
ABSTRACT
The timely and precise duplication of cellular DNA is essential for maintaining genome integrity and is thus tightly-regulated. During mitosis and G1, the Origin Recognition Complex (ORC) binds to future replication origins, coordinating with multiple factors to load the minichromosome maintenance (MCM) complex onto future replication origins as part of the pre-replication complex (pre-RC). The pre-RC machinery, in turn, remains inactive until the subsequent S phase when it is required for replication fork formation, thereby initiating DNA replication. Multiple myeloma SET domain-containing protein (MMSET, a.k.a. WHSC1, NSD2) is a histone methyltransferase that is frequently overexpressed in aggressive cancers and is essential for normal human development. Several studies have suggested a role for MMSET in cell-cycle regulation; however, whether MMSET is itself regulated during cell-cycle progression has not been examined. In this study, we report that MMSET is degraded during S phase in a cullin-ring ligase 4-Cdt2 (CRL4Cdt2) and proteasome-dependent manner. Notably, we also report defects in DNA replication and a decreased association of pre-RC factors with chromatin in MMSET-depleted cells. Taken together, our results suggest a dynamic regulation of MMSET levels throughout the cell cycle, and further characterize the role of MMSET in DNA replication and cell-cycle progression.
Introduction
Precise duplication of genomic DNA is essential to maintain genome stability and prevent genetic abnormalities associated with cancer and other diseases. Accordingly, DNA replication includes an ordered and highly-regulated series of steps, both before and during synthesis (S) phase.Citation1 In preparation for S phase, DNA replication origins are generated in a process called replication licensing that occurs during late mitosis and G1. During this process, the origin recognition complex (ORC) is recruited to specific genomic sites where it binds and recruits the ATPase cell division cycle 6 (Cdc6) and chromatin licensing and DNA replication factor 1 (Cdt1), forming the pre-replication complex (pre-RC) which in turn facilitates the loading of the heterohexameric minichromosome maintenance 2–7 (MCM2-7) complex onto chromatin.Citation2-4 Once S phase begins, the MCM complex is activated to serve as the replicative helicase in coordination with Cdc45 and GINS (Go, Ichi, Nii, and San; 5,1,2,3), unwinding DNA at the replication fork.Citation5,6 The replication fork is then loaded with Proliferating cell nuclear antigen (PCNA), a sliding processivity clamp for DNA synthesis in coordination with the replicative polymerases Polδ or Polϵ.Citation7 Once replication initiates at a given origin, the MCM helicase is displaced ahead of the replication fork and is therefore never associated with newly-replicated DNA.Citation3
Importantly, in order to prevent multiple rounds of replication in a single cell cycle, replication licensing factors are degraded or inhibited during S phase, thereby limiting replication initiation to previously-established replication origins.Citation3,8-10 A well-described mechanism for the degradation of licensing proteins during S phase has been elucidated for the ubiquitin E3 ligase complex Cullin-ring ligase 4-Cdt2 (CRL4Cdt2).Citation11 This complex utilizes a cullin scaffold (Cul4), an adaptor protein (DNA damage-binding protein 1, Ddb1), and Cdt2 which is a DCAF (Ddb1- and Cul4-associated factor) that directly binds to Ddb1 and provides substrate specificity.Citation11-16 Importantly, the S-phase specific degradation of multiple licensing factors including Cdt1, the cyclin-dependent kinase inhibitor p21, and the H4K20 methyltransferase SET domain-containing protein 8 (Set8) have been shown to be degraded by this pathway.Citation15,17-20 The majority of CRL4Cdt2 substrates bind to chromatin-bound PCNA specifically during S phase; these PCNA-bound substrates are then recognized by the substrate receptor Cdt2, recruiting the CRL4Cdt2 complex to polyubiquitinate the substrate, thereby targeting the substrate for proteasome-mediated degradation.Citation11 Notably, inhibition of CRL4Cdt2, or stabilization of its substrates, during S phase results in aberrant rounds of re-replication in a single cell-cycle, underscoring the importance of CRL4Cdt2 in regulating replication licensing factors.Citation4,12,Citation20-23
MMSET (Multiple myeloma SET domain-containing protein), also known as Wolf-Hirschhorn syndrome candidate 1 (WHSC1) and Nuclear SET domain-containing protein 2 (NSD2), is a histone methyltransferase with multiple implications in human diseases. MMSET's role in cancers was first described in the t(4;14) subgroup of multiple myelomas, which make up 15–20% of all multiple myelomas, result in MMSET overexpression, and correlate with extremely-poor prognosis.Citation24-26 Importantly, MMSET is considered the principle oncogene driving tumorigenesis in multiple myelomas harboring t(4;14) translocations.Citation25,27,28 MMSET is also overexpressed in many cancers in which its expression levels are associated with tumor aggressiveness.Citation29-31 Recently, a recurrent MMSET hyper-activating mutation (E1099K) which effectively mimics MMSET overexpression was reported in lymphoid malignancies, further reinforcing a role for MMSET in oncogenesis.Citation32,33 Beyond an association with cancers, MMSET has also been linked to the developmental disorder Wolf-Hirschhorn syndrome; in these patients, one copy of MMSET is absent due to a deletion of the subtelomeric region of the short arm of chromosome 4. Patients affected by this syndrome suffer from mental retardation, severe growth delays, cranio-facial dysgenesis, and other developmental effects.Citation34-38 Consistently, MMSET heterozygous mice have been shown to have severe growth defects and Wolf-Hirschhorn-like features, while homozygous MMSET−/- mice die shortly after birth due to fatal growth retardation.Citation39 Understanding MMSET's cellular functions is critical to elucidate MMSET's roles in development as well as the tumor aggressiveness correlating with MMSET overexpression in many cancers.
In this study, we examined the cell-cycle-dependent regulation of MMSET protein levels, and found MMSET levels fluctuate dynamically during cell-cycle progression. We further show that MMSET is ubiquitinated and degraded by the proteasome, and MMSET degradation is downstream of the CRL4Cdt2 ubiquitination pathway. Furthermore, like many CRL4Cdt2 substrates, MMSET interacts with PCNA and is degraded during S phase in a PCNA-dependent manner. Notably, we also report replication defects and pre-replication complex assembly abnormalities in MMSET-depleted cells, potentially suggesting a role for MMSET upstream of pre-RC assembly. Taken together, our findings elucidate the dynamic regulation of MMSET expression throughout the cell cycle and further describe the importance of MMSET in normal DNA replication and cell-cycle progression.
Results
MMSET levels are dynamically regulated during the cell-cycle and MMSET is degraded by the ubiquitin-proteasome pathway during S phase
To examine whether MMSET may be regulated during cell-cycle progression, we synchronized U2OS cells at the G1-S border, released the cells, and prepared whole-cell lysates at various time points to analyze MMSET protein levels by immunoblot. As can be seen in , MMSET levels were lower in G1-S synchronized cells compared to unsynchronized cells. Notably, MMSET levels were also further reduced as S phase progressed, and then increased again at later time points (). To further confirm the reduction in MMSET levels as cells enter S phase, we also examined MMSET expression at the single-cell level. Asynchronous cells were plated on coverslips, fixed, and stained for MMSET and cyclin E1 as a marker for the G1-S transition Citation40 (). Consistent with our observations in , cyclin E1+ cells had lower MMSET expression compared to cyclin E1− cells (). Taken together, these findings suggest that MMSET levels are dynamically regulated during the cell cycle, with a decrease in MMSET expression observed starting at the G1-S transition and decreasing further as S phase progresses.
Figure 1. MMSET is regulated in a cell-cycle-dependent manner and degraded by the ubiquitin-proteasome pathway during S phase. (A) Asynchronous U2OS cells or cells synchronized at the G1-S border were released into S phase for the indicated times. Lysates were collected for each time point and blotted for the indicated proteins. (B) Asynchronous U2OS cells were plated on coverslips, fixed, and stained for either Cyclin E1 (red) or MMSET (green). Nuclei were visualized with 4',6-Diamidino-2-Phenylindole, Dihydrochloride (DAPI, blue). Scale bar=10 μm. (C) U2OS cells were synchronized at the G1-S border by double-thymidine block and released into S phase for the indicated times either in the absence (top) or presence (bottom) of MG132. Lysates were collected at each time point and blotted for the indicated proteins. (D) His-pulldowns were performed under denaturing conditions using lysates from HEK293 cells transfected with the indicated plasmids and synchronized in S phase using the serum starvation and release method. Purified species were subjected to SDS-PAGE and immunoblotted with the indicated antibodies. Arrowhead and a bar indicate monoubiquitinated and polyubiquitinated MMSET, respectively. Data shown are representative of 3 independent experiments.
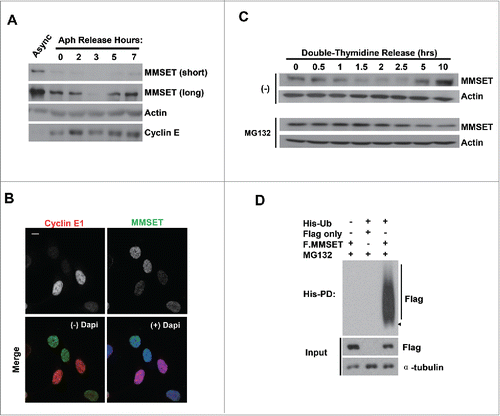
We next sought to identify the molecular mechanism regulating this decrease in MMSET levels. To test whether MMSET is degraded by the ubiquitin-proteasome pathway during S phase, we synchronized cells in G1-S and released them into fresh media either in the absence or presence of the proteasome-inhibitor MG132. After release into S phase, whole-cell lysates from both groups were collected and MMSET levels were examined by immunoblot (). While the control group confirmed the MMSET decrease during S phase, MMSET levels were stabilized in the presence of the proteasome inhibitor MG132. This suggests that MMSET is degraded during S phase in a proteasome-dependent manner. Next, we examined whether MMSET is also ubiquitinated during S phase. We transfected cells with His-tagged ubiquitin along with Flag-only or Flag-tagged MMSET plasmid, and synchronized the cells in S phase. Prior to harvest, MG132 was added and His-ubiquitinated species were pulled down under denaturing conditions (). Indeed, MMSET was polyubiquitinated in S-phase-synchronized cells. Taken together, we conclude that MMSET levels are dynamically-regulated throughout the cell cycle and MMSET undergoes ubiquitination and proteasome-mediated degradation during cell-cycle progression.
CRL4Cdt2 Promotes MMSET degradation
Based on our finding that MMSET is degraded by the ubiquitin-proteasome pathway during S phase, we next sought to examine potential pathways involved in MMSET's regulation. The cell-cycle regulator cullin-ring ligase 4-Cdt2 (CRL4Cdt2) is an E3 ubiquitin ligase complex that promotes the S-phase-specific degradation of several substrates including Cdt1, p21, and N-lysine methyltransferase SETD8 (Set8).Citation15,17-20 Interestingly, we observed an interaction between MMSET and the CRL4Cdt2 substrate specificity factor Cdt2 (). To test for a potential significance of this interaction, we next asked whether MMSET levels may be regulated by the CRL4Cdt2 pathway. We examined MMSET levels during S phase after overexpressing Cdt2. Notably, Cdt2 overexpression resulted in a reduction in MMSET levels in S-phase-synchronized cells (). To further confirm this finding, we fixed Flag-Cdt2 transfected cells for immunofluorescence staining of Flag (Cdt2) and endogenous MMSET (). Consistent with our findings by immunoblot (), we observed decreased MMSET levels in cells overexpressing Flag-Cdt2. As a reciprocal confirmation that MMSET is negatively regulated by CRL4Cdt2, we also examined MMSET levels during S phase in cells depleted of Cdt2. As shown in , depletion of Cdt2 with 2 different siRNAs increased MMSET levels in S-phase-synchronized cells, further suggesting a role for the CRL4Cdt2 pathway in MMSET's S-phase degradation. Importantly, we did not observe a dramatic change in cell-cycle profile following Cdt2 depletion in our transient knockdown system (Fig. S1), ruling out the possibility of indirect cell-cycle effects affecting MMSET levels.
Figure 2. MMSET is degraded by CRL4Cdt2. (A) HEK293T cells were transfected with the plasmids denoted above each lane. Lysates were prepared 48 hours post transfection, immunoprecipitated with anti-Flag, and examined for MMSET. Arrowheads denote Flag-YFP (lower) and Flag-Cdt2 (upper). (B) HEK293T cells were transfected with the plasmids denoted above each lane and synchronized in S phase by the serum starvation and release method. Lysates were prepared and examined for the indicated proteins. The numbers beneath the blots provide the densitometric ratio of MMSET signal to Actin, setting the value for the control group as 1. (C) HEK293T cells were transfected as in B, fixed, and stained for either Cdt2 (Flag, red) or MMSET (green). Nuclei were visualized with 4',6-Diamidino-2-Phenylindole, Dihydrochloride (DAPI, blue). Scale bar= 10 μm. (D) HEK293T cells were treated with the indicated siRNAs for 24 hours, followed by an aphidicolin block to synchronize cells into S phase. Cells were released into S phase for 4 hours in the presence of cycloheximide and lysates were prepared and examined for the indicated proteins. The numbers beneath the blots provide densitometric ratio of MMSET or Cdt2 signal to Tubulin, setting the value for the control group as 1. *Denotes a non-specific band detected by the Cdt2 antibody. (E) His-pull-downs were performed under denaturing conditions using lysates from HEK293 cells transfected with the indicated plasmids and synchronized in S phase using the serum starvation and release method. Purified species were subjected to SDS-PAGE and immunoblotted with the indicated antibodies. Arrowhead and a bar indicate monoubiquitinated and polyubiquitinated MMSET, respectively. (F) His-pull-downs were performed under denaturing conditions using lysates from HEK293 cells transfected with the indicated plasmids and synchronized in S phase using the serum starvation and release method. Purified species were subjected to SDS-PAGE and immunoblotted with the indicated antibodies. Arrowhead and a bar indicate monoubiquitinated and polyubiquitinated MMSET, respectively. Data shown are representative of 3 independent experiments.
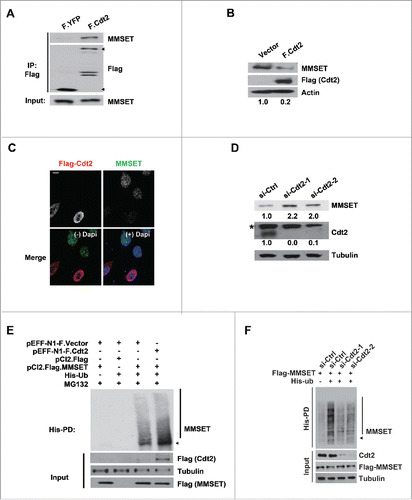
Given our finding that MMSET is ubiquitinated in S-phase-synchronized cells, we next wondered whether MMSET ubiquitination may also be regulated in a CRL4Cdt2-dependent manner. To examine this possibility, we transfected cells with His-ubiquitin and Flag-MMSET in combination with either Flag-only or Flag-Cdt2 plasmids. We then synchronized cells by serum starvation and release into S phase, treated cells with MG132, and performed His pull-downs under denaturing conditions as in (). We detected an increase in MMSET ubiquitination following Cdt2 overexpression, consistent with a role for CRL4Cdt2 in promoting MMSET ubiquitination. Furthermore, we detected reduced MMSET ubiquitination following Cdt2 knockdown (). Taken together, we conclude that CRL4Cdt2 promotes the ubiquitination and degradation of MMSET in cells.
MMSET interacts with PCNA through its N terminus and PCNA depletion promotes MMSET stability
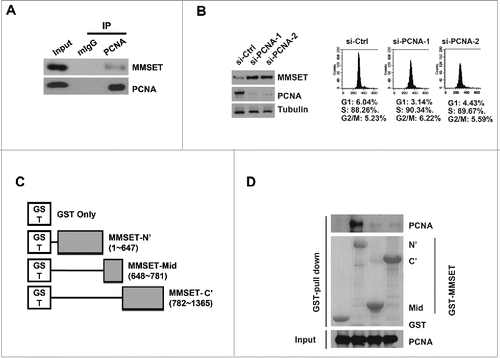
Many CRL4Cdt2 substrates, including Cdt1, p21, and Set8, utilize PCNA binding as a ubiquitin- and proteasome-dependent degradation signal at replication forks.Citation15,17-20 The PCNA-bound substrate is recognized by CRL4Cdt2 and thereby targeted for ubiquitination and proteasome-mediated degradation during S-phase.Citation11 However, reports of CRL4Cdt2 substrates degraded in a PCNA-independent manner have also recently emerged.Citation41,42 We therefore next asked whether MMSET's degradation downstream of CRL4Cdt2 could be occurring in a PCNA-dependent manner. Substrates degraded in a PCNA-dependent manner are typically found to co-immunoprecipitate with both Cdt2 and PCNA.Citation43 Therefore, we first examined whether we could detect an interaction between MMSET and PCNA. Indeed, we confirmed that endogenous PCNA co-immunoprecipitated with endogenous MMSET in asynchronous lysates (). This result suggested that MMSET could be degraded downstream of the CRL4Cdt2-PCNA ubiquitination pathway. To further examine whether MMSET may be degraded by the CRL4Cdt2-PCNA pathway, we next examined whether MMSET levels are also stabilized by PCNA depletion. To address this, we treated cells with either control or PCNA-targeting siRNAs followed by synchronization in S phase (, right panels). MMSET levels were then examined by immunoblot (, left panels). Indeed, we observed an increase in MMSET levels following PCNA knockdown in S-phase synchronized cells. This result suggests that MMSET's degradation by CRL4Cdt2 may be dependent upon PCNA, similar to the degradation mechanism observed in the canonical CRL4Cdt2 substrate pathway.Citation11 This result was somewhat unexpected, as MMSET lacks a canonical PCNA-interaction degron motif that is typical of substrates degraded by the PCNA-CRL4Cdt2 ubiquitination pathway.Citation11 We therefore next decided to delineate the region on MMSET responsible for the observed MMSET-PCNA interaction. We prepared 3 non-overlapping fragments of MMSET fused with glutathione S-transferase (GST) at the N-terminus (). The ability of these fragments to interact with recombinant PCNA was then tested using a GST-MMSET pull-down assay (). While GST alone, MMSET-mid, and MMSET-C’ showed no interaction with PCNA, the MMSET-N’ fragment successfully pulled down PCNA. Taken together, we conclude that MMSET is degraded in a PCNA-dependent manner during S phase, and MMSET interacts directly with PCNA through MMSET's N terminus.
Figure 3. MMSET interacts with PCNA via its N-terminus and PCNA depletion promotes MMSET stability. (A) U2OS cells were immunoprecipitated with PCNA or mouse IgG and the resulting samples were examined for MMSET or PCNA by western blot. (B) Left: T98G cells were transfected with the indicated siRNAs and synchronized in S phase by the serum starvation and release method. Lysates were prepared 48 hours later and examined for the indicated proteins. Right: cell-cycle profiles of the samples. (C) Schematic representation of MMSET fragments used for the GST pull-down assay. (D) A GST-pull down assay was carried out between purified PCNA and GST-MMSET fragments. Coomassie blue staining shows GST fusion proteins used in the pull-down assay (middle). Data shown are representative of 3 independent experiments.
MMSET is required for normal S-phase progression
We next wondered the functional significance of MMSET levels during the cell cycle. MMSET deregulation is well established in cancers and has been associated with defects in cell proliferation and viability in various cell types.Citation44-49 Given our finding that MMSET is degraded during S phase, we decided to examine the effect of depleting MMSET on cell-cycle profile. We transfected cells with either control or 2 different MMSET-targeting siRNAs, stained DNA with propidium iodide (PI), and examined cell-cycle profiles by flow cytometry (). Notably, we observed a significant accumulation of S phase cells following MMSET depletion ().
Figure 4. MMSET is required for normal cell-cycle progression. (A) HeLa cells were treated with the indicated siRNAs and, 48 hours later, cells were fixed and examined for DNA content by flow cytometry. (B) Cell-cycle profiles from A were analyzed and S phase was plotted for each condition. Error bars indicate SEM. *P < 0.05. (C) HCT116 cells stably expressing MMSET shRNA were reconstituted with WT MMSET or the MMSET deletion mutant (648-1356) and the indicated proteins were examined by western blot. (D) A subset of cells from C was examined for DNA content and BrdU incorporation using flow cytometry. (E) HeLa cells were treated with the indicated siRNAs, synchronized at the G1-S border using aphidicolin, and total cell extracts (TCE) or chromatin and non-chromatin enriched fractionations were prepared 48 hours post-transfection. The indicated proteins were examined by western blot. The numbers beneath the blots provide the densitometric ratio of ORC2, MCM2, or MCM7 signal to H3, setting the value for the control group as 100. Data shown are representative (A, C, D, and E) or an average of 3 independent experiments (B).
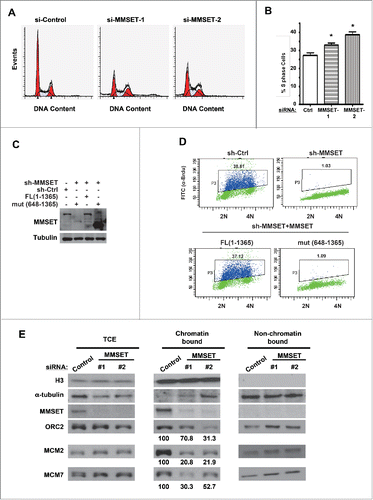
Multiple scenarios could potentially cause an increase in cells with S-phase DNA content. One potential scenario is slower or stalled S-phase progression due to defects in DNA replication.Citation50 To examine whether MMSET depletion may affect DNA replication, we examined MMSET's effect on the ability of cells to incorporate the nucleotide analog bromodeoxyuridine (BrdU), a marker of active DNA replication. Cells depleted of MMSET were pulse-labeled with BrdU for 30 minutes, and BrdU-incorporating cells were detected by flow cytometry. For control cells, 38.61% of cells incorporated DNA during the pulse period under these experimental conditions (, top left). Remarkably, the majority of MMSET-depleted cells failed to incorporate BrdU during the same period ( and Fig. S3A, B). Reconstituting these cells with WT MMSET, but not mutant MMSET (648-1365) that was depleted of the region required for PCNA interaction, restored BrdU incorporation (, bottom panels). Taken together, we conclude that MMSET is required for normal DNA replication.
MMSET promotes pre-RC component association with chromatin
The cell-cycle regulation of MMSET expression by the CRL4Cdt2 pathway as well as the defective DNA replication observed in MMSET-depleted cells suggested an interesting potential role for MMSET in cell-cycle control. During the initial replication licensing step occurring in mitosis/G1, the origin recognition complex (ORC) binds to replication origin sites where it recruits Cdt1 and Cdc6 in order to load the minichromosome maintenance (MCM) complex onto chromatin. The complex formed (pre-RC) mediates formation of replication forks once S-phase begins and is therefore required for DNA replication initiation. Interestingly, many substrates regulated by CRL4Cdt2 have important roles in this tightly-regulated process of replication licensing, and are then degraded during S phase to prevent aberrant re-replication.Citation4,12,Citation20-23 In addition, the replication defects after MMSET depletion are similar to those observed following pre-RC component depletion.Citation51,52 We therefore hypothesized that, like other CRL4Cdt2 substrates, MMSET may also be important for the replication licensing process.
To determine this, we examined the association of pre-RC component proteins with chromatin in G1-S synchronized cells. We compared the chromatin association of ORC and MCM complex component proteins in control-transfected and MMSET-suppressed cells. After depleting MMSET, we synchronized cells at the G1-S border and fractionated the cells into chromatin-bound and non-chromatin-bound portions or total cell extracts (TCE) (). Notably, although total levels of MCM2, MCM7, and ORC2 were unaffected by MMSET knockdown (, left panel (TCE)), we observed a reduction in chromatin-bound levels for each of these pre-RC components (, middle panel (chromatin bound)). Importantly, MMSET depletion did not affect the ability of cells to be synchronized at G1-S in our experimental system (Fig. S2), ruling out the possibility of indirect cell-cycle effects on chromatin loading following MMSET depletion. To further confirm this result, we synchronized cells in G1 and examined the pre-RC component association with chromatin. As shown in Fig. S3C, MMSET depletion resulted in a reduction of the chromatin loading of the pre-RC components (MCM2, MCM7, and ORC2) in G1 cells, while the total levels for these proteins were unaffected by MMSET depletion. Taken together, we conclude that MMSET is important for the physical association of several pre-RC components with chromatin, although MMSET does not affect the total expression levels of these pre-RC component proteins.
Discussion
Previous reports have shown that depleting known chromatin-licensing factors in G1-S synchronized cells causes a decreased association of pre-RC components with chromatin.Citation20 Thus, our finding that MMSET is required for the association of pre-RC components with chromatin in G1-S synchronized cells could suggest a role for MMSET upstream of pre-RC assembly. Notably, this could, among other factors, contribute to the replication and proliferation defects observed following MMSET depletion (). Previous studies have suggested a role for MMSET in regulating the cell cycle and cell viability through transcriptional regulation.Citation44-46,Citation48 Although we could not completely exclude the possibility that MMSET regulates pre-RC assembly through its role in transcription regulation, the observation that MMSET does not affect the expression level of the affected replication factors () supports a potentially transcription-independent role for MMSET in promoting pre-RC assembly. We also show that MMSET levels are tightly regulated during the cell cycle, consistent with an important role for MMSET in cell-cycle progression. Importantly, we found that MMSET is negatively regulated downstream of CRL4Cdt2 and PCNA during S phase. Notably, similar to other proteins important for pre-RC assembly,Citation4,12,Citation20-23 MMSET's S-phase degradation reported here might be important for preventing unwanted replication licensing once S phase begins. Taken together, our observations help to explain both the tight regulation of MMSET levels during cell-cycle progression and the cell-cycle deregulation observed upon MMSET depletion.
During mitosis and G1, the ORC and MCM complexes bind to future replication origin sites, forming the pre-RC where replication will initiate during the subsequent S phase. Considering the potential importance of MMSET in pre-RC assembly, an important resulting question is the underlying mechanism. Given that MMSET is a histone methyltransferase, one tempting possibility is that MMSET-mediated histone methylation promotes loading of the ORC complex at replication origin sites. Indeed, replication origins have previously been reported to be associated with several chromatin modifiers such as human acetylase binding to ORC1 (HBO1, also known as KAT7 and MYST2), the histone methyltransferase Set8, and the chromatin-remodeling complex sucrose nonfermenting 2 homolog (SNF2H), which directly modify histones or chromatin structure to promote pre-RC assembly.Citation20,53,54 Set8, for example, has been shown to associate with replication origins and provide a chromatin environment conducive to ORC binding by promoting H4K20 monomethylation.Citation20 The bromo adjacent homology (BAH) domain of ORC1, in turn, has been shown to directly bind H4K20 dimethylation, a mark that requires Set8-mediated H4K20 monomethylation as a prerequisite.Citation55 Furthermore, H4K20 monomethylation is dynamically regulated during the cell cycle, with lower levels during S phase, correlating with reduced Set8 expression.Citation56,57 Based on the findings presented here, it will be very important and interesting to examine whether MMSET exhibits a similar effect through direct methylation of histones at origin sites, and whether MMSET's S-phase-specific degradation also correlates with lower levels of its downstream methylation marks during DNA synthesis. In addition to a direct role in histone methylation at replication origins, it is also plausible that MMSET could physically associate with or directly methylate the ORC complex itself, thereby promoting ORC localization to replication origins. Finally, a non-mutually-exclusive possibility is that MMSET may promote the binding of other currently unknown licensing factors to origins which in turn promote the recruitment of the ORC complex. Future work elucidating the mechanism by which MMSET supports pre-RC assembly will provide important insight into this process.
In this study we also found that MMSET depletion resulted in an accumulation of cells with S-phase DNA content (). Interestingly, we also observed a defect in BrdU incorporation in MMSET-depleted cells (). One important remaining question is whether the defect in DNA replication after depleting MMSET results in the observed accumulation of S phase cells and, if so, the mechanism by which this is occurring. It is possible that MMSET depletion causes replication stress, activating an S-phase checkpoint and thereby preventing S-phase progression. Because we also observed a defect in the chromatin association of both ORC and MCM complex components, it is tempting to speculate that MMSET-depleted cells may enter S phase with insufficient origins licensed, experience replication stress, and thereby undergo such an S-phase checkpoint. This in turn would explain the observed accumulation of cells with S-phase DNA content as well as the defect in BrdU incorporation after MMSET depletion (). In support of this model, depletion of components of the ORC complex has been shown to cause a similar accumulation of S-phase cells which exhibit a BrdU incorporation defect.Citation51,52 One important caveat is that, while these previous studies demonstrated the effects of overall depletion of ORC components,Citation51,52 we show that defects in ORC chromatin association following MMSET depletion correlate with a similar cell-cycle phenotype, while total ORC component protein levels do not change. Despite this, the similar phenotypes observed upon depleting ORC components Citation51,52 and depleting MMSET could be suggestive of an important role for MMSET-mediated pre-RC formation in maintaining normal cell-cycle progression. Future studies further elucidating the signaling pathways involved will be very interesting.
We also report here that MMSET is degraded by the ubiquitin-proteasome pathway during S-phase and MMSET's ubiquitination is mediated by the E3 ubiquitin ligase CRL4Cdt2. To date, the degradation of CRL4Cdt2 substrates has been found to occur either in coordination with PCNA Citation11 or independent of PCNA.Citation41,42 Together, the endogenous interaction of MMSET with PCNA, as well as the increased MMSET levels upon PCNA depletion, suggests that MMSET may be degraded via the PCNA-CRL4Cdt2 pathway. Substrates reported to be degraded by CRL4Cdt2 in a PCNA-dependent manner typically contain a PCNA interacting protein (PIP) degron motif [Q-X-X-(L/V/I/M)-T-D-(F/Y)-(F/Y)-X-X-X-(K/R)], which allows substrate interaction with chromatin-bound PCNA and subsequent recognition by CRL4Cdt2 prior to substrate ubiquitination.Citation11 Surprisingly, MMSET lacks a conserved PIP degron motif. Previously, other non-canonical PCNA binding motifs have been identified, such as the KA-box.Citation58 It will therefore be very interesting to address whether MMSET could be degraded by the PCNA-CRL4Cdt2 axis through a non-canonical PCNA interaction domain. Notably, we found a direct interaction of MMSET with PCNA via MMSET's N-terminus using a GST pull-down approach (). Future studies further examining the motif on MMSET responsible for this interaction will be very interesting.
Deregulation of MMSET has been well-described in cancer and human developmental disorders due to MMSET overexpression or insufficiency, respectively. This work provides a framework for further insight into both disease states. Given the important role for MMSET in replication origin assembly described here, we speculate that the overexpression of MMSET reported in multiple cancer types Citation29-31 may result in deregulated replication and genomic instability. Notably, overexpression or failure to degrade licensing proteins prior to S phase in many cases results in additional rounds of replication and genomic instability.Citation4,20,22,23 It will thus be useful to examine whether high-levels of MMSET overexpression or expression of a non-degradable MMSET mutant are sufficient to induce re-replication and resulting genomic instability in cancer cells. Given the observation that MMSET has an important role in DNA replication, it will be interesting to determine whether MMSET deficiency contributes to the replication defects observed in WHS patient cell lines,Citation59,60 and whether the developmental defects in patients suffering from WHS may be due to defects in the same underlying replication pathway. Taken together, our results elucidate a role for MMSET in supporting normal DNA replication, demonstrate the dynamic regulation of MMSET during cell-cycle progression, and provide further insight into potential molecular mechanisms underlying MMSET's roles in human disease.
Materials and methods
Plasmids and transfections
Full-length MMSET was cloned into the pCI2 plasmid in-frame with FLAG or pLenti6.3 in frame with FLAG for transient transfection or stable overexpression, respectively. Plasmid transfections were performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions or using polyethylenimine (PEI) following standard transfection procedures. siRNAs were purchased from Dharmacon and transfected using Oligofectamine (Invitrogen) according to the manufacturer's instructions. MMSET was targeted using MMSET siRNA-1 (5′-AGGGATCGGAAGAGTCTTCAA-3′) and MMSET siRNA-2 (5’-CCATGAGGCTTGTGTGAAA-3′). PCNA was targeted using PCNA siRNA-1 (5′-GGAGGAAGCTCTTACCATA-3′) and PCNA siRNA-2 (5′-GCCGAGATCTCAGCCATAT-3′). Cdt2 was targeted using Cdt2 siRNA-1 (5′-GAATTATACTGCTTATCGA-3′) and Cdt2 siRNA-2 (5′-ATACAAGAGTGACTCTATA-3′).
Cell culture, drug treatments, and synchronization
U2OS (ATCC, Cat No. HTB-96) and HCT-116 (ATCC, Cat No. CCL-247) cells were cultured in McCoy's 5A Medium (HyClone, Cat No. SH3020001) supplemented with 10% fetal bovine serum (Atlantic Biologicals, Cat No. S11150). HEK293 (ATCC, Cat No. CRL-1573) and HeLa (ATCC, Cat No CCL-2) cells were cultured in Dulbecco's Modified Eagle Medium (Invitrogen, Cat. No. 11965-092) supplemented with 10% fetal bovine serum.
MG132, aphidicolin, thymidine, and cycloheximide were purchased from Sigma and used at 20 μM, 4 μM, 2 mM, and 100 μg/mL, respectively. Synchronization at the G1-S border was performed by aphidicolin treatment for 24 hours, or a double-thymidine block in which cells were treated with thymidine for 16 hours, released for 8 hours, and treated with a second thymidine block for an additional 16 hours prior to release. To synchronize cells in S phase, serum starvation was performed by treating cells with 0.1% FBS for 48 hours followed by release into normal culture medium for 16–18 hours. To synchronize cells in G1 phase, cells were treated with a Thymidine-Nocodazole block (2 mM Thymidine for 24 h, release 3 h, 100 ng/ml nocodazole for 12 h) and a 6 h release into G1 phase.
Antibodies
Antibodies for MMSET (ab75359, mouse monoclonal) and Histone H3 (ab1791, rabbit polyclonal) were purchased from Abcam. An anti-MMSET antibody (NBP1-82633, rabbit polyclonal) was purchased from Novus for co-staining in immunofluorescence. Anti-α-tubulin (T9026, mouse monoclonal), anti-FLAG (clone M2, F1804, mouse monoclonal), and anti-Actin (A2228, mouse monoclonal) antibodies were purchased from Sigma. Anti-ORC2 (4736S, rat monoclonal), anti-MCM2 (D7G11, rabbit monoclonal), and anti-MCM7 (D10A11, rabbit monoclonal) antibodies were purchased from Cell Signaling. Antibody for Cyclin E1 (3327, rabbit monoclonal) was purchased from Epitomics. Anti-PCNA (clone PC10, sc-56, mouse monoclonal) antibody was purchased from Santa Cruz. Anti-Cdt2 (ABS500, rabbit polyclonal) was purchased from Millipore and anti-BrdU-FITC (11202693001, mouse) antibody was purchased from Roche.
Immunoprecipitations and western blots
Cells were lysed with cell lysis buffer (25 mM HEPES, 25 mM NaCl, 1 mM EDTA, 0.5 mM CaCl2, 0.5% NP40) containing protease inhibitors and 1.0 mg of clarified whole cell lysates was incubated overnight at 4°C with antibody or IgG as indicated. Complexes were recovered by incubating with protein G-Sepharose (Amersham Biosciences) for 2 hours at 4°C. Immunocomplexes were then washed with cell lysis buffer and recovered by boiling with 40 μL SDS loading buffer. Eluted proteins were resolved by SDS-PAGE and transferred to Immobilon-P membranes (Millipore) for western blotting.
Chromatin Fractionation
Chromatin-enriched fractions were obtained by separating lysates into Triton X-100 soluble and insoluble fractions essentially as previously described.Citation61,62 Briefly, trypsinized cell pellets were washed with PBS and then resuspended in cytoskeleton (CSK) buffer (10 mM PIPES, pH 6.8, 100 mM NaCl, 1.5 mM MgCl2, 300 mM sucrose) containing 0.5% Triton X-100, 1 mM ATP, 1 mM dithiothreitol, and protease inhibitors for 20 minutes on ice. Lysates were then spun down at 1000 x g for 5 minutes and the soluble supernatant was saved as the non-chromatin enriched fraction. The insoluble chromatin-enriched pellet was washed once with CSK buffer and then resuspended in 100 μL CSK buffer followed by 100 μL 2x SDS sample buffer.
GST and His Pull-downs
GST-conjugated MMSET fragments used here include: MMSET-N′ (aa 1–647); MMSET-Mid (aa 648–781); and MMSET-C′ (aa 782–1365). GST Pull-downs were performed as previously described.Citation63 Briefly, 40 μg of each GST fusion protein was bound to glutathione (GSH)-agarose slurry in pull-down buffer (PB: 1 M HEPES, pH 7.2, 50 mM CH3CO2K, 1 mM EDTA, 200 mM D-sorbitol, 0.1% Triton X-100, 1 mM PMSF, 10 μg/ml of leupeptin, and 5 μg/ml of aprotinin) for 60 min at 4°C. The resin was then washed once in PB, and incubated with 1.0 mg of clarified whole-cell lysate for 5 hours at 4°C. Resulting protein complexes were washed twice with PB, eluted in 40 μl of SDS-sample buffer, and resolved by SDS-PAGE.
His pull-downs were performed essentially as previously described,Citation64 with a few modifications. Briefly, cells were transfected with His-tagged ubiquitin followed by serum starvation for 48 hours and a 16-hour release into S phase. Six hours prior to harvest, cells were treated with 20 μM MG132. Cells were then harvested with 500 μL of Urea lysis buffer (8 M Urea, 0.1 M NaH2PO4, 0.1 M Tris pH 8.0, 0.05% Tween 20, 0.01 M Imidazole), pulse sonicated 10 times at one second intervals, and incubated at 4°C for 30 minutes with end-to-end rotation. Lysates were pelleted for 10 minutes at 4°C and lysates were incubated with pre-washed cobalt metal affinity resin (Clontech) overnight at 4°C. Resin was then washed 2 times with Urea Wash Buffer (8 M Urea, 0.1 M NaH2PO4, 0.1 M Tris pH 8.0, 0.05% Tween 20, 0.02 M Imidazole) followed by 2 washes with Native Wash Buffer (0.1 M NaH2PO4, 0.1 M Tris pH 8.0, 0.05% Tween 20, 0.02 M Imidazole) and then boiled with loading dye. Eluted proteins were resolved by SDS-PAGE and transferred to Immobilon-P membranes (Millipore).
Flow cytometry
For cell-cycle analyses, cells were fixed with 70 percent ethanol and stained with propidium iodide following standard protocols. DNA content was examined using a FACSCanto II flow cytometer (BD Biosciences) and analyzed with ModFit (Verity Software). For analysis of BrdU incorporation, cells were pulsed with 10 μM bromodeoxyuridine for 30 minutes followed by trypsinization, PBS wash, and fixation with ice cold 70% ethanol. Cells were then resupended in 2N HCl, 0.5% Triton X-100 and incubated for 30 minutes at room temperature. Next, cells were resuspended in 0.1 M Na2B4O7 (pH 8.5), spun down, and resuspended in antibody incubation buffer (0.2% BSA, 1% Tween 20 in phosphate buffered saline) containing Anti-Bromodeoxyuridine-Fluorescein (Roche) for 30 minutes in the dark at room temperature. Cells were then washed with PBS and resuspended in PI staining solution containing RNase A for 1 hour at 4°C in the dark prior to running on a FACSCanto II flow cytometer (BD Biosciences) and analysis with FlowJo (Tree Star).
Immunofluorescence
Cells previously plated on coverslips were fixed with 4% paraformaldehyde in PBS and stained with the indicated antibodies followed by Alexa Fluor 488 or rhodamine-conjugated secondary antibodies. Nuclei were counterstained with 4',6-Diamidino-2-Phenylindole, Dihydrochloride
(DAPI). Images were obtained using a LSM-710 laser scanning confocal microscope (Carl Zeiss, Inc.) and Zen2009 software.
Statistical analysis
All statistical analyses in this study were performed using a 2-tailed Student's t test.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
1121323_Supplemental_Material.pdf
Download PDF (272.5 KB)Acknowledgments
We thank the members of the Lou lab for helpful discussion and technical advice throughout this project.
Funding
DE and HH acknowledge fellowship funding from Mayo Graduate School. This work was supported in part by NIH grants CA130996, CA189666, and CA148940.
References
- Blow JJ, Gillespie PJ. Replication licensing and cancer–a fatal entanglement? Nat Rev Cancer 2008; 8:799–806; PMID:18756287; http://dx.doi.org/10.1038/nrc2500
- Mendez J, Stillman B. Perpetuating the double helix: molecular machines at eukaryotic DNA replication origins. BioEssays 2003; 25:1158–67; PMID:14635251; http://dx.doi.org/10.1002/bies.10370
- Blow JJ, Dutta A. Preventing re-replication of chromosomal DNA. Nat Rev Mol Cell Biol 2005; 6:476–86; PMID:15928711; http://dx.doi.org/10.1038/nrm1663
- Arias EE, Walter JC. Strength in numbers: preventing rereplication via multiple mechanisms in eukaryotic cells. Gen Dev 2007; 21:497–518; PMID:17344412; http://dx.doi.org/10.1101/gad.1508907
- Moyer SE, Lewis PW, Botchan MR. Isolation of the Cdc45/Mcm2-7/GINS (CMG) complex, a candidate for the eukaryotic DNA replication fork helicase. Proc Natl Acad Sci U S A 2006; 103:10236–41; PMID:16798881; http://dx.doi.org/10.1073/pnas.0602400103
- Ilves I, Petojevic T, Pesavento JJ, Botchan MR. Activation of the MCM2-7 helicase by association with Cdc45 and GINS proteins. Mol Cell 2010; 37:247–58; PMID:20122406; http://dx.doi.org/10.1016/j.molcel.2009.12.030
- Moldovan GL, Pfander B, Jentsch S. PCNA, the maestro of the replication fork. Cell 2007; 129:665–79; PMID:17512402; http://dx.doi.org/10.1016/j.cell.2007.05.003
- Wohlschlegel JA, Dwyer BT, Dhar SK, Cvetic C, Walter JC, Dutta A. Inhibition of eukaryotic DNA replication by geminin binding to Cdt1. Science 2000; 290:2309–12; PMID:11125146; http://dx.doi.org/10.1126/science.290.5500.2309
- Tada S, Li A, Maiorano D, Mechali M, Blow JJ. Repression of origin assembly in metaphase depends on inhibition of RLF-B/Cdt1 by geminin. Nat Cell Biol 2001; 3:107–13; PMID:11175741; http://dx.doi.org/10.1038/35055000
- Li X, Zhao Q, Liao R, Sun P, Wu X. The SCF(Skp2) ubiquitin ligase complex interacts with the human replication licensing factor Cdt1 and regulates Cdt1 degradation. J Biol Chem 2003; 278:30854–8; PMID:12840033; http://dx.doi.org/10.1074/jbc.C300251200
- Havens CG, Walter JC. Mechanism of CRL4(Cdt2), a PCNA-dependent E3 ubiquitin ligase. Gen Dev 2011; 25:1568–82; PMID:21828267; http://dx.doi.org/10.1101/gad.2068611
- Jin J, Arias EE, Chen J, Harper JW, Walter JC. A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol Cell 2006; 23:709–21; PMID:16949367; http://dx.doi.org/10.1016/j.molcel.2006.08.010
- Lee J, Zhou P. DCAFs, the missing link of the CUL4-DDB1 ubiquitin ligase. Mol Cell 2007; 26:775–80; PMID:17588513; http://dx.doi.org/10.1016/j.molcel.2007.06.001
- O'Connell BC, Harper JW. Ubiquitin proteasome system (UPS): what can chromatin do for you?. Curr Opin Cell Biol 2007; 19:206–14; PMID:17314036; http://dx.doi.org/10.1016/j.ceb.2007.02.014
- Arias EE, Walter JC. PCNA functions as a molecular platform to trigger Cdt1 destruction and prevent re-replication. Nat Cell Biol 2006; 8:84–90; PMID:16362051; http://dx.doi.org/10.1038/ncb1346
- Zhong W, Feng H, Santiago FE, Kipreos ET. CUL-4 ubiquitin ligase maintains genome stability by restraining DNA-replication licensing. Nature 2003; 423:885–9; PMID:12815436; http://dx.doi.org/10.1038/nature01747
- Abbas T, Sivaprasad U, Terai K, Amador V, Pagano M, Dutta A. PCNA-dependent regulation of p21 ubiquitylation and degradation via the CRL4Cdt2 ubiquitin ligase complex. Gen Dev 2008; 22:2496–506; PMID:18794347; http://dx.doi.org/10.1101/gad.1676108
- Nishitani H, Lygerou Z, Nishimoto T. Proteolysis of DNA replication licensing factor Cdt1 in S-phase is performed independently of geminin through its N-terminal region. J Biol Chem 2004; 279:30807–16; PMID:15138268; http://dx.doi.org/10.1074/jbc.M312644200
- Nishitani H, Shiomi Y, Iida H, Michishita M, Takami T, Tsurimoto T. CDK inhibitor p21 is degraded by a proliferating cell nuclear antigen-coupled Cul4-DDB1Cdt2 pathway during S phase and after UV irradiation. J Biol Chem 2008; 283:29045–52; PMID:18703516; http://dx.doi.org/10.1074/jbc.M806045200
- Tardat M, Brustel J, Kirsh O, Lefevbre C, Callanan M, Sardet C, Julien E. The histone H4 Lys 20 methyltransferase PR-Set7 regulates replication origins in mammalian cells. Nat Cell Biol 2010; 12:1086–93; PMID:20953199; http://dx.doi.org/10.1038/ncb2113
- Sansam CL, Shepard JL, Lai K, Ianari A, Danielian PS, Amsterdam A, Hopkins N, Lees JA. DTLCDT2 is essential for both CDT1 regulation and the early G21487;M checkpoint. Gen Dev 2006; 20:3117–29; PMID:17085480; http://dx.doi.org/10.1101/gad.1482106
- Kim Y, Starostina NG, Kipreos ET. The CRL4Cdt2 ubiquitin ligase targets the degradation of p21Cip1 to control replication licensing. Gen Dev 2008; 22:2507–19; PMID:18794348; http://dx.doi.org/10.1101/gad.1703708
- Lovejoy CA, Lock K, Yenamandra A, Cortez D. DDB1 maintains genome integrity through regulation of Cdt1. Mol Cell Biol 2006; 26:7977–90; PMID:16940174; http://dx.doi.org/10.1128/MCB.00819-06
- Chesi M, Nardini E, Lim RS, Smith KD, Kuehl WM, Bergsagel PL. The t(4;14) translocation in myeloma dysregulates both FGFR3 and a novel gene, MMSET, resulting in IgHMMSET hybrid transcripts. Blood 1998; 92:3025–34; PMID:9787135
- Keats JJ, Maxwell CA, Taylor BJ, Hendzel MJ, Chesi M, Bergsagel PL, Larratt LM, Mant MJ, Reiman T, Belch AR, et al. Overexpression of transcripts originating from the MMSET locus characterizes all t(4;14)(p16;q32)-positive multiple myeloma patients. Blood 2005; 105:4060–9; PMID:15677557; http://dx.doi.org/10.1182/blood-2004-09-3704
- Keats JJ, Reiman T, Belch AR, Pilarski LM. Ten years and counting: so what do we know about t(4;14)(p16;q32) multiple myeloma. Leukemia & lymphoma 2006; 47:2289–300; PMID:17107900; http://dx.doi.org/10.1080/10428190600822128
- Keats JJ, Reiman T, Maxwell CA, Taylor BJ, Larratt LM, Mant MJ, Belch AR, Pilarski LM. In multiple myeloma, t(4;14)(p16;q32) is an adverse prognostic factor irrespective of FGFR3 expression. Blood 2003; 101:1520–9; PMID:12393535; http://dx.doi.org/10.1182/blood-2002-06-1675
- Santra M, Zhan F, Tian E, Barlogie B, Shaughnessy J, Jr. A subset of multiple myeloma harboring the t(4;14)(p16;q32) translocation lacks FGFR3 expression but maintains an IGHMMSET fusion transcript. Blood 2003; 101:2374–6; PMID:12433679; http://dx.doi.org/10.1182/blood-2002-09-2801
- Kassambara A, Klein B, Moreaux J. MMSET is overexpressed in cancers: link with tumor aggressiveness. Biochem Biophys Res commun 2009; 379:840–5; PMID:19121287; http://dx.doi.org/10.1016/j.bbrc.2008.12.093
- Hudlebusch HR, Santoni-Rugiu E, Simon R, Ralfkiaer E, Rossing HH, Johansen JV, Jorgensen M, Sauter G, Helin K. The histone methyltransferase and putative oncoprotein MMSET is overexpressed in a large variety of human tumors. Clin Cancer Res; 17:2919–33; PMID:21385930; http://dx.doi.org/10.1158/1078-0432.CCR-10-1302
- Hudlebusch HR, Skotte J, Santoni-Rugiu E, Zimling ZG, Lees MJ, Simon R, Sauter G, Rota R, De Ioris MA, Quarto M, et al. MMSET is highly expressed and associated with aggressiveness in neuroblastoma. Cancer Res 2011; 71:4226–35; PMID:21527557; http://dx.doi.org/10.1158/0008-5472.CAN-10-3810
- Oyer JA, Huang X, Zheng Y, Shim J, Ezponda T, Carpenter Z, Allegretta M, Okot-Kotber CI, Patel JP, Melnick A, et al. Point mutation E1099K in MMSET(NSD2 enhances its methyltranferase activity and leads to altered global chromatin methylation in lymphoid malignancies. Leukemia 2014; 28:198–201; PMID:23823660; http://dx.doi.org/10.1038/leu.2013.204
- Jaffe JD, Wang Y, Chan HM, Zhang J, Huether R, Kryukov GV, Bhang HE, Taylor JE, Hu M, Englund NP, et al. Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nat Gen 2013; 45:1386–91; PMID:24076604; http://dx.doi.org/10.1038/ng.2777
- Hirschhorn K, Cooper HL, Firschein IL. Deletion of short arms of chromosome 4-5 in a child with defects of midline fusion. Humangenetik 1965; 1:479–82; PMID:5895684
- Wright TJ, Clemens M, Quarrell O, Altherr MR. Wolf-Hirschhorn and Pitt-Rogers-Danks syndromes caused by overlapping 4p deletions. Am J Med Gen 1998; 75:345–50; PMID:9482639; http://dx.doi.org/10.1002/(SICI)1096-8628(19980203)75:4%3c345::AID-AJMG2%3e3.0.CO;2-P
- Wright TJ, Ricke DO, Denison K, Abmayr S, Cotter PD, Hirschhorn K, Keinanen M, McDonald-McGinn D, Somer M, Spinner N, et al. A transcript map of the newly defined 165 kb Wolf-Hirschhorn syndrome critical region. Hum Mol Gen 1997; 6:317–24; PMID:9063753; http://dx.doi.org/10.1093/hmg/6.2.317
- Stec I, Wright TJ, van Ommen GJ, de Boer PA, van Haeringen A, Moorman AF, Altherr MR, den Dunnen JT. WHSC1, a 90 kb SET domain-containing gene, expressed in early development and homologous to a Drosophila dysmorphy gene maps in the Wolf-Hirschhorn syndrome critical region and is fused to IgH in t(4;14) multiple myeloma. Hum Mol Gen 1998; 7:1071–82; PMID:9618163; http://dx.doi.org/10.1093/hmg/7.7.1071
- Bergemann AD, Cole F, Hirschhorn K. The etiology of Wolf-Hirschhorn syndrome. TrendsGen 2005; 21:188–95; PMID:15734578; http://dx.doi.org/10.1016/j.tig.2005.01.008
- Nimura K, Ura K, Shiratori H, Ikawa M, Okabe M, Schwartz RJ, Kaneda Y. A histone H3 lysine 36 trimethyltransferase links Nkx2-5 to Wolf-Hirschhorn syndrome. Nature 2009; 460:287–91; PMID:19483677; http://dx.doi.org/10.1038/nature08086
- Ohtsubo M, Theodoras AM, Schumacher J, Roberts JM, Pagano M. Human cyclin E, a nuclear protein essential for the G1-to-S phase transition. Molecular and cellular biology 1995; 15:2612–24; PMID:7739542; http://dx.doi.org/10.1128/MCB.15.5.2612
- Li Y, Jaramillo-Lambert A, Hao J, Yang Y, Zhu W. The stability of histone acetyltransferase general control non-derepressible (Gcn) 5 is regulated by Cullin4-RING E3 ubiquitin ligase. J Biol Chem 2011; 286:41344–52; PMID:21987584; http://dx.doi.org/10.1074/jbc.M111.290767
- Huh J, Piwnica-Worms H. CRL4(CDT2) targets CHK1 for PCNA-independent destruction. Mol Cell Biol 2013; 33:213–26; PMID:23109433; http://dx.doi.org/10.1128/MCB.00847-12
- Centore RC, Havens CG, Manning AL, Li JM, Flynn RL, Tse A, Jin J, Dyson NJ, Walter JC, Zou L. CRL4(Cdt2)-mediated destruction of the histone methyltransferase Set8 prevents premature chromatin compaction in S phase. Mol Cell 2010; 40:22–33; PMID:20932472; http://dx.doi.org/10.1016/j.molcel.2010.09.015
- Marango J, Shimoyama M, Nishio H, Meyer JA, Min DJ, Sirulnik A, Martinez-Martinez Y, Chesi M, Bergsagel PL, Zhou MM, et al. The MMSET protein is a histone methyltransferase with characteristics of a transcriptional corepressor. Blood 2008; 111:3145–54; PMID:18156491; http://dx.doi.org/10.1182/blood-2007-06-092122
- Brito JL, Walker B, Jenner M, Dickens NJ, Brown NJ, Ross FM, Avramidou A, Irving JA, Gonzalez D, Davies FE, et al. MMSET deregulation affects cell cycle progression and adhesion regulons in t(4;14) myeloma plasma cells. Haematologica 2009; 94:78–86; PMID:19059936; http://dx.doi.org/10.3324/haematol.13426
- Martinez-Garcia E, Popovic R, Min DJ, Sweet SM, Thomas PM, Zamdborg L, Heffner A, Will C, Lamy L, Staudt LM, et al. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood 2011; 117:211–20; PMID:20974671; http://dx.doi.org/10.1182/blood-2010-07-298349
- Kuo AJ, Cheung P, Chen K, Zee BM, Kioi M, Lauring J, Xi Y, Park BH, Shi X, Garcia BA, et al. NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol cell 2011; 44:609–20; PMID:22099308; http://dx.doi.org/10.1016/j.molcel.2011.08.042
- Toyokawa G, Cho HS, Masuda K, Yamane Y, Yoshimatsu M, Hayami S, Takawa M, Iwai Y, Daigo Y, Tsuchiya E, et al. Histone lysine methyltransferase Wolf-Hirschhorn syndrome candidate 1 is involved in human carcinogenesis through regulation of the Wnt pathway. Neoplasia 2011; 13:887–98; PMID:22028615; http://dx.doi.org/10.1593/neo.11048
- Ezponda T, Popovic R, Shah MY, Martinez-Garcia E, Zheng Y, Min DJ, Will C, Neri A, Kelleher NL, Yu J, et al. The histone methyltransferase MMSETWHSC1 activates TWIST1 to promote an epithelial-mesenchymal transition and invasive properties of prostate cancer. Oncogene 2013; 32:2882–90; PMID:22797064; http://dx.doi.org/10.1038/onc.2012.297
- Jorgensen S, Elvers I, Trelle MB, Menzel T, Eskildsen M, Jensen ON, Helleday T, Helin K, Sorensen CS. The histone methyltransferase SET8 is required for S-phase progression. J Cell Biol 2007; 179:1337–45; PMID:18166648; http://dx.doi.org/10.1083/jcb.200706150
- Prasanth SG, Prasanth KV, Siddiqui K, Spector DL, Stillman B. Human Orc2 localizes to centrosomes, centromeres and heterochromatin during chromosome inheritance. EMBO J 2004; 23:2651–63; PMID:15215892; http://dx.doi.org/10.1038/sj.emboj.7600255
- Prasanth SG, Prasanth KV, Stillman B. Orc6 involved in DNA replication, chromosome segregation, and cytokinesis. Science 2002; 297:1026–31; PMID:12169736; http://dx.doi.org/10.1126/science.1072802
- Miotto B, Struhl K. HBO1 histone acetylase activity is essential for DNA replication licensing and inhibited by Geminin. Mol Cell 2010; 37:57–66; PMID:20129055; http://dx.doi.org/10.1016/j.molcel.2009.12.012
- Sugimoto N, Yugawa T, Iizuka M, Kiyono T, Fujita M. Chromatin remodeler sucrose nonfermenting 2 homolog (SNF2H) is recruited onto DNA replication origins through interaction with Cdc10 protein-dependent transcript 1 (Cdt1) and promotes pre-replication complex formation. J Biol Chem 2011; 286:39200–10; PMID:21937426; http://dx.doi.org/10.1074/jbc.M111.256123
- Kuo AJ, Song J, Cheung P, Ishibe-Murakami S, Yamazoe S, Chen JK, Patel DJ, Gozani O. The BAH domain of ORC1 links H4K20me2 to DNA replication licensing and Meier-Gorlin syndrome. Nature 2012; 484:115–9; PMID:22398447; http://dx.doi.org/10.1038/nature10956
- Rice JC, Nishioka K, Sarma K, Steward R, Reinberg D, Allis CD. Mitotic-specific methylation of histone H4 Lys 20 follows increased PR-Set7 expression and its localization to mitotic chromosomes. Gen Dev 2002; 16:2225–30; PMID:12208845; http://dx.doi.org/10.1101/gad.1014902
- Tardat M, Murr R, Herceg Z, Sardet C, Julien E. PR-Set7-dependent lysine methylation ensures genome replication and stability through S phase. J Cell Biol 2007; 179:1413–26; PMID:18158331; http://dx.doi.org/10.1083/jcb.200706179
- Xu H, Zhang P, Liu L, Lee MY. A novel PCNA-binding motif identified by the panning of a random peptide display library. Biochem 2001; 40:4512–20; PMID:11284708; http://dx.doi.org/10.1021/bi010103+
- Kerzendorfer C, Colnaghi R, Abramowicz I, Carpenter G, O'Driscoll M. Meier-Gorlin syndrome and Wolf-Hirschhorn syndrome: two developmental disorders highlighting the importance of efficient DNA replication for normal development and neurogenesis. DNA Repair 2013; 12:637–44; PMID:23706772; http://dx.doi.org/10.1016/j.dnarep.2013.04.016
- Kerzendorfer C, Hannes F, Colnaghi R, Abramowicz I, Carpenter G, Vermeesch JR, O'Driscoll M. Characterizing the functional consequences of haploinsufficiency of NELF-A (WHSC2) and SLBP identifies novel cellular phenotypes in Wolf-Hirschhorn syndrome. Human molecular genetics 2012; 21:2181–93; PMID:22328085; http://dx.doi.org/10.1093/hmg/dds033
- Deng M, Li F, Ballif BA, Li S, Chen X, Guo L, Ye X. Identification and functional analysis of a novel cyclin e/cdk2 substrate ankrd17. J Biol Chem 2009; 284:7875–88; PMID:19150984; http://dx.doi.org/10.1074/jbc.M807827200
- Braden WA, Lenihan JM, Lan Z, Luce KS, Zagorski W, Bosco E, Reed MF, Cook JG, Knudsen ES. Distinct action of the retinoblastoma pathway on the DNA replication machinery defines specific roles for cyclin-dependent kinase complexes in prereplication complex assembly and S-phase progression. Mol Cell Biol 2006; 26:7667–81; PMID:16908528; http://dx.doi.org/10.1128/MCB.00045-06
- Ham H, Guerrier S, Kim J, Schoon RA, Anderson EL, Hamann MJ, Lou Z, Billadeau DD. Dedicator of cytokinesis 8 interacts with talin and Wiskott-Aldrich syndrome protein to regulate NK cell cytotoxicity. J Immunol 2013; 190:3661–9; http://dx.doi.10.4049/jimmunol.1202792
- Yanagiya A, Suyama E, Adachi H, Svitkin YV, Aza-Blanc P, Imataka H, Mikami S, Martineau Y, Ronai ZA, Sonenberg N. Translational homeostasis via the mRNA cap-binding protein, eIF4E. Mol Cell 2012; 46:847–58; PMID:22578813; http://dx.doi.org/10.1016/j.molcel.2012.04.004