
Abstract
Hypoxia-inducible factor 1 (HIF-1) transcriptionally promotes production of adenosine triphosphate (ATP) whereas AMPK senses and regulates cellular energy homeostasis. A histone deacetylase (HDAC) activity has been proven to be critical for HIF-1 activation but the underlying mechanism and its role in energy homesostasis remain unclear. Here, we demonstrate that HIF-1 activation depends on a cytosolic, enzymatically active HDAC5. HDAC5 knockdown impairs hypoxia-induced HIF-1α accumulation and HIF-1 transactivation, whereas HDAC5 overexpression enhances HIF-1α stabilization and nuclear translocation. Mechanistically, we show that Hsp70 is a cytosolic substrate of HDAC5; and hyperacetylation renders Hsp70 higher affinity for HIF-1α binding, which correlates with accelerated degradation and attenuated nuclear accumulation of HIF-1α. Physiologically, AMPK-triggered cytosolic shuttling of HDAC5 is critical; inhibition of either AMPK or HDAC5 impairs HIF-1α nuclear accumulation under hypoxia or low glucose conditions. Finally, we show specifically suppressing HDAC5 is sufficient to inhibit tumor cell proliferation under hypoxic conditions. Our data delineate a novel link between AMPK, the energy sensor, and HIF-1, the major driver of ATP production, indicating that specifically inhibiting HDAC5 may selectively suppress the survival and proliferation of hypoxic tumor cells.
Introduction
Hypoxia-inducible factor-1 (HIF-1) is the master transcription factor that promotes tissue angiogenesis and cellular energy homeostasis in response to hypoxia.Citation1-3 HIF-1 activation has been associated with normal development, ischemic disorders, tumorigenesis, infectious diseases and neurodegenerative disorders.Citation4-8 While HIF-1 activation alters multiple cellular processes, at cellular level, a major direct biological effect of HIF-1 activation is enhanced glycolysis which provides rapid supply of ATP.
AMP-activated protein kinase (AMPK), a conserved serine/threonine kinase, is an energy sensor of cells.Citation9,10 AMPK is a heterotrimeric enzyme consisting of α, β, and γ subunits; binding of AMP to AMPK-γ subunit exposes the catalytic domain on AMPK-α, thus activating AMPK. A cascade of AMPK-triggered phosphorylation events eventually inhibits ATP-consuming anabolic processes, stimulates ATP-producing catabolic processes and initiates autophagy.Citation11 The activation of AMPK under hypoxic or low glucose conditions has been reported.Citation12-14 However, a mechanistic link and functional interaction between AMPK activation and HIF-1 activation remain elusive.
HIF-1 is found in all metazoan as a heterodimeric transcription factor whose activity is functionally determined by the HIF-1α subunitCitation15 and association with coactivator p300/CBP. Both the protein level and the activity of HIF-1α are regulated by O2 -dependent pathways.Citation16-18 Mature HIF-1α protein is hydroxylated by a family of prolyl hydroxylases in the presence of O2, and the hydroxylated HIF-1α is ubiquitinated by an E3 ubiquitin-ligase complex containing the von Hippel-Lindau protein (VHL)Citation19-22 and subsequently degraded by the proteasome.Citation23 Citation24,25 In addition, HIF-1α transactivation activity is interrupted by another O2-dependent hydroxylase FIH-1,Citation26 which hydroxylates HIF-1α at an asparagine residue (N803), thus suppressing HIF-1α interaction with coactivator p300/CBP.Citation27,28
The observation that inhibition of HDAC activity triggers HIF-1α degradation and represses HIF-1 function Citation29,30 suggests that HIF-1 activation is subject to additional regulation. Previously, we reported that HDACI (histone deacetylase inhibitor) triggered O2-independent destabilization of HIF-1α.Citation29,31 Since Hsp70 and Hsp90 interact with HIF-1α,Citation32,33 and Hsp90 inhibitors also induce proteasomal degradation of HIF-1α in the absence of O2, it is suggested that posttranslational processing of nascent HIF-1α to its mature conformation requires the actions of molecular chaperones.Citation29,33,34 However, the functional interplay between HDACs and molecular chaperones is unclear; and the biological significance of this layer of regulation remains elusive.
The HDACIs that suppress HIF-1 include trichostatin A (TSA), SAHA (vorinostat) and LAQ824.Citation29,30 These HDACIs non-selectively inhibit all classes I and II HDACs, including HDAC1–9.Citation35,36 HDAC1 and HDAC2 are nuclear deacetylases that deacetylate histones. HDAC3 can be located in both the nucleus and the cytosol, forming complexes with other HDACs.Citation37,38 HDAC4 and 5 are closely related homologues, both shuttling between the nucleus and the cytosol.Citation39,40 HDAC6 deacetylates tubulin and binds misfolded proteins and dynein motors,Citation41,42 which may regulate the intracellular trafficking of misfolded proteins. While a consensus is that an HDAC activity is required for HIF-1α stabilization and HIF-1 activation,Citation29,30 the precise member of the HDAC involved, the underlying molecular mechanisms, and the biological significance remain unclear. As HDAC family members have diverse characteristics and function, identifying the specific HDAC required for HIF-1 is the critical step for the understanding of the underlying mechanisms and the biological significance of the deacetylase-dependent regulation.
In this report, we start with the identification of HDAC5, a member of Class IIa HDACs, as the specific HDAC member that facilitates the stabilization and nuclear accumulation of HIF-1α. We demonstrate that deacetylation of the cytosolic molecular chaperone Hsp70 by HDAC5 promotes HIF-1α interaction with Hsp90 and facilitates the rapid nuclear accumulation of HIF-1α, and show that AMPK-mediated cytosolic translocation of HDAC5 is an active cellular response to hypoxia and low glucose stresses, which facilitates HIF-1 activation.
Results
HDAC5 knockdown induces proteasome-dependent HIF-1α degradation
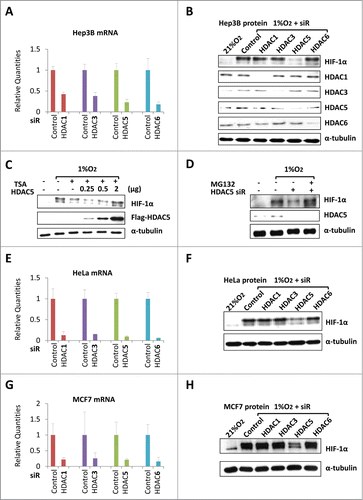
To address which HDAC is needed for HIF-1α stability, we used siRNA-based screening. We examined the effect of individually knockdown of HDAC1 (Class Ia), HDAC3 (Class Ib), HDAC5 (Class IIa) and HDAC6 (Class IIb) on hypoxic accumulation of HIF-1α. We validated the efficiency of siRNA knockdown, showing that HDAC siRNA specifically and significantly decreased the targeted mRNA () and proteins (). However, only HDAC5 knockdown remarkably impaired hypoxic accumulation of HIF-1α (), suggesting that hypoxic accumulation of HIF-1α requires HDAC5.
Figure 1. HDAC5 knockdown induces HIF-1α degradation. (A) Confirmation of HDAC knockdown in Hep3B. 42 h after siRNAs transfection, total RNA were collected and qRT-PCR was performed to determine the mRNA levels of HDACs. siR: siRNA.(B) HDAC5 knockdown attenuates hypoxia-triggered HIF-1α accumulation. Hep3B transfected with siRNA were exposed to 1% O2 for 6 h. Protein levels of HIF-1α, HDACs and α-tubulin (loading control) were determined by Western. (C) HDAC5 overexpression rescues TSA-induced HIF-1α degradation. Hep3B cells were transfected with 0.25, 0.5, 2 μg of Flag-HDAC5 and cultured in 1% O2 with TSA (300 nM) or DMSO (control) for 6 h. HIF-1α and Flag-HDAC5 were detected by Western blotting. (D) MG132 blocks HDAC5 knockdown-caused decrease of HIF-1α. Hep3B were transfected with HDAC5 or control siRNA, exposed to 1% O2 in the presence or absence of MG132 (5 μM) for 6 h. (E-H) HDAC5 knockdown blocks HIF-1α accumulation in HeLa (E-F) and MCF7 (G-H) cells. Procedures are the same as in (A) and (B). The mRNA levels of target genes were examined by qRT-PCR, and HIF-1α levels determined by Western blotting.
Previously we reported that SAHA and TSA trigger proteasome-dependent degradation of HIF-1α but do not block de novo translation of HIF-1α.Citation29 If TSA destabilizes HIF-1α through inhibiting HDAC5, overexpression of HDAC5 should be able to protect HIF-1α from TSA-induced degradation. To test this hypothesis, we treated cells overexpressing Flag-HDAC5 with TSA, and found that HDAC5 prevented TSA-induced decrease of HIF-1α levels in a dose-dependent manner (). As TSA induces proteasome-dependent HIF-1α degradation,Citation29 we next asked if the reduction of HIF-1α levels caused by HDAC5 knockdown requires the proteasome activity. We performed HDAC5 knockdown and examined HIF-1α levels in the presence of MG132, a proteasome inhibitor. We observed that in the presence of MG132, HDAC5 knockdown failed to reduce HIF-1α protein levels (). Thus, impaired hypoxic accumulation of HIF-1α in HDAC5 knockdown cells involves an accelerated proteasome degradation, recapitulating the HDACI effects on HIF-1α stability. These data indicate that HDAC5 knockdown impairs hypoxic stabilization of HIF-1α.
To further investigate whether the role of HDAC5 on HIF-1α accumulation is cell-type specific, we performed HDAC knockdown in HeLa and MCF7 cells. The efficiency of knockdown of each individual HDAC in HeLa and MCF7 was confirmed (); only HDAC5 knockdown effectively suppressed HIF-1α levels (). These data indicate that HDAC5-facilitated HIF-1α stabilization is a general mechanism existing in different cell types.
HDAC5 specific inhibitor LMK235 impairs hypoxic accumulation of HIF-1α by ubiquitination-independent pathway
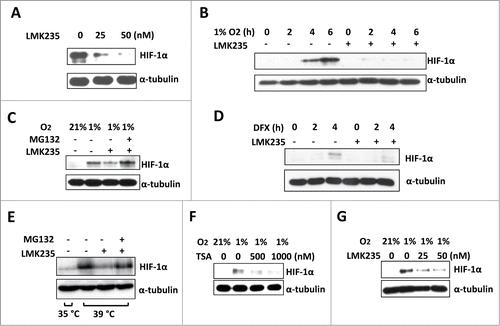
A small molecule HDAC5 specific inhibitor LMK235 (IC50 for HDAC5: 4.22 nM; IC50 of TSA for HDAC5: 520 nM) has been recently developed.Citation43 We treated Hep3B with increasing concentrations of LMK235, and found that 25 nM LMK235 was sufficient to reduce the steady-state HIF-1α levels in hypoxic cells (). Moreover, in the presence of LMK235, the time-dependent hypoxic accumulation of HIF-1α was impaired (). Similar effects were observed within HeLa and MCF7 cells (not shown). MG132 blocked LMK235-induced reduction of HIF-1α (), indicating HDAC5 activity protects HIF-1α from proteasome degradation. In addition, LMK235 was able to reduce HIF-1α accumulated by desferrioxamine (DFX), a hydroxylase inhibitor which inhibits HIF-1α hydroxylation (), suggesting LMK235-mediated HIF-1α degradation is hydroxylation-independent. To determine whether LMK235-triggered HIF-1α degradation is a ubiquitination-independent process as observed with other HDACIs,Citation29 we cultured TS20 cells, which carry a temperature sensitive ubiquitin activating enzyme (E1) caused by 2 mutations.Citation44 The restrictive temperature (39°C) inactivates E1, causing HIF-1α accumulation. LMK235 effectively induced HIF-1α degradation even E1 was inactivated, and this degradation was blocked by MG132 (). To determine if HDAC5 facilitates hypoxic accumulation of HIF-1α in non-tumor cells, we treated H9c2, immortalized cardiomyocytes generated from normal rat heart, with TSA and LMK235. We found that both effectively blocked HIF-1α accumulation (), suggesting that HDAC5 also facilitates HIF-1α accumulation in non-tumor cells. Taken together, these data indicate that specifically inhibiting HDAC5 causes ubiquitination-independent, proteasome-mediated degradation of HIF-1α. These data corroborate that lack of HDAC5 activity induces ubiquitination-independent, proteasome-dependent degradation of HIF-1α.
Figure 2. HDAC5 specific inhibitor LMK235 impairs hypoxic accumulation of HIF-1α by ubiquitination-independent pathway. (A) Dose dependent effects of LMK235 on HIF-1α. Hep3B cells were treated with 0, 25 or 50 nM of LMK235 and exposed to 1% O2 for 6 h prior to analysis. (B) Effects of LMK235 on hypoxia-induced accumulation of HIF-1α. Hep3B cells were cultured with DMSO as control (−) or 50 nM of LMK235 (+) in 1% O2 for 0 to 6 h, HIF-1α were examined by Western blotting. (C) MG132 blocks LMK235-triggered reduction of HIF-1α. Hep3B cells were cultured in 1% O2 for 6 h in the presence of 25 nM of LMK235, with or without MG132 (5 μM). (D) LMK235-induced degradation of HIF-1α is hydroxylation-independent. Hep3B cells were treated with DFX (100 μM) for 0 to 4 h in the presence or absence of LMK235 (25 nM). (E) LMK235-triggers ubiquitination-indepdendent degradation of HIF-1α. TS20 cells with temperature sensitive ubiquitin activating enzyme E1 were cultured at 39°C for 6 h with or without LMK235. The LMK235-triggered degradation was blocked by MG132 (5 μM). (F-G) Dose-dependent effect of HDACIs on HIF-1α in cardiomyocyte. H9c2 cells were cultured with 0, 500 or 1000 nM of TSA (F), or 0, 25 or 50 nM of LMK235 (G), and exposed to 1% O2 for 6 h prior to analysis for HIF-1α.
HDAC5 knockdown inhibits hypoxic stimulation of HIF-1-dependent transactivation
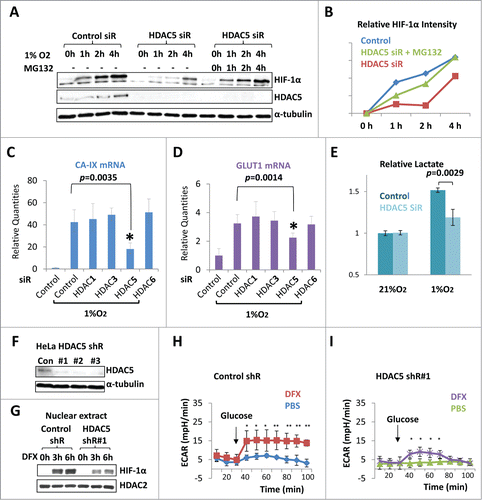
Since HDAC5 knockdown slowed down hypoxic accumulation of HIF-1α (), we examined if HDAC5 knockdown impairs HIF-1-dependent transcription. Carbonic anhydrase 9 (CA-IX) and glucose transporter 1 (GLUT1) are 2 well-known metabolic enzymes mainly regulated by HIF-1 transactivation activity. We sought to examine the mRNA levels of CA-IX and GLUT1 in each HDAC-specific knockdown cells (). 6 h of hypoxia overtly increased their expression levels. However, only HDAC5 knockdown significantly blunted the hypoxic upregulation of CA-IX (p = 0.0035, ) and GLUT1 (p = 0.0014, ). Increased glycolysis and lactate formation best demonstrate the physiological function of HIF-1 activation. To test whether HDAC5 knockdown affects glycolysis, we measured the lactate yield of cells cultured in 21% or 1% O2. Hypoxia stimulated glycolysis and lactate production (), however, HDAC5 knockdown cells showed approximately 30% reduction in hypoxia-stimulated lactate production (). We generated HDAC5 knockdown HeLa cells with lentiviral-shRNA and confirmed the knockdown effects (). Since the Seahorse metabolic analyzer is not compatible with true hypoxia, we used DFX to stimulate HIF-1. HDAC5 knockdown suppressed DFX-triggered accumulation of HIF-1α () and lactate production, which is indicated by the extracellular acidification rate (ECAR) (). Taken together, these data indicate that HDAC5 is required for optimal hypoxic activation of HIF-1 target genes.
Figure 3. HDAC5 knockdown attenuates hypoxia-induced HIF-1α accumulation and impairs hypoxic activation of HIF-1 function. (A) Effect of HDAC5 knockdown on HIF-1α accumulation in response to hypoxia. Hep3B cells were transfected with HDAC5 or control siRNA. After 44 h, cells were exposed to 1% O2 for 0 to 4 h in the presence or absence of MG132 (5 μM). HIF-1α levels at each time point were determined. (B) Quantification of data in (A). The HIF-1α levels in were quantified by ImageJ software and normalized to α-tubulin. (C-D) HDAC5 knockdown represses the expression of HIF-1 target genes. 42 h after transfection with indicated siRNAs, MCF7 cells were exposed to 1% O2 for 6 h. The mRNA levels of CA-IX and GLUT1 were determined by qRT-PCR as indicators of HIF-1 function. (E) HDAC5 knockdown represses hypoxia-stimulated lactate fermentation. HeLa Cells transfected with control or HDAC5 siRNA were cultured for 48 h in 21% or 1% O2. Media were collected, lactate levels measured and normalized to cell numbers. (F-G) HDAC5 knockdown by shRNA represses nuclear accumulation of HIF-1α. HeLa cells transduced with control or HDAC5 shRNA were cultured with hydroxylase inhibitor DFX for 0-6 h prior to nuclear extract preparation. HDAC2 was detected as a control. (H-I) HDAC5 knockdown represses HIF-1-stimulated lactate formation. Control or HDAC5 shRNA transduced HeLa cells (20,000) were seeded into Seahorse XF24 microplates. Cells were treated with DFX for 4 h to activate HIF-1. The ECAR was recorded (* p < 0.05, **p < 0.01, shR: shRNA).
Cytosolic deacetylase activity of HDAC5 is required for HIF-1α stabilization
HDAC5 dynamically shuttles between the nucleus and the cytosol.Citation40 HDAC5 performs its transcriptional repressive function by interacting with other nuclear partners independent of its intrinsic deacetylase activity.Citation45 To investigate whether the intrinsic deacetylase activity is required for HDAC5 to stabilize HIF-1α, we constructed a Flag-HDAC5 mutant deficient for deacetylase activity. Two mutations in the catalytic domain of HDAC4 (C669/H675A) were reported to completely abolish its deacetylase activity.Citation46 Since the catalytic domains of HDAC4 and HDAC5 are highly conserved (), mutating the conserved residues C698/H704 of HDAC5 to alanine was expected to abolish HDAC5 deacetylase activity. We confirmed that the HDAC5-C698/H704A mutant was unable to deacetylate α-tubulin compared with WT HDAC5 (). We next used the C698/H704A mutant and tested its ability to facilitate HIF-1α stabilization. Western blotting revealed that the HDAC5-C698/H704A mutant was unable to stabilize HIF-1α (), indicating that the deacetylase activity is required for HDAC5 to stabilize HIF-1α.
Figure 4. The cytosolic HDAC5 deacetylase activity is required for HIF-1α stabilization. (A) The schematic structures of HDAC5 and 4. (B) HDAC5-C698A/H704A mutant is deficiency of deacetylase. Hep3B cells were transfected with 2 μg of vectors, HDAC5-C698A/H704A or wt-HDAC5. (C) Deacetylase activity of HDAC5 is required for HIF-1α accumulation. Hep3B cells were transfected with HA-HIF-1α and 2 μg of vector, HDAC5-C698A/H704A or WT-Flag-HDAC5. HIF-1α and Flag-HDAC5 were examined. (D) Schematic structure of cytosolically and nuclearly localized HDAC5 mutants. Mutations were introduced in NLS (S278/279A) or NES (L1092A). (E) Subcellular localization of HDAC5 mutants. Hep3B cells were transfected with 2 μg of Flag-HDAC5-WT, HDAC5-L1092A or HDAC5-S278/279A. (F) The cytosolic HDAC5 is sufficient to stabilize HIF-1α. Cells were transfected with 2 μg of HA-HIF-1α and 2 μg of vector, Flag-HDAC5-WT, Flag-HDAC5-S278/279A (cytosol) or Flag-HDAC5-L1092A (nucleus). HIF-1α and Flag-HDAC5 were examined by Western blotting.
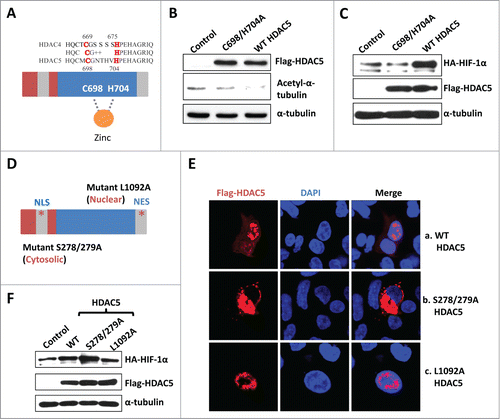
HDAC5 contains both a nuclear localization signal (NLS) and a nuclear export signal (NES), which regulate HDAC5 shuttling between the nucleus and the cytosol.Citation40 Mutating S278/279 to alanine blocks HDAC5 nuclear localization, whereas the L1092A mutation blocks nuclear export.Citation40,47 To determine whether HDAC5 stabilizes HIF-1α through a nuclear or cytosolic function, we generated the S278/279A and L1092A mutants () and confirmed their subcellular localization (, full images see Fig. S1). WT flag-HDAC5 was detected in both the nucleus and the cytosol (a) whereas the HDAC5-S278/279A mutant was found only in the cytosol (); and the HDAC5 L1092A mutant was observed predominantly in the nucleus (). Overexpression of the nuclearly localized HDAC5 mutant barely increased the HIF-1α levels, whereas overexpression of the cytosolically localized HDAC5 mutant significantly increased HIF-1α protein levels compared to control (), indicating that the cytosolically localized HDAC5 facilitates HIF-1α stabilization.
HDAC5 facilitates nuclear accumulation of HIF-1α
We next explored how the cytosolic activity of HDAC5 might affect the stability and activity of HIF-1α, a nuclear protein. As a transcription factor, nuclear localization of mature HIF-1α is critical for its transactivation activity. After translation, nascent HIF-1α must undergo post-translational processes in the cytosol prior to entering the nucleus (). Previously, we demonstrated that HDACIs promoted HIF-1α-Hsp70 interaction but impaired HIF-1α-Hsp90 interaction, suggesting HDACIs may disrupt HIF-1α folding process thus generating unstable, misfolded HIF-1α.Citation29 Expression of recombinant proteins often overwhelms the endogenous folding capacity, providing a model to study the role of HDAC5 in the posttranslational processing of HIF-1α. To investigate whether HDAC5 activity facilitates the processing of nascent HIF-1α into mature conformation thus being nuclear import-competent, we examined the effect of HDAC5 on the stability and nuclear localization of co-transfected HA-HIF-1α. We found that overexpressed HA-HIF-1α was mainly present in the cytosolic fractions (), consistent with a non-functional status. However, when HDAC5 was co-overexpressed, the majority of HA-HIF-1α was observed in the nuclear fraction. Immunofluorescent staining of HIF-1α and HDAC5 revealed that in cells lacking overexpressed HDAC5, HA-HIF-1α was mostly localized in the cytosol (); whereas in cells with overexpressed HDAC5, HA-HIF-1α was predominantly observed in the nucleus (). To address whether HDAC5-enhanced nuclear localization of HIF-1α has a functional consequence, we measured the mRNA levels of HIF-1 target genes CA-IX and GLUT1 (). The mRNA level of HIF-1α was not affected by HDAC5 co-transfection (). Importantly, the mRNA levels of CA-IX and GLUT1 were not significantly increased by HIF-1α overexpression alone (), but were significantly enhanced by the co-transfection of both HDAC5 and HIF-1α (). To further determine the importance of HDAC5 for nuclear accumulation of endogenous HIF-1α, we exposed cells to hypoxia or DFX in the presence or absence of LMK235. Immunofluorescent staining () showed that endogenous HIF-1α induced by either hypoxia or DFX was predominantly nuclear, consistent with sufficient folding capacity with normal HDAC5 activity (). In the presence of LMK235, a relatively weak signal of HIF-1α was detected, which was mainly in the cytosol (). Taken together, these data indicate that the cytosolic HDAC5 activity facilitates the nuclear localization of HIF-1α, thereby promoting functional activation of HIF-1.
Figure 5. HDAC5 facilitates nuclear accumulation and potentiates the transactivation activity of exogenously expressed HIF-1α. (A) Posttranslational processes of HIF-1α. The cytosolic HIF-1α represents the portion undergoing posttranslational processes, while nuclear HIF-1α represents the mature form. (B-D) HDAC5 promotes nuclear localization of overexpressed HA-HIF-1α and potentiates its transactivation activity. Hep3B cells were transfected with 2 μg of HA-HIF-1α and 2 μg of flag-HDAC5. Cytosolic and nuclear extracts were separated and examined (B). Cells were stained with anti-Flag, anti-HIF-1α and DAPI. A representative microimage showing a cell expressing HIF-1α alone and a cell expressing both HIF-1α and HDAC5 in the same microscopic field was presented (C). The mRNA levels were determined by qRT-PCR (D). (F) HDAC5 inhibition blocks HIF-1α nuclear localization. Hep3B cells were cultured in 1% O2 or with DFX (100 μM) for 6 h in the presence or absence of 25 nM of LMK235.
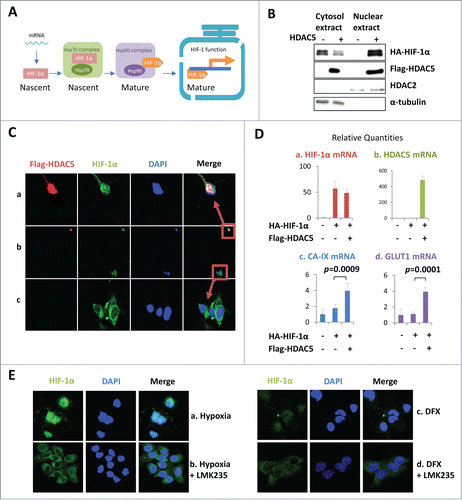
HDAC5 deacetylates Hsp70 and enhances Hsp90-HIF-1α interaction
HIF-1α is a client protein of the Hsp70-Hsp90 molecular chaperone system, and HDACIs promote HIF-1α-Hsp70 interaction but reduce HIF-1α-Hsp90 interaction.Citation29 To determine if HDAC5 regulates the dynamic interaction of HIF-1α with Hsp70 and Hsp90, we co-immunoprecipited HIF-1α, and examined the effects of HDAC5 knockdown on the interaction of HIF-1α with Hsp90 and Hsp70. HDAC5 knockdown resulted in decreased Hsp90 and increased Hsp70 in the HIF-1α complexes (). Overexpressing HDAC5 resulted in approximately 80% more Hsp90 but 30% less Hsp70 co-precipitated with HIF-1α (). These data suggest that HDAC5 facilitates HIF-1α transfer from the Hsp70 complex to the Hsp90 complex.
Figure 6 (See previous page). HDAC5 deacetylates Hsp70, attenuates Hsp70-HIF-1α interaction but enhances Hsp90-HIF-1α interaction. (A) Hep3B cells were treated as indicated and lysates were immunoprecipitated with anti-HIF-1α. The same blot was firstly used to detect Hsp90 and Hsp70, and then HIF-1α. MG132 (5 μM) was added to balance HIF-1α levels. (B) Hep3B cells were co-transfected with 2 μg of HA-HIF-1α and 2 μg of flag-HDAC5. Cell lysates were immunoprecipitated with anti-HIF-1α followed by detecting coimmunoprecipitated Hsp90 and Hsp70. (C) Hep3B cells were transfected with control or HDAC5 siRNA and exposed to 1% O2 in the presence of MG132 (5 μM) for 6 h. Cell lysates were immunoprecipitated with anti-acetyl-lysine antibody. Precipitated Hsp90 and Hsp70 were detected sequentially by Western blotting. (D-E) HDAC5 deacetylates Hsp70 in cultured cells. D. Hep3B cells were co-transfected with flag-HDAC5 and Flag-Hsp70, or Flag-HDAC3 and Flag-Hsp70. 5% of cell lysates were used to detect indicated proteins (left), and remaining cell lysates were immunoprecipitated with anti-Hsp70 or non-specific IgG. Acetyl-K and Flag-Hsp70 were detected sequentially. E. Hep3B cells were transfected with wt-HDAC5 or HDAC5C698/H704A mutant. Cell lysates were immunoprecipitated with anti-Hsp70 followed by examining Acetyl-K proteins and Hsp70. (F) HDAC5 directly deacetylates Hsp70 in vitro. Hep3B cells were transfected with Flag-Hsp70. Flag-Hsp70 was purified by immunoprecipitation with anti-flag antibody, and incubated with or without 60 ng of recombinant GST-HDAC5 (rHDAC5) at 37 °C for 30 min in deacetylase buffer. Acetylated Hsp70 and total Hsp70 were detected sequentially. (G) Overexpression of Hsp70 impairs HIF-1α accumulation, which is reversed by HDAC5. Cells were transfected with 2 μg of HA-HIF-1α. In addition, the following plasmids were cotransfected: (1) 2 μg control vector; (2) 1 μg Flag-Hsp70, 1 μg control vector; (3) 1 μg Flag-HDAC5, 1 μg control vector; and (4) 1 μg Flag-Hsp70 and 1 μg of Flag-HDAC5. (H) Schematic representing Hsp70 acetylation sites. (I) HDAC5 deacetylates Hsp70 at K559/561. From left to right, Hep3B cells were transfected with: 1. control siRNA+Hsp70 WT; 2. HDAC5 siRNA+Hsp70 WT; 3. control siRNA+Hsp70 K559/561R; 4. HDAC5 siRNA+Hsp70 K559/561R. Cells were exposed to 1% O2 for 6 h. Cell lysates were immunoprecipitated with anti-acetyl-K. (J) Deacetylation of Hsp70 at K559/561 enhances the stabilization of HIF-1α. Cells were transfected with empty vectors, Hsp70 or K559/561R as indicated and exposed to 1% O2 for 6 h. Hsp70 was immunoprecipitated with anti-Flag and associated HIF-1α was detected by Western blotting.
Since all HIF-1α, Hsp90 and Hsp70 have been reported to be acetylated proteins,Citation42,48-51 we next asked if HDAC5 deacetylates one or more of these proteins. HDAC5 knockdown did not significantly affect the total levels of Hsp90, Hsp70 or HIF-1α in the presence of MG132 (, Fig. S2A, right panel), and HIF-1α acetylation levels did not seem to be affected by HDAC5 knockdown either (Fig. S2). A search for K-acetylated proteins from total lysates of HDAC knockdown cells revealed a 70KD candidate (Fig. S2B), and further studies confirmed that HDAC5 knockdown resulted an overt increase of acetylated Hsp70 (), indicating that acetylated Hsp70 is a cytosolic substrate of HDAC5. Next, we co-transfected Flag-Hsp70 with either HDAC5 or HDAC3 into Hep3B cells, immunoprecipitated Hsp70 and examined the acetylation status of Hsp70. We observed that overexpression of HDAC5, but not HDAC3, reduced Hsp70 acetylation (). Compared with wt HDAC5, HDAC5-C698/H704A, the inactive mutant, failed to reduce the acetylation levels of endogenous Hsp70 (). To test if Hsp70 is directly deacetylated by HDAC5 or a deacetylase activity associated with HDAC5, we used purified recombinant HDAC5 to treat Flag-Hsp70 purified from Hep3B cells, and this in vitro treatment apparently reduced Flag-Hsp70 acetylation (), suggesting HDAC5 directly deacetylates Hsp70. Taken together, these data show that HDAC5 deacetylates Hsp70 in cultured cells and in vitro, indicating that acetylated Hsp70 is likely a bona fide cytosolic substrate of HDAC5.
To further investigate the interplay between Hsp70 and HDAC5 on HIF-1α stability, we co-transfected Hsp70 with HIF-1α, or together with HDAC5 (). We found that overexpression of Hsp70 decreased HIF-1α levels; however, co-overexpression of HDAC5, which induces Hsp70 deacetylation, prevented HIF-1α degradation caused by Hsp70 overexpression (), further confirming that deregulated interaction between Hsp70 and HIF-1α triggers HIF-1α degradation.
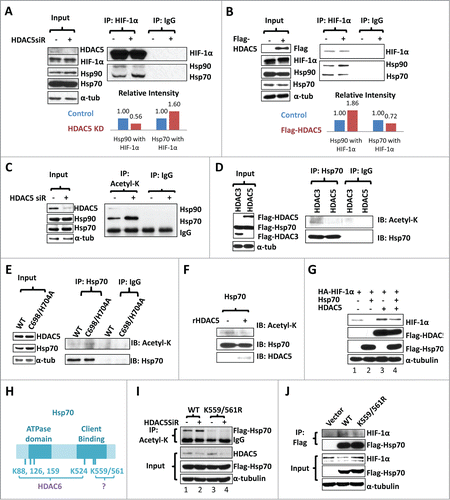
A recent study with mass spectrometry shows that Hsp70 is acetylated at 6 different lysyl residues.Citation49 Four sites (K88, 126, 159, 524) are subject to deacetylation by HDAC6,Citation49 while the deacetylase modifying K559 and K561, which are located at the client binding domain of Hsp70, remains unknown (). To determine if acetylation of K559/561 affect Hsp70 binding to HIF-1α, we created the K559/561R mutant. Compared with wt Hsp70, the K559/561R mutant showed reduced acetylation levels, which were not affected by HDAC5 knockdown (). Consistently, the K559/561R mutant showed a decreased affinity for HIF-1α (). These data suggest that HDAC5 deacetylates acetylated K559/561 of Hsp70, and that K559/561 acetylation deregulates HIF-1α-Hsp70 interaction.
Cytosolic shuttling of HDAC5 triggered by AMPK is critical for rapid HIF-1α accumulation
AMPK phosphorylates HDAC5 on Serine 259/498, which regulates HDAC5 shuttling to the cytosol Citation40. Glucose depletion and hypoxia activate AMPK Citation12-14(Fig. S3A). We found hypoxia was sufficient to enhance cytosolic levels of endogenous HDAC5 (), and this cytosolic localization was blocked by Leptomycin B (LMB), a general inhibitor of protein nuclear export (). Glucose starvation Citation52 and exposure to AICAR, a specific AMPK activator also promoted cytosolic translocation of HDAC5 (). Compound C, a specific AMPK inhibitor, blocked HDAC5 nuclear export and HIF-1α accumulation, so did LMB (). In addition, we generated HDAC5 S259/498A, which cannot be phosphorylated by AMPK,Citation40,53 thus being resistant to cytosolic translocation by AMPK (Figure S3B). Upon glucose deprivation, wt HDAC5 enhanced the accumulation of HIF-1α while the HDAC5-S259/498A mutant showed no effect (). Moreover, in normal conditions, the cytosolic HDAC5 mutant S278/279A stabilized HIF-1α more effectively than wt HDAC5, which is only partially localized to the cytosol (). Under AMPK-activated conditions (hypoxia or glucose starvation) that shift wt HDAC5 to the cytosol, wt HDAC5 and the S278/279A mutant stabilize HIF-1α with similar efficiency, supporting the notion that the cytosolic translocation of HDAC5 facilitates HIF-1α stabilization.
Figure 7. AMPK-triggered cytosolic shuttling of HDAC5 facilitates HIF-1α accumulation. (A) Effect of AMPK on HDAC5 subcellular localization. HeLa cells with stable Flag-HDAC5 were established and cultured in with 0.25 mM AICAR, or in 1% O2 for 6 h, or in glucose depleted medium (0 mM Glc) for 12 h. Cells were stained with anti-Flag and DAPI. (B) AMPK inhibition impairs hypoxic accumulation of HIF-1α. Hep3B cells were exposed to 1% O2 for 0–4 h in the presence of Compound C (Comp C, 20 μM) or DMSO. (C) Blocking nuclear export reduces hypoxic stabilization of HIF-1α. Hep3B cells were exposed to 1% O2 for 6 h in the presence or absence of LMB (20 ng/ml). (D) AMPK-facilitated HIF-1α stabilization depends on cytosolic localization of HDAC5. Hep3B cells were transfected with 2 μg of HA-HIF-1α and 2 μg of WT or the AMPK-resistant HDAC5 S259/498A mutant. Cells were cultured in glucose-free medium for 12 h. (E) AMPK enhances the ability of HDAC5 to stabilize HIF-1α. Hep3B cells were transfected with 2 μg of HA-HIF-1α and 2 μg of wt HDAC5 or cytosol-localized HDAC5 mutant (S278/279A). Cells were cultured in 21% or 1% O2 for 6 h, or in glucose free media for 12 h. (F-H) Hypoxia upregulates HDAC5 expression. Hep3B cells were cultured in 21% or 1% O2 for 6 h. HDAC5 and HIF-1α protein levels were determined by Western blotting (F). The mRNA levels of CA-IX (G: as positive control) and HDAC5 (H) were determined by qRT-RCR.
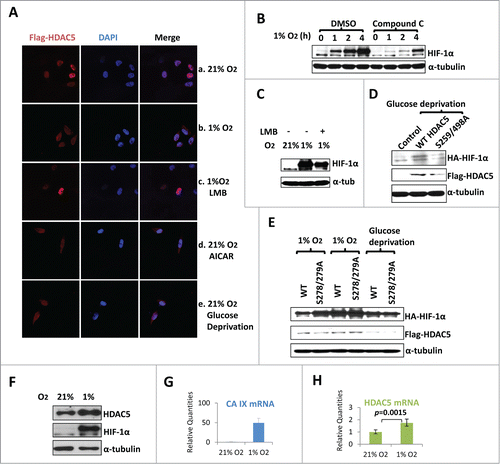
Among normal tissues, HDAC5 is highly expressed in cardiac muscle and brain, 2 tissues that are highly sensitive to hypoxia.Citation54 We asked if hypoxia also affects HDAC5 expression in addition to the AMPK-triggered cytosolic localization. After exposing Hep3B cells to 1% O2 for 6 h, HDAC5 expression was upregulated at both protein and mRNA levels (), further supporting a role of HDAC5 in cellular response to hypoxia.
Taken together, these data demonstrate that hypoxia-triggered cytosolic translocation of HDAC5 and upregulation of HDAC5 promote rapid nuclear accumulation of functional HIF-1α, and AMPK activation represents a physiological signaling pathway that facilitates HIF-1 activation via promoting the cytosolic translocation of HDAC5.
HDAC5 inhibition preferentially suppresses tumor cell proliferation under hypoxic conditions
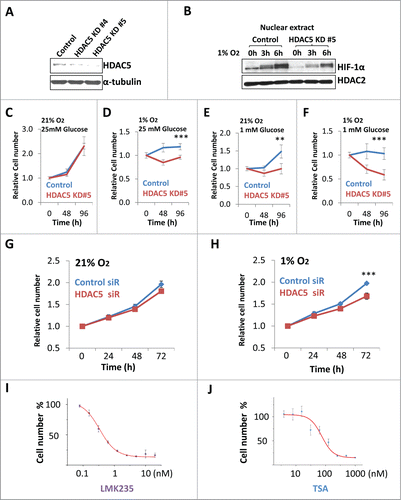
The AMPK-HDAC5 pathway represents a critical adaptive response to support cell survival and proliferation under hypoxic conditions. We next asked if inhibiting HDAC5 is sufficient to specifically suppress tumor cell proliferation under hypoxic conditions. Using CRISPR-based genome editing,Citation55 we deleted one allele of HDAC5 from Hep3B cells (), and Hep3B (HDAC5 +/−) cells showed slower nuclear accumulation of HIF-1α after exposure to hypoxia (). When cultured in normal culture conditions, Hep3B (HDAC5 +/−) cells proliferated at a rate similar to the parental Hep3B (). When cultured in 1% O2 or 1 mM glucose, Hep3B cells proliferate at reduced rate, but (HDAC5 +/−) cells proliferated at a rate range from extremely slow to total arrest (). While Hep3B cells survived with 1 mM glucose under hypoxia up to 96 h, significant numbers of Hep3B (HDAC5 +/−) cells died (). Similarly, siRNA-based HDAC5 knockdown impaired HeLa proliferation, particularly under hypoxic conditions (Fig. 8G, H). Finally, to test if HDACI-induced suppression of tumor cell proliferation is correlated to inhibiting HDAC5, we compared the effects of TSA, the general HDAC inhibitor and the HDAC5 specific inhibitor LMK235 on Hep3B cell proliferation in 1% O2 and analyzed the correlation between the IC50 for cell proliferation and IC50 for deacetylase activities (). LMK235, with a low IC50 for HDAC5 (4.22 nM) and higher IC50 for HDAC1 (320 nM) or HDAC6 IC50 (56 nM), gave an IC50 of 0.4 nM for Hep3B cell proliferation (). TSA, which has a higher IC50 (520 nM) for HDAC5 and a low IC50 for HDAC1 (0.4 nM), HDAC3 (1.0 nM) and HDAC6 (2.0 nM),Citation43, Citation56 showed an IC50 of 87 nM for Hep3B cell proliferation (). Similar results were obtained when HeLa cells or MCF7 cells were used (data not shown). These data suggest that HDACIs inhibit tumor cell proliferation is largely by suppressing HDAC5.
Figure 8 (See previous page). Inhibition of HDAC5 suppresses tumor cell proliferation under hypoxic conditions. (A) CRISPR-based knockdown of HDAC5 in Hep3B. (B) HDAC5 knockdown in Hep3B cells attenuates nuclear accumulation of HIF-1α. Control Hep3B or HDAC5 KD#5 cells were exposed to 1% O2 for 0, 3 and 6 h prior to nuclear extract preparation. HDAC2 were used as loading control. (C-F) Effects of HDAC5 knockdown on Hep3B cell survival and proliferation under hypoxic and/or low glucose conditions. HDAC5-KD or control Hep3B cells were seeded into 96-well microplates, each condition with 6 replicates. One microplate was measured 4 h after seeding. Other microplates were incubated in 21% or 1% O2 with indicated glucose concentrations and cell numbers were determined after 48 and 96 h culture. Both HDAC5-KD lines were tested and results are similar; and data from KD#5 cells are shown. (G, H) SiRNA-based HDAC5 knockdown represses HeLa cell proliferation under hypoxic conditions. HeLa cells were treated with HDAC5 siRNA or control siRNA. After 24 h and seeded into 96-well microplates, allowing 8 replicates for each experimental condition. One microplate was measured 4 h after seeding. The other microplates were cultured in 21% (G) or 1% (H) O2. Cell numbers were determined at 24, 48 or 72 h. (I, J) The inhibitory efficacy of HDACIs on tumor cell proliferation positively correlates to HDAC5 inhibition. Hep3B cells were seeded into 96 well microplates and cultured with various concentrations of LMK235 (I) or TSA (J) for indicated time. Cell proliferation inhibition was expressed as a percentage of the cell numbers obtained, with DMSO-treated negative control samples as 100%.
Discussion
Tissue hypoxia plays a regulatory role in normal physiology such as development and aging, and a pathological role in many diseases including cancer, heart disease, stroke, neurodegeneration and inflammation.Citation4-8 Activated by tissue hypoxia, HIF-1 and AMPK pathway are the 2 most important regulators of cellular responses to hypoxia, maintaining energy homeostasis in hypoxic cells. While biochemical mechanisms underlying the activation of HIF-1 by hypoxia and the activation of AMPK pathway by ATP shortage are well-known, a physiological and biochemical interaction between these 2 pathways was not clear. The observation that both HDAC inhibitors and Hsp90 inhibitors trigger O2-independent, ubiquitination-independent degradation HIF-1α suggests an additional mechanism involved in HIF-1 activation. In this study, we used siRNA to screen representative members of HDACs and found that specifically knockdown of HDAC5 induces HIF-1α degradation and suppresses HIF-1 activation. Importantly, we show that Hsp70 is a cytosolic substrate of HDAC5, and its deacetylation is enhanced by metabolic stresses (low ATP status caused by hypoxia or low glucose) that induce cytosolic translocation of HDAC5. These findings directly link AMPK, the cellular energy sensor to HIF-1, the regulator of glucose metabolism and ATP production.
HDAC5 has transcription repressive function in the nucleus, but its cytosolic function was less clear. HDAC5 suppresses transcription by interacting with other transcription regulators, a mechanism that does not depend on its intrinsic deacetylase activity.Citation45 HDAC5 has weak enzymatic activity on nuclear substrates such as histonesCitation37; thus its enzymatic activity was considered not important for biological function. While HDAC5 has been known to shuttle between the cytosol and the nucleus for a long time, nuclear export of HDAC5 was considered a passive sequestration mechanism which relieves the transcription suppression of HDAC5.Citation40,57-59 Our study for the first time shows that the AMPK-triggered cytosolic translocation of HDAC5 represents an important mechanism for cells to cope with hypoxic stress, demonstrating an active role of HDAC5 deacetylase activity in the cytosol, which facilitates cell adaptation to metabolic stresses. Recently HDAC5 was reported to translocate to cytosol in injured neurons thereby promoting axon regeneration,Citation60 which also supports an active role of HDAC5 in the cytosol in response to cell stresses.
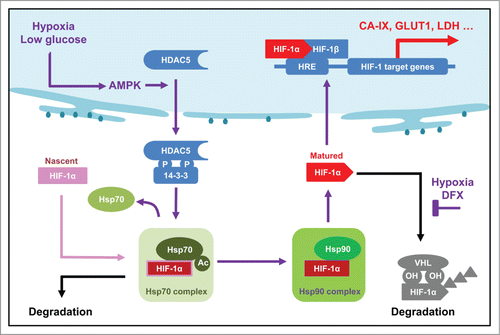
Our data indicate that a cytosolic HDAC5 activity facilitates HIF-1α stabilization and rapid nuclear accumulation in response to metabolic stresses. These data provide a clear mechanistic understanding of the findings that HDACIs trigger hydroxylation-independent, VHL-independent, proteasomal degradation of HIF-1α,Citation29 which is summarized in . As a transcription factor, HIF-1α is a nuclear protein, and its stability is physiologically regulated by O2. However, like most other proteins, de novo HIF-1α is translated and undergoes posttranslational processes in the cytosol prior to assuming its functional native conformation. It was proposed that nascent HIF-1α first binds to Hsp70 complex and then is delivered to Hsp90 complex to complete the maturation processes, which requires a deacetylase activity.Citation29 This study identifies HDAC5 as the deacetylase activity which directly deacetylates Hsp70 in the cytosol and regulates HIF-1α maturation, illustrating the mechanistic details. In the updated model shown in , lack of cytosolic HDAC5 deacetylase could cause hyperacetylation of Hsp70, thereby deregulating Hsp70-HIF-1α interaction, leading to an increase of misfolded HIF-1α in the cytosol and subsequent degradation. As such, lack of cytosolic HDAC5 deacetylase activity impairs the nuclear accumulation of HIF-1α and the eventual activation of HIF-1. Additional evidence from other independent studies that support this novel concept includes: 1) Hsp70 acetylation is enhanced by HDACIs Citation50,51; 2) Hsp70 ϵ-acetylation at multiple lysyl residues has been confirmed by mass spectrometry Citation49; 3) A physical interaction between HDAC5 and Hsp70 has been shown by multiple groups independently Citation61 and confirmed in this study (not shown); and 4) Deacetylated Hsp70 could enhance molecular chaperone client stability.Citation50,51
Figure 9. Updated model showing the role of AMPK-HDAC5 in HIF-1 activation as a response to hypoxia. Hypoxia-induced decrease of ATP level is couple to an increase of AMP levels, which activates AMPK. HDAC5 is phosphorylated by AMPK and exported to cytosol, where it deacetylate Hsp70, facilitates the transfer of Hsp70 associated HIF-1α to Hsp90 complex to complete the maturation process. Therefore, lack of cytosolic HDAC5 causes hyperacetylation of Hsp70, thereby deregulating Hsp70-HIF-1α interaction, leading to an increase of pre-mature HIF-1α in the cytosol and an inhibition of nuclear accumulation of HIF-1α.
Hypoxia-induced activation of HIF-1 has been well known, and hypoxia also activates AMPK.Citation12-14 AMPK was found to repress HIF-1α translation through inhibiting the mTORC1 signaling pathway.Citation62 Our findings directly link AMPK-induced HDAC5 nuclear exporting to HIF-1α folding and stabilization, establishing a new cross-talk between cellular energy sensing pathway and the HIF-1-promoted ATP generating metabolic pathway. While AMPK activation triggered by hypoxia or glucose starvation globally suppresses protein translation by inhibiting mTORC1, the increased efficiency of molecular chaperone system to process HIF-1α ensures a rapid nuclear accumulation of functional HIF-1α under metabolic stresses. This regulation may be dispensable for cells in normal physiological conditions, whereas it may become critical in cells undergoing frequent and severe stresses and demanding for increase of ATP production through glycolysis.
Since the original report that ARD1 acetylates HIF-1α at K532 and HDACI blocks its deacetylation thus triggering VHL-mediated ubiquitination of HIF-1α,Citation48 the HIF-1α acetylation triggered VHL-dependent degradation model, which shares great similarity with the hydroxylation-initiated VHL-dependent degradation,Citation20 has become a paradigm. Even though this model has been subsequently challenged by reports from independent researchers,Citation63-65 HDAC1 and HDAC4 were reported to directly deacetylate HIF-1α,Citation66,67 following the originally established paradigm. In this study, we provide new evidence to support that the HDACI-induced HIF-1α degradation is a ubiquitination-independent process which does not involve a change of HIF-1α acetylation; instead, HDAC5 facilitates HIF-1α maturation and stabilization by deacetylating Hsp70, thus providing new insight into the deacetylase-regulated stabilization of HIF-1α and probably other client proteins of Hsp70/Hsp90.
Despite the fact that both homologues may be involved in HIF-1α stabilization, HDAC4 and HDAC5 are unlikely to be functionally redundant. Their tissue-specific expression pattern varies dramatically; HDAC4 is highly expressed in myeloid whereas HDAC5 is highly expressed in the cardiac muscle and brain, which are the tissues most sensitive to fluctuation of O2 levels.Citation54 Furthermore, hypoxia specifically up-regulates HDAC5, but not HDAC4. In addition, HDAC5 knockout mice developed cardiac hypertrophy, which may be a consequence of deregulated developmental adaptation to cardiac hypoxia.Citation54 Transgenic overexpression of HIF-1α attenuated the cardiac hypertrophy caused by HDAC5 knockout,Citation68 proving a functional interplay between HDAC5 and HIF-1 with a genetic approach.
It remains unclear whether HDAC5 has other cytosolic substrates in addition to α-tubulin and Hsp70. Recent studies have shown that acetyl CoA levels affect the acetylation status of cytosolic metabolic enzymes thus regulating their activities.Citation69-74 Considering acetyl CoA levels are directly related to the fuel metabolism and biosynthetic activities, it would be interesting to ask whether cytosolic HDAC5 activity regulates the acetylation status of cytosolic metabolic enzymes. Moreover, it remains unclear if the cytosolic acetyl CoA level affects the acetylation status of Hsp70. Nevertheless, our findings suggest that AMPK-promoted HDAC5 nuclear export may represent an important cellular response to metabolic stresses which maintains the bioenergetic homeostasis.
In summary, our findings reported here demonstrate that the cytosolic HDAC5 deacetylates Hsp70, which is required for the rapid nuclear accumulation of HIF-1α and functional activation of HIF-1 complex. Thus, AMPK-induced HDAC5 cytosolic translocation plays an active role in cellular adaptation to hypoxia, and perhaps other metabolic stresses as well, by maintaining energy homeostasis.
Experimental Procedures
Chemicals and special reagents
Reagents chemicals were purchased from Fisher Scientific (Waltham, MA) or Sigma-Aldrich (St. Louis, MO). TSA #P9971e was from BioMol (Plymouth Meeting, PA). LMK235 (4830) was from Tocris Bioscience (Bristol, United Kingdom). AICAR (A9978), Leptomycin B (L2913) and Compound C (p5499) were from Sigma-Aldrich.
Cell lines, cell proliferation and lactate assays
Hep3B, HeLa and MCF7 cells were obtained from ATCC. H9c2 cells were a kind gift from Dr. P. Lelkes (Temple Univ). Cell culture media and reagents were purchased from Life Technologies. Hypoxic treatments were carried out by directly incubating cells in hypoxia work station (Ruskinn Technology Limited, UK). Cell proliferation was assayed with CyQUANT®NF-Cell Proliferation Kit (Life Technologies). Lactate concentration was measured with Lactate Assay Kit (Eton Bioscience, San Diego, CA).
RNA interference and CRISPR genome editing
HDAC1 (s73), HDAC3 (s16687), HDAC4 (s18839), HDAC5 (s19463), HDAC6 (s19459) and control (22400105) siRNAs were purchased from Life Technologies. Transfections were performed with Lipofectamine 2000 reagents (Life Technologies) following manufacturer's procedures. pGIPZ lentiviral empty shRNA control, shHDAC5-1 (V2LHS_68644), shHDAC5-2 (V2LHS_68645) and shHDAC5-3 (V2LHS_200875) were from Open Biosystems. Lentivirus was prepared with Trans-Lentiviral Packaging System (Fisher Scientific) in HEK293T cells. For lentivirus infection, cells were cultured with lentivirus and polyberene for 24 h and changed to regular medium. Stable knockdown cells were selected by puromycin. For CRISPR, target primers were designed by using online CRISPR design tool (http://crispr.mit.edu/), and engineered into pX330 plasmid by following the procedures from Zhang lab (http://www.genome-engineering.org/crispr). Sequences are detailed in Table S1. Hep3B cells were co-transfected with 800 ng of pX300 and 100 ng of pcDNA3.0 which facilitates selection with G418 (1 mg/ml). Selected individual colonies (HDAC5+/-) were expanded, screened by Western blotting, and confirmed by genomic PCR.
RNA extraction and qRT-PCR
Total RNA was extracted with Qiagen RNeasy kit (Valencia, CA). Complement DNA was synthesized using Superscript II Reverse Transcriptase (Life Technologies) with random hexamers. qRT-PCR was performed with Taqman primers and StepOnePlus Real-Time PCR System (Applied Biosystems, Life Technologies). β-actin was set as control. Taqman primers HDAC1 (Hs02621185_s1), HDAC3 (Hs00187320_m1), HDAC5 (Hs00608366_m1), HDAC6 (Hs00195869_m1), CA-IX (Hs00154208_m1), GLUT1 (Hs00892681_m1) and β-actin (Hs99999903_m1) were used.
DNA recombination
Mutagenesis primers were designed by QuikChange Primer Design tool online (http://www.genomics.agilent.com). All primers used are listed in Table S1. Restriction enzymes and other enzymes used in DNA recombination were from Life Technologies, Fisher Scientific or La Roche (Indianapolis, IN). Vector pcDNA3.0 was purchased from Life Technologies, and pcDNA3.0 n-flag was constructed by replacing 5’-ggtaccgagctc-3′ with 5′-gccaccatggactataaggacgatgacgatgacgacaagccgggc-3′. HDAC5 (NM_005474) cDNA was purchased from Origene (Rockville, MD), and flag-HDAC5 was constructed by inserting an EcoRV-XbaI fragment amplified from PCR into the pcDNA3.0 n-flag. Flag-HDAC4 (plasmid 13821) was purchased from Addgene (Cambridge, MA).Citation75 BamHI-EcoRI HDAC3 fragment was generated by PCR and then inserted into pcDNA3.0 n-flag. Hsp70 cDNA was generated by PCR and inserted into pcDNA3.0 n-flag BamHI-XhoI sites. Cytosolically localized HDAC5 (flag-HDAC5 S278/279A), nucleus localized HDAC5 (flag-HDAC5 L1092A), AMPK resistant HDAC5 (flag-HDAC5 S259/498A) and cytosollically localized HDAC4 (Flag-HDAC4 S265/266A) were constructed using QuikChange® II Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA). Catalytically inactive HDAC5 (flag-HDAC5 C698A/H704A) was constructed by Mutagenex Inc., (Piscataway, NJ). All constructs were confirmed by sequencing.
Antibodies, immunoprecipitation and western
The antibodies and sources are: Anti-HIF-1α polyclonal antibody (NB100-519): Novus (Littleton, CO); Anti-HDAC4 (#2072), anti-HDAC5 (#2082), anti-AMPKα (#2532) and anti-phospho-AMPKα(Thr172) (#2535S): Cell Signaling Technology (Danvers, MA); Anti-acetyl-K: Cell Signaling Technology (#9441) and Stressgen (#12210519); Anti-flag (050M6000) and anti-α-tubulin (081M4861), horseradish peroxidase-coupled secondary antibodies: Sigma-Aldrich. For Western blotting, cells were lysed in urea buffer as described previously.Citation65 For immunoprecipitation, cells were lysed with IP buffer (50 mM Tris–HCl, 150 mM NaCl, 1% Triton-X-100, 5 mM EDTA, 50 mM NaF, 0.1 mM Na3VO4, and protease inhibitor cocktail) and immunoprecipitated as described previously.Citation76
Immunofluorescent Cell Imaging
Cells were cultured in chamber slides, fixed with 4% formaldehyde in 1 x PBS for 6 min, and permeabilized with 0.2% Triton X-100 in PBS for 6 min. The slides were incubated with anti-Flag (1:200)/anti-HIF-1α. Alexa Fluor 594 goat anti-mouse IgG (Life technologies), Alexa Fluor 488 goat anti-rabbit IgG was used to visualize the antigens. All slides were mounted with SlowFade® Gold with DAPI (Life Technologies). The slides were imaged under Olympus (Center Valley, PA) FluoView 1000 Confocal Microscope.
Statistical analysis
For qRT-PCR, data were analyzed by StepOne software with ΔΔCT method. The relative quantities (RQ) value equals 2−ΔΔCT and the ± error bars represent the 95% confidence intervals. The converted form 2−CT which more accurately depicts the individual variations was used to perform the student's t-test and a p value < 0.01 was considered statistically significant. For cell proliferation IC50 assay, data was analyzed by Origin 8.0 software and fitted by sigmoidal model, error bars represent the 95% confidence intervals.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author Contributions
NS initiated the concept and working model, supervised the research and participated in data organization, manuscript preparation. SC performed most of the experiments and participated in discussion, data organization and manuscript preparation. CW established the shRNA-based knockdown cell lines used in this study. Other authors performed some of the experiments and provided technical support to SC.
1055426_suplpemental_files.zip
Download Zip (1.1 MB)Acknowledgments
We thank Dr. J. Caro (Cardeza Foundation, Thomas Jefferson University), Dr. G. Stein (Vermont Cancer Center) and Dr. D. Dhanasekaran (Stephenson Cancer Center) for constructive advices. We thank our colleagues Drs. T. Gidalevitz, J. Bethea, V. Bracchi-Ricard (Drexel Univ) for helpful discussion and critical reviewing the manuscript prior to submission. We thank Dr. P. Lelkes (Temple University) for the gift of H9c2 cells.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Funding
This work is supported in part by grant R01-CA129494 (to NS) from NCI, National Institutes of Health (NIH). SC is a recipient of a PhD dissertation fellowship from Drexel University.
Related Research Data
References
- Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer 2003; 3:721-32; PMID:13130303; http://dx.doi.org/10.1038/nrc1187
- Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell 2012; 148:399-408; PMID:22304911; http://dx.doi.org/10.1016/j.cell.2012.01.021
- Kaelin WG Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell 2008; 30:393-402; PMID:18498744; http://dx.doi.org/10.1016/j.molcel.2008.04.009
- Semenza GL. Hypoxia-inducible factor 1 and cardiovascular disease. Ann Rev Physiol 2014; 76:39-56; PMID:23988176; http://dx.doi.org/10.1146/annurev-physiol-021113-170322
- Takubo K, Goda N, Yamada W, Iriuchishima H, Ikeda E, Kubota Y, Shima H, Johnson RS, Hirao A, Suematsu M, et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell 2010; 7:391-402; PMID:20804974; http://dx.doi.org/10.1016/j.stem.2010.06.020
- Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E, Johnson RS, Goldrath AW. Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nat Immunol 2013; 14:1173-82; PMID:24076634; http://dx.doi.org/10.1038/ni.2714
- Keith B, Simon MC. Hypoxia-inducible factors, stem cells, and cancer. Cell 2007; 129:465-72; PMID:17482542; http://dx.doi.org/10.1016/j.cell.2007.04.019
- Zhang Z, Yan J, Chang Y, ShiDu Yan S, Shi H. Hypoxia inducible factor-1 as a target for neurodegenerative diseases. Cur Med Chem 2011; 18:4335-43; PMID:21861815; http://dx.doi.org/10.2174/092986711797200426
- Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 2012; 13:251-62; PMID:22436748; http://dx.doi.org/10.1038/nrm3311
- Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol 2011; 13:1016-23; PMID:21892142; http://dx.doi.org/10.1038/ncb2329
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13:132-41; PMID:21258367; http://dx.doi.org/10.1038/ncb2152
- Mungai PT, Waypa GB, Jairaman A, Prakriya M, Dokic D, Ball MK, Schumacker PT. Hypoxia triggers AMPK activation through reactive oxygen species-mediated activation of calcium release-activated calcium channels. Mol Cell Biol 2011; 31:3531-45; PMID:21670147; http://dx.doi.org/10.1128/MCB.05124-11
- Laderoute KR, Amin K, Calaoagan JM, Knapp M, Le T, Orduna J, Foretz M, Viollet B. 5′-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol Cell Biol 2006; 26:5336-47; PMID:16809770; http://dx.doi.org/10.1128/MCB.00166-06
- Jibb LA, Richards JG. AMP-activated protein kinase activity during metabolic rate depression in the hypoxic goldfish, Carassius auratus. J Exp Biol 2008; 211:3111-22; PMID:18805810; http://dx.doi.org/10.1242/jeb.019117
- Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. P Natl Acad Sci USA 1995; 92:5510-4; PMID:7539918; http://dx.doi.org/10.1073/pnas.92.12.5510
- Kaelin WG Jr. The von Hippel-Lindau protein, HIF hydroxylation, and oxygen sensing. Biochem Biophys Res Commun 2005; 338:627-38; PMID:16153592; http://dx.doi.org/10.1016/j.bbrc.2005.08.165
- Lando D, Gorman JJ, Whitelaw ML, Peet DJ. Oxygen-dependent regulation of hypoxia-inducible factors by prolyl and asparaginyl hydroxylation. Eur J Biochem 2003; 270:781-90; PMID:12603311; http://dx.doi.org/10.1046/j.1432-1033.2003.03445.x
- Semenza GL. Hydroxylation of HIF-1: oxygen sensing at the molecular level. Physiol (Bethesda) 2004; 19:176-82; PMID:15304631; http://dx.doi.org/10.1152/physiol.00001.2004
- Yu F, White SB, Zhao Q, Lee FS. HIF-1alpha binding to VHL is regulated by stimulus-sensitive proline hydroxylation. P Natl Acad Sci USA 2001; 98:9630-5; PMID:11504942; http://dx.doi.org/10.1073/pnas.181341498
- Epstein ACR, Gleadle JM, McNeill LA, Hewitson KS, O'Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, et al. C-elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001; 107:43-54; PMID:11595184; http://dx.doi.org/10.1016/S0092-8674(01)00507-4
- Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 2001; 292:464-8; PMID:11292862; http://dx.doi.org/10.1126/science.1059817
- Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001; 292:468-72; PMID:11292861; http://dx.doi.org/10.1126/science.1059796
- Koh MY, Spivak-Kroizman TR, Powis G. HIF-1 regulation: not so easy come, easy go. Trends Biochem Sci 2008; 33:526-34; PMID:18809331; http://dx.doi.org/10.1016/j.tibs.2008.08.002
- Huang LE, Gu J, Schau M, Bunn HF. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. P Natl Acad Sci U S A 1998; 95:7987-92; PMID:NOT_FOUND; http://dx.doi.org/10.1073/pnas.95.14.7987
- Salceda S, Caro J. Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem 1997; 272:22642-7; PMID:9278421; http://dx.doi.org/10.1074/jbc.272.36.22642
- Mahon PC, Hirota K, Semenza GL. FIH-1: a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev 2001; 15:2675-86; PMID:11641274; http://dx.doi.org/10.1101/gad.924501
- Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science 2002; 295:858-61; PMID:11823643; http://dx.doi.org/10.1126/science.1068592
- Sang N, Fang J, Srinivas V, Leshchinsky I, Caro J. Carboxyl-terminal transactivation activity of hypoxia-inducible factor 1 alpha is governed by a von Hippel-Lindau protein-independent, hydroxylation-regulated association with p300/CBP. Mol Cell Biol 2002; 22:2984-92; PMID:11940656; http://dx.doi.org/10.1128/MCB.22.9.2984-2992.2002
- Fath DM, Kong X, Liang D, Lin Z, Chou A, Jiang Y, Fang J, Caro J, Sang N. Histone deacetylase inhibitors repress the transactivation potential of hypoxia-inducible factors independently of direct acetylation of HIF-alpha. J Biol Chem 2006; 281:13612-9; PMID:16543236; http://dx.doi.org/10.1074/jbc.M600456200
- Qian DZ, Kachhap SK, Collis SJ, Verheul HM, Carducci MA, Atadja P, Pili R. Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1 alpha. Cancer Res 2006; 66:8814-21; PMID:16951198; http://dx.doi.org/10.1158/0008-5472.CAN-05-4598
- Liang D, Kong X, Sang N. Effects of histone deacetylase inhibitors on HIF-1. Cell Cycle 2006; 5:2430-5; PMID:17102633; http://dx.doi.org/10.4161/cc.5.21.3409
- Isaacs JS JY, Mimnaugh EG, Martinez A, Cuttitta F, Neckers LM. Hsp90 regulates a von Hippel Lindau-independent hypoxia-inducible factor-1 alpha-degradative pathway. J Biol Chem 2002; 277:29936-44; PMID:12052835; http://dx.doi.org/10.1074/jbc.M204733200
- Minet E, Mottet D, Michel G, Roland I, Raes M, Remacle J, Michiels C. Hypoxia-induced activation of HIF-1: role of HIF-1alpha-Hsp90 interaction. FEBS Lett 1999; 460:251-6; PMID:10544245; http://dx.doi.org/10.1016/S0014-5793(99)01359-9
- Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol 2010; 11:515-28; PMID:20531426; http://dx.doi.org/10.1038/nrm2918
- Chen S, Sang N. Histone deacetylase inhibitors: the epigenetic therapeutics that repress hypoxia-inducible factors. J Biomed Biotech 2011; 2011:197946
- Yang XJ, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene 2007; 26:5310-8; PMID:17694074; http://dx.doi.org/10.1038/sj.onc.1210599
- Fischle W, Dequiedt F, Hendzel MJ, Guenther MG, Lazar MA, Voelter W, Verdin E. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol Cell 2002; 9:45-57; PMID:11804585; http://dx.doi.org/10.1016/S1097-2765(01)00429-4
- Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol 2008; 9:206-18; PMID:18292778; http://dx.doi.org/10.1038/nrm2346
- Yang XJ, Gregoire S. Class II histone deacetylases: from sequence to function, regulation, and clinical implication. Mol Cell Biol 2005; 25:2873-84; PMID:15798178; http://dx.doi.org/10.1128/MCB.25.8.2873-2884.2005
- McKinsey TA, Zhang CL, Olson EN. Identification of a signal-responsive nuclear export sequence in class II histone deacetylases. Mol Cell Biol 2001; 21:6312-21; PMID:11509672; http://dx.doi.org/10.1128/MCB.21.18.6312-6321.2001
- Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP. HDAC6 is a microtubule-associated deacetylase. Nature 2002; 417:455-8; PMID:12024216; http://dx.doi.org/10.1038/417455a
- Kovacs JJ, Murphy PJ, Gaillard S, Zhao X, Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt WB, Yao TP. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell 2005; 18:601-7; PMID:15916966; http://dx.doi.org/10.1016/j.molcel.2005.04.021
- Marek L, Hamacher A, Hansen FK, Kuna K, Gohlke H, Kassack MU, Kurz T. Histone deacetylase (HDAC) inhibitors with a novel connecting unit linker region reveal a selectivity profile for HDAC4 and HDAC5 with improved activity against chemoresistant cancer cells. J Med Chem 2013; 56:427-36; PMID:23252603; http://dx.doi.org/10.1021/jm301254q
- Lao T, Chen S, Sang N. Two mutations impair the stability and function of ubiquitin-activating enzyme (E1). J Cell Physiol 2012; 227:1561-8; PMID:21678405; http://dx.doi.org/10.1002/jcp.22870
- Lahm A, Paolini C, Pallaoro M, Nardi MC, Jones P, Neddermann P, Sambucini S, Bottomley MJ, Lo Surdo P, Carfi A, et al. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. P Natl Acad Sci USA 2007; 104:17335-40; PMID:17956988; http://dx.doi.org/10.1073/pnas.0706487104
- Bottomley MJ, Lo Surdo P, Di Giovine P, Cirillo A, Scarpelli R, Ferrigno F, Jones P, Neddermann P, De Francesco R, Steinkuhler C, et al. Structural and functional analysis of the human HDAC4 catalytic domain reveals a regulatory structural zinc-binding domain. J Biol Chem 2008; 283:26694-704; PMID:18614528; http://dx.doi.org/10.1074/jbc.M803514200
- Greco TM, Yu F, Guise AJ, Cristea IM. Nuclear import of histone deacetylase 5 by requisite nuclear localization signal phosphorylation. Mol Cell Proteomics 2011; 10:M110 004317; PMID:21081666; http://dx.doi.org/10.1074/mcp.M110.004317
- Jeong JW, Bae MK, Ahn MY, Kim SH, Sohn TK, Bae MH, Yoo MA, Song EJ, Lee KJ, Kim KW. Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell 2002; 111:709-20; PMID:12464182; http://dx.doi.org/10.1016/S0092-8674(02)01085-1
- Yang Y, Fiskus W, Yong B, Atadja P, Takahashi Y, Pandita TK, Wang HG, Bhalla KN. Acetylated hsp70 and KAP1-mediated Vps34 SUMOylation is required for autophagosome creation in autophagy. P Natl Acad Sci USA 2013; 110:6841-6; PMID:23569248; http://dx.doi.org/10.1073/pnas.1217692110
- Wang Y, Wang SY, Zhang XH, Zhao M, Hou CM, Xu YJ, Du ZY, Yu XD. FK228 inhibits Hsp90 chaperone function in K562 cells via hyperacetylation of Hsp70. Biochem Biophys Res Commun 2007; 356:998-1003; PMID:17397803; http://dx.doi.org/10.1016/j.bbrc.2007.03.076
- To KK, Robey R, Zhan Z, Bangiolo L, Bates SE. Upregulation of ABCG2 by romidepsin via the aryl hydrocarbon receptor pathway. Mol Cancer Res 2011; 9:516-27; PMID:21357443; http://dx.doi.org/10.1158/1541-7786.MCR-10-0270
- Towler MC, Hardie DG. AMP-activated protein kinase in metabolic control and insulin signaling. Cir Res 2007; 100:328-41; PMID:17307971; http://dx.doi.org/10.1161/01.RES.0000256090.42690.05
- McGee SL, van Denderen BJ, Howlett KF, Mollica J, Schertzer JD, Kemp BE, Hargreaves M. AMP-activated protein kinase regulates GLUT4 transcription by phosphorylating histone deacetylase 5. Diabetes 2008; 57:860-7; PMID:18184930; http://dx.doi.org/10.2337/db07-0843
- Chang SR, McKinsey TA, Zhang CL, Richardson JA, Hill JA, Olson EN. Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol Cell Biol 2004; 24:8467-76; PMID:15367668; http://dx.doi.org/10.1128/MCB.24.19.8467-8476.2004
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013; 339:819-23; PMID:23287718; http://dx.doi.org/10.1126/science.1231143
- Shi P, Scott MA, Ghosh B, Wan D, Wissner-Gross Z, Mazitschek R, Haggarty SJ, Yanik MF. Synapse microarray identification of small molecules that enhance synaptogenesis. Nat Comm 2011; 2:510; PMID:22027590; http://dx.doi.org/10.1038/ncomms1518
- McGee SL, Sparling D, Olson AL, Hargreaves M. Exercise increases MEF2- and GEF DNA-binding activity in human skeletal muscle. FASEB J 2006; 20:348-9; PMID:16368714
- Zhao JX, Yue WF, Zhu MJ, Du M. AMP-activated protein kinase regulates beta-catenin transcription via histone deacetylase 5. J Biol Chem 2011; 286:16426-34; PMID:21454484; http://dx.doi.org/10.1074/jbc.M110.199372
- McKinsey TA, Zhang CL, Olson EN. Activation of the myocyte enhancer factor-2 transcription factor by calcium/calmodulin-dependent protein kinase-stimulated binding of 14-3-3 to histone deacetylase 5. P Natl Acad Sci USA 2000; 97:14400-5; PMID:11114197; http://dx.doi.org/10.1073/pnas.260501497
- Cho Y, Cavalli V. HDAC5 is a novel injury-regulated tubulin deacetylase controlling axon regeneration. EMBO J 2012; 31:3063-78; PMID:22692128; http://dx.doi.org/10.1038/emboj.2012.160
- McGee SL, Hargreaves M. Histone modifications and skeletal muscle metabolic gene expression. Clin Exp Pharm Physiol 2010; 37:392-6; PMID:19793100; http://dx.doi.org/10.1111/j.1440-1681.2009.05311.x
- Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer 2009; 9:563-75; PMID:19629071; http://dx.doi.org/10.1038/nrc2676
- Arnesen T, Kong X, Evjenth R, Gromyko D, Varhaug JE, Lin Z, Sang N, Caro J, Lillehaug JR. Interaction between HIF-1α (ODD) and hARD1 does not induce acetylation and destabilization of HIF-1α. FEBS Lett 2005; 579:6428-32; PMID:16288748; http://dx.doi.org/10.1016/j.febslet.2005.10.036
- Bilton R, Mazure N, Trottier E, Hattab M, Déry M-A, Richard DE, Pouysségur J, Brahimi-Horn MC. Arrest-defective-1 protein, an acetyltransferase, does not alter stability of hypoxia-inducible factor (HIF)-1α and is not induced by hypoxia or HIF. J Biol Chem 2005; 280:31132-40; PMID:15994306; http://dx.doi.org/10.1074/jbc.M504482200
- Kong X, Lin Z, Liang D, Fath D, Sang N, Caro J. Histone deacetylase inhibitors induce VHL and ubiquitin-independent proteasomal degradation of hypoxia-inducible factor 1alpha. Mol Cell Biol 2006; 26:2019-28; PMID:16507982; http://dx.doi.org/10.1128/MCB.26.6.2019-2028.2006
- Yoo YG, Kong, G, Lee M-O. Metastasis-associated protein 1 enhances stability of hypoxia-inducible factor-1a protein by recruiting histone deacetylase 1. EMBO J 2006; 25:1231-41; PMID:16511565; http://dx.doi.org/10.1038/sj.emboj.7601025
- Geng H, Harvey CT, Pittsenbarger J, Liu Q, Beer TM, Xue C, Qian DZ. HDAC4 protein regulates HIF1alpha protein lysine acetylation and cancer cell response to hypoxia. J Biol Chem 2011; 286:38095-102; PMID:21917920; http://dx.doi.org/10.1074/jbc.M111.257055
- Xue W, Cai L, Tan Y, Thistlethwaite P, Kang YJ, Li X, Feng W. Cardiac-specific overexpression of HIF-1{alpha} prevents deterioration of glycolytic pathway and cardiac remodeling in streptozotocin-induced diabetic mice. Am J Pathol 2010; 177:97-105; PMID:20566749; http://dx.doi.org/10.2353/ajpath.2010.091091
- Cai L, Sutter BM, Li B, Tu BP. Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol Cell 2011; 42:426-37; PMID:21596309; http://dx.doi.org/10.1016/j.molcel.2011.05.004
- Jiang W, Wang S, Xiao M, Lin Y, Zhou L, Lei Q, Xiong Y, Guan KL, Zhao S. Acetylation regulates gluconeogenesis by promoting PEPCK1 degradation via recruiting the UBR5 ubiquitin ligase. Mol Cell 2011; 43:33-44; PMID:21726808; http://dx.doi.org/10.1016/j.molcel.2011.04.028
- Lv L, Li D, Zhao D, Lin R, Chu Y, Zhang H, Zha Z, Liu Y, Li Z, Xu Y, et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell 2011; 42:719-30; PMID:21700219; http://dx.doi.org/10.1016/j.molcel.2011.04.025
- Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, Yao J, Li H, Xie L, Zhao W, Yao Y, et al. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 2010; 327:1004-7; PMID:20167787; http://dx.doi.org/10.1126/science.1179687
- Zhang T, Wang S, Lin Y, Xu W, Ye D, Xiong Y, Zhao S, Guan KL. Acetylation negatively regulates glycogen phosphorylase by recruiting protein phosphatase 1. Cell Metab 2012; 15:75-87; PMID:22225877; http://dx.doi.org/10.1016/j.cmet.2011.12.005
- Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, et al. Regulation of cellular metabolism by protein lysine acetylation. Science 2010; 327:1000-4; PMID:20167786; http://dx.doi.org/10.1126/science.1179689
- Fischle W, Emiliani S, Hendzel MJ, Nagase T, Nomura N, Voelter W, Verdin E. A new family of human histone deacetylases related to Saccharomyces cerevisiae HDA1p. J Biol Chem 1999; 274:11713-20; PMID:10206986; http://dx.doi.org/10.1074/jbc.274.17.11713
- Stiehl DP, Fath DM, Liang D, Jiang Y, Sang N. Histone deacetylase inhibitors synergize p300 autoacetylation that regulates its transactivation activity and complex formation. Cancer Res 2007; 67:2256-64; PMID:17332356