
ABSTRACT
Progression of cell cycle is associated with changes in K+ channel expression and activity. In this study, we report that Kv10.1, a K+ channel that increases cell proliferation and tumor growth, is regulated at the transcriptional level by the pRb/E2F1 pathway. De-repression of E2F1 by HPV-E7 oncoprotein leads to increased expression of Kv10.1. In proliferating cells, E2F1 transcription factor binds directly to the Kv10.1 promoter during (or close to) G2/M, resulting in transient expression of the channel. Importantly, this happens not only in cancer cells but also in non-transformed cells. Lack of Kv10.1 in both cancer and non-transformed cells resulted in prolonged G2/M phase, as indicated by phosphorylation of Cdk1 (Y15) and sustained pRb hyperphosphorylation. Our results strongly suggest that Kv10.1 expression is coupled to cell cycle progression and facilitates G2/M progression in both healthy and tumor cells.
KEYWORDS:
Introduction
Uncontrolled proliferation, one of the hallmarks of cancer,Citation1 reflects failures in the machinery that controls cell cycle progression. This machinery relies on the interplay between gene expression, cytoskeletal reorganization and chromosomal rearrangement.Citation2,3 The order and interdependence of cell cycle events is determined by periodic transcription of genes that guarantee an accurate cell division.Citation4
Potassium channels are involved in cell proliferation and cell cycle control.Citation5 Altered K+ channel expression contributes to pathological proliferation, and their overexpression has been associated with tumor growth.Citation6 Although different studies show that the expression and function of K+ channels vary along cell cycle, the mechanisms controlling ion channel expression during the cell cycle remain elusive. In proliferating cells, K+ permeability determines the periodic changes of the resting membrane potential associated with each of the phases of the cell cycle.Citation7 K+ channels also play a role in cell volume changes required for the completion of mitosis,Citation8 and might influence signal transduction elicited by cell cycle checkpoints through protein-protein interactions or phospholipid clustering that modulates mitogenic signaling.Citation9
KV10.1 (KCNH1, Eag1) is one of the best-studied K+ channels in the context of aberrant proliferation. It is widely expressed in the human brain but practically undetectable in peripheral tissues. KV10.1 is frequently up-regulated in human cancers, where it contributes to tumor progression.Citation10,11 Transformed cells and tumors expressing KV10.1 appear to acquire selective advantages, such as increased migrationCitation12 and tumor vascularizationCitation13 that favor tumor progression. Although several studies implicate cell-cycle signaling cascades, the mechanisms triggering Kv10.1 up-regulation are still not understood. In serum-starved breast cancer MCF-7 cells, Kv10.1 expression is upregulated as cells are induced to enter into the cell cycle.Citation14 Viral oncoproteins HPV-E6 and HPV-E7 that respectively target p53 and the growth suppressor Retinoblastoma protein (pRb), trigger transcription of Kv10.1 mRNA in primary keratinocytes.Citation15 Interestingly, the promoter region of Kv10.1 contains an E2F1-responsive element.Citation16 Since E2F1 transcription factor is released upon inactivation of pRb and regulates gene expression during G1/S and G2/M transitions,Citation17–19 we hypothesized that Kv10.1 expression might be temporally regulated along the cell cycle by the pRb/E2F1 growth control pathway.
To test whether Kv10.1 expression is under the influence of the cell cycle through the pRb/E2F1 pathway, we abolished the binding of the E2F1 transcription factor to the promoter region of Kv10.1, reduced the pRb suppression on E2F1 through HPV-E7 overexpression, and examined the consequences in the Kv10.1 promoter activity as well as protein expression in thymidine synchronized HeLa cells. Our results demonstrate that E2F1 drives periodic Kv10.1 expression through direct binding to the promoter. E2F1 preferentially binds to Kv10.1 promoter toward G2/M, resulting in restricted expression of the channel during a short time window in G2/M in cancer cells, and interestingly also in normal proliferating cells. To elucidate whether the expression of Kv10.1 has an influence on cell cycle progression, we analyzed the cell cycle of Kv10.1-depleted HeLa cells and mouse embryonic fibroblasts (MEFs) from Kv10.1 knockout mice. Lack of Kv10.1 resulted in delayed progression through mitosis, as there was an increase in inhibition of Cdk1 and the reconstitution of pRb repression was delayed. These results indicate that regulation of Kv10.1 by the pRb/E2F1 pathway contributes to the accuracy of cell division.
Results
Periodic expression of Kv10.1 along the cell cycle
In order to elucidate whether Kv10.1 expression is associated to the cell cycle, we synchronized exponentially growing HeLa cells – which endogenously express Kv10.1 – to the G1/S border using a double thymidine block protocol. The cells were then allowed to progress into the cell cycle, and we assessed Kv10.1 expression levels along with other cell cycle-regulated genes every 4 h, from the G1/S transition to mitosis and entrance to the next cell cycle, as determined by studying the cell cycle distribution by flow cytometry ().
Figure 1. Periodic expression of Kv10.1 along the cell cycle. A. HeLa cells were synchronized with a double thymidine block and released into fresh medium. Total cell lysates were prepared at the indicated time points and endogenous Kv10.1 was precipitated. Analysis by SDS-PAGE and Western blotting using anti-Kv10.1, anti-cyclin A2 and anti-Cyclin B1 showed that Kv10.1 expression changes along the cell cycle, with a peak expression between 8 and 12 h corresponding to G2/M. Calnexin and Actin were used as loading controls. B. Asynchronous HeLa cells were labeled with anti-Cyclin B1, anti- p-Histone H3 (Ser 28) and anti-Kv10.1. Cells in G2, as evidenced by cytoplasmic Cyclin B1 signal, as well as mitotic cells (nuclear Cyclin B1 and p-H3 Ser 28) showed Kv10.1 reactivity at the plasma membrane. C. Cyclin B1-positive cells were localized to the proliferative compartment, at the bottom and sides of the crypt. Kv10.1 positive cells were also found in the proliferative compartment of the colon crypt. White arrows indicate Kv10.1 and Cyclin B1 expressing cells. Scale bar 20 µm.
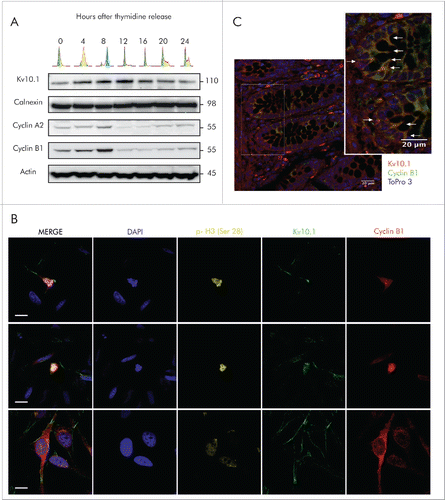
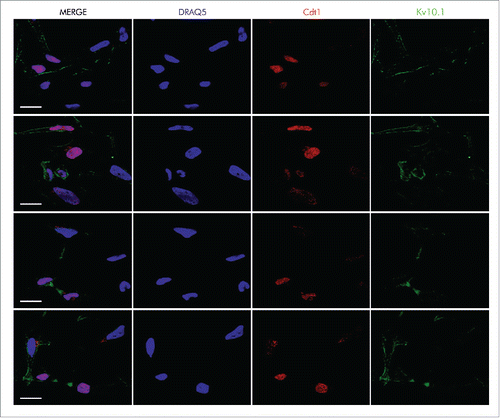
Expression of Cyclin A2 and Cyclin B1 increased gradually and both accumulated as expected when the majority of cells reached the G2/M phases 8 h after release from block. We observed a similar pattern in Kv10.1 expression, with the exception that it was maintained until the completion of mitosis and by the beginning of the next cycle at 12 h and fell thereafter (). Expression of Kv10.1 during cell cycle progression was also validated by immunocytochemistry in asynchronous and exponentially growing HeLa cells. Kv10.1 staining was evident in the plasma membrane of cells entering G2 phase, distinguishable by cytoplasmic Cyclin B1 expression, as well as in mitotic cells, which show nuclear localization of Cyclin B1 and phosphorylation of Histone H3 ( and S1). Kv10.1 was also enriched at the cytoplasmic bridge of cells undergoing cytokinesis, and was still present in cells entering the new cell cycle in G1 phase, as determined by Cdt1 expression, which accumulates in G1Citation20 (). Together, these results indicate that expression of Kv10.1 is periodic during the cell cycle, peaks at G2/M and falls again as cells enter into the new cycle.
Figure 2. Kv10.1 can be detected at the cytoplasmic bridge in cells undergoing cytokinesis, and it is also present on the cell surface of cells at G1 phase, as determined by Cdt1 expression. HeLa cells were labeled with anti- Cdt1 and anti-Kv10.1. Scale bar 20 µm.
Previous studies indicated that Kv10.1 expression is abundant in human brain, but failed to detect the channel in healthy non-neuronal tissue,Citation21 except in restricted cell populations, which correspond to terminal developmental stages of different cell lineages.Citation10
Given that Kv10.1 expression is temporally correlated with expression of Cyclins A2 and B1, we hypothesized that Kv10.1 expression could also be found in healthy non-neuronal tissues, yet only in cells progressing through G2/M, which are very scarce in somatic tissues. Intestinal epithelium offers a convenient model to test this hypothesis, because stem cells located at the bottom of colonic crypts give rise to proliferating progenitor cells, which are spatially constrained to the lower third of the crypt, and later differentiate and migrate from the bottom of the crypt to the surface of the epithelium.Citation22–24 Immunohistochemical analysis on normal human colon paraffin sections using Kv10.1 and Cyclin B1 antibodies revealed localization of both signals to the deeper areas of the crypt. Kv10.1-positive cells in the proliferative compartment of the colonic crypt were also Cyclin B1-positive (), indicating that Kv10.1 expression is also enriched during G2/M progression in non-transformed cells. Strongly positive cells outside the epithelium probably correspond to plasma cells, which typically stain strongly positive for Kv10.1.Citation10
Kv10.1 expression in G2/M depends on pRb/E2F1 activity
Apart from genes specific for G1/S, E2F1 also controls a substantial number of genes normally expressed at G2/M.Citation17,18,25 The promoter region of Kv10.1 contains an E2F1-responsive element,Citation16 suggesting a possible transcriptional regulation by E2F1 during cell cycle progression. To elucidate whether Kv10.1 is regulated during the cell cycle by E2F1, we used luciferase reporter assays. The core promoter of Kv10.1, a fragment spanning from positions −630 to +114 that contains the E2F1 responsive element (), cloned into the pGL3 luciferase reporter vector was used to assess promoter activity in synchronized HeLa cells progressing through the cell cycle (this construct was a gift from Prof. Zhiguo Wang, and corresponds to the one reported in ref. Citation16). Firefly luciferase activity under the control of the Kv10.1 promoter varied significantly with time and followed the same periodic pattern as endogenous Kv10.1 protein levels (). As the cells passed through the G1/S border (0 h) and progressed through S phase (4 h), Kv10.1 promoter activity increased and reached a peak at the time of the G2/M transition (8 h). Then, activity levels declined gradually as the cells completed mitosis (12 h) and proceeded into the next cycle.
Figure 3. Kv10.1 expression in G2/M depends on pRb/E2F1 activity. A. Schematic of human Kv10.1 promoter indicating E2F1 responsive element upstream of the transcription starting site (TSS). B. Luciferase activity driven by Kv10.1 promoter (black trace) showed a peak activity during G2/M transition (One-Way ANOVA, P < 0.0001). Mutation of E2F1 responsive element (red trace) abolished promoter activity (Two-way ANOVA. Changes in expression levels with time after release P < 0.0001, mutated vs. wild type Kv10.1 promoter P < 0.0001). C. HPV-E7 overexpression increased Kv10.1 promoter activity (Two-way ANOVA. Changes in expression levels with time after release P < 0.01, effect of HPV-E7 overexpression P < 0.0001). D. Analysis by SDS-PAGE and Western blotting using anti-Kv10.1, anti-pRb and anti-E2F1 showed that Kv10.1 expression profile is delayed from E2F1 peak expression. E. E2F1 binding to endogenous Kv10.1 promoter (One-Way ANOVA, P < 0.0001) and Cyclin A2 promoter (One-Way ANOVA, P < 0.001) during G2/M transition (8 h). Cyclin A2, an E2F1 target gene regulated at the G2/M transition was used here as a positive control. Fold enrichment was calculated relative to GAPDH promoter signal. GAPDH is a non-E2F1 regulated gene. All experiments were performed at least 3 times.
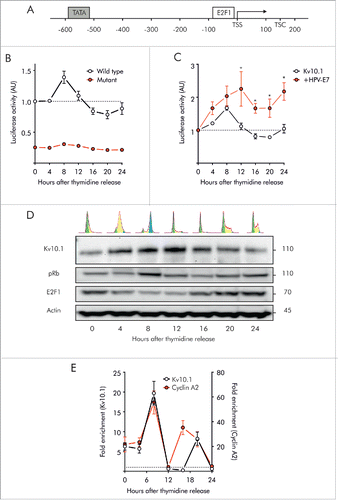
To further demonstrate the direct regulation of Kv10.1 by E2F1, we abolished the E2F1 binding site in the core promoter region of Kv10.1 by performing base-substitution mutations.Citation16 Along all the cell cycle (), luciferase activity driven by the mutant promoter was significantly reduced compared to the wild type (WT) Kv10.1 promoter.
The influence of E2F1 on Kv10.1 transcriptional activity was further confirmed by HPV-E7 overexpression (). HPV-E7, which disrupts pRb suppression on E2F1,Citation26,27 is known to increase endogenous Kv10.1 expression.Citation15 HPV-E7 overexpression significantly increased luciferase activity driven by the Kv10.1 promoter. Overexpression of E2F1 as well as HPV-E7 in HeLa cells also enhanced Kv10.1 at the protein level (Fig. S2), indicating transcriptional control of Kv10.1 by the pRb/E2F1 pathway. Taken together, these results strongly suggest that E2F1 is directly responsible for the periodic expression of Kv10.1 along the cell cycle.
Interaction of E2F1 with the Kv10.1 promoter occurs during G2/M
Periodic expression of E2F1 reaches a peak in G1/S, when the suppression by pRb is reduced. In contrast, Kv10.1 expression peak is shifted by several hours, reaching the maximum in G2/M (). To validate whether E2F1 directly binds to the Kv10.1 promoter in vivo, we conducted ChIP experiments pulling down E2F1 in synchronized HeLa cells, and checked the presence of the endogenous Kv10.1 promoter region covering the predicted E2F1 binding site using qRT-PCR on the immunoprecipitated fraction. The Cyclin A2 promoter was used as a control because it is an E2F1-regulated gene normally transcribed during G2/M.Citation19,28
We found that E2F1 interacts with both Kv10.1 and Cyclin A2 promoters in a cell-cycle dependent manner (). As the cells passed through the G1/S border and S phase (0 h and 4 h respectively), binding of E2F1 to Kv10.1 and cyclin A2 promoters was less prominent. The interaction between E2F1 and the Kv10.1 promoter was significantly increased when the cells reached the G2/M transition. As expected, the interaction between E2F1 and the Cyclin A2 promoter was also significantly increased at that time. As soon as mitosis was completed and the cells re-entered the cell cycle (12 h), E2F1 binding to either Kv10.1 or Cyclin A2 promoters was dramatically diminished. A second, weaker peak of E2F1 binding was detected at later time points on both Kv10.1 (20 h) and Cyclin A2 promoters (16 and 20 h). This could be explained by imperfect synchronization resulting in a second wave of cells reaching G2/M at a later point, or because of the large expression levels of E2F1 between 16 h and 20 h.
Kv10.1 depletion interferes with cell cycle progression in HeLa cells
To determine whether the time-dependent expression of Kv10.1 in G2/M has an impact on the cell cycle, we performed siRNA knockdown of Kv10.1 in HeLa cells and analyzed the expression levels of Cyclin proteins essential for cell cycle progression (). Cyclins A2 and B1, which are critical for G2/M transition, were more abundant after Kv10.1 depletion, while the expression of Cyclin D1 and Cyclin E1, expressed in G1 and S phase respectively, was not significantly altered.
Figure 4. Kv10.1 depletion disrupts cell cycle progression in HeLa cells. A. Western blotting showed upregulated expression of Cyclins A2 and B1 upon Kv10.1 knockdown. Expression of Cyclins D1 and E1 was comparable between knockdown and control. B-D. HeLa cells transfected with siRNA control (black trace) and Kv10.1 siRNA (red trace) were synchronized with a double thymidine block and released into fresh medium. Cells were harvested at the indicated time points for FACS analysis of cell cycle profile. B. Percentage of cells in G0/G1 phase. C. Percentage of cells in S phase. D. Percentage of cells in G2/M phase. HeLa cells accumulated at G2/M upon Kv10.1 knockdown. All experiments were performed at least 3 times.
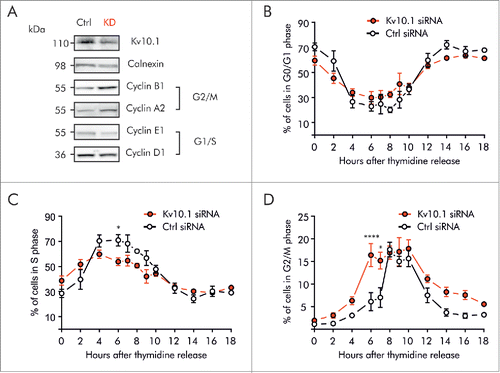
To further validate whether accumulation of Cyclins resulted from prolonged arrest in any phase of the cell cycle, we used synchronized cultures. HeLa cells transfected with siRNA against Kv10.1 were synchronized in G1/S by double thymidine block. After synchronization, cells were released into the cell cycle for 18 h (, S3). FACS analysis revealed that Kv10.1 depletion did not increase the fraction of cells in G1 and S phase (). By contrast, the peak of cells progressing through G2/M was clearly broader in Kv10.1 siRNA transfected cells than in control cells (), indicating that the cell population spends longer time in G2/M.
To understand whether this alteration in cell cycle progression results from premature entry in G2 or rather a delayed exit from mitosis, western blot analysis of major cell cycle regulatory proteins was performed in synchronized control and Kv10.1 siRNA-transfected cells. We monitored the periodic expression of Cyclin D1 and Cyclin E1, which form complexes with the corresponding CDKs in G1 and S phases to phosphorylate pRb, and also determined the overall phosphorylation of pRb protein, which induces the transition from G1 to S phase (). Knockdown of Kv10.1 did not alter the time of accumulation of Cyclins D1 and E1 after release, indicating that Kv10.1 does not affect the entry and progression through G1 and S phases. In contrast, although the levels of phosphorylated pRb rose at the same time in control cells and Kv10.1-depleted cells, pRb remained in hyperphosphorylated state for longer time in cells lacking Kv10.1, suggesting that exit from mitosis is delayed in Kv10.1-depleted cells.
Figure 5. Kv10.1 depletion alters the periodicity of key G2/M regulatory proteins. HeLa cells were synchronized with a double thymidine block and released into fresh medium. Total cell lysates from siRNA control and Kv10.1 siRNA-transfected cells were prepared at the indicated time points. A. Analysis by SDS-PAGE and Western blotting of Cyclin D1 and E1 did not show differences. Levels of phosphorylated pRb rose at the same time in siRNA control and Kv10.1 siRNA transfected cells. However, pRb remained in hyperphosphorylated state for longer time in cells lacking Kv10.1. Densitograms from the blots corresponding to pRb Ser 780, pRb Ser 795 and pRb Ser 807–811 are plotted in the lower part of the panel. B. Western blotting showed that degradation of Cyclin A2 and B1 was delayed in Kv10.1 knockdown cells, as well as the dephosphorylation of pCdk1 (Y15). All experiments were performed at least 3 times.
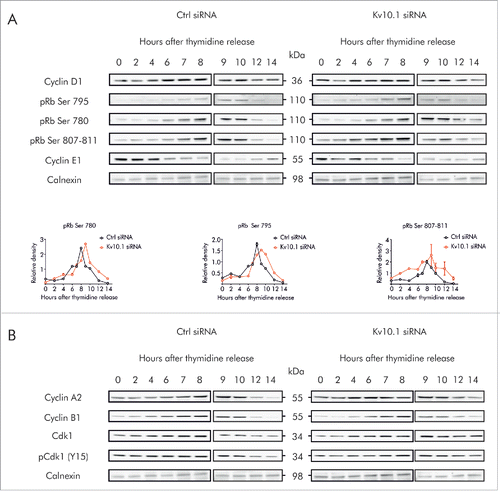
To further identify the molecular consequences of Kv10.1 depletion during G2/M progression, we monitored the degradation of Cyclin A2 and Cyclin B1, which occur during prometaphase and metaphase, respectively (). In control HeLa cells, Cyclin A2 reached maximal accumulation 9 h after release from thymidine block, and decayed thereafter. Degradation of Cyclin A2 was notably delayed after Kv10.1 knockdown. The degradation of Cyclin B1 was also slightly delayed in Kv10.1 knockdown cells. Activation of Cyclin A2/Cdk1 and Cyclin B1/Cdk1, required for entrance and progression of G2/M, occurs through dephosphorylation of Cdk1 (Y15). In control HeLa cells, dephosphorylation of Cdk1 occurred between 10 and 12 h after thymidine block. In Kv10.1-knockdown cells, phosphorylated (inactive) Cdk1 persisted for at least 14 h after release. Together, these results confirm that disruption of the periodic expression of Kv10.1 in HeLa cells leads to a delayed G2/M progression.
Absence of Kv10.1 alters cell cycle progression in mouse embryonic fibroblasts
To elucidate if the effect of Kv10.1 on the cell cycle progression of HeLa cells also happens in non-transformed cells, we used primary mouse embryonic fibroblasts (MEFs) from Kv10.1 knockout (KO MEFs)Citation29 and wild type (WT MEFs) mice. Like Kv10.1-depleted HeLa cells (), non-synchronized KO MEFs showed increased G2/M cyclins (A2 and B1). However, Cyclin D1 expression was also up-regulated (). Together, these results indicate that cell cycle progression is also altered in KO MEFs.
Figure 6. Kv10.1 absence alters cell cycle progression in mouse embryonic fibroblasts. A. Western blotting showed upregulated expression of Cyclins D1, A2 and B1 in Kv10.1 knockout MEFs. Expression of Cyclin E1 was comparable between knockout and wild type. (B-D) Wild type MEFs (black trace) and knockout MEFs (red trace) were synchronized after 72 h of serum starvation and released into fresh medium. MEFs were harvested at the indicated time points for FACS analysis of cell cycle status. B. Percentage of cells in G0/G1 phase. C. Percentage of cells in S phase. D. Percentage of cells in G2/M phase. Knockout MEFs accumulated at G2/M. All experiments were performed at least 3 times.
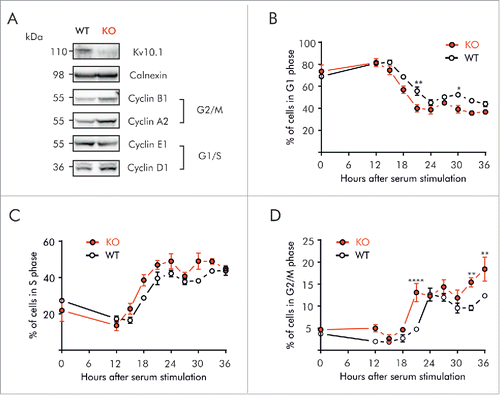
To determine whether accumulation of Cyclins resulted from cell cycle arrest, MEFs were partially synchronized in G0/G1 by serum starvation for 72 h, and then allowed to progress through the cell cycle for 36 h. Cell cycle distributions at the given times are shown in Figure S4. At the moment of release, both WT and Knockout MEFs were preferentially at G0/G1 (68.9 ± 2.08 vs. 73.5 ± 10.11%). After serum re-introduction, WT as well as KO MEFs had completed G1 and progressed into S phase at 15 h (), distribution of cells progressing into S phase did not show major differences between WT and KO, indicating that G1/S transition is not affected in MEFs after Kv10.1 depletion. These results were confirmed at the molecular level (), since both WT and KO MEFs expressed low levels of Cyclin D1 and E1 at time 0. After serum re-stimulation, accumulation of Cyclins D1 and E1 followed the same pattern in WT and KO MEFs, and the rise in pRb phosphorylation occurred between 15 and 18 h in both groups, indicating a normal G1/S transition.
Figure 7. Kv10.1 absence alters the periodicity of key G2/M regulatory proteins. MEFs were synchronized after 72 h of serum starvation and released into fresh medium. Total cell lysates from wild type and Kv10.1 knockout MEFs were prepared at the indicated time points. A. Analysis by SDS-PAGE and Western blotting of Cyclin D1 and E1 did not show differences. Levels of phosphorylated pRb rose at the same time in wild type and Kv10.1 knockout MEFs. However, pRb remained in hyperphosphorylated state for longer time in cells lacking Kv10.1. Densitograms from the blots corresponding to pRb Ser 780, pRb Ser 795 and pRb Ser 807–811 are plotted in the lower part of the panel. B. Western blotting showed increased levels of the inhibitory phosphorylation (Y15) on Cdk1. All experiments were performed at least 3 times.
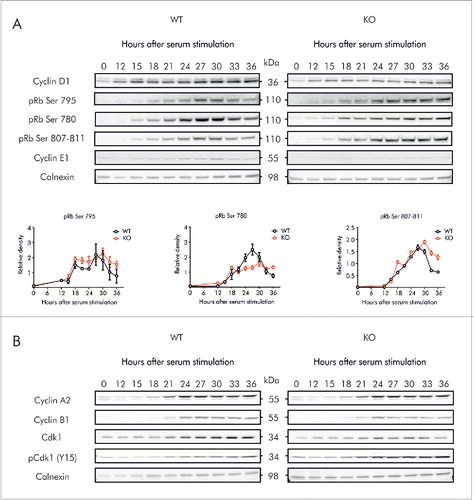
MEFs started to progress into G2/M at 21 h, as revealed by DNA content determination by FACS analysis (), and also by the rise of the expression levels of Cyclins A2 and B1 (). As observed in Kv10.1 depleted HeLa cells, KO MEFs also accumulated in G2/M (). Consistent with prolonged mitosis, pRb remained for longer time in hyperphosphorylated state in KO MEFs (). Although degradation of Cyclin A2 and B1 followed the same pattern in WT and KO, the levels of their catalytic partner Cdk1 were lower in KO MEFs (), and there was also an increase in the levels of the inhibited form pCdk1 (Y15), which would result in an inhibition of mitosis, pointing again toward abnormal mitosis progression in Kv10.1 depleted cells, as determined for HeLa cells.
Discussion
We report here that Kv10.1 expression is coupled to the cell cycle in both cancer and non-transformed cells. Kv10.1 transcription is directly regulated by the pRb/E2F1 pathway during G2/M, resulting in transient expression of Kv10.1 that contributes to progression through G2/M in HeLa cells and MEFs.
Although E2F1 is released from pRb repression in G1/S, the interaction with the Kv10.1 promoter occurs mainly toward G2/M (), a phenomenon also seen in the Cyclin A2 promoter, known to be an E2F1 target expressed during G2/M.Citation19,28 This observation suggests that additional components determine the time of E2F1 interaction with Kv10.1 promoter. Chromatin modifying proteins such as histone deacetylases and acetyl-transferases may play a role in the cell cycle-dependent expression of E2F1-responsive genes.Citation28 Consistently, high acetylation of histones in the Kv10.1 promoter is associated with upregulation of Kv10.1 in tumor-derived cells.Citation30 Thus, histone acetylation during G2/M could help to maintain the Kv10.1 promoter available for E2F1 binding.
Our observations can also explain why Kv10.1 is often upregulated in cancer cell lines and clinical tumor specimens (e.g. refs. Citation10,30–38), because the pRb/E2F1 complex is disrupted in almost all cancer cells.Citation39 Kv10.1 expression has been reported in a broad variety of tumor types, most of which also show pRb loss of function.Citation10,40–45 Aberrant Kv10.1 expression could thus simply be due to the deregulation of upstream signaling cascades. Nevertheless, our results also show that Kv10.1 plays an active role in cell cycle progression in both cancer and non-transformed cells. In HeLa cells, Kv10.1 depletion leads to delayed G2/M progression, indicating that channel expression at the end of the cell cycle facilitates G2/M completion. A delayed end of G2/M also in MEFs is suggested by the longer presence of hyperphosphorylated pRb and increased levels of phosphorylated Cdk1 (Y15) in KO cells.
Cyclin D1 levels were unaltered in HeLa cells after knockdown of Kv10.1 (), but increased in the KO MEFs with respect to wild type (). This could be due to the expression of HPV proteins in HeLa cells, which maintain certain basal activity of E2F1 that could increase cyclin D1.Citation46 MEFs would have a lower constitutive basal activity, and this can explain a requirement of Kv10.1 also to progress through G1, as it was described for MCF7 breast cancer cells,Citation47 which are also HPV-negative.Citation48
We also show that the tumor-specific expression of Kv10.1 (outside the CNS) is only apparent, and cells in normal tissue (at least in colon) also express the channel during a limited period of time. End-differentiated tissues show very low proliferation rates and therefore cells in G2/M are very scarce. Any method aiming to determine expression in whole normal tissues would therefore miss expression in such restricted population of cells, explaining why extra-cerebral tissues are negative for Kv10.1 expression. An increase in the proliferative fraction of cells, such as it occurs in tumors, would allow detection using tissue as starting material. Constitutive expression of Kv10.1 in the brain could in turn obey to the role of E2F1 in neurons, where it acts as a cell-cycle suppressor rather than as an activator.Citation49
The intimate molecular mechanism of action of K+ channels in G2/M still remains elusive. In heterologous expression systems, activation of the mitosis-promoting factor induces changes in the electrophysiological properties of rat Kv10.1.Citation50,51 These changes might be a way to regulate the fluctuations in membrane potential along G2/M.Citation52 Furthermore, Kv10.2, the closest relative to Kv10.1, has been proposed to regulate the changes in cellular volume prior to mitosis via its action on intracellular osmolarity,Citation8 which may reflect another stage for dynamic regulation of G2/M progression due to K+ flow.
Besides ion permeation, protein-protein interactions might also determine the importance of Kv10.1 during G2/M. Kv10.1 has been reported to influence cell proliferation and tumorigenesis in the absence of K+ permeation.Citation13,53
In conclusion, our results show that expression of Kv10.1 is linked to cell cycle progression as a downstream effector of the pRb/E2F1 pathway that facilitates G2/M progression in both healthy and tumor cells.
Materials and methods
Cell culture
HeLa cells (ACC 57) were cultured in RPMI 1640 (GIBCO/Invitrogen) medium supplemented with 10 % FCS (PAA) at 37°C in a 5 % CO2 incubator.
Wild type and Kv10.1 knockout embryos were dissected 13 d after detection of vaginal plugs in the pregnant females. The head and internal organs were removed and the carcasses were minced and digested in Trypsin-EDTA/DNase (Invitrogen) at 37°C for 25 min. The suspension was homogenized with a cannula and filtered with a cell strainer (Falcon). Then, cells were resuspended in DMEM supplemented with 10 % FCS. After centrifugation the supernatant was discarded and the Mouse Embryonic Fibroblasts (MEFs) were cultured on a 10 cm dish in 10 ml DMEM with 10 % FCS until confluence.
Cell cycle synchronization and FACS analysis
G0/G1 synchronization was performed as described in ref. (Citation54): 2 × 106 passage 1 MEFs were plated in a 15 cm dish containing DMEM with 10 % FCS for 2 d until confluence was reached. The cells were then washed with PBS and incubated for 72 h at 37°C in FCS-free DMEM. For release into the cell cycle, cells were washed with PBS, detached by trypsin incubation, counted and resuspended in DMEM containing 10 % FCS at 1.5 × 106 cells per 10 cm dish for each time point.
For cell synchronization at G1/S transition, passage 8–10 HeLa cells were seeded in 6-well plates, treated with 2 mM thymidine for 18 h, washed twice with PBS, and grown in fresh medium without thymidine. After 9 h, the cells were treated again with 2 mM thymidine for 17 h. Synchronized cells were released by washing twice with PBS and addition of fresh medium.
Cell cycle distribution was determined by flow cytometry analysis of nuclear DNA content (FACS Aria flow cytometer, BD Biosciences). Cells cultured on dishes were trypsinized, washed twice with PBS and fixed in 70 % cold ethanol. The fixed cells were incubated with RNase for 10 minutes to digest RNA, and stained with propidium iodide (cycleTEST PLUS DNA Reagent Kit, BD Biosciences, http://www.bdbiosciences.com/dna-reagent-kit/p/340242).
RNA interference and overexpression
For Kv10.1 knockdown experiments in HeLa cells, a validated Kv10.1 siRNA sequence TACAGCCATCTTGGTCCCTTA (Quiagen)Citation11 and the non-targeting siRNA control sequence (Ambion, https://www.thermofisher.com/catalog/product/AM4635) were transfected at 30 nM final concentration using Lipofectamine RNAiMAX (Invitrogen) following the indications of the manufacturer. For E2F1 and HPV E7 overexpression, DNA plasmids 408 pSG5L HA E2F1 Addgene p10736 https://www.addgene.org/Plasmid10736/,Citation55 p1324 HPV-16 E7 Addgene p8643 https://www.addgene.org/Plasmid8643/ Citation56 and pGL3 5′UTR-KCNH1Citation16 were transfected (1 µg DNA/200.000 cells) using Lipofectamine 2000 (Invitrogen).
Site-directed mutagenesis and dual luciferase assay
Mutations on the E2F1 binding site of the Kv10.1 promoter were generated using QuikChange II Site- Directed Mutagenesis Kit (Agilent Technologies) following the manufacturer's instructions. Primers used to create mutations on the E2F1 binding sequence on Kv10.1 promoter were 5′-CGCAGGGAGGGAGGATCGTCGAGGGCGCGAGGGT and 5′-ACCCTCGCGCCCTCGACGATCCTCCCTCCCTGCG.
HeLa cells were synchronized at the G1/S transition by thymidine block, washed twice to remove thymidine excess, and at the desired time points cells were co-transfected using Lipofectamine 2000 (Invitrogen) with the Firefly and Renilla (pRL-CMV (Promega) luciferase reporter plasmids. Twenty-four h after transfection cells were harvested and activities of Firefly and Renilla luciferases were analyzed using the DualGlo luciferase kit (Promega) following the manufacturer's instructions.
Chromatin Immunoprecipitation
HeLa cells were grown on 15 cm2 dishes and synchronized by double thymidine block as described. At the desired time points, cells were cross-linked in 1 % formaldehyde solution (Sigma) at room temperature for 10 minutes, and the reaction was quenched with 125 mM glycine. Cell lysis was performed in 150 mM NaCl, 20 mM EDTA pH 8, 50 mM Tris pH 8, 0.5 % Nonidet P-40 (NP-40), 1 % Triton X-100 and complete protease inhibitor cocktail (Roche). The cell lysate was centrifuged at 12000 xg for 2 minutes at 4°C, and the resulting nuclear pellet was resuspended in 300 µL of 50 mM Tris pH 8, 10 mM EDTA, 1 % SDS and protease inhibitors, incubated at 4°C for 15 minutes with constant rotation, and sonicated (Biorupter Plus, Diagenode; 30 cycles at 30 seconds ON/OFF at high power). The sheared chromatin was diluted by adding 900 µL of 150 mM NaCl, 1 % NP-40, 0.5 % sodium deoxycholate, 0.1 % SDS, 50 mM Tris-HCl pH 8, 20 mM EDTA and protease inhibitors. Twenty µg of chromatin was pre-cleared with 20 µL of ChIP-Grade Protein G Magnetic Beads (Cell Signaling Technology) for 1 hour with constant rotation at 4°C. For the input sample, 10 % of the supernatant was removed, transferred to a new tube and stored at −20°C until further use. For immunoprecipitation, pre-cleared chromatin was incubated with 10 µg of anti-E2F1 rabbit antibody (Cell Signaling Technology http://www.cellsignal.com/antibody/3742) at 4°C with constant rotation overnight. Ten µg Normal Rabbit IgG (Cell Signaling Technology http://www.cellsignal.com/rabbit-igg/2729) were added to negative control samples.
Immunocomplexes were isolated using 30 µL of ChIP- Grade Protein G Magnetic Beads (Cell Signaling Technology http://www.cellsignal.com/products//9006) at 4°C for 2 h with constant rotation. The resulting pellet was resuspended in 150 µL of elution buffer (5 Mm Tris/HCl, pH 8.5), and incubated for 30 minutes at 65°C with gentle vortexing to elute chromatin from antibody/Protein G Magnetic Beads. All samples (including the input) were incubated with 0.1 µg/µL RNAse A (Sigma; 45 min at 37°C). Cross-links were reversed by 2 h incubation at 65°C in 200 mM NaCl and 0.2 µg/ µL of Proteinase K (Ambion, Life technologies). DNA purification was performed using DNA spin columns (Cell Signaling Technology 11137S), and DNA was eluted in 60 µL of elution buffer. The samples were then ready to be amplified by qRT-PCR.
Quantitative real time PCR
Quantitative PCR was performed using SYBR Green PCR master mix (Roche). Primers flanking the E2F1 site of the Kv10.1 and Cyclin A2Citation57 promoters were: hKv10.1-Fw 5′-CGAGGGTAGCAGCCAGA and hKv10.1-Rv 5′-CTGGCGCGGCTTCTTAC, and hCyclin A2-Fw 5′-CTGCTCAGTTTCCTTTGGTTTACC and hCyclin A2-Rv 5′-CAAAGACGCCCAGAGATGCAG. GAPDH was used as a control non-E2F1 regulated gene, hGAPDH-Fw- 5′-CCGGGAGAAGCTGAGTCATG and hGAPDH-Rv 5′- TTTGCGGTGGAAATGTCCTT.Citation58 Amplification was carried out in a LightCycler 480 (Roche Applied Science) detection system for 29 cycles at 95°C for 30 s, 40 s at 72°C, followed by a final 5 minutes extension at 72°C. Quantification was done using the ΔΔCt method,Citation59 using GAPDH for normalization and immunoprecipitation with non-specific IgG as negative control.
Immunoblotting
Cells were collected, washed in PBS and lysed on ice for 30 min in 1 % Triton X- 100, 50 mM Tris-HCl, 300 mM NaCl, 5 mM EDTA and protease inhibitors. Lysates were cleared by centrifugation for 15 min at 16000 xg at 4°C. 700 µg of total protein precleared for 1 hour at 4°C with protein G Magnetic Beads (New England BioLabs https://www.neb.com/products/s1430s) were incubated for 1 hour with 3µg anti-Kv10.1 monoclonal antibody (Kv10.1–33.mAb; 4°C), and the complexes were pulled down by incubation with 30 µL protein G magnetic beads for 1 hour.
SDS–PAGE was performed using 3–8 % or 4–12 % polyacrylamide gels (Thermo Fisher Scientific). Bands were blotted on nitrocellulose membrane (GE Healthcare), and the membrane was probed with the primary antibodies (listed in Supplementary Tables), followed by incubation with their respective HRP-conjugated secondary antibodies (GE Healthcare). Washes were performed in TBS (20 mM Tris, 150 mM NaCl) containing 0.05 % Tween (TBS-T). The signal was detected as chemiluminescence (Millipore).
Immunostaining
For immunocytochemistry, cells were plated on poly-L-lysine-coated coverslips (Menzel), fixed using 10 % formalin solution (Sigma) at 4°C for 10 minutes, washed 3 times with PBS, and permeabilized with 0.5 % Triton X-100 (Sigma) in PBS for 5 minutes. Cells were washed 3 times with PBS containing 0.05 % Tween 20 (Sigma), blocked with 10 % BSA (Sigma) in PBS-T for 1 hour, and incubated with primary antibodies mouse anti- Kv10.1 (1:1000,Citation10 and rabbit anti-Cyclin B1 (1:100; Cell Signaling http://www.cellsignal.com/antibody/4138) overnight at 4°C. Secondary antibody Alexa Fluor 488 anti-rabbit (1:1000; Thermofisher https://www.thermofisher.com/Secondary-Antibody-Polyclonal/A-11008) and Alexa Fluor 546 anti-mouse (1:1000; Thermofisher https://www.thermofisher.com/Secondary-Antibody-Polyclonal/A-11003) incubation was performed at room temperature for 1 h. Nuclei were counter- stained with 1:1000 dilution of TO-PRO-3 (Invitrogen) in PBS-T.
For immunohistochemistry, tissue sections were deparaffined in xylene and rehydrated by ethanol series (100, 90, 70, and 0 %) for 5 minutes each step. Afterwards, the tissue sections were incubated for 30 minutes at 90°C in 10 mM citrate buffer for antigen retrieval. Then, the sections were allowed to cool down for 1 h at room temperature, and washed with TBST. Blocking was performed using 10 % BSA (Sigma) in TBST. The tissue sections were incubated overnight at 4°C with primary antibodies mouse anti-Kv10.1 dilution 1:100,Citation10 and rabbit anti-Cyclin B1 (1:200; Acris https://www.acris-antibodies.com/cyclin-b1-ta590439.htm). Secondary antibodies were Alexa Fluor 488 anti-rabbit (1:1000; Thermofisher https://www.thermofisher.com/Secondary-Antibody-Polyclonal/A-11008) and Alexa Fluor 546 anti-mouse (1:1000; Thermofisher https://www.thermofisher.com/Secondary-Antibody-Polyclonal/A-11003) (1 hour at RT). Confocal images were taken using a LSM 510 Meta laser scanning confocal microscope (Zeiss), ZEN (Zeiss) software was used for image acquisition, and imaging processing was done using the image analysis software FIJI (Schindelin 2008).
Statistical analysis
All experiments were performed at least 3 times. Comparison between treatments at different time points were performed using 2-way ANOVA. Data are represented by mean values and SEM.
Abbreviations
| HPV | = | human papilloma virus |
| Kv10.1 | = | voltage-gated potassium channel protein, subfamily 10, member 1 |
| pRb | = | retinoblastoma protein. |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
DU and LAP designed research, DU and NM carried out experiments, DU, NM and LAP analyzed results. RU contributed critical materials. DU and LAP wrote the manuscript, with inputs from the rest of the authors.
1138187_Supplemental_Material.pdf
Download PDF (1.9 MB)Acknowledgments
We would like to thank Prof. Walter Stühmer for continued support and for critical comments to the manuscript. U. Kutzke and B. Heidrich provided technical assistance. We thank Dr. A. Barrantes (Göttingen) for help with tissue samples, Dr. M. Hennion, Dr. S. Bonn and Prof. A. Fischer (Göttingen) for help with ChIP assays; Prof. Z. Wang (Montreal) for the pGLEag1 reporter construct, and Adam Tomczak for helpful suggestions and comments on the manuscript.
References
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646–74; PMID:21376230; http://dx.doi.org/10.1016/j.cell.2011.02.013.
- Rieder CL. Mitosis in vertebrates: the G2/M and M/A transitions and their associated checkpoints. Chromosome Res 2011; 19:291–306; PMID:21194009; http://dx.doi.org/10.1007/s10577-010-9178-z.
- Foster DA, Yellen P, Xu L, Saqcena M. Regulation of G1 cell cycle progression: distinguishing the restriction point from a nutrient-sensing cell growth checkpoint(s). Genes Cancer 2010; 1:1124–31; PMID:21779436; http://dx.doi.org/10.1177/1947601910392989.
- Haase SB, Wittenberg C. Topology and control of the cell-cycle-regulated transcriptional circuitry. Genetics 2014; 196:65–90; PMID:24395825; http://dx.doi.org/10.1534/genetics.113.152595.
- Urrego D, Tomczak AP, Zahed F, Stühmer W, Pardo LA. Potassium channels in cell cycle and cell proliferation. Philos Trans R Soc London Ser B 2014; 369:20130094; http://dx.doi.org/10.1098/rstb.2013.0094.
- Pardo LA, Stühmer W. The roles of K+ channels in cancer. Nat Rev Cancer 2014; 14:39–48; PMID:24336491; http://dx.doi.org/10.1038/nrc3635.
- Blackiston DJ, McLaughlin KA, Levin M. Bioelectric controls of cell proliferation: ion channels, membrane voltage and the cell cycle. Cell Cycle 2009; 8:3519–28; http://dx.doi.org/10.4161/cc.8.21.9888.
- Huang X, Dubuc AM, Hashizume R, Berg J, He Y, Wang J, Chiang C, Cooper MK, Northcott PA, Taylor MD, et al. Voltage-gated potassium channel EAG2 controls mitotic entry and tumor growth in medulloblastoma via regulating cell volume dynamics. Genes Dev 2012; 26:1780–96; http://dx.doi.org/10.1101/gad.193789.112.
- Zhou Y, Wong C-O, Cho K-j, van der Hoeven D, Liang H, Thakur DP, Luo J, Babic M, Zinsmaier KE, Zhu MX, et al. Membrane potential modulates plasma membrane phospholipid dynamics and K-Ras signaling. Science 2015; 349:873–6; PMID:26293964; http://dx.doi.org/10.1126/science.aaa5619.
- Hemmerlein B, Weseloh RM, de Queiroz FM, Knötgen H, Sánchez A, Rubio ME, Martin S, Schliephacke T, Jenke M, Radzun H-J, et al. Overexpression of Eag1 potassium channels in clinical tumours. Mol Cancer 2006; 5:41; PMID:17022810; http://dx.doi.org/10.1186/1476-4598-5-41.
- Weber C, Mello de Queiroz F, Downie BR, Suckow A, Stuhmer W, Pardo LA. Silencing the activity and proliferative properties of the human EagI potassium channel by RNA interference. J Biol Chem 2006; 281:13030–7; PMID:16537547; http://dx.doi.org/10.1074/jbc.M600883200.
- Hammadi M, Chopin V, Matifat F, Dhennin-Duthille I, Chasseraud M, Sevestre H, Ouadid-Ahidouch H. Human ether a-gogo K+ channel 1 (hEag1) regulates MDA-MB-231 breast cancer cell migration through Orai1-dependent calcium entry. J Cell Physiol 2012; 227:3837–46; PMID:22495877; http://dx.doi.org/10.1002/jcp.24095.
- Downie BR, Sánchez A, Knötgen H, Contreras-Jurado C, Gymnopoulos M, Weber C, Stühmer W, Pardo LA. Eag1 expression interferes with hypoxia homeostasis and induces angiogenesis in tumors. J Biol Chem 2008; 283:36234–40; PMID:18927085; http://dx.doi.org/10.1074/jbc.M801830200.
- Borowiec AS, Hague F, Harir N, Guenin S, Guerineau F, Gouilleux F, Roudbaraki M, Lassoued K, Ouadid-Ahidouch H. IGF-1 activates hEAG K+ channels through an Akt-dependent signaling pathway in breast cancer cells: role in cell proliferation. J Cell Physiol 2007; 212:690–701; PMID:17520698; http://dx.doi.org/10.1002/jcp.21065.
- Diaz L, Ceja-Ochoa I, Restrepo-Angulo I, Larrea F, Avila-Chavez E, Garcia-Becerra R, Borja-Cacho E, Barrera D, Ahumada E, Gariglio P, et al. Estrogens and human papilloma virus oncogenes regulate human ether-a-go-go-1 potassium channel expression. Cancer Res 2009; 69:3300–7; PMID:19351862; http://dx.doi.org/10.1158/0008-5472.CAN-08-2036.
- Lin H, Li Z, Chen C, Luo X, Xiao J, Dong D, Lu Y, Yang B, Wang Z. Transcriptional and post-transcriptional mechanisms for oncogenic overexpression of ether à go-go K+ channel. PloS one 2011; 6:e20362; PMID:21655246; http://dx.doi.org/10.1371/journal.pone.0020362.
- Ishida S, Huang E, Zuzan H, Spang R, Leone G, West M, Nevins JR. Role for E2F in control of both DNA replication and mitotic functions as revealed from DNA microarray analysis. Mol Cell Biol 2001; 21:4684–99; PMID:11416145; http://dx.doi.org/10.1128/MCB.21.14.4684-4699.2001.
- Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA, Dynlacht BD. E2F integrates cell cycle progression with DNA repair, replication, and G2/M checkpoints. Genes Dev 2002; 16:245–56; PMID:11799067; http://dx.doi.org/10.1101/gad.949802.
- Zhu W, Giangrande PH, Nevins JR. E2Fs link the control of G1/S and G2/M transcription. EMBO J 2004; 23:4615–26; PMID:15510213; http://dx.doi.org/10.1038/sj.emboj.7600459.
- Nishitani H, Taraviras S, Lygerou Z, Nishimoto T. The human licensing factor for DNA replication Cdt1 accumulates in G1 and is destabilized after initiation of S-phase. J Biol Chem 2001; 276:44905–11; PMID:11555648; http://dx.doi.org/10.1074/jbc.M105406200.
- Pardo LA, del Camino D, Sánchez A, Alves F, Brüggemann A, Beckh S, Stühmer W. Oncogenic potential of EAG K+ channels. EMBO J 1999; 18:5540–7; PMID:10523298; http://dx.doi.org/10.1093/emboj/18.20.5540.
- Kosinski C, Li VS, Chan AS, Zhang J, Ho C, Tsui WY, Chan TL, Mifflin RC, Powell DW, Yuen ST, et al. Gene expression patterns of human colon tops and basal crypts and BMP antagonists as intestinal stem cell niche factors. Proc Natl Acad Sci USA 2007; 104:15418–23; PMID:17881565; http://dx.doi.org/10.1073/pnas.0707210104.
- Humphries A, Wright NA. Colonic crypt organization and tumorigenesis. Nat Rev Cancer 2008; 8:415–24; PMID:18480839; http://dx.doi.org/10.1038/nrc2392.
- Shaker A, Rubin DC. Intestinal stem cells and epithelial-mesenchymal interactions in the crypt and stem cell niche. Transl Res 2010; 156:180–7; PMID:20801415; http://dx.doi.org/10.1016/j.trsl.2010.06.003.
- Polager S, Ginsberg D. E2F mediates sustained G2 arrest and down-regulation of Stathmin and AIM-1 expression in response to genotoxic stress. J Biol Chem 2003; 278:1443–9; PMID:12446714; http://dx.doi.org/10.1074/jbc.M210327200.
- Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer 2010; 10:550–60; PMID:20592731; http://dx.doi.org/10.1038/nrc2886.
- Darnell GA, Schroder WA, Antalis TM, Lambley E, Major L, Gardner J, Birrell G, Cid-Arregui A, Suhrbier A. Human papillomavirus E7 requires the protease calpain to degrade the retinoblastoma protein. J Biol Chem 2007; 282:37492–500; PMID:17977825; http://dx.doi.org/10.1074/jbc.M706860200.
- Takahashi Y, Rayman JB, Dynlacht BD. Analysis of promoter binding by the E2F and pRB families in vivo: distinct E2F proteins mediate activation and repression. Genes Dev 2000; 14:804–16; PMID:10766737.
- Ufartes R, Schneider T, Mortensen LS, de Juan Romero C, Hentrich K, Knoetgen H, Beilinson V, Moebius W, Tarabykin V, Alves F, et al. Behavioural and functional characterization of Kv10.1 (Eag1) knockout mice. Hum Mol Genet 2013; 22:2247–62; PMID:23424202; http://dx.doi.org/10.1093/hmg/ddt076.
- Menendez ST, Villaronga MA, Rodrigo JP, Alvarez-Teijeiro S, Garcia-Carracedo D, Urdinguio RG, Fraga MF, Pardo LA, Viloria CG, Suarez C, et al. Frequent aberrant expression of the human ether à go-go (hEAG1) potassium channel in head and neck cancer: pathobiological mechanisms and clinical implications. J Mol Med (Berl) 2012; 90:1173–84; PMID:22466864; http://dx.doi.org/10.1007/s00109-012-0893-0.
- Patt S, Preussat K, Beetz C, Kraft R, Schrey M, Kalff R, Schonherr K, Heinemann SH. Expression of ether a go-go potassium channels in human gliomas. Neurosci Lett 2004; 368:249–53; PMID:15364405; http://dx.doi.org/10.1016/j.neulet.2004.07.001.
- de Queiroz FM, Suarez-Kurtz G, Stühmer W, Pardo LA. Ether a go-go potassium channel expression in soft tissue sarcoma patients. Mol Cancer 2006; 5:42; PMID:17022811; http://dx.doi.org/10.1186/1476-4598-5-42.
- Ding XW, Luo HS, Jin X, Yan JJ, Ai YW. Aberrant expression of Eag1 potassium channels in gastric cancer patients and cell lines. Med Oncol 2007; 24:345–50; PMID:17873312; http://dx.doi.org/10.1007/s12032-007-0015-y.
- Ding XW, Wang XG, Luo HS, Tan SY, Gao S, Luo B, Jiang H. Expression and prognostic roles of Eag1 in resected esophageal squamous cell carcinomas. Dig Dis Sci 2008; 53:2039–44; PMID:18080766; http://dx.doi.org/10.1007/s10620-007-0116-7.
- Ding XW, Yan JJ, An P, Lu P, Luo HS. Aberrant expression of ether a go-go potassium channel in colorectal cancer patients and cell lines. World J Gastroenterol 2007; 13:1257–61; PMID:17451210; http://dx.doi.org/10.3748/wjg.v13.i8.1257.
- Asher V, Khan R, Warren A, Shaw R, Schalkwyk GV, Bali A, Sowter HM. The Eag potassium channel as a new prognostic marker in ovarian cancer. Diagn Pathol 2010; 5:78; PMID:21138547; http://dx.doi.org/10.1186/1746-1596-5-78.
- del Pliego MG, Aguirre-Benitez E, Paisano-Ceron K, Valdovinos-Ramirez I, Rangel-Morales C, Rodriguez-Mata V, Solano-Agama C, Martin-Tapia D, de la Vega MT, Saldoval-Balanzario M, et al. Expression of Eag1 K+ channel and ErbBs in human pituitary adenomas: cytoskeleton arrangement patterns in cultured cells. Int J Clin Exp Pathol 2013; 6:458–68; PMID:23413122.
- Agarwal JR, Griesinger F, Stühmer W, Pardo LA. The potassium channel Ether à go-go is a novel prognostic factor with functional relevance in acute myeloid leukemia. Mol Cancer 2010; 9:18; PMID:20105281; http://dx.doi.org/10.1186/1476-4598-9-18.
- Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell 2002; 2:103–12; PMID:12204530; http://dx.doi.org/10.1016/S1535-6108(02)00102-2.
- Xue Q, Sano T, Kashiwabara K, Oyama T, Nakajima T. Aberrant expression of pRb, p16, p14ARF, MDM2, p21 and p53 in squamous cell carcinomas of lung. Jpn J Cancer Res 2001; 92:285–92; PMID:11267938; http://dx.doi.org/10.1111/j.1349-7006.2001.tb01093.x.
- Mettus RV, Rane SG. Characterization of the abnormal pancreatic development, reduced growth and infertility in Cdk4 mutant mice. Oncogene 2003; 22:8413–21; PMID:14627982; http://dx.doi.org/10.1038/sj.onc.1206888.
- Carriere C, Gore AJ, Norris AM, Gunn JR, Young AL, Longnecker DS, Korc M. Deletion of Rb accelerates pancreatic carcinogenesis by oncogenic Kras and impairs senescence in premalignant lesions. Gastroenterol 2011; 141:1091–101; http://dx.doi.org/10.1053/j.gastro.2011.05.041.
- Ertel A, Dean JL, Rui H, Liu C, Witkiewicz AK, Knudsen KE, Knudsen ES. RB-pathway disruption in breast cancer: differential association with disease subtypes, disease-specific prognosis and therapeutic response. Cell Cycle 2010; 9:4153–63; PMID:20948315; http://dx.doi.org/10.4161/cc.9.20.13454.
- Witkiewicz AK, Cox DW, Rivadeneira D, Ertel AE, Fortina P, Schwartz GF, Knudsen ES. The retinoblastoma tumor suppressor pathway modulates the invasiveness of ErbB2-positive breast cancer. Oncogene 2013; 30:3980–91.
- Cen L, Carlson BL, Schroeder MA, Ostrem JL, Kitange GJ, Mladek AC, Fink SR, Decker PA, Wu W, Kim JS, et al. p16-Cdk4-Rb axis controls sensitivity to a cyclin-dependent kinase inhibitor PD0332991 in glioblastoma xenograft cells. Neuro-oncology 2012; 14:870–81; PMID:22711607; http://dx.doi.org/10.1093/neuonc/nos114.
- Lee RJ, Albanese C, Fu M, D'Amico M, Lin B, Watanabe G, Haines GK, Siegel PM, Hung M-C, Yarden Y, et al. Cyclin D1 is required for transformation by activated Neu and is induced through an E2F-dependent signaling pathway. Mol Cell Biol 2000; 20:672–83; PMID:10611246; http://dx.doi.org/10.1128/MCB.20.2.672-683.2000.
- Ouadid-Ahidouch H, Le Bourhis X, Roudbaraki M, Toillon RA, Delcourt P, Prevarskaya N. Changes in the K+ current-density of MCF-7 cells during progression through the cell cycle: Possible involvement of a h-ether.a-gogo K+ channel. Recept Channels 2001; 7:345–56; PMID:11697078.
- Heng B, Glenn WK, Ye Y, Tran B, Delprado W, Lutze-Mann L, Whitaker NJ, Lawson JS. Human papilloma virus is associated with breast cancer. Br J Cancer 2009; 101:1345–50; PMID:19724278; http://dx.doi.org/10.1038/sj.bjc.6605282.
- Wang L, Wang R, Herrup K. E2F1 works as a cell cycle suppressor in mature neurons. J Neurosci 2007; 27:12555–64; PMID:18003834; http://dx.doi.org/10.1523/JNEUROSCI.3681-07.2007.
- Brüggemann A, Stühmer W, Pardo LA. Mitosis-promoting factor-mediated suppression of a cloned delayed rectifier potassium channel expressed in Xenopus oocytes. Proc Natl Acad Sci USA 1997; 94:537–42; PMID:9012819; http://dx.doi.org/10.1073/pnas.94.2.537.
- Pardo LA, Brüggemann A, Camacho J, Stühmer W. Cell cycle-related changes in the conducting properties of r-eag K+ channels. J Cell Biol 1998; 143:767–75; PMID:9813096; http://dx.doi.org/10.1083/jcb.143.3.767.
- Wonderlin WF, Woodfork KA, Strobl JS. Changes in membrane potential during the progression of MCF-7 human mammary tumor cells through the cell cycle. J Cell Physiol 1995; 165:177–85; PMID:7559799; http://dx.doi.org/10.1002/jcp.1041650121.
- Hegle AP, Marble DD, Wilson GF. A voltage-driven switch for ion-independent signaling by ether-à-go-go K+ channels. Proc Natl Acad Sci USA 2006; 103:2886–91; PMID:16477030; http://dx.doi.org/10.1073/pnas.0505909103.
- Malek NP, Sundberg H, McGrew S, Nakayama K, Kyriakides TR, Roberts JM. A mouse knock-in model exposes sequential proteolytic pathways that regulate p27Kip1 in G1 and S phase. Nature 2001; 413:323–7; PMID:11565035; http://dx.doi.org/10.1038/35095083.
- Sellers WR, Novitch BG, Miyake S, Heith A, Otterson GA, Kaye FJ, Lassar AB, Kaelin WG, Jr. Stable binding to E2F is not required for the retinoblastoma protein to activate transcription, promote differentiation, and suppress tumor cell growth. Genes Dev 1998; 12:95–106; PMID:9420334; http://dx.doi.org/10.1101/gad.12.1.95.
- Munger K, Phelps WC, Bubb V, Howley PM, Schlegel R. The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primary human keratinocytes. J Virol 1989; 63:4417–21; PMID:2476573.
- Docquier A, Augereau P, Lapierre M, Harmand PO, Badia E, Annicotte JS, Fajas L, Cavailles V. The RIP140 gene is a transcriptional target of E2F1. PloS one 2012; 7:e35839; PMID:22629304; http://dx.doi.org/10.1371/journal.pone.0035839.
- Shema E, Tirosh I, Aylon Y, Huang J, Ye C, Moskovits N, Raver-Shapira N, Minsky N, Pirngruber J, Tarcic G, et al. The histone H2B-specific ubiquitin ligase RNF20/hBRE1 acts as a putative tumor suppressor through selective regulation of gene expression. Genes Dev 2008; 22:2664–76; PMID:18832071; http://dx.doi.org/10.1101/gad.1703008.
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−DDCT method. Methods 2001; 25:402–8; PMID:11846609; http://dx.doi.org/10.1006/meth.2001.1262.