycle.Citation49 Senescence may then act as a barrier for the expansion of cells that undergo mitotic slippage and a mechanism for tumor suppression in response to drugs targeting mitosis. It is not entirely clear whether S22A-progerin-induced senescence requires the p16/RB pathway. S22A-progerin induced senescence in HeLa cells that express the RB inhibitor E7, suggesting that RB is not essential. On the other hand in GSEA of our microarray data obtained form U-2 OS cells, there is a clear signature of E2F genes downregulation that matched data published for Ras-induced senescence (Supplemental table II). This suggests that E2Fs genes are repressed, a task normally associated to RB pathway activation. It has been proposed that the p53 pathway is sufficient to trigger senescence in the absence of RB and that both p53 and RB should be disabled to bypass ras-induced senescence.Citation50–53 The CDK inhibitor p21 can mediate RB-independent E2F repression and senescence in cells where lamin A is disabled.Citation53 However, U2-OS cells expressing a shRNA against p53 express little p21 and senesced in response to S22A-progerin. A senescence pathway acting independently of both RB and p53 is very interesting for cancer therapies because these tumor suppressors are often inactivated in advanced tumors. Further mechanistic understanding on how a lamin A nuclear cage and altered prophase force cells into senescence may help to identify druggable targets to induce senescence in advanced tumors with mutations in the p53 and RB tumor suppressor pathways.
Senescence is a promising endpoint for anticancer therapies not only due to its capability to halt cell growth but also for its ability to engage the immune system.Citation54,55 However, senescence can have both pro-tumor and anti-tumor effects and more work is needed to find treatments that avoid the pro-tumor activity of senescent cells. Otherwise, the good anticancer activity of a progerin nuclear cage may anticipate novel cancer therapies acting by providing mechanical barriers for cell cycling. Such physical barriers might work despite the mutations present in cancer cells, and therefore could be effective for advanced tumors.
Materials and Methods
Cell culture and reagents
Human non-small cells lung carcinoma H 1299 (CRL-5803, ATCC), human osteosarcoma U-2 OS cells (HTB-96, ATCC), human cervix carcinoma HeLa cells (CCL−2, ATCC) were cultured in Dulbecco's modified Eagle's medium (Wisent, St-Bruno, QC) supplemented with 10% fetal bovine serum (FBS) (Wisent). Human prostate carcinoma PC-3 cells (CRL-1435, ATCC) were cultured in RPMI-1640 medium (Wisent) supplemented with 10% fetal bovine serum (FBS) (Invitrogen) and 1% L-glutamine (Wisent). Crystal violet retention assay used for cell proliferation analysis and senescence associated β-galactosidase assay were described before.Citation51 For cell proliferation the data is expressed as relative absorbance of crystal violet extracted from the cells and diluted in 10% acetic acid.
Retroviral vectors and retroviral-mediated gene transfer
Retroviral vectors pLPC and MLP are from S.W. Lowe. pRetroSuper-shp53 and pRetroSuper-shGFP were described by Voorhoeve and Agami.Citation56 pLPC wtlaminA, pLPC L647RlaminA and pLPC progerin were constructed by subcloning wild type or mutated laminA from pMXIH-hygro vectors kindly provided by Dr. B. Kennedy. pLPC L647RlaminACS, pLPC S22AlaminACS and pLPC progerinCS were generated by substitution of cysteine in CaaX-motif by serine (TGC→TCC) using PCR. S22A point mutation was introduced into progerin by site-directed mutagenesis using forward primer 5′-AGCTCCACTCCGCTGGCACCCACCCGCATCACC-3′ and reverse primer 5′-GGTGATGCGGGTGGGTGCCAGCGGAGTGGAGCT-3′. Hairpin ZMPSTE24/FACE1 (shZMPSTE24) was designed against the sequence 5′-GATCATGGATTCTGAAACATT-3′ of the human ZMPSTE24 gene. Retroviral-mediated gene transfers were done as in.Citation57
Mice
For xenografts, BALB/c 5-wk-old nude mice were injected subcutaneously in both flanks with 106 H 1299 cells expressing pLPC-Vector or S22A-progerin. Cells were resuspended in 50 µL of PBS mixed with 50 µL of Matrigel (BD Biosciences) at 4°C. Tumor formation was evaluated 3 times per week over a period of 31 d after which the mice were euthanized.
Immunoblotting
To prepare total protein lysates, cells were collected by trypsinization, washed with phosphate-buffered saline (PBS), lysed in sodium dodecyl sulfate (SDS) sample buffer (60 mM Tris-HCl, pH 6.8, 10% glycerol, 2% SDS, and 5% β-mercaptoethanol), and boiled for 5 min. For Western blotting, 25 μg of total protein were separated on SDS-polyacrylamide gel electrophoresis gels and transferred to Immobilon-P membranes (Millipore, Bedford, MA). We used the following primary antibodies: anti-Cyclin B1 (sc-245, 1:200, Santa Cruz Biotechnology), anti-Cyclin A (sc-596, 1:200, Santa Cruz Biotechnology), anti-gamma H2AX (phospho S139) (ab11174, 1:1000, Abcam), anti-LaminA/C (636, sc-7292, 1:1000, Santa Cruz Biotechnology), anti-Lamin A/C (2032, 1:1000, Cell Signaling), anti-phospho-Ser22Lamin A (2026, 1:1000, Cell Signaling), anti-phospho-Ser10Histone H3 (06–570, 1:1000, Millipore), anti-phospho-Thr180/Tyr182p38 MAPK (4511, 1:1000, Cell Signaling), anti-phospho-Ser536NFkB p65 (3033, 1:1000, Cell Signaling), anti-p21 (sc-397, 1:500, Santa Cruz Biotechnology), anti-p53 (DO-1) (sc-126, 1:1000, Santa Cruz Biotechnology), anti-p21 (556431, 1:1000, BD PharMingen), anti-p16 (F-12) (sc-1661, 1:500, Santa Cruz Biotechnology), anti-Tubulin (T5168, 1:5,000, Sigma). Signals were revealed after incubation with anti-mouse or anti-rabbit secondary antibodies coupled to peroxidase (Amersham Pharmacia, Amersham, United Kingdom) by using enhanced chemiluminescence (Amersham Pharmacia).
Fluorescence microscopy
Cells were plated on coverslips at least 24 h prior to fixation with 4% paraformaldehyde in PBS for 15 minutes at room temperature. After washing with PBS, cells were permeabilized using 0.2% Triton X-100 in PBS–3% bovine serum albumin (BSA) solution for 5 min. The cells were then washed 3 times with PBS–3% BSA and incubated for 1–2 h at room temperature with the primary antibody: anti-lamin A/C (636) (sc-7292, 1:500, Santa Cruz Biotechnology), anti-Tubulin (T5168, 1:1000, Sigma), anti-phospho-H2AX (JBW301, 1:200, Upstate Biotechnologies), anti-PML (A301–167A, 1:200, Bethyl), anti-progerin (ab66587, 1:100, Abcam), anti-phospho-Ser22LaminA (2026, 1:200, Cell Signaling) diluted in PBS/BSA. Next, cells were washed 3 times in PBS/BSA and incubated with the appropriate secondary antibody (1:2000, AlexaFluor 488 goat anti-mouse, AlexaFluor 568 goat anti-mouse, or AlexaFluor 488 goat anti-rabbit, Molecular Probes-Invitrogen) for 1h at room temperature. Finally, cells were rinsed 3 times with PBS alone and once with PBS containing 300 nM DAPI for 10 minutes. Images from independent fields were captured with a fluorescence microscope and processed with Metamorph.
Immunofluorescence from paraffin embedding samples
Tumor sections from xenografts were deparaffinised and rehydrated. Antigen epitope retrieval was performed by heating for 25 min at 95°C in Sodium-Citrate Buffer (10 mM Sodium Citrate, 0.05% Tween 20, pH 6.0). Samples were permeabilized with Triton 0.5%-PBS solution for 15 min and blocked with 3% BSA, 5% Goat Serum, 0.1% Tween20-PBS solution for 1 h. Then, they were incubated with mouse monoclonal anti-Lamin A/C (1:200, sc-7292, Santa Cruz Biotechnology) at 4ºC overnight. The next day, samples were washed in PBS and incubated with goat anti-mouse AlexaFluor 568 (1:1000, A-11031, Molecular Probes, Invitrogen). For mounting, we used ProLong Gold Antifade Reagent with DAPI (P36931, Invitrogen).
Transmission electron microscopy
Cells were fixed in 1% glutaraldehyde for 30 min at room temperature (RT) and washed 2 times with PBS. Then, the cells were incubated with 1% osmium tetroxide solution for 1h at RT, followed by extensive washing with PBS, dehydration with ethanol, and embedding in LR White resin. 70nm sections were cut, stained with 4% uranyl acetate for 2min, and examined in a FEI Tecnai 12 electron microscope (FEI, Eindhoven, The Netherlands), operating at 80 kV.
Real-Time PCR, microarray and bioinformatic analysis of microarray data
Real-Time PCR and Microarray analysis were done as previously described.Citation58 Briefly, RNA was extracted with Trizol 4 d after infection of U-2 OS cells with wild type lamin A or S22A-progerin. Total RNA was sent to Genome Quebec (McGill University) for cRNA amplification and subsequent hybridization on GeneChIP Human Gene 2.1 ST Array Affymetrix DNA Chip. Data were analyzed using the Affymetrix Softwares Expression Console and Transcriptome Analysis Console, http://www.affymetrix.com. Data is available at GEO (GSE51349).
Statistics
Statistical analysis was performed using SPSS software 16.0 (SPSS, Inc.). The nonparametric Kruskal-Wallis test was used to show significant differences for tumor growth in mice.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
1053671_supplemental_files.docx
Download MS Word (34 MB)Acknowledgments
We thank Drs. L. Brakier-Gingras, Damien D'Amours and B. Kennedy for critical reading and reagents. We are in debt to Dr. Antonio Nanci, Faculty of Dentistry, University of Montreal, for electron microscopy.
Funding
This work was supported by grants from CIHR and the Progeria Research Foundation to GF. FL is supported by FRQS (Fonds de Recherche Santé Quebec. MA-A is supported by CONACYT (Consejo Nacional de Ciencia y Tecnologia, Mexico). GF is a FRQS “chercheur national.”
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Related Research Data
References
- Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest 2013; 123:966-72; PMID:23454759
- Baker DJ, Jeganathan KB, Cameron JD, Thompson M, Juneja S, Kopecka A, Kumar R, Jenkins RB, de Groen PC, Roche P, et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet 2004; 36:744-9; PMID:15208629
- Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011; 479:232-6; PMID:22048312; http://dx.doi.org/10.1038/nature10600
- De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer M, et al. Lamin a truncation in Hutchinson-Gilford progeria. Science 2003; 300:2055; PMID:12702809; http://dx.doi.org/10.1126/science.1084125
- Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003; 423:293-8; PMID:12714972; http://dx.doi.org/10.1038/nature01629
- Hennekam RC. Hutchinson-Gilford progeria syndrome: review of the phenotype. Am J Med Genet A 2006; 140:2603-24; PMID:16838330; http://dx.doi.org/10.1002/ajmg.a.31346
- Liu B, Wang J, Chan KM, Tjia WM, Deng W, Guan X, Huang JD, Li KM, Chau PY, Chen DJ, et al. Genomic instability in laminopathy-based premature aging. Nat Med 2005; 11:780-5; PMID:15980864
- Benson EK, Lee SW, Aaronson SA. Role of progerin-induced telomere dysfunction in HGPS premature cellular senescence. J Cell Sci 2010; 123:2605-12; PMID:20605919
- Krishnan V, Chow MZ, Wang Z, Zhang L, Liu B, Liu X, Zhou Z. Histone H4 lysine 16 hypoacetylation is associated with defective DNA repair and premature senescence in Zmpste24-deficient mice. Proc Natl Acad Sci U S A 2011; 108:12325-30; PMID:21746928
- Shumaker DK, Dechat T, Kohlmaier A, Adam SA, Bozovsky MR, Erdos MR, Eriksson M, Goldman AE, Khuon S, Collins FS, et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc Natl Acad Sci U S A 2006; 103:8703-8; PMID:16738054
- Cao K, Capell BC, Erdos MR, Djabali K, Collins FS. A lamin A protein isoform overexpressed in Hutchinson-Gilford progeria syndrome interferes with mitosis in progeria and normal cells. Proc Natl Acad Sci U S A 2007; 104:4949-54; PMID:17360355
- Goldman RD, Shumaker DK, Erdos MR, Eriksson M, Goldman AE, Gordon LB, Gruenbaum Y, Khuon S, Mendez M, Varga R, et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A 2004; 101:8963-8; PMID:15184648
- Broers JL, Ramaekers FC, Bonne G, Yaou RB, Hutchison CJ. Nuclear lamins: laminopathies and their role in premature ageing. Physiol Rev 2006; 86:967-1008; PMID:16816143; http://dx.doi.org/10.1152/physrev.00047.2005
- Glynn MW, Glover TW. Incomplete processing of mutant lamin A in Hutchinson-Gilford progeria leads to nuclear abnormalities, which are reversed by farnesyltransferase inhibition. Hum Mol Genet 2005; 14:2959-69; PMID:16126733
- Shalev SA, De Sandre-Giovannoli A, Shani AA, Levy N. An association of Hutchinson-Gilford progeria and malignancy. Am J Med Genet A 2007; 143A:1821-6; PMID:17618517
- Broers JL, Machiels BM, van Eys GJ, Kuijpers HJ, Manders EM, van Driel R, Ramaekers FC. Dynamics of the nuclear lamina as monitored by GFP-tagged A-type lamins. J Cell Sci 1999; 112 ( Pt 20):3463-75; PMID:10504295
- Peter M, Nakagawa J, Doree M, Labbe JC, Nigg EA. In vitro disassembly of the nuclear lamina and M phase-specific phosphorylation of lamins by cdc2 kinase. Cell 1990; 61:591-602; PMID:2188731; http://dx.doi.org/10.1016/0092-8674(90)90471-P
- Peter M, Heitlinger E, Haner M, Aebi U, Nigg EA. Disassembly of in vitro formed lamin head-to-tail polymers by CDC2 kinase. EMBO J 1991; 10:1535-44; PMID:1851086
- Heald R, McKeon F. Mutations of phosphorylation sites in lamin A that prevent nuclear lamina disassembly in mitosis. Cell 1990; 61:579-89; PMID:2344612; http://dx.doi.org/10.1016/0092-8674(90)90470-Y
- Hennekes H, Nigg EA. The role of isoprenylation in membrane attachment of nuclear lamins. A single point mutation prevents proteolytic cleavage of the lamin A precursor and confers membrane binding properties. J Cell Sci 1994; 107 ( Pt 4):1019-29; PMID:8056827
- Mallampalli MP, Huyer G, Bendale P, Gelb MH, Michaelis S. Inhibiting farnesylation reverses the nuclear morphology defect in a HeLa cell model for Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A 2005; 102:14416-21; PMID:16186497; http://dx.doi.org/10.1073/pnas.0503712102
- Yang SH, Chang SY, Ren S, Wang Y, Andres DA, Spielmann HP, Fong LG, Young SG. Absence of progeria-like disease phenotypes in knock-in mice expressing a non-farnesylated version of progerin. Hum Mol Genet 2011; 20:436-44; PMID:21088111; http://dx.doi.org/10.1093/hmg/ddq490
- Pendas AM, Zhou Z, Cadinanos J, Freije JM, Wang J, Hultenby K, Astudillo A, Wernerson A, Rodriguez F, Tryggvason K, et al. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat Genet 2002; 31:94-9.; PMID:11923874
- Barrowman J, Hamblet C, George CM, Michaelis S. Analysis of prelamin A biogenesis reveals the nucleus to be a CaaX processing compartment. Mol Biol Cell 2008; 19:5398-408; PMID:18923140; http://dx.doi.org/10.1091/mbc.E08-07-0704
- Gruber J, Lampe T, Osborn M, Weber K. RNAi of FACE1 protease results in growth inhibition of human cells expressing lamin A: implications for Hutchinson-Gilford progeria syndrome. J Cell Sci 2005; 118:689-96; PMID:15671064; http://dx.doi.org/10.1242/jcs.01652
- Akakura S, Nochajski P, Gao L, Sotomayor P, Matsui S, Gelman IH. Rb-dependent cellular senescence, multinucleation and susceptibility to oncogenic transformation through PKC scaffolding by SSeCKS/AKAP12. Cell Cycle 2010; 9:4656-65; PMID:21099353; http://dx.doi.org/10.4161/cc.9.23.13974
- Spano A, Monaco G, Barni S, Sciola L. Cisplatin treatment of NIH/3T3 cultures induces a form of autophagic death in polyploid cells. Histol Histopathol 2008; 23:717-30; PMID:18366010
- Joe AK, Liu H, Suzui M, Vural ME, Xiao D, Weinstein IB. Resveratrol induces growth inhibition, S-phase arrest, apoptosis, and changes in biomarker expression in several human cancer cell lines. Clin Cancer Res 2002; 8:893-903; PMID:11895924
- Sudo T, Nitta M, Saya H, Ueno NT. Dependence of paclitaxel sensitivity on a functional spindle assembly checkpoint. Cancer Res 2004; 64:2502-8; PMID:15059905; http://dx.doi.org/10.1158/0008-5472.CAN-03-2013
- Medina I, Carbonell J, Pulido L, Madeira SC, Goetz S, Conesa A, Tarraga J, Pascual-Montano A, Nogales-Cadenas R, Santoyo J, et al. Babelomics: an integrative platform for the analysis of transcriptomics, proteomics and genomic data with advanced functional profiling. Nucleic Acids Res 2010; 38:W210-3; PMID:20478823; http://dx.doi.org/10.1093/nar/gkq388
- Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 2010; 5:99-118; PMID:20078217
- Cao K, Blair CD, Faddah DA, Kieckhaefer JE, Olive M, Erdos MR, Nabel EG, Collins FS. Progerin and telomere dysfunction collaborate to trigger cellular senescence in normal human fibroblasts. J Clin Invest 2011; 121:2833-44; PMID:21670498
- Gosselin K, Martien S, Pourtier A, Vercamer C, Ostoich P, Morat L, Sabatier L, Duprez L, T'Kint de Roodenbeke C, Gilson E, et al. Senescence-associated oxidative DNA damage promotes the generation of neoplastic cells. Cancer Res 2009; 69:7917-25; PMID:19826058
- Gerace L, Blobel G. The nuclear envelope lamina is reversibly depolymerized during mitosis. Cell 1980; 19:277-87; PMID:7357605; http://dx.doi.org/10.1016/0092-8674(80)90409-2
- Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol 2011; 12:385-92; PMID:21527953; http://dx.doi.org/10.1038/nrm3115
- Andreassen PR, Lohez OD, Lacroix FB, Margolis RL. Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in G1. Mol Biol Cell 2001; 12:1315-28; PMID:11359924; http://dx.doi.org/10.1091/mbc.12.5.1315
- Panopoulos A, Pacios-Bras C, Choi J, Yenjerla M, Sussman MA, Fotedar R, Margolis RL. Failure of cell cleavage induces senescence in tetraploid primary cells. Mol Biol Cell 2014; 25:3105-18; PMID:25143403; http://dx.doi.org/10.1091/mbc.E14-03-0844
- Uetake Y, Sluder G. Prolonged prometaphase blocks daughter cell proliferation despite normal completion of mitosis. Curr Biol 2010; 20:1666-71; PMID:20832310
- Harrington EA, Bebbington D, Moore J, Rasmussen RK, Ajose-Adeogun AO, Nakayama T, Graham JA, Demur C, Hercend T, Diu-Hercend A, et al. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat Med 2004; 10:262-7; PMID:14981513; http://dx.doi.org/10.1038/nm1003
- Chuprin A, Gal H, Biron-Shental T, Biran A, Amiel A, Rozenblatt S, Krizhanovsky V. Cell fusion induced by ERVWE1 or measles virus causes cellular senescence. Genes Dev 2013; 27:2356-66; PMID:24186980
- Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci U S A 2010; 107:9660-4; PMID:20457898
- Luderus ME, den Blaauwen JL, de Smit OJ, Compton DA, van Driel R. Binding of matrix attachment regions to lamin polymers involves single-stranded regions and the minor groove. Mol Cell Biol 1994; 14:6297-305; PMID:8065361
- Lohka MJ, Maller JL. Induction of nuclear envelope breakdown, chromosome condensation, and spindle formation in cell-free extracts. J Cell Biol 1985; 101:518-23; PMID:3926780
- Gehrig K, Cornell RB, Ridgway ND. Expansion of the nucleoplasmic reticulum requires the coordinated activity of lamins and CTP:phosphocholine cytidylyltransferase α. Mol Biol Cell 2008; 19:237-47; PMID:17959832
- Malhas A, Goulbourne C, Vaux DJ. The nucleoplasmic reticulum: form and function. Trends Cell Biol 2011; 21:362-73; PMID:21514163
- McClintock D, Gordon LB, Djabali K. Hutchinson-Gilford progeria mutant lamin A primarily targets human vascular cells as detected by an anti-Lamin A G608G antibody. Proc Natl Acad Sci U S A 2006; 103:2154-9; PMID:16461887; http://dx.doi.org/10.1073/pnas.0511133103
- Scaffidi P, Misteli T. Lamin A-dependent nuclear defects in human aging. Science 2006; 312:1059-63; PMID:16645051; http://dx.doi.org/10.1126/science.1127168
- Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L, Sharpless NE. Ink4a/Arf expression is a biomarker of aging. J Clin Invest 2004; 114:1299-307; PMID:15520862; http://dx.doi.org/10.1172/JCI22475
- Kung AL, Sherwood SW, Schimke RT. Cell line-specific differences in the control of cell cycle progression in the absence of mitosis. Proc Natl Acad Sci U S A 1990; 87:9553-7; PMID:2263610; http://dx.doi.org/10.1073/pnas.87.24.9553
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997; 88:593-602; PMID:9054499; http://dx.doi.org/10.1016/S0092-8674(00)81902-9
- Ferbeyre G, de Stanchina E, Querido E, Baptiste N, Prives C, Lowe SW. PML is induced by oncogenic ras and promotes premature senescence. Genes Dev 2000; 14:2015-27; PMID:10950866
- Chan HM, Narita M, Lowe SW, Livingston DM. The p400 E1A-associated protein is a novel component of the p53 –>p21 senescence pathway. Genes Dev 2005; 19:196-201; PMID:15655109; http://dx.doi.org/10.1101/gad.1280205
- Moiseeva O, Bourdeau V, Vernier M, Dabauvalle MC, Ferbeyre G. Retinoblastoma-independent regulation of cell proliferation and senescence by the p53-p21 axis in lamin A /C-depleted cells. Aging Cell 2011; 10:789-97; PMID:21535365; http://dx.doi.org/10.1111/j.1474-9726.2011.00719.x
- Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011; 479:547-51; PMID:22080947; http://dx.doi.org/10.1038/nature10599
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007; 445:656-60; PMID:17251933; http://dx.doi.org/10.1038/nature05529
- Voorhoeve PM, Agami R. The tumor-suppressive functions of the human INK4A locus. Cancer Cell 2003; 4:311-9; PMID:14585358; http://dx.doi.org/10.1016/S1535-6108(03)00223-X
- Moiseeva O, Mallette FA, Mukhopadhyay UK, Moores A, Ferbeyre G. DNA Damage Signaling and p53-dependent Senescence after Prolonged β-Interferon Stimulation. Mol Biol Cell 2006; 17:1583-92; PMID:16436515; http://dx.doi.org/10.1091/mbc.E05-09-0858
- Moiseeva O, Deschenes-Simard X, St-Germain E, Igelmann S, Huot G, Cadar AE, Bourdeau V, Pollak MN, Ferbeyre G. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-kappaB activation. Aging Cell 2013; 12:489-98; PMID:23521863; http://dx.doi.org/10.1111/acel.12075
- Bhaumik D, Scott GK, Schokrpur S, Patil CK, Orjalo AV, Rodier F, Lithgow GJ, Campisi J. MicroRNAs miR-146a/b negatively modulate the senescence-associated inflammatory mediators IL-6 and IL-8. Aging (Albany NY) 2009; 1:402-11; PMID:20148189