
A defining feature of eukaryotic cells is the presence of a membrane-bound organelle, the nucleus, that surrounds the genetic material during most of a cell's life cycle.Citation1 The basic function of this organelle is to physically separate nuclear transactions from other metabolic activities that occur in the cytoplasm. The importance of an optimal cytoplasmic-nuclear barrier is well illustrated by the fact that mutations in lamin A—a structural component of the nuclear lamina—can cause accelerated aging disorders such as the Hutchinson-Gilford (HG) progeria syndrome.Citation2
The presence of a physical barrier between the genome and the cytoplasm raises a major regulatory challenge during cell division. Indeed, to effectively segregate their genome, mitotic cells must attach chromosomes on microtubules emanating from a cytoplasmic structure, the centrosome. The presence of a nuclear envelope separating chromosomes from centrosomes can prevent the access of microtubules to chromosomes. Most eukaryotic cells have resolved this problem by directing Cdk1 (cyclin-dependent kinase 1) to phosphorylate serine 22 of lamin A during mitosis, thus allowing disassembly of the nuclear envelope.Citation3 Interestingly, the relationship between lamin A mutations found in HG progeria syndrome and the regulatory circuits leading to nuclear disassembly in mitosis are not well understood. A recent study by Moiseeva and colleaguesCitation4 has shed new light on this question and revealed an unexpected link between the post-translational modifications of lamin A and the induction of cellular senescence.
Previous work in the field has established that expression of progerin (i.e., the unprocessed variant of lamin A found in HS progeria syndrome) is capable of inducing senescence in normal human cells, but has limited ability to promote the same process in tumor cells defective in p53 activity.Citation5,6 The reason why tumor cells showed resistance to progerin-induced senescence was unclear, which prompted Moiseeva and colleagues to test whether including a Cdk1 phosphorylation site mutation (S22A) in progerin would lead to synergistic effects on cell proliferation and senescence (). The rationale for those experiments was that loss of S22 phosphorylation would lead to higher levels of progerin accumulation in the nuclear lamina, thereby potentiating the effect of progerin on cells. As predicted, expression of S22A-progerin led to a very strong induction of senescence in tumor cells, accompanied by a corresponding reduction in cell proliferation and diminished tumorigenesis in mouse xenograph experiments. Tumors formed from S22A-progerin-expressing cells were also considerably smaller than those formed by control cells in inoculation experiments. Importantly, the induction of senescence by S22A-progerin was insensitive to the p53 status of the parental cells. These results show that preventing the Cdk1 phosphorylation of progerin potentiates the ability of this protein to engage tumor cells in the senescence program.
Figure 1. The lamin A cage traps tumor cells in a p53-independent senescence program. A schematic representation of structural elements defective in S22A-Progerin is shown at the top of the figure. The lower part of the figure illustrates the cytological consequences of S22A-Progerin expression in human cells. Note how expression of S22A-Progerin engages tumor cells in a p53-independent senescence pathway.
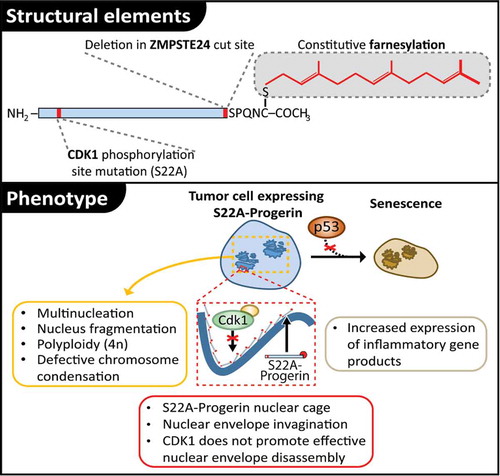
The mechanistic basis for the increased ability of S22A-progerin to induce senescence is very interesting. Microscopic examination of cells expressing this mutant protein show that they contain large invaginations of the nuclear envelope and form a much thicker nuclear lamina than cells expressing the phospho-proficient form of progerin (). It was proposed by the authors that this thick lamin A “cage” surrounding the nucleus acts as a mechanical obstacle that interferes with normal chromosome dynamics during cell division. Consistent with this view, expression of S22A-progerin prevented chromosome condensation in prometaphase, and led to nucleus fragmentation, multinucleation, and polyploidy (). Ultimately, the lamin A cage acts as a “trap” that forces cells into the senescence program.
The therapeutic implications of the discovery of a p53-independent senescence trap pathway are important since cells often loose p53 activity during tumorigenesis. In this context, finding compounds that would stimulate a S22A-progerin-like senescence pathway might be a promising new approach to treat advanced tumors in humans. This work also raises a number of fascinating questions for cell biologists. For instance, how might the lamin A cage prevent chromosome condensation is unclear since the process is normally initiated by Cdk1 in prophase and can occur in species where the nuclear envelope remains intact in mitosis.Citation7 Also, what other aspects of chromosome dynamics might be affected by S22A-progerin is unclear. Finally, the effectors of the senescence pathway induced by the lamin A cage remain to be discovered. Further work will be required to resolve these interesting questions and decipher how the formation of the lamin A cage can trap cells in senescence without the help of p53.
References
- Stewart CL, et al. Science 2007; 318:1408–12; PMID:18048680; http://dx.doi.org/10.1126/science.1142034
- Schreiber KH, et al. Cell 2013; 152:1365–75; PMID:23498943; http://dx.doi.org/10.1016/j.cell.2013.02.015
- Heald R, McKeon F. Cell 1990; 61:579–89; PMID:2344612; http://dx.doi.org/10.1016/0092-8674(90)90470-Y
- Moiseeva O, et al. Cell Cycle 2015; 14(15):2408-21; PMID:26029982; http://dx.doi.org/10.1080/15384101.2015.1053671
- Huang S, et al. Exp Cell Res 2008; 314:82–91; PMID:17870066; http://dx.doi.org/10.1016/j.yexcr.2007.08.004
- Kudlow BA, et al. Mol Biol Cell 2008; 19:5238–48; PMID:18843043; http://dx.doi.org/10.1091/mbc.E08-05-0492
- Robellet X, et al. Genes Dev 2015; 29:426–39; PMID:25691469; http://dx.doi.org/10.1101/gad.253294.114