
ABSTRACT
The mitochondrial unfolded protein response (UPRmt) is involved in numerous diseases that have the common feature of mitochondrial dysfunction. However, its pathophysiological relevance in the context of hypoxia/reoxygenation (H/R) in endothelial cells remains elusive. Previous studies have demonstrated that acetylcholine (ACh) protects against cardiomyocyte injury by suppressing generation of mitochondrial reactive oxygen species (mtROS). This study aimed to explore the role of UPRmt in endothelial cells during H/R and to clarify the beneficial effects of ACh. Our results demonstrated that H/R triggered UPRmt in endothelial cells, as evidenced by the elevation of heat shock protein 60 and LON protease 1 protein levels, and resulted in release of mitochondrial pro-apoptotic proteins, including cytochrome C, Omi/high temperature requirement protein A 2 and second mitochondrial activator of caspases/direct inhibitor of apoptosis-binding protein with low PI, from the mitochondria to cytosol. ACh administration markedly decreased UPRmt by inhibiting mtROS and alleviating the mitonuclear protein imbalance. Consequently, ACh alleviated the release of pro-apoptotic proteins and restored mitochondrial ultrastructure and function, thereby reducing the number of terminal deoxynucleotidyl transferase mediated dUTP-biotin nick end labeling (TUNEL)-positive cells. Intriguingly, 4-diphenylacetoxy-N-methylpiperidine methiodide, a type-3 muscarinic ACh receptor (M3AChR) inhibitor, abolished the ACh-elicited attenuation of UPRmt and TUNEL positive cells, indicating that the salutary effects of ACh were likely mediated by M3AChR in endothelial cells. In conclusion, our studies demonstrated that UPRmt might be essential for triggering the mitochondrion-associated apoptotic pathway during H/R. ACh markedly suppressed UPRmt by inhibiting mtROS and alleviating the mitonuclear protein imbalance, presumably through M3AChR.
Introduction
Ischemia/reperfusion (I/R) injury is a critical factor in the pathogenesis of tissue injury following myocardial infarction, multiple organ failure and other acute ischemic events.Citation1 Endothelial cells seem to be particularly susceptible during reperfusion, as they are the first to undergo apoptosis within 5 min of reperfusion in isolated rat hearts.Citation2 Mitochondria are essential eukaryotic organelles that participate in processes such as cellular energy production, cell signaling and apoptosis.Citation3-5 A reduction in mitochondrial permeability transition and reactive oxygen species (ROS) generation during reperfusion reduces infarct size in isolated mice hearts.Citation6 However, the role of endothelial mitochondria in I/R injury has been neglected. Recent studies have revealed that endothelial mitochondria not only contribute to ATP generation but also play a role in regulating the homeostatic triangle of nitric oxide, ROS, and calcium ions under normal conditions.Citation7 Endothelial mitochondria may be a potential therapeutic target in I/R injury.
Mitochondria have a specific set of chaperones involved in importing, refolding, and preventing aggregation of proteins. Perturbation of the protein-folding process activates the mitochondrial unfolded protein response (UPRmt) by inducing expression of mitochondrial chaperones, such as heat shock protein 60 (HSP60), and proteases, such as LON protease1 (LONP1).Citation8,9 Notably, excessive ROS can induce oxidative damage to DNA, lipids, and proteins, leading to their misfolding and aggregation in mitochondria, thus activating the UPRmt.Citation10 The mitochondrion is the main location of ROS production.Citation10,11 Whether inhibition of the production of mitochondrial ROS (mtROS) contributes to regulation of the UPRmt has not been reported. Previous studies have suggested that the UPRmt is involved in numerous diseases that share the common pathogenetic mechanism of mitochondrial dysfunction, including spastic paraplegia, Parkinson disease, Friedreich's ataxia and cancer.Citation12 However, the role of the UPRmt in cardiovascular diseases has not been elucidated. Although mild UPRmt can overcome the initial insult and have a beneficial impact,Citation13,14 prolonged or high-dose activation, in fact, is harmful and maladaptive for homeostasis.Citation15 The UPRmt has recently been implicated as a mechanism of cell death, which may be related to the upregulation of the stress response transcription factor CHOP.Citation16 Nevertheless, the role of the UPRmt in reperfusion-induced vascular endothelial cell death has not been characterized.
Cardiovascular diseases are accompanied by increased sympathetic and suppressed vagal activity, and therefore improvement of vagal activity appears to be a promising therapeutic strategy.Citation17 Previous studies have demonstrated that vagal stimulation significantly attenuated cardiac mtROS production, mitochondrial depolarization, and swelling during I/R.Citation18 In an in vitro study using primary-cultured cardiomyocytes, acetylcholine (ACh), the major neurotransmitter of the vagal nerve, prevented reoxygenation-induced collapse in the mitochondrial transmembrane potential by inhibiting permeability transition pore opening.Citation19 Recent studies in our laboratory have suggested that ACh inhibits mitochondrial morphological abnormalities and improves mitochondrial biogenesis and function in cardiomyocytes subjected to hypoxia/reoxygenation (H/R).Citation20 However, whether ACh-mediated endothelial protection is related to mitochondria regulation remains unclear. To test this hypothesis, we used human umbilical vein endothelial cells (HUVECs) suffering from H/R and investigated the role of ACh in modulating the UPRmt and subsequent mitochondrion-dependent apoptotic signals, with a focus on mitonuclear imbalance and mitochondrial ROS production.
Results
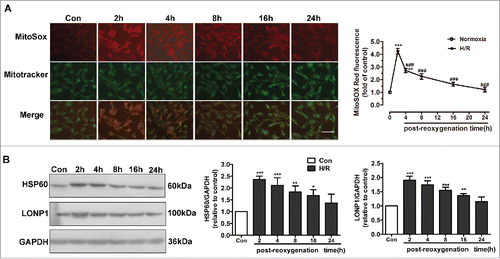
mtROS formation and UPRmt occurred during H/R in endothelial cells
Excessive ROS induce oxidative damage to DNA, lipids, and proteins, leading to their misfolding and aggregation in mitochondria. Accumulation of misfolded and aggregated proteins in mitochondria triggers the UPRmt. The mitochondrion is the main location of ROS production. Thus, we used MitoSOX Red to detect changes in mtROS levels. mtROS levels were significantly elevated after H/R and peaked at 2 h (). We determined the time course of changes in UPRmt markers after reoxygenation. HUVECs were subjected to hypoxia (1% O2, 8 h), followed by reoxygenation for 2, 4, 8, or 16 h. As shown in , compared with the control group, the expression of HSP60, a mitochondrion-located molecular chaperone, was upregulated in response to H/R and peaked at 2 h. The protein kinase LONP1, another marker of the UPRmt, was also increased significantly at 2 h in the context of H/R (). Therefore, a reoxygenation time of 2 h was used in subsequent experiments.
Figure 1. UPRmt and mtROS generation are involved in H/R-induced endothelial injury. (A) mtROS production in HUVECs was detected using different time courses of reoxygenation. In the mitochondrial ROS assay, MitoSOX Red fluorescence colocalized with that of MitoTracker Green. Scale bar, 50 μm. (B) HUVEC lysates after different time courses of reoxygenation were immunoblotted using antibodies against HSP60 and LONP1. GAPDH served as the internal control. Quantitative analysis of HSP60 and LONP1 expression during different post-reoxygenation time courses. Open bar, normoxia; filled bar, H/R. The data expressed as mean ± SEM in each bar graph represent the average of 4 independent experiments. *P < 0.05 vs. Con; **P < 0.01 vs. Con; ***P < 0.001 vs. Con; ###P < 0.001 vs. H/R.
N-acetyl cysteine decreased H/R-induced mtROS and UPRmt and protected endothelial mitochondria
We further explored the effect of N-acetyl cysteine (NAC) on mtROS levels and the expression of UPRmt markers (). As shown in , H/R of HUVECs led to increased mtROS generation. NAC pretreatment was associated with significantly decreased mtROS levels. We determined the protein levels of UPRmt markers after NAC treatment (). The increased expression of HSP60, LONP1, CHOP and caseinolytic protease1 (CLPP1) induced by H/R injury was significantly downregulated by treatment with NAC.
Figure 2. NAC treatment inhibited the UPRmt and protected mitochondrial structure and function against H/R injury. (A) NAC administration reduced mitochondrial ROS levels in the context of H/R. (B) NAC treatment inhibited the upregulation of HSP60, LONP1, CLPP1 and CHOP expression induced by H/R. (C) Transmission electron microscopy indicated that NAC prevented the ultrastructural changes triggered by H/R in HUVECs, particularly the changes in mitochondrial area, perimeter, Feret's diameter and volume density. Magnification, 40000×; scale bar, 500 nm. The number of mitochondria is shown in the circle. (D) Intracellular ATP levels after NAC pretreatment were determined using ATPlite. (E) The H/R-induced increase in TUNEL-positive cells was inhibited by NAC treatment. Scale bar, 50 μm. Open bar, normoxia; filled bar, H/R. The data expressed as mean ± SEM in each bar graph represent the average of 4 independent experiments. *P < 0.05 vs. Con; **P < 0.01 vs. Con; ***P < 0.001 vs. Con; #P < 0.05 vs. H/R; ##P < 0.01 vs. H/R; ###P < 0.001 vs. H/R.
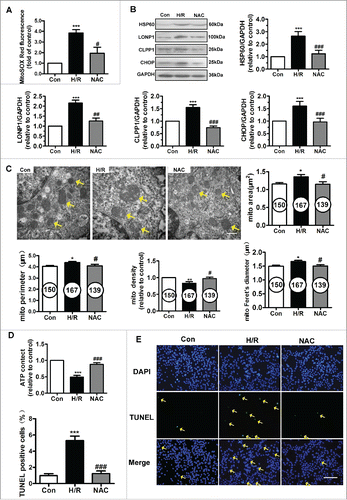
Mitochondrial function and ultrastructure were assessed. H/R-injured mitochondria exhibited swelling and disordered cristae, as evidenced by an increase in the mitochondrial area, Feret's diameter and perimeter and by a decrease in the mitochondrial volume density (). NAC restored the fine structure of mitochondria. ATP content in HUVECs was measured using an ATP bioluminescent assay. As shown in , H/R induced depletion of cellular ATP reserves, and NAC administration partially restored energy generation in HUVECs. Furthermore, we performed a TUNEL assay in the context of H/R. As shown in , NAC treatment significantly reduced the number of H/R-induced TUNEL-positive HUVECs. These results suggest correlations among mtROS, UPRmt and mitochondrial function and ultrastructure.
ACh decreased the H/R-induced UPRmt and attenuated the mitonuclear protein imbalance in HUVECs
The effects of ACh on the UPRmt and mtROS formation induced by H/R in endothelial cells are shown in . The elevated mtROS formation and increased HSP60 and LONP1 expression in H/R were ameliorated by ACh (10−7–10−5M) in a dose-dependent manner, indicating that ACh attenuated H/R-induced ROS formation and UPRmt. Therefore, 10−6M ACh was used in subsequent experiments.
Figure 3. ACh administration decreased H/R-induced mtROS levels and UPRmt via M3AChR. (A) ACh diminished H/R-induced mtROS levels in a dose-dependent manner. In the mitochondrial ROS assay, MitoSOX Red fluorescence was colocalized with that of MitoTracker Green. Scale bar, 50 μm. (B and C) ACh inhibited the expression of HSP60 and LONP1 in a dose-dependent manner. Open bar, normoxia; filled bar, H/R. (D) ACh increased the reduction in the nuclear DNA-encoded ATP5A/mitochondrial DNA-encoded MTCO1 ratio. (E) ACh treatment decreased the upregulation of HSP60 and LONP1 expression induced by H/R. The beneficial effect of ACh was abolished by the M3AChR antagonist 4-DAMP (10−6M). Open bar, normoxia; filled bar, H/R. The data expressed as mean ± SEM in each bar graph represent the average of 4 independent experiments. ***P < 0.001 vs. Con; ##P < 0.01 vs. H/R. ###P < 0.001 vs. H/R; &&P < 0.01 vs. H/R + ACh; &&&P < 0.001 vs. H/R + ACh.
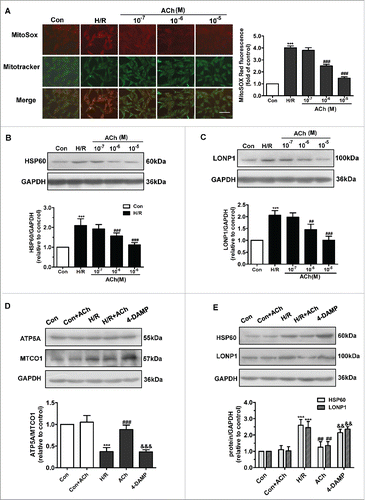
We assessed the mitonuclear protein imbalance by determining the expression of ATP5A (encoded by nuclear DNA) and MTCO1 (encoded by mitochondrial DNA) (). The ATP5A/MTCO1 ratio decreased when cells were subjected to H/R, while ACh administration prevented this imbalance. Furthermore, as shown in , ACh significantly weakened the increase in HSP60 and LONP1 levels induced by H/R in endothelial cells. To identify the ACh receptor playing the predominant role in ACh-mediated suppression of the UPRmt and mitonuclear protein imbalance, we used 4-diphenylacetoxy-N-methylpiperidinemethiodide (4-DAMP), an M3AChR inhibitor; the protective effects of ACh were reversed by treatment with 4-DAMP.
ACh inhibited mitochondrion-dependent apoptotic signals via M3AChR
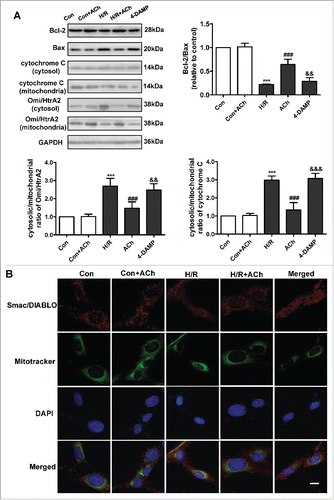
As shown in , ACh increased expression of the anti-apoptotic factor Bcl-2 and upregulated the Bcl-2/Bax ratio during the reoxygenation period. We also evaluated cytochrome C and Omi/high temperature requirement protein A 2 (Omi/HtrA2) release from mitochondria. In the control group, relatively low levels of cytochrome C and Omi/HtrA2 were released from mitochondria into the cytosol. After H/R, cytochrome C and Omi/HtrA2 levels were significantly increased in the cytosol and decreased in the mitochondria. Administration of ACh inhibited the release of cytochrome C and Omi/HtrA2 into the cytosol. We also examined the subcellular locations of second mitochondrial activator of caspases/direct inhibitor of apoptosis binding protein of low PtdIns (Smac/DIABLO) by confocal imaging (). In the control group, Smac/DIABLO remained in the mitochondria. After H/R stimulation, Smac/DIABLO was released from mitochondria into the cytosol, which was inhibited by ACh treatment; these effects of ACh were reversed by treatment with 4-DAMP. ACh treatment alone had no significant effect on the apoptotic signaling cascade in the control cells.
Figure 4. ACh treatment suppressed the mitochondrial pathway of apoptosis. (A) ACh upregulated the Bcl-2/Bax ratio and suppressed cytochrome C and Omi/HtrA2 release, and these effects of ACh were abrogated by treatment with 4-DAMP. Quantification is shown in the bar graph. (B) ACh decreased the release of Smac/DIABLO from mitochondria into the cytosol. Scale bar, 10 μm. Open bar, normoxia; filled bar, H/R. The data expressed as mean ± SEM in each bar graph represent the average of 4 independent experiments. ***P < 0.001 vs. Con; ###P < 0.001 vs. H/R; &&P < 0.01 vs. H/R + ACh; &&&P < 0.001 vs. H/R + ACh.
ACh restored mitochondrial morphology and function via M3AChR
We evaluated the effect of ACh on mitochondrial ultrastructure in HUVECs by transmission electron microscopy. As depicted in , endothelial cells subjected to H/R showed swelling and crista deconstruction. Quantitative analysis showed that the mitochondrial area, diameter and perimeter were increased, and mitochondrial volume density was decreased in the context of H/R. In addition, these changes could be prevented by ACh treatment. 4-DAMP–treated cells showed severe swelling and disordered cristae, indicating that ACh prevented the mitochondrial ultrastructural changes triggered by H/R via M3AChR in endothelial cells.
Figure 5. ACh treatment preserved mitochondrial morphology and function via M3AChR. (A) Ultrastructural changes in mitochondria in HUVECs. Magnification, 40000×; scale bar, 500 nm. Mitochondrial area, volume density, Feret's diameter and perimeter are shown in (B). (C) ACh treatment prevented the H/R-induced decrease in ATP production. Open bar, normoxia; filled bar, H/R. The data expressed as mean ± SEM in each bar graph represent the average of 4 independent experiments. *P < 0.05 vs. Con; **P < 0.01 vs. Con; ***P < 0.001 vs. Con; #P < 0.05 vs. H/R. ##P < 0.01 vs. H/R; &P < 0.05 vs. H/R + ACh; &&P < 0.01 vs. H/R + ACh.
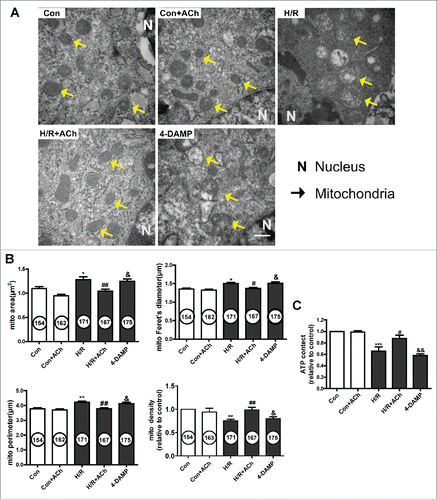
To further assess the effect of ACh on mitochondrial function, ATP production was determined using an ATP bioluminescent assay (). ATP production was significantly reduced in the H/R versus control cells, and ACh treatment prevented the inhibition of ATP generation. The protective effect of ACh on mitochondrial function was abolished by 4-DAMP treatment.
Knockdown of M3AChR blocked the inhibitory effect of ACh on the UPRmt
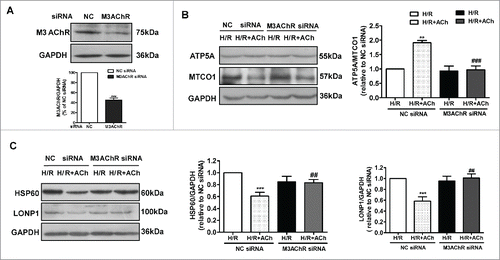
To determine whether M3AChR is associated with the ACh-mediated suppression of the UPRmt during H/R, we suppressed M3AChR expression in endothelial cells by siRNA transfection. As shown in , M3AChR expression was downregulated by its corresponding siRNA. Next, we assessed the role of M3AChR in inhibition of mitonuclear protein imbalance and suppression of the UPRmt by ACh in H/R-injured HUVECs ( and 6C). M3AChR-depleted cells displayed a reduced ATP5A/MTCO1 ratio and higher HSP60 and LONP1 levels compared with the NC siRNA group in the presence of ACh. These results support a role for M3AChR in the ACh-mediated inhibition of mitonuclear protein imbalance and alleviation of the UPRmt.
Figure 6. Knockdown of M3AChR blocked the beneficial effects of ACh during H/R. (A) Silencing efficiency of M3AChR siRNA. Cells were transfected with siRNA followed by H/R. (B) ACh administration restored the mitonuclear protein balance, and M3AChR siRNA abolished the protective effects. Changes in HSP60 and LONP1 expression (C) were determined after application of M3AChR/NC siRNA with or without ACh. The data expressed as mean ± SEM in each bar graph represent the average of 4 independent experiments. **P < 0.01 vs. NC siRNA group; ***P < 0.001 vs. NC siRNA group; ##P < 0.01 vs. ACh-treated NC siRNA group; ###P < 0.001 vs. ACh-treated NC siRNA group.
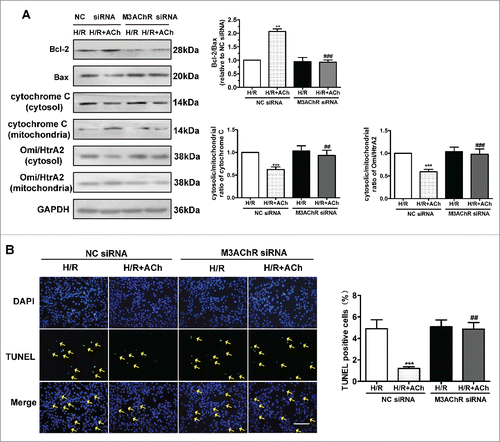
Knockdown of M3AChR blocked the antiapoptotic effects of ACh. We clarified whether M3AChR is responsible for the anti-apoptotic effects of ACh during H/R. As shown in , compared with the NC siRNA group, M3AChR knockdown reduced expression of the antiapoptotic factor Bcl-2, downregulated the Bcl-2/Bax ratio, and prevented the downregulated release of cytochrome C and Omi/HtrA2 from mitochondria into the cytosol. Moreover, apoptosis was evaluated by TUNEL staining. As shown in , suppression of M3AChR expression by siRNA transfection abolished the antiapoptotic effect of ACh. These data support a role for M3AChR in ACh-mediated inhibition of the UPRmt and apoptosis in H/R-induced endothelial injury.
Figure 7. Knockdown of M3AChR abrogated the anti-apoptotic effect of ACh. (A) Representative immunoblots and quantitative analysis of the Bcl-2/Bax ratio, cytochrome C and the Omi/HtrA2 ratio after application of M3AChR/NC siRNA with or without ACh. (B) TUNEL-positive cells were enumerated after application of M3AChR/NC siRNA with or without ACh. Scale bar, 50 μm. The data expressed as mean ± SEM in each bar graph represent the average of 4 independent experiments. **P < 0.01 vs. NC siRNA group; ***P < 0.001 vs. NC siRNA group; ##P < 0.01 vs. ACh-treated NC siRNA group; ###P < 0.001 vs. ACh-treated NC siRNA group.
Discussion
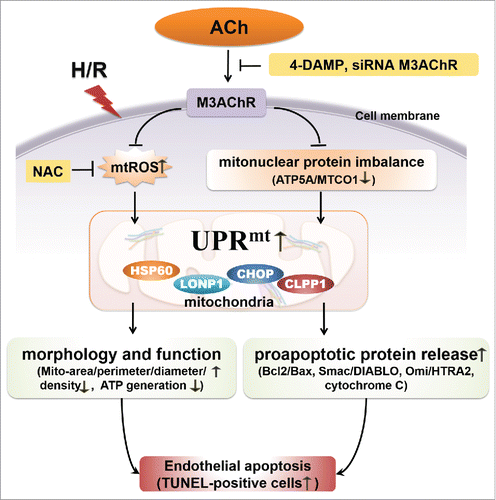
The results of this study show that ACh suppresses UPRmt caused by H/R, which results in preservation of mitochondrial morphology and function, thus alleviating endothelial cell apoptosis (). The first interesting finding was that H/R increased mtROS generation and created an imbalance between nuclear-encoded (ATP5A) and mitochondrial-encoded protein (MTCO1) in HUVECs, which triggered the UPRmt. Secondly, ACh treatment reduced mtROS formation and increased the ATP5A/MTCO1 ratio, thereby significantly inhibiting the UPRmt and the release of cytochrome C, Omi/HtrA2 and Smac/DIABLO from mitochondria. Intriguingly, ACh prevented the cellular ultrastructural changes, enhanced ATP generation and diminished apoptosis triggered by H/R. Furthermore, 4-DAMP or M3AChR siRNA abrogated the ACh-mediated effects, suggesting that ACh exerted beneficial effects likely via M3AChR. Taken together, these results suggest that the UPRmt may be a novel molecular target for endothelial protection, and mitochondrial protection conferred by ACh is crucial for alleviation of I/R injury in endothelial cells.
Figure 8. Proposed schematic of the mechanism by which ACh protects the endothelium against H/R injury. ACh decreases expression of the UPRmt by inhibiting mtROS formation and suppressing the mitonuclear protein imbalance during H/R, presumably through M3AChR. Consequently, ACh reduces the release of pro-apoptotic proteins (Smac/DIABLO, Omi/HtrA2, cytochrome C) and preserves endothelial mitochondrial ultrastructure and function, thus reducing the number of TUNEL-positive cells.
UPRmt activation upon mitochondrial stress is intrinsically related to perturbation of proteostasis in the mitochondria, and proteostasis is challenged when expression of electron transport chain (ETC) subunits is absent or reduced.Citation21 ETC dysfunction is involved in I/R injury of the liver and heart.Citation22,23 This may imply a link between the UPRmt and H/R-induced injury. Our findings showed that, in response to H/R stimuli, the expression of HSP60 and LONP1 in endothelial cells increased significantly, indicating that H/R triggered the UPRmt in vascular endothelial cells. Thus, it is conceivable that modulating the UPRmt may be a potential target for I/R injury. Numerous factors can induce the UPRmt, including ROS overproduction,Citation9 mitonuclear protein imbalance,Citation14 and perturbation of the protein quality control network.Citation24 Runkel et al. reported that ROS generated by paraquat constitutes a toxic activity that provokes the UPRmt in Caenorhabditis elegans, and the ROS scavenger NAC substantially reduced the level of HSP-6, an essential component of the UPRmt.Citation25 In our study, the H/R-mediated induction of the UPRmt was inhibited by NAC, which is in agreement with previous reports. Furthermore, we found that NAC administration restored mitochondrial morphology and function and subsequently alleviated endothelial cell apoptosis. These results indicate that ROS play an important role in mediating the UPRmt, and inhibition of ROS may be beneficial to H/R-injured HUVECs. Another important factor that triggers the UPRmt is a mitonuclear protein imbalance.Citation14 Houtkooper et al. demonstrated that mitochondrial ribosomal protein S5 knockdown triggers a mitonuclear protein imbalance, reducing mitochondrial respiration and activating the UPRmt.Citation14 Consistent with these results, we found a decreased ATP5A/MTCO1 ratio concomitant with robust activation of the UPRmt. These data suggest that both mtROS and mitonuclear protein imbalance lead to activation of the UPRmt in endothelial cells subjected to H/R.
The mitochondrion is the central organelle in the intrinsic pathway of apoptosis. A previous study showed that renal I/R injury down-regulated the Bcl-2/Bax ratio and altered mitochondrial membrane permeability, leading to release of cytochrome C and eventually cell death.Citation26 In agreement with previous studies, our findings suggested that, in response to H/R stimuli, the upregulation of UPRmt paralleled a decrease in the Bcl-2/Bax ratio, resulting in increased release of cytochrome C, Smac/DIABLO, and Omi/HtrA2. UPRmt-induced apoptosis is likely due to upregulated expression of CHOP in the context of H/R. CHOP plays a central role in UPRmt signaling and is involved in endoplasmic reticulum stress-induced apoptosis.Citation27,28 Moreover, He et al. suggested that accumulation of unfolded proteins within mitochondria contributes to mitochondrial permeability transition, an early event in apoptosis.Citation29 These findings support a correlation between the UPRmt and mitochondrion-dependent apoptotic pathway in the present study. Additionally, mitochondrial structure and function are key mediators apoptosis in myocardial I/R.Citation30 Recent evidence showed that reduced mitochondrial ATP generation favored MPTP opening and led to early apoptosis in a murine model.Citation31 In agreement with this, our findings showed decreased ATP generation and increased apoptosis in H/R-injured HUVECs. Moreover, endothelial mitochondria in the context of H/R displayed marked swelling and disorganized cristae. These results suggest that H/R-induced apoptosis in endothelial cells is due in part to dysfunction and structural abnormalities of mitochondria.
Cardiovascular disease is accompanied by an autonomic imbalance that is almost always characterized by both increased sympathetic activity and suppressed vagal activity.Citation32-34 Improved vagal activity through vagal nerve stimulation or the neurotransmitter ACh has been reported to elicit beneficial effects in various cardiovascular diseases. A previous study indicated that in a swine model of I/R, vagal stimulation significantly reduced infarct size, improved ventricular function and attenuated mitochondrial swelling.Citation35 Our laboratory recently suggested that ACh benefits the endothelium by suppressing endoplasmic reticulum stress and inhibiting pro-apoptotic signaling.Citation36 However, little information is available on the effect of ACh on the regulation of UPRmt-related apoptosis. Importantly, this study indicated that ACh attenuated the UPRmt as evidenced by decreased HSP60 and LONP1 expression, inhibited the mitochondrion-related apoptotic pathway and preserved mitochondrial structure and function, leading to a reduced number of TUNEL-positive cells. These results suggested that inhibition of the UPRmt was responsible for the ACh-mediated endothelial protection, and that mitochondria are vital for improvement of vagal function-induced endothelium protection. Furthermore, Miao et al. demonstrated that ACh inhibits mtROS generation, thus protecting cardiomyocytes against H/R-induced cell injury.Citation37 In the current study, both mtROS overproduction and mitonuclear protein imbalance may have triggered the UPRmt in endothelial cells. ACh treatment inhibited mtROS generation and mitonuclear protein imbalance, thereby attenuating activation of the UPRmt in endothelial cells during H/R, indicating that ACh alleviated H/R-induced UPRmt by inhibiting mtROS and mitonuclear protein imbalance.
We further determined whether ACh acted via muscarinic or nicotinic AChR to exert its beneficial effects. A previous study in our laboratory suggested that activation of M3AChR with choline alleviated I/R-induced vascular injury in a rat model.Citation38 More recently, our laboratory has revealed that ACh suppresses mitochondrial calcium overload and protects endothelial cells by acting on M3AChR.Citation39 In our study, the M3AChR-specific antagonist 4-DAMP, as well as M3AChR siRNA, abrogated ACh-mediated inhibition of the UPRmt and apoptosis, implying that the effect of ACh on the UPRmt was mediated mainly through M3AChR. However, the effects of ACh on the UPRmt in vivo in experimental animals require further investigation. Intriguingly, our findings also showed that 4-DAMP abolishes ACh-mediated preservation of mitochondrial structure and function during H/R. Therefore, we speculate that ACh exerts protective effects on endothelial mitochondria mainly through M3AChR.
Collectively, our study showed that ACh suppressed the generation of mtROS and alleviated mitonuclear protein imbalance, thereby inhibiting the UPRmt and preserving the ultrastructure and function of mitochondria to prevent H/R-induced, mitochondrion-dependent apoptosis. Furthermore, these favorable ACh-mediated effects in endothelial cells likely involved M3AChR. The UPRmt may be an essential mechanism in I/R injury, and the complexity of the UPRmt is only beginning to be revealed. The present study also provides novel insight into the benefits of ACh in endothelial cells subjected to I/R injury. As it may represent an Achilles' heel of I/R, a full understanding of the UPRmt is critical for effective targeting of the mitochondrial network to improve the treatment of I/R injury and other cardiovascular diseases.
Materials and methods
Cell line
HUVECs were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in Ham's F12K medium (Macgene Biotech Co., Ltd, Beijing, China) with 0.03 mg/ml endothelial cell growth supplement (Macgene Biotech Co., Ltd), 10% fetal bovine serum (FBS; Hyclone, Logan, UT, USA), 100 μg/ml streptomycin (Sigma, St. Louis, MO, USA) and 100 U/ml penicillin (Sigma). Cells were maintained at 37°C in a humidified 5% CO2 atmosphere and used at passages 4-8 in all experiments.
Cell culture
To achieve a quiescent state, the medium was replaced with serum-free Ham's F12K (FBS was eliminated) once cell growth reached 80% confluence, and cells were re-incubated for 12 h prior to all subsequent experiments. Then, the medium was replaced with a modified ischemia-mimetic solution (in mM: NaCl, 135; KCl, 8; NaH2PO4, 0.33; MgCl2, 0.5; HEPES, 5.0; CaCl2, 1.8; and lactate, 20, pH 6.80) and transferred into a hypoxic incubator (1% O2, 5% CO2, and 94% N2) for 8 h.36 After hypoxia, the medium was replaced with fresh serum-free F12K medium containing the appropriate drugs, and cells were incubated under normoxia for 2 h. The cells were divided at random into the following 6 treatment groups: (1) Con: culture with F12K under a normoxic atmosphere; (2) ACh: treatment with ACh (10−6 M) alone and culture for 2 h; (3) H/R: hypoxia for 8 h followed by reoxygenation for 2 h; (4) H/R + ACh group: administration of ACh (10−6 M) at the beginning of reoxygenation; (5) H/R + NAC: ROS scavenger antioxidant NAC (10−3 M) applied 2 h before H/R; and (6) H/R + 4-DAMP + ACh: M3AChR antagonist 4-DAMP (10−6 M) co-applied with ACh for 2 h before H/R. These agents were obtained from Sigma.
Western blot analysis
After treatment, RIPA lysis buffer containing 1 mM phenylmethylsulfonyl fluoride was used to extract total proteins from HUVECs. After centrifuging the lysates at 13000 rpm for 15 min at 4°C, the supernatants were collected as the protein fractions. A bicinchoninic acid protein assay kit (Beyotime Biotech) was used to estimate protein concentrations. For Western blot analysis, total proteins were mixed with SDS sample buffer and boiled at 100°C for 10 min. Equal amounts of protein (30 μg) were separated by 10% SDS-polyacrylamide gels and electrotransferred to PVDF membranes (Millipore). The membranes were blocked in 5% nonfat milk in Tris-buffered saline containing 0.1% Tween 20 (TBST) for 1 h at room temperature and incubated overnight with the relevant primary antibodies at 4°C. The following primary antibodies were used: GAPDH (diluted 1:5000; Sinopept, Beijing, China), CHOP (diluted 1:500; Signalway Antibody, Pearland, TX, USA), HSP60 (diluted 1:1000; Cell Signaling Technology), LONP1 (diluted 1:200; Santa Cruz Biotechnology, Santa Cruz, CA, USA), CLPP1 (diluted 1:200; Santa Cruz Biotechnology), ATP5A (diluted 1:500; Signalway Antibody, Pearland, TX, USA), MTCO1 (diluted 1:1000; Abcam,), Omi/HtrA2 (diluted 1:1000; Abcam), Bcl-2 (diluted 1:1000; Cell Signaling Technology), Bax (diluted 1:1000; Cell Signaling Technology), and M3AChR (diluted 1:1000; Millipore). TBST (1×) was used to wash away the primary antibodies. The membranes were incubated with the appropriate secondary antibodies for 40 min at room temperature, washed, and visualized for signal using chemiluminescence reagent.
Measurement of mitochondrial ROS levels
mtROS levels were measured using MitoSOX Red (Invitrogen, Carlsbad, CA, USA), a fluorescent probe specific for superoxide anion. Briefly, cells were incubated with MitoSOX Red (5 µmol/L) for 30 min at 37°C and then washed twice with phosphate-buffered saline (PBS). After fixation of HUVECs in 4% paraformaldehyde in PBS for 30 min at room temperature and washing twice with PBS, MitoTracker Green (200 nM, Beyotime) was used to label mitochondria for 30 min at 37°C,Citation37 followed by washing with PBS before visualization. Fluorescence images were captured using a laser confocal microscope (Nikon C2, Nikon, Tokyo, Japan). MitoSOX Red fluorescence was detected by excitation at 510 nm and emission at 580 nm. MitoTracker Green fluorescence was detected by excitation at 490 nm and emission at 516 nm.
Isolation of cytosolic and mitochondrial fractions
HUVECs were washed with PBS and collected by centrifugation at 1000 rpm for 5 min at room temperature. Subsequently, the supernatants were harvested for mitochondria and cytosol isolation using a cell mitochondria isolation kit, as follows. Cells were rinsed gently with pre-chilled PBS and then centrifuged at 600 g for 5 min at 4°C. Mitochondria isolation reagent (provided in the kit) was added to the precipitates. The lysate was transferred to a glass homogenizer and stirred by forcefully passing the cells, followed by centrifugation at 600 g for 10 min at 4°C. The supernatant was further centrifuged at 11,000 g for 10 min at 4°C, and mitochondria were collected from the precipitates. The remaining supernatant was centrifuged at 12,000 g for 10 min at 4°C, and supernatant was collected as the cytosolic protein fraction. Protein concentrations were determined using NanoPhotometer P-330 (Implen, Germany).Citation19,40
Transmission electron microscopy
For electron microscopy, HUVECs were fixed at 4°C with 2.5% glutaraldehyde in 0.1 M phosphate buffer, pH 7.4 (phosphate buffer) for 2 h. The cells were washed in the same buffer and then post-fixed in 1% osmium tetroxide in 0.1 M phosphate buffer for 2 h.Citation41 Samples were dehydrated using an alcohol gradient and propylene oxide, prior to embedding in epoxy resin and cutting into ultrathin sections using glass knives on a LKB-V ultramicrotome according to standard procedure. The sections were double-stained with uranyl acetate and lead citrate,Citation36 and electron microphotographs were taken using a transmission electron microscope (H-7650; Hitachi, Tokyo, Japan).
TUNEL staining
HUVEC apoptosis was assessed using a DeadEnd Fluorometric TUNEL system (Promega, Madison, WI, USA) according to the manufacturer's protocol. Briefly, cells were seeded onto collagen-coated coverslips, and then TUNEL reaction mixture was added after fixation with 4% paraformaldehyde (Sigma). Nuclei were stained using DAPI.Citation36 The staining was imaged using a fluorescence microscope (TE-2000U, Nikon, Japan). Apoptotic cells in 10 fields (randomly chosen) in each group were counted to calculate the apoptotic index: apoptotic index = positive cells/ (positive cells + negative cells) × 100%.Citation42
ATP generation
Intracellular ATP content was determined using a bioluminescent assay kit (Beyotime, Nanjing, China) according to the manufacturer's instructions. Briefly, cell lysates were collected and then centrifuged at 12,000 g for 10 min at 4°C. After addition of the supernatant to detection reagent (provided in the kit), the ATP content was determined using a multimode microplate reader with a luminescence luminometer (FLUOstar Omega, BMG Labtech, Germany) and normalized to the protein concentration. Standardization was carried out using known quantities of ATP provided with the kit (5, 10, 15, 20, and 25 pmole) and determined in parallel.
Immunofluorescence and confocal microscopy
Following treatment, the cells were incubated with MitoTracker Green to label mitochondria for 30 min at 37°C. Cells were then washed with PBS twice, fixed with 4% paraformaldehyde in PBS for 20 min at room temperature and permeabilized in 0.3% Triton X-100 in PBS.Citation43 Fixed cells were blocked with 5% bovine serum albumin for 1 h at room temperature and stained with antibodies against Smac/DIABLO (1:250) (Cell Signaling Technology) for 1 h at 37°C. After washing with PBS 3 times, the cells were incubated with a FITC-conjugated anti-rabbit secondary antibody for 1 h at 37°C.Citation44 Nuclei were stained with DAPI for 5 min at room temperature. Images were captured using a confocal microscope.
Small interfering RNA targeting M3AChR
The siRNAs specific for M3AChR and the negative control siRNAs (NC) were synthesized by Shanghai GenePharma Co. Ltd. (Shanghai, China). According to the manufacturer's protocol, HUVECs were transfected with 100 nM of each siRNA using Lipofectamine 2000 transfection reagent (Invitrogen) for 6 h after reaching 70-80% confluence in 6-well plates. Then, the medium was replaced with fresh F12K, and cells were incubated for a further 48 h.36 Thereafter, HUVECs were collected for subsequent experiments. Western blotting was used to monitor the efficiency of siRNA-mediated M3AChR knockdown after transfection.
Statistical analysis
Data are expressed as means ± SEM. The software package GraphPad Prism version 5.01 (GraphPad Software; San Diego, CA, USA) was used to perform all statistical analyses. Differences among groups were assessed by one-way ANOVA followed by Tukey's post hoc test. Student's t-test was used to assess differences between two groups. P < 0.05 was considered to indicate statistical significance.Citation45
Abbreviations
| ACh | = | acetylcholine |
| CHOP | = | C/EBP homologous protein |
| CLPP1 | = | Caseinolytic protease1 |
| 4-DAMP | = | 4-diphenylacetoxy-N-methylpi-peridine methiodide |
| DAPI | = | 4,6-diamidino-2-phenylindole |
| PBS | = | phosphate-buffered saline |
| UPRmt | = | mitochondrial unfolded protein response |
| GAPDH | = | glyceraldehyde 3-phospharte dehydrogenase |
| HSP60 | = | heat shock protein 60 |
| HtrA2 | = | high temperature requirement protein A 2 |
| H/R | = | hypoxia and reoxygenation |
| LONP1 | = | LON protease1 |
| mtROS | = | mitochondrial reactive oxygen species |
| M3AChR | = | type-3 muscarinic acetycholine receptor |
| Smac/DIABLO | = | second mitochondrial activator of caspases/direct inhibitor of apoptosis binding protein of low PI |
| TUNEL | = | terminal deoxynucleotidyl transferase mediated dUTP-biotin nick end labeling |
Disclosure of potential conflicts of interest
The authors declare no conflict of interest.
Authorship Contributions
Man Xu, Wei-Jin Zang and Ming Zhao designed the research study; Man Xu, Xue-Yuan Bi, Xi He, performed the research; Man Xu, Xue-Yuan Bi, and Xiao-Jiang Yu analyzed the data; Man Xu, Xi He and Wei-Jin Zang wrote the paper; Wei-Jin Zang acquired funding for the research. All authors read and approved the final manuscript.
The English in this document has been checked by at least 2 professional editors, both native speakers of English. For a certificate, please see: http://www.textcheck.com/certificate/inZDGo
Acknowledgments
We appreciate the technical support and materials from the electron microscope center of Xi'an Jiaotong University.
Funding
This work is supported by National Natural Science Foundation of China (Major International Joint Research Project, No. 81120108002; General Project, No. 81473203), and Specialized Research Fund for the Doctoral Program of Higher Education (No. 20130201130008).
References
- Feng Y, Hu L, Xu Q, Yuan H, Ba L, He Y, Che H. Cytoprotective role of alpha-1 antitrypsin in vascular endothelial cell under hypoxia/reoxygenation condition. J Cardiovasc Pharmacol 2015; 66:96-107; PMID:25815674; http://dx.doi.org/10.1097/FJC.0000000000000250
- Scarabelli T, Stephanou A, Rayment N, Pasini E, Comini L, Curello S, Ferrari R, Knight R, Latchman D. Apoptosis of endothelial cells precedes myocyte cell apoptosis in ischemia/reperfusion injury. Circulation 2001; 104:253-6; PMID:11457740; http://dx.doi.org/10.1161/01.CIR.104.3.253
- Blackstone NW. The impact of mitochondrial endosymbiosis on the evolution of calcium signaling. Cell Calcium 2015; 57:133-9; PMID:25481706; http://dx.doi.org/10.1016/j.ceca.2014.11.006
- Saraste M. Oxidative phosphorylation at the fin de siecle. Science 1999; 283:1488-93; PMID:10066163; http://dx.doi.org/10.1126/science.283.5407.1488
- Wang C, Youle RJ. The role of mitochondria in apoptosis. Annu Rev Genetics 2009; 43:95-118; PMID:19659442; http://dx.doi.org/10.1146/annurev-genet-102108-134850
- Laura Valls-Lacalle IBEM, Marisol Ruiz-Meana MFAR, García-Dorado. Succinate dehydrogenase inhibition with malonate during reperfusion reduces infarct size by preventing mitochondrial permeability transition. Cardiovasc Res 2015; 109: 374-384; PMID:26705364; http://dx.doi.org/10.1093/cvr/cvv279.
- Davidson SM. Endothelial mitochondria and heart disease. Cardiovasc Res 2010; 88:58-66; PMID:20558442; http://dx.doi.org/10.1093/cvr/cvq195
- Maity S, Basak T, Bhat A, Bhasin N, Ghosh A, Chakraborty K, Sengupta S. Cross-compartment proteostasis regulation during redox imbalance induced ER stress. Proteomics 2014; 14:1724-36; PMID:24838640; http://dx.doi.org/10.1002/pmic.201300449
- Yoneda T, Benedetti C, Urano F, Clark SG, Harding HP, Ron D. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J Cell Sci 2004; 117:4055-66; PMID:15280428; http://dx.doi.org/10.1242/jcs.01275
- Papa L, Germain D. SirT3 Regulates the Mitochondrial Unfolded Protein Response. Mol Cell Biol 2014; 34:699-710; PMID:24324009; http://dx.doi.org/10.1128/MCB.01337-13
- Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell 2012; 48:158-67; PMID:23102266; http://dx.doi.org/10.1016/j.molcel.2012.09.025
- Haynes CM, Ron D. The mitochondrial UPR - protecting organelle protein homeostasis. J Cell Sci 2010; 123:3849-55; PMID:21048161; http://dx.doi.org/10.1242/jcs.075119
- Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 2011; 144:79-91; PMID:21215371; http://dx.doi.org/10.1016/j.cell.2010.12.016
- Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, Auwerx J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 2013; 497:451-7; PMID:23698443; http://dx.doi.org/10.1038/nature12188
- Moullan N, Mouchiroud L, Wang X, Ryu D, Williams EG, Mottis A, Jovaisaite V, Frochaux MV, Quiros PM, Deplancke B, Houtkooper RH, Auwerx J. Tetracyclines disturb mitochondrial function across eukaryotic models: A call for caution in biomedical research. Cell Rep 2015; 10: 1681-1691; PMID:25772356; http://dx.doi.org/10.1016/j.celrep.2015.02.034
- Siegelin MD, Dohi T, Raskett CM, Orlowski GM, Powers CM, Gilbert CA, Ross AH, Plescia J, Altieri DC. Exploiting the mitochondrial unfolded protein response for cancer therapy in mice and human cells. J Clin Invest 2011; 121:1349-60; PMID:21364280; http://dx.doi.org/10.1172/JCI44855
- Sanyal SN, Wada T, Yamabe M, Anai H, Miyamoto S, Shimada T, Ono K. Cardiac autonomic nerve abnormalities in chronic heart failure are associated with presynaptic vagal nerve degeneration. Pathophysiology 2012; 19:253-60; PMID:22921612; http://dx.doi.org/10.1016/j.pathophys.2012.07.004
- Shinlapawittayatorn K, Chinda K, Palee S, Surinkaew S, Thunsiri K, Weerateerangkul P, Chattipakorn S, KenKnight BH, Chattipakorn N. Low-amplitude, left vagus nerve stimulation significantly attenuates ventricular dysfunction and infarct size through prevention of mitochondrial dysfunction during acute ischemia-reperfusion injury. Heart Rhythm 2013; 10:1700-7; PMID:23933295; http://dx.doi.org/10.1016/j.hrthm.2013.08.009
- Katare RG, Ando M, Kakinuma Y, Arikawa M, Handa T, Yamasaki F, Sato T. Vagal nerve stimulation prevents reperfusion injury through inhibition of opening of mitochondrial permeability transition pore independent of the bradycardiac effect. J Thorac Cardiovasc Surg 2009; 137:223-31; PMID:19154929; http://dx.doi.org/10.1016/j.jtcvs.2008.08.020
- Sun L, Zhao M, Yang Y, Xue RQ, Yu X, Liu JK, Zang WJ. Acetylcholine attenuates hypoxia/reoxygenation injury by inducing mitophagy through PINK1/Parkin signal pathway in H9c2 cells. J Cell Physiol 2016:1171-81; PMID:26465230; http://dx.doi.org/10.1002/jcp.25215
- Mottis A, Jovaisaite V, Auwerx J. The mitochondrial unfolded protein response in mammalian physiology. Mamm Genome 2014; 25:424-33; PMID:24898297; http://dx.doi.org/10.1007/s00335-014-9525-z
- Strifler G, Tuboly E, Szél E, Kaszonyi E, Cao C, Kaszaki J, Mészáros A, Boros M, Hartmann P. Inhaled methane limits the mitochondrial electron transport chain dysfunction during experimental liver ischemia-reperfusion injury. PLoS One 2016; 11:e146363; PMID:26741361; http://dx.doi.org/10.1371/journal.pone.0146363
- Aluri HS, Simpson DC, Allegood JC, Hu Y, Szczepanek K, Gronert S, Chen Q, Lesnefsky EJ. Electron flow into cytochrome c coupled with reactive oxygen species from the electron transport chain converts cytochrome c to a cardiolipin peroxidase: role during ischemia-reperfusion. Biochim Biophys Acta 2014; 1840:3199-207; PMID:25092653; http://dx.doi.org/10.1016/j.bbagen.2014.07.017
- Schleit J, Johnson SC, Bennett CF, Simko M, Trongtham N, Castanza A, Hsieh EJ, Moller RM, Wasko BM, Delaney JR, et al. Molecular mechanisms underlying genotype-dependent responses to dietary restriction. Aging Cell 2013; 12:1050-61; PMID:23837470; http://dx.doi.org/10.1111/acel.12130
- Runkel ED, Liu S, Baumeister R, Schulze E. Surveillance-activated defenses block the ROS - induced mitochondrial unfolded protein response. PLoS Genet 2013; 9:e1003346; PMID:23516373; http://dx.doi.org/10.1371/journal.pgen.1003364
- Lin M, Li L, Li L, Pokhrel G, Qi G, Rong R, Zhu T. The protective effect of baicalin against renal ischemia-reperfusion injury through inhibition of inflammation and apoptosis. BMC Complement Altern Med 2014; 14:19; PMID:24417870; http://dx.doi.org/10.1186/1472-6882-14-19
- Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. A mitochondrial specific stress response in mammalian cells. EMBO J 2002; 21:4411-9; PMID:12198143; http://dx.doi.org/10.1093/emboj/cdf445
- Hu F, Liu F. Mitochondrial stress: A bridge between mitochondrial dysfunction and metabolic diseases? Cell Signal 2011; 23:1528-33; PMID:21616143; http://dx.doi.org/10.1016/j.cellsig.2011.05.008
- He L, Lemasters JJ. Regulated and unregulated mitochondrial permeability transition pores: a new paradigm of pore structure and function? FEBS Lett 2002; 512:1-7; PMID:11852041; http://dx.doi.org/10.1016/S0014-5793(01)03314-2
- Yu E, Mercer J, Bennett M. Mitochondria in vascular disease. Cardiovasc Res 2012; 95:173-82; PMID:22392270; http://dx.doi.org/10.1093/cvr/cvs111
- Kokoszka JE, Coskun P, Esposito LA, Wallace DC. Increased mitochondrial oxidative stress in the Sod2 (+/−) mouse results in the age-related decline of mitochondrial function culminating in increased apoptosis. Proc Natl Acad Sci U S A 2001; 98:2278-83; PMID:11226230; http://dx.doi.org/10.1073/pnas.051627098
- Abboud FM. The Walter B. Cannon Memorial Award Lecture, 2009. Physiology in perspective: The wisdom of the body. In search of autonomic balance: the good, the bad, and the ugly. Am J Physiol Regul Integr Comp Physiol 2010; 298:R1449-67; PMID:20219871; http://dx.doi.org/10.1152/ajpregu.00130.2010
- Esler M. The sympathetic nervous system through the ages: from Thomas Willis to resistant hypertension. Exp Physiol 2011; 96:611-22; PMID:21551268; http://dx.doi.org/10.1113/expphysiol.2011.052332
- He X, Zhao M, Bi X, Sun L, Yu X, Zhao M, Zang W. Novel strategies and underlying protective mechanisms of modulation of vagal activity in cardiovascular diseases. Br J Pharmacol 2015; 172:5489-500; PMID:25378088; http://dx.doi.org/10.1111/bph.13010
- De Ferrari GM. Vagal Stimulation in Heart Failure. J Cardiovasc Transl Res 2014; 7:310-20; PMID:24500409; http://dx.doi.org/10.1007/s12265-014-9540-1
- Bi XY, He X, Xu M, Zhao M, Yu XJ, Lu XZ, Zang WJ. Acetylcholine ameliorates endoplasmic reticulum stress in endothelial cells after hypoxia/reoxygenation via M3 AChR-AMPK signaling. Cell Cycle 2015; 14:2461-72; PMID:26066647; http://dx.doi.org/10.1080/15384101.2015.1060383
- Miao Y, Zhou J, Zhao M, Liu J, Sun L, Yu X, He X, Pan X, Zang W. Acetylcholine attenuates hypoxia/ reoxygenation-induced mitochondrial and cytosolic ROS formation in H9c2 cells via M2 acetylcholine receptor. Cell Physiol Biochem 2013; 31:189-98; PMID:23407103; http://dx.doi.org/10.1159/000343360
- Lu XZ, Bi XY, He X. Zhao M, Xu M, Yu XJ, Zhao ZH, Zang WJ. Activation of M 3 cholinoceptors attenuates vascular injury ischaemia/reperfusion by inhibiting the Ca+/calmodulin-dependent protein kinase II pathway. Br J Pharmac 2015; 172:5619-33; PMID:25953628; http://dx.doi.org/10.1111/bph.13183
- He X, Bi XY, Lu XZ, Zhao M, Yu XJ, Sun L, Xu M, Wier WG, Zang WJ. Reduction of mitochondria-endoplasmic reticulum interactions by acetylcholine protects human umbilical vein endothelial cells from hypoxia/reoxygenation injury. Arterioscler Thromb Vasc Biol 2015; 35:1623-34; PMID:25977565; http://dx.doi.org/10.1161/ATVBAHA.115.305469
- Wang XX, Li YB, Yao HJ, Ju RJ, Zhang Y, Li RJ, Yu Y, Zhang L, Lu WL. The use of mitochondrial targeting resveratrol liposomes modified with a dequalinium polyethylene glycol-distearoylphosphatidyl ethanolamine conjugate to induce apoptosis in resistant lung cancer cells. Biomaterials 2011; 32:5673-87; PMID:21550109; http://dx.doi.org/10.1016/j.biomaterials.2011.04.029
- Yoshii SR, Kishi C, Ishihara N, Mizushima N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem 2011; 286:19630-40; PMID:21454557; http://dx.doi.org/10.1074/jbc.M110.209338
- Yu L, Li Q, Yu B, Yang Y, Jin Z, Duan W, Zhao G, Zhai M, Liu L, Yi D, Chen M, Yu S. Berberine attenuates myocardial ischemia/reperfusion injury by reducing oxidative stress and inflammation response: role of silent information regulator 1. Oxid Med Cell Longev 2016; 2016:1689602; PMID:26788242; http://dx.doi.org/10.1155/2016/1689602
- Liu JJ, Huang N, Lu Y, Zhao M, Yu XJ, Yang Y, Yang YH, Zang WJ. Improving vagal activity ameliorates cardiac fibrosis induced by angiotensin II: in vivo and in vitro. Sci Rep 2015; 5:17108; PMID:26596640; http://dx.doi.org/10.1038/srep17108
- Ghavami S, Kerkhoff C, Chazin WJ, Kadkhoda K, Xiao W, Zuse A, Hashemi M, Eshraghi M, Schulze-Osthoff K, Klonisch T, Los M. S100A8/9 induces cell death via a novel, RAGE-independent pathway that involves selective release of Smac/DIABLO and Omi/HtrA2. Biochim Biophys Acta 2008; 1783:297-311; PMID:10860880; http://dx.doi.org/10.1016/j.bbamcr.2007.10.015
- Gallerne C, Prola A, Lemaire C. Hsp90 inhibition by PU-H71 induces apoptosis through endoplasmic reticulum stress and mitochondrial pathway in cancer cells and overcomes the resistance conferred by Bcl-2. Biochim Biophy Acta 2013; 1833:1356-66; PMID:23485394; http://dx.doi.org/10.1016/j.bbamcr.2013.02.014