
ABSTRACT
Cdt1 is rapidly degraded by CRL4Cdt2 E3 ubiquitin ligase after UV (UV) irradiation. Previous reports revealed that the nucleotide excision repair (NER) pathway is responsible for the rapid Cdt1-proteolysis. Here, we show that mismatch repair (MMR) proteins are also involved in the degradation of Cdt1 after UV irradiation in the G1 phase. First, compared with the rapid (within ∼15 min) degradation of Cdt1 in normal fibroblasts, Cdt1 remained stable for ∼30 min in NER-deficient XP-A cells, but was degraded within ∼60 min. The delayed degradation was also dependent on PCNA and CRL4Cdt2. The MMR proteins Msh2 and Msh6 were recruited to the UV-damaged sites of XP-A cells in the G1 phase. Depletion of these factors with small interfering RNAs prevented Cdt1 degradation in XP-A cells. Similar to the findings in XP-A cells, depletion of XPA delayed Cdt1 degradation in normal fibroblasts and U2OS cells, and co-depletion of Msh6 further prevented Cdt1 degradation. Furthermore, depletion of Msh6 alone delayed Cdt1 degradation in both cell types. When Cdt1 degradation was attenuated by high Cdt1 expression, repair synthesis at the damaged sites was inhibited. Our findings demonstrate that UV irradiation induces multiple repair pathways that activate CRL4Cdt2 to degrade its target proteins in the G1 phase of the cell cycle, leading to efficient repair of DNA damage.
KEYWORDS:
Introduction
To maintain genome integrity, cells correctly replicate DNA once per cell cycle and respond to genotoxic insults by triggering a DNA damage response.Citation1,2 Ubiquitin ligases are involved in DNA metabolic pathways to preserve genome integrity. CRL4Cdt2, a member of the Cullin4-RING ubiquitin ligase (CRL4) family, is activated both in the S phase and after DNA damage.Citation3 CRL4 comprises DNA damage-binding protein 1 (DDB1), Cul4, and Rbx1, and DDB1 acts as an adaptor protein connecting Cul4-Rbx1 to a family of substrate receptors called DDB1 and Cul4 associated factors (DCAFs).Citation4-6 Cdc10-dependent transcript 2 (Cdt2) is a DCAF protein that forms CRL4Cdt2.
CRL4Cdt2 was originally identified as a ubiquitin ligase that acts during the S phase for the DNA replication-licensing factor Cdc10-dependent transcript 1 (Cdt1).Citation4-6 Before DNA replication is initiated, Cdt1 functions with Cdc6 to load MCM2–7 DNA helicase on origin recognition complex-bound origins, and replication origins are licensed for replication.Citation2,7-9 When DNA replication is initiated, CRL4Cdt2 is activated to ubiquitinate Cdt1 protein for degradation, thus preventing re-replication of DNA in the same cell cycle. This process is dependent on chromatin-bound proliferating cell nuclear antigen (PCNA). PCNA, identified as an accessory factor for DNA polymerases, is loaded at the replication forks, and supports the function of various molecules for chromatin duplication.Citation10 Cdt1 also associates with PCNA, but is recognized by CRL4Cdt2 for ubiquitination and proteolysis.Citation11-13 The p21, Set8, and thymine DNA glycosylase proteins are degraded by the same pathway as Cdt1.Citation14-21 Analyses of target proteins led to the identification of a common sequence, called the PIP-degron, which comprises a canonical PIP box and conserved amino acids within and downstream of the PIP box; Q-x-x-[I/L/M/V]-T-D-[F/Y]-[F/Y]-x-x-[K/R]-[K/R] (PIP box is underlined) in higher eukaryotic cells.Citation22-24
When cells are exposed to irradiation or other DNA-damaging reagents, Cdt1 is degraded via the same PCNA- and CRL4Cdt2 -dependent pathways.Citation25-30 Exposure of cells to UV (UV) irradiation induces helix-distorting DNA lesions, such as cyclobutane pyrimidine dimers (CPDs) and 6–4 pyrimidine-pyrimidone photoproducts. Upon UV irradiation, Cdt1 is rapidly degraded within minutes, primarily dependent on the nucleotide excision repair (NER) process,Citation31,32 based on the finding that Cdt1 degradation is attenuated when xeroderma pigmentosum (XP)-related proteins are inactivated or depleted. NER is a versatile system for repairing UV-induced DNA lesions.Citation33-35 Many XP-related proteins are involved in this process. The DCAF protein DDB2 was originally identified as part of a UV-damaged DNA binding protein (UV-DDB) complex, comprising DDB1 and DDB2. CRL4DDB2 binds to CPDs and (6–4)-photoproducts, ubiquitinates XPC, and initiates NER.Citation36,37 The strand containing the UV-photoproduct is excised by NER factors such as XPA, XPG, and XPF-ERCC1.Citation35 PCNA is loaded on the resulting gap 3’-OH terminus-containing intermediates by the aid of replication factor C with a large subunit RFC1 (RFC1-RFC) to facilitate the recruitment of DNA polymerases.Citation38,39 The thus-loaded PCNA connects Cdt1 to CRL4Cdt2 for degradation.Citation31,32
MMR functions during DNA replication in the S phase, first recognizing mismatches in newly synthesized strands mediated primarily by Msh2/Msh6, and then excising and degrading the strand beyond the mismatch by the function of PCNA, RFC1-RFC, Mlh1/Pmas2, and EXO1 exonuclease, and later resynthesizing the excised tract.Citation40,41 Recent reports revealed that some MMR proteins, called noncanonical MMR, also function outside of the S phase.Citation42,43 When serum-starved cells or confluent cells, i.e., cells in the G0/G1 phase, are exposed to various oxidizing or alkylating agents, MMR proteins act to mono-ubiquitinate PCNA and recruit translesion synthesis (TLS) polymerases.Citation42,43 Therefore, MMR functions not only during the S phase, but also outside of the S phase independently of DNA replication.
When cells initiate NER or the noncanonical MMR response outside of the S phase, replicative DNA polymerases and TLS polymerases are recruited to the damaged chromatin sites.Citation42-45 Some data show that degradation of PIP-degron–containing proteins is important for recruiting polymerases to the damaged sites.Citation46,47
In the present study, we further examined UV-induced degradation of Cdt1. The rapid Cdt1 degradation was attenuated in XP-A cells as previously reported, but we observed that Cdt1 was degraded later (within ∼60 min). We demonstrated that even in the absence of NER function in XP-A cells, MMR factors were recruited to UV-damaged sites to degrade Cdt1. The NER-independent recruitment of MMR proteins reveals another aspect of the MMR response to UV irradiation outside of the S phase, in the G1 phase.
Results
UV-induced Cdt1 degradation was delayed in XP-A cells compared with normal cells
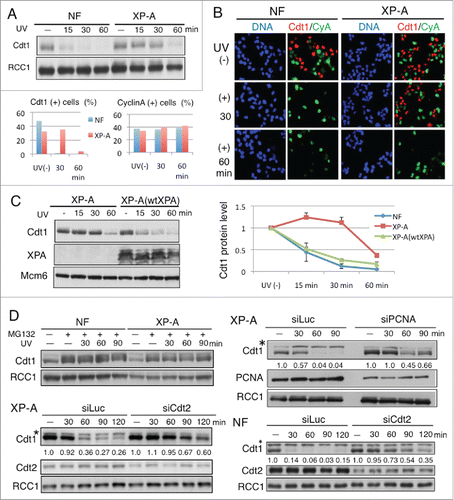
Previous reports indicated that exposure to UV irradiation leads to rapid degradation of Cdt1 depending on the NER activity.Citation31,32 In those studies, however, the Cdt1 protein levels were examined for only 15 min or 30 min after UV irradiation. Here, we examined Cdt1 levels in an NER-defective cell line, XPA-deficient XP2OSSV (XP-A), at a later time-point. As reported previously, while Cdt1 was mostly degraded within ∼15 min in normal fibroblasts (NF), Cdt1 was stably detected at 30 min in XP-A cells (). We consistently observed, however, that Cdt1 was degraded ∼60 min after UV irradiation in XP-A cells (). Immunofluorescence analysis showed the same results. When asynchronously growing cells were stained with antibodies for Cdt1 and Cyclin A, Cdt1 was detected in G1-phase cell nuclei, while it was degraded in the S to G2 phases and thus absent in Cyclin A-positive cells.Citation12 When the cells were exposed to UV, the Cdt1 signal disappeared at 30 min in NF. In contrast, in XP-A cells, Cdt1 remained stable at 30 min and was then degraded at 60 min (). There was no change in the CyclinA levels after UV irradiation. To confirm that the above-described observations were due to an XPA mutation, we examined Cdt1 degradation after adding back the wild-type XPA gene into XP-A cells (XP-A [wtXPA]). In this cell line, similar to NF, Cdt1 was degraded in 15 min, indicating that the wild-type XPA gene rescued the mutation in XP-A cells (). The delayed degradation of Cdt1 in XP-A cells also occurs through the ubiquitin-proteasome pathway mediated by CRL4Cdt2, because proteasome inhibitor MG132 treatment or knockdown of either PCNA or Cdt2 inhibited Cdt1 degradation ().
Figure 1. Cdt1 degradation in XP-A cells after UV irradiation is delayed and it is also dependent on PCNA and CRL4Cdt2. (A) Normal fibroblasts (NF) and XP-A cells were irradiated with UV at 5 J/m2 and collected at the indicated time-points for Western blotting. RCC1 was used as a loading control. (B) Cells grown on coverslips were irradiated as in (A) and fixed for staining with antibodies for Cdt1 and Cyclin A (CyA). Cells positive for Cdt1 (+) or Cyclin A (+) were counted at the indicated time-points and their frequency was plotted (%) for both NF and XP-A cells. (C) XP-A cells and XP-A cells complemented with the wild-type XPA gene (wtXPA) were UV-irradiated (5 J/m2) and collected at the indicated time-points for Western blotting (left panel). Comparison of the time-course of Cdt1 degradation in XP-A, XP-A(wtXOA), and NF after UV irradiation (right panel; means and standard deviation of 3 independent experiments are shown.) (D) The delayed Cdt1 degradtion in XP-A cells is also dependent on ubiquitin-proteasome pathway mediated by CRL4Cdt2. NF or XP-A cells were incubated with MG132, 10 min later irradiated with UV at 5 J/m2 and collected at the indicated time-point (min) for Western blotting (left upper panel). Cultures of cells were transfected with the indicated siRNAs twice and incubated for a total of 3 d. Cells were irradiated with UV at 5 J/m2 and collected at the indicated time-points (min) for Western blotting. Cdt1 protein levels were measured and its relative amounts, normalized to RCC1, were shown. siRNA for luciferase was used as a control (siLuc). Asterisks indicate a non-specific band.
PCNA, Cdt2, and Cdt1 accumulated at UV-damaged sites in XP-A cells, but with delayed kinetics as compared with NF
To investigate whether Cdt1 was also degraded at the UV-damaged sites in XP-A cells, we performed a micropore UV irradiation assay. In this assay, UV was used to locally irradiate the nuclei through a microporous membrane. The irradiated sites were visualized after staining with antibodies for CPDs. The accumulation of PCNA, Cdt2, and Cdt1 at the UV-irradiated sites was examined by co-staining with antibodies for each protein and CPD. We first monitored the accumulation of PCNA. When asynchronously growing non-irradiated cells were pre-treated with detergent and stained with antibodies for PCNA, only S-phase cells were identified as heavily stained cells ( - triangles), while most of the PCNA-negative cells were identified as G1-phase cells.Citation48 After local UV irradiation, CPD foci were observed in all cell phases irrespective of the cell type (). In NF, PCNA foci-containing cells, mostly representing G1-phase cells, were detected 30 min post-irradiation. PCNA foci were colocalized with the CPD antibody, indicating that PCNA was recruited to the UV-irradiated sites, as previously reported.Citation29 The levels of PCNA staining at the foci remained almost the same for 90 min in NF (). In contrast, the PCNA levels in XP-A cells were very low at 30 min. The PCNA staining levels then gradually increased, and at 60 min when Cdt1 was degraded, PCNA accumulation was more clearly detected, but still at lower levels than in NF (). Consistent with the finding that introduction of the XPA gene to XP-A cells recovered the rapid Cdt1 degradation, PCNA staining levels at the CPD sites in XP-A(wtXPA) cells recovered to the levels found in NF (). We then examined the accumulation of Cdt1 at the CPD sites. For this analysis, proteasome inhibitor MG132 was included to prevent Cdt1 degradation. Cdt1 staining was clearly detected at 30 min and its levels increased thereafter in NF (). In parallel with the accumulation of PCNA at the UV-irradiated sites in XP-A cells, the accumulation of Cdt1 increased ( and Supplemental Fig. S1). Similar to PCNA, however, Cdt1 recruitment was delayed in XP-A cells and the signals were weaker than those in NF (). Cdt2 was also recruited to the CPD sites, but delayed like Cdt1 in XP-A cells (Supplemental Fig. S2).
Figure 2. PCNA, Cdt2, and Cdt1 accumulate at the UV-irradiated sites in XP-A cells, but later than in normal fibroblasts. (A) Asynchronously growing normal fibroblasts (NF) and XP-A cells were covered with a membrane with 5-μm pores, irradiated locally with UV (100 J/m2; micropore assay), and incubated for the indicated times. The cells were fixed and stained with antibodies for CPD and PCNA. Bar, 10 μm. The white triangles in UV (-) indicate S phase cells (see text). (B) PCNA intensity was measured using ImageJ at the CPD spots as shown in (A) (> 25 spots for each time-point) and mean values (arbitrary) were plotted. (C) Asynchronously growing NF, XP-A, and XP-A(wtXPA) cells were treated as in (A), incubated for 30 min, and stained with antibodies for CPD and PCNA. (D) The intensity of PCNA at the CPD sites in (C) was measured using ImageJ (30 spots/cell line) and mean values (arbitrary) are shown. (E) Asynchronously growing cells were treated as in (A) and stained with antibodies for CPD and Cdt1. To visualize Cdt1 proteins, MG132 was added to the medium 10 min before UV irradiation. Bar, 5 μm. Cells were examined for Cdt1 staining at CPD sites (> 100 sites for each time-point) and the frequency of Cdt1-positive sites is shown (%) (left panel). Cdt1 intensity of cells was also measured and mean values are shown (arbitrary) (right panel).
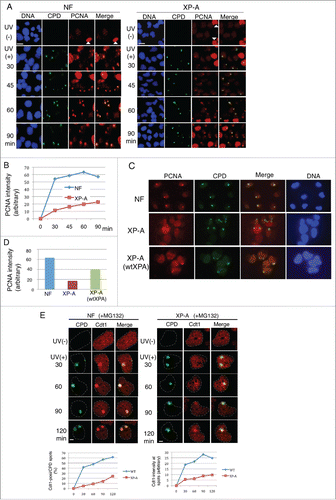
Because XPA is required to excise the damaged DNA and create the 3'OH terminus where PCNA is loaded, we predicted that PCNA loading is defective in XP-A cells. Our findings, however, suggested that even in XP-A cells, PCNA was recruited to the UV-damaged sites in the G1 phase, where Cdt1 was recruited and degraded by CRL4Cdt2. The fact that the levels of accumulated PCNA were lower in XP-A cells than in NF is consistent with the observation that Cdt1 was degraded at later time-points in XP-A cells.
MMR proteins are involved in Cdt1 degradation in XP-A cells
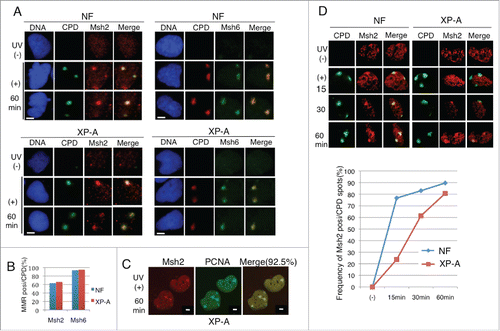
Because PCNA was recruited to UV-irradiated sites even in XP-A cells, we examined whether other DNA damage repair pathways could function to recruit PCNA and degrade Cdt1. Base excision repair (BER) and MMR are other major repair pathways. In the BER pathway, the damaged base is removed by DNA glycosylases, releasing an abasic site that is recognized by Ape1, an apurinic/apyrimidinic endonuclease.Citation49 Msh2 and Msh6 form heterodimers with each other and play a fundamental role in MMR during the S phase by recognizing mismatches.Citation40,41 In addition, recent reports revealed that Msh2 and Msh6 also function outside of the S phase, responding to various DNA-damaging reagents.Citation42,43 Thus, we examined the recruitment of several types of repair proteins, Ape1 as a BER protein, and Msh2 and Msh6 as MMR proteins, at the CPD sites 60 min after UV irradiation. Because Cdt1 degradation after UV irradiation is a G1-phase event, we synchronized the cells in the G1 phase and irradiated them locally with UV. In NF, both Msh2 and Msh6 were recruited to the UV-irradiated sites (). Similarly, both Msh2 and Msh6 proteins were detected in XP-A cells at the CPD sites 60 min after UV irradiation at a frequency similar to that in NF (). In locally irradiated XP-A cells, Msh2 was colocalized at the PCNA foci at a frequency greater than 90% (). In contrast, Ape1 recruitment to CPD sites was not detected by normal or confocal microscopy analysis in either XP-A cells or HeLa cells (Supplemental Fig. S3B and S3C), suggesting that the BER pathway might not be involved in UV-induced DNA damage repair. We compared the kinetics of Msh2 accumulation at the CPD sites between NF and XP-A cells. In NF, Msh2 was detected in more than 70% of cells at 15 min post UV-irradiation. In XP-A cells, however, the frequency of Msh2 recruitment and its signal intensity were low at 15 min, and then gradually increased to almost the level observed in NF at 60 min ().
Figure 3. Msh2 and Msh6 proteins were recruited to the UV-irradiated sites in XP-A cells. (A) Cells synchronized in the G1 phase were locally irradiated (+) or not (−) through a membrane with 5-μm pores. Cells were incubated for 1 h, pre-extracted, and fixed for staining with CPD and Msh2 or Msh6 antibodies. (B) Cells were examined for Msh2 or Msh6 staining at CPD sites. Frequency of CPD sites that were positive for Msh2 or Msh6 are shown (%) (> 100 CPD sites were examined). (C) XP-A cells treated as in (A) were co-stained with Msh2 and PCNA. 92.5% of CPD sites were positive for Msh2 (62/67 CPD sites). (D) Cells synchronized in the G1 phase were locally irradiated with UV (+) and fixed for staining with antibodies for CPD and Msh2 at the indicated time-points. (−): not irradiated with UV. Cells were examined for CPD and Msh2, and the frequency of cells co-stained with CPD and Msh2 was plotted. Bar, 5 μm.
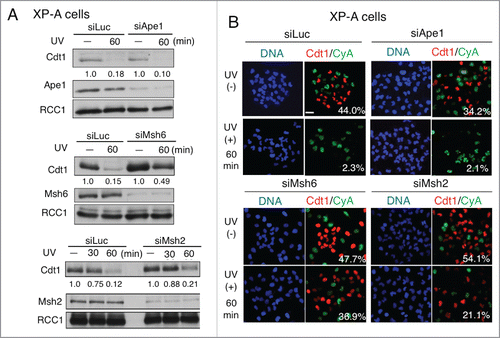
Because Msh2 and Msh6 proteins were recruited to the UV-irradiated sites even in XP-A cells, we thought that MMR proteins were engaged in the delayed Cdt1 degradation. To investigate this possibility, we knocked down each protein in XP-A cells and examined the effect on Cdt1 degradation. Consistent with the fact that Ape1 was not recruited to the CPD sites, Cdt1 was degraded in Ape1-depleted cells at 60 min, similar to control cells, based on immunoblotting analysis (). In contrast, Msh6 depletion resulted in Cdt1 stabilization at 60 min post-irradiation in XP-A cells (). Msh2 depletion had the same effect, but a smaller effect than Msh6 depletion. Immunofluorescence analysis confirmed that depletion of either Msh2 or Msh6, but not Ape1, prevented Cdt1 degradation in XP-A cells (). We obtained the same results with different small interfering RNAs (siRNAs) for Msh2 (_#2) and Msh6 (_#2) (Supplemental Figure S4). These findings suggested that MMR mediated the degradation of Cdt1 in XP-A cells.
Figure 4. MMR is involved in the Cdt1 degradation in XP-A cells. (A) XP-A cells were transfected twice with the siRNAs for Ape1, Msh2, Msh6, or luciferase (siLuc) as a control. Three days later, half of the cell cultures were irradiated with UV at 5 J/m2 and incubated for the indicated period of time. Cells were collected at the indicated time-points for Western blotting with antibodies for Cdt1, Msh2, Msh6, Ape1, and RCC1. Cdt1 protein levels were measured and its relative amounts were shown. (B) XP-A cells were treated as in (A) and fixed and stained with antibodies for Cdt1 and Cyclin A (CyA). Cells were examined for Cdt1 staining and the frequency of Cdt1-positive cells is shown (%). Bar, 10 μm.
The MMR pathway also contributes to degrade Cdt1 in normal cell lines
To examine whether MMR acts to degrade Cdt1 when XPA is absent or whether MMR is involved in Cdt1 degradation independently of XPA, we investigated Cdt1 degradation in NF after knocking down XPA and/or Msh6. Depletion of XPA in NF delayed the degradation of Cdt1, as expected. Co-depletion with Msh6 led to further stabilization of Cdt1, as when Msh6 was depleted in XP-A cells (). We obtained the same results using another cell line, U2OS cells (). In this cell line, co-depletion of XPA and Msh6 led to high stabilization of Cdt1. Furthermore, depletion of Msh6 alone stabilized Cdt1 in both NF and U2OS cells to levels similar to those observed in XPA-depleted cells (). These findings indicated that MMR-mediated degradation of Cdt1 was not a specific finding in XP-A cells, but rather that the MMR pathway is involved in Cdt1 degradation independently of the NER pathway in normal cells after UV-irradiation, and this occurs in the G1 phase of the cell cycle.
Figure 5. MMR is involved in Cdt1 degradation in normal cells. (A) Normal fibroblasts (NF) were transfected with the indicated siRNAs and 3 d later, UV irradiated (25 J/m2) and collected at the indicated time-points. Cdt1 protein levels were normalized with Mcm6 protein and mean values and standard deviation of 3 independent experiments are shown. (B) U2OS cells were treated as in (A) and analyzed. (means and standard deviation of 3 independent experiments are shown). (C) Interaction of Cdt2–3xFLAG with MMR proteins. Cdt2–3xFLAG–expressing stable U2OS cells were isolated, and examined for expression levels of Cdt2–3xFLAG (3xFLAG) and endogenous Cdt2 (end.) by Western blotting with Cdt2 antibody. U2OS cells and Cdt2–3xFLAG–expressing U2OS cells were treated with MG132 for 10 min and UV irradiated (25 J/m2) (+) or not (−). After incubation for 30 min, the cells were collected, fixed with paraformaldehyde, and immunoprecipitated with anti-FLAG as described in the Materials and Methods. Precipitates were examined for the indicated proteins. The band intensities for Msh2, Mas6 and PCNA were measured and normalized to UV(−). Means and standard deviation of 3 independent experiments are shown for Mas2 and Mas6. For PCNA, the mean of 2 independent assays is shown. Asterisk indicates Msh2 bands remaining after re-blotting with anti Cdt2 antibody.
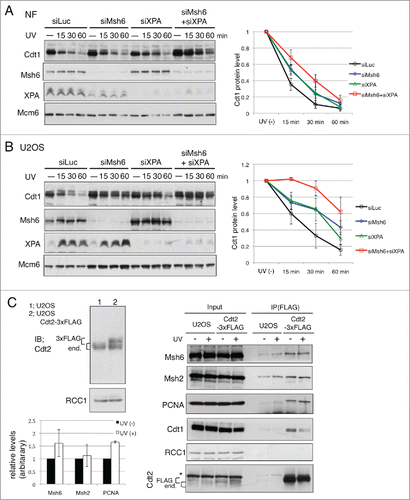
To investigate how MMR contributes to Cdt1 degradation, we examined the interaction between CRL4Cdt2 and MMR proteins. Because our Cdt2 antibody did not efficiently immunoprecipitate Cdt2 protein, we overexpressed Cdt2–3×FLAG and performed a pull-down assay after crosslinking treatment to stabilize the protein complexes on chromatin. In the Cdt2–3×FLAG immunoprecipitates, both PCNA and Msh6 protein were detected (Supplemental Fig. S5). We then isolated U2OS cells stably expressing Cdt2–3×FLAG at a level comparable to that of endogenous Cdt2 and examined the interaction with or without UV irradiation. Cdt2–3×FLAG was efficiently precipitated irrespective of UV irradiation. In the Cdt2–3×FLAG precipitates, not only PCNA but also both Msh2 and Msh6 proteins were detected. Their levels increased slightly after UV irradiation (), suggesting that MMR proteins and CRL4Cdt2 interact at the UV-damaged sites for Cdt1 degradation. The control protein RCC1 was not co-precipitated.
Cdt1 degradation is important for DNA repair synthesis
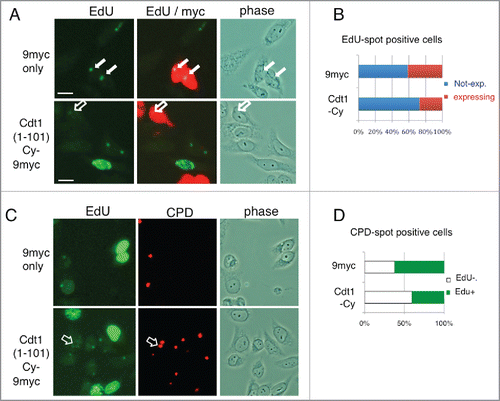
As Cdt1 was rapidly degraded and DNA polymerases were recruited to the DNA damage sites,Citation44 Cdt1 degradation should be important for repairing damaged DNA. To address this notion, we overexpressed Cdt1 and examined whether it inhibited repair DNA synthesis at the locally UV-irradiated sites. The DNA synthesis was monitored for 30 min based on the incorporation of 5-ethynyl-2-deoxyuridine (EdU). When non-irradiated HeLa cells were incubated with EdU, only S-phase cells were identified and detected because they had heavily labeled nuclei (Supplemental Fig. S6). After local UV irradiation, EdU-positive spot signals were detected and colocalized with CPD, indicating DNA repair synthesis at the UV-irradiated sites in cells at stages other than S-phase. We then examined EdU labeling after overexpressing Cdt1(1–101)-Cy-9myc or 9myc-only as a control. To prevent SCF-Skp2–mediated degradation of Cdt1, Cy mutation was included, which inhibits CyclinA-CDK binding and the subsequent phosphorylation required for the SCF-Skp2 interaction.Citation12 Transfection frequencies for both clones were ∼40%. Compared with 9myc-only–expressing cells, we observed cells with reduced or no-EdU incorporation in Cdt1(1–101)-Cy-9myc transfected cultures (; open arrows indicate a Cdt1(1–101)-Cy-9myc expressing cell with low EdU incorporation), suggesting that high expression of Cdt1(1–101)-Cy-9myc inhibited repair. To confirm this notion, we selected cells positive for EdU spots, and then examined whether each cell expressed the transfected genes. The frequency of cells expressing transfected genes was calculated for each gene. In the 9myc-only transfected cell culture, ∼40% of cells with EdU-positive spots were also positive for the myc signal, similar to the transfection frequency. On the other hand, in the Cdt1(1–101)-Cy-9myc transfected cell culture, the frequency of cells expressing Cdt1(1–101)-Cy-9myc was reduced to less than 30% (). In a different analysis, we examined whether cells with CPD spots were positive or negative for EdU incorporation in the Cdt1(1–101)-Cy-9myc or the 9myc-only transfected cell cultures. We found cells with CPD spots, but with no or reduced EdU incorporation (). The frequency of such cells was higher in Cdt1(1–101)-Cy-9myc transfected cells than in 9myc-only transfected cells (). These findings suggest that high expression of Cdt1-Cy-9myc inhibited repair synthesis.
Figure 6. Cdt1 degradation is important for DNA repair synthesis. (A) HeLa cells were transfected with Cdt1(1–101)-Cy-9myc or 9myc only. The next day, cells were locally irradiated with UV, incubated in the presence of EdU for 30 min, and fixed for staining with antibodies for myc and Alexa Fluor 488 azide to detect EdU as supplied by the manufacturer. The open arrow indicates Cdt1(1–101)-Cy-9myc expressing cells with low EdU staining, while the filled arrows indicate 9myc-only–expressing cells with EdU spot signals. (B) EdU spot-positive cells in (A) were examined for expression of Cdt1(1–101)-Cy-9myc (Cdt1-Cy) or 9myc-only, and the frequency of cells not expressing (blue) or expressing (red) (%) is shown for each transfected cell culture. The mean of 2 independent assays is shown. (C) HeLa cells were treated as described in A, and stained for EdU and CPD. Open arrows indicate CPD sites with low or negative EdU signals. (D) CPD spot-positive cells in (C) were examined for EdU incorporation, and the frequency of EdU-positive (+; green) or -negative or low (-; white) cells (%) are shown for each transfected cell culture. The mean of 2 independent assays is shown.
Discussion
Cdt1 is rapidly degraded when genomic DNA is damaged by UV irradiation. Previous studies reported that NER is involved in Cdt1 degradation after UV irradiation. Here we demonstrated that MMR factors also contribute to Cdt1 degradation after UV irradiation in the G1 phase.
Cells respond to various types of DNA damage in a cell-cycle specific manner.Citation1 As Cdt1 is present only during the G1 phase, analysis of Cdt1 degradation after DNA damage provides a useful means for investigating the DNA damage response in the G1 phase. As presented here, examination of NER-deficient XP-A cells revealed that the MMR pathway also functions in degrading Cdt1 after UV irradiation. Although Msh2 recruitment was not detected immediately after irradiation in XP-A cells in a previous report,Citation50 we noticed that Msh2 and Msh6 as well as PCNA were recruited and detected at the irradiated sites in XP-A cells approximately 60 min after irradiation, around the time when Cdt1 was degraded. Depletion of MMR proteins in XP-A cells prevented Cdt1 degradation. We demonstrated that the MMR protein-mediated degradation was not specific to the XP-A cell line used in this study. A contribution of MMR factors to Cdt1 degradation after UV irradiation was also revealed using normal fibroblasts (NFs) and U2OS cells. Depletion of XPA in these cells delayed the degradation of Cdt1, as in XP-A cells. Upon co-depletion of Msh6, Cdt1 was further stabilized. Of note, Msh6 depletion alone delayed Cdt1 degradation as much as in XPA-depleted cells in both NFs and U2OS cells. These findings demonstrate that, like the NER pathway, the MMR pathway also mediates Cdt1 degradation after UV irradiation in the G1 phase.
How does MMR contribute to Cdt1 degradation after UV irradiation in the G1 phase? MMR is normally involved in the replication process by repairing replication errors formed on nascent DNA strands during the S phase.Citation40,41 The Msh2 protein is recruited to the UV-irradiated sites soon after irradiation in an NER-dependent manner,Citation50 implying that MMR proteins also function to correct mismatches during NER repair synthesis. On the other hand, the recruitment of MMR proteins to UV-damaged sites was also observed in the G1 phase, and this occurred in the absence of XP-A (). Recent reports indicated that MMR proteins also function outside of the S phase, known as a noncanonical MMR.Citation43 The Msh2-Msh6 heterodimers recognize not only mismatches but also many DNA adducts, such as cisplatin, O6-methylguanine, 2-hydroxyadenine, and 8-oxoguanine lesions in vitro.Citation42,51-53 Exposure of serum-starved or confluent cells, that is, G0/G1-phase cells, to DNA damaging agents by oxidation or alkylation promotes PCNA mono-ubiquitination on chromatin depending on the MMR protein.Citation42,43 The observation that treatment with methyl-nitro-nitrosoguanidine causes degradation of p21, another CRL4Cdt2 substrate, depending on the MMR proteins in the G1 phaseCitation54 suggests that p21 degradation is dependent on a noncanonical function of MMR. Thus, we speculate that noncanonical MMR responded to UV-induced DNA lesions that caused Cdt1 degradation. UV irradiation, in addition to producing the main bipyrimidine photoproducts, provokes oxidation reactions, which produce 8-hydroxydeoxyguanosine,Citation55 although the UV-C used in the present study produces less reactive oxygen species than UV-A or UV-B. It is possible that MMR proteins respond to CPD or (6–4)-photoproducts, as in vitro experiments using naked DNA demonstrated that MMR proteins interact with thymine-dimer-containing DNA.Citation56,57 Though the interaction was very weak, such lesions could be recognized by MMR proteins when present in the form of nucleosomes. We predict that the noncanonical MMR responds to such UV-induced lesions in the G1 phase, leading to the formation of a single-strand gap to load PCNA in G0/G1-phase cells, or that PCNA was first recruited to the lesion sites through direct interaction with Msh2-Msh6, because Msh6 has a PIP-box, and thus CRL4Cdt2 is activated for Cdt1 degradation. Consistently, co-immunoprecipitation of Cdt2–3×FLAG with Msh2 and Msh6 proteins was detected after UV irradiation.
While Cdt1 destruction after the initiation of DNA replication is important to prevent re-replication, the physiologic roles of Cdt1 degradation after DNA damage are not well known. We previously demonstrated that M-phase cells are resistant to UV irradiation-mediated degradation of Cdt1, but when released into the G1 phase, Cdt1 was degraded and origin licensing was not established.Citation58 Those cells are more likely to arrest in the G1 phase and survive than cells irradiated in the G1 phase.Citation58 On the other hand, in the G1 phase, origin licensing is already established and thus Cdt1 degradation would not affect licensing. Cdt1 is recruited to and associated with PCNA; thus, highly expressed Cdt1 protein would mask PCNA to inhibit the repair process. In this study, we showed that high Cdt1 expression prevented repair synthesis after UV irradiation (). Such an effect is similar to the inhibition of DNA replication by the expression of p21.Citation59,60 Both replicative DNA polymerases (pol) delta and pol epsilon, and TLS pol kappa are recruited to UV-damaged sites via NER,Citation44 and pol eta is recruited to UV-damaged sites outside of the S phase and independently of NER.Citation45 Consistently, high expression of Cdt1 or a PIP-degron Cdt1 mutant prevents recruitment of the TLS DNA polymerases pol kappa and pol eta to UV-damaged sites.Citation46 Similarly, expression of another CRL4Cdt2 target, helicase FBH1, impairs the recruitment of DNA pol eta.Citation47 These results are in accordance with our observations. Although these findings may represent a dominant negative effect of ectopic expression of PIP-degron peptide proteins, it is probable that CRL4Cdt2-mediated rapid degradation of Cdt1 and other targets facilitates DNA repair. In addition, degradation of Cdt1 in the G1 phase may help to prevent re-replication. Some population of cells irradiated with UV in the G1 phase enter the S phase,Citation58 and such cells are expected to undergo replication stalling due to the ongoing repair synthesis or DNA damage. In those situations, Cdt1 degradation might be compromised and origins just fired could be re-licensed. The removal of Cdt1 in the G1 phase could thus help to circumvent such a situation.
Our data revealed that multiple pathways are engaged in Cdt1 degradation after UV irradiation. In addition to NER, MMR responds to UV-induced lesions at least in the G1 phase. Both pathways may function additively to repair the lesions or compete with each other. In the case of UV-irradiated mice, defects in NER and MMR additively contribute to skin tumorigenesis.Citation61,62 Because noncanonical MMR appears to be mutagenic, such a response is predicted to enhance genome instability in patients with XP after UV exposure. Thus, it is important to know how MMR is activated outside of the S phase following UV irradiation.
Materials and methods
Cell culture
Normal fibroblasts, XP2OSSV (XP-A) cells, XP2OSSV cells complemented with FLAG-tagged wild-type XPA cDNA, U2OS cells, and HeLa cells were cultured in Dulbecco's modified Eagle's medium with 10% fetal bovine serum, 1% penicillin-streptomycin and 5% CO2. U2OS cells stably expressing Cdt2–3×FLAG were isolated similarly to the HEK293-Cdt2–3×FLAG–expressing cells.Citation27 To obtain G1-phase () cells, mitotic cells were collected after tapping the plate, washed with medium by centrifuging at 1,000 rpm for 1 min, and cultured for 3 h to release into the G1 phase. To obtain mitotic cells, cells were plated and cultured for 24 h, incubated in the presence of 2 mM thymidine for 20 h, washed with phosphate-buffered saline (PBS) twice and once with medium, and cultured in the presence of 40 ng/mL nocodazole for 14 h. Proteasome inhibitor MG132 was used at 25 μM. To analyze the DNA content, flow-cytometry was performed as described previously.Citation12 Cdt1(1-101)-Cy-9myc and 9myc only expression plasmids were described.Citation12
Antibodies, western blotting, and immunofluorescence
For Western blotting, whole cell lysates were prepared by lysing cell pellets directly in sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis buffer. For immunofluorescence, cells were fixed in 4% paraformaldehyde (WAKO) for 10 min, washed with PBS, permeabilized in 0.15–0.25% TritonX-100 in PBS, and stained with the indicated antibodies. To visualize chromatin-bound proteins, cells were pre-extracted with PBS containing 0.5% Triton X-100, and fixed in 4% paraformaldehyde solution. As secondary antibodies, Alexa Fluor 488-conjugated anti-mouse, anti-goat, or anti-rabbit antibodies, and Alexa Fluor 594-conjugated anti-mouse or anti-rabbit antibodies were used with Hoechst 33258 to visualize DNA. The following antibodies were used: Cdt1 (rabbit, described in ref. Citation63), Cdt2 (rabbit, described in ref. Citation17), PCNA (rabbit, kindly provided by Dr. T Tsurimoto, Santa Cruz Biotechnology, sc-7907; mouse, Santa Cruz Biotechnology, PC10), CPD (mouse, Cosmo Bio, TDM-2), Cyclin A (mouse, Neomarkers, Ab-6), Msh2 (rabbit, Santa Cruz Biotechnology, sc-494), Msh6 (goat, Santa Cruz Biotechnology, sc-1243), Ape1 (rabbit, Novus Biologicals), myc (mouse, Millipore; rabbit, Santa Cruz Biotechnology), Mcm6 (goat, Santa Cruz Biotechnology, sc-9843) and RCC1 (described in Citationref. 12).
RNA-interference knockdown experiments
Double-stranded RNAs were transfected at 100 nM using HiPerFect (Qiagen). Twenty-four hours after the first transfection, a second transfection was performed and the cells were cultured for 2 more days. The following siRNAs were made by Dharmacon: PCNA; CGGUGACACUCAGUAUGUCdTdT, Cdt2; CCAGGAGGUGAUAAACUUUdTdT, and siRNA for siLuc, known as GL2, used as a control siRNA. The siRNAs for Msh2, Msh6, and Ape1 were purchased from Invitrogen.
Micropore UV irradiation assay
The assay was performed as described previously.Citation29 Cells were cultured on glass coverslips in a 35-mm dish and incubated for 24 to 48 h. To perform micropore UV irradiation, the cells were washed twice with PBS, and covered with an Isopore polycarbonate membrane filter (Millipore) with a 5-µm pore size. The PBS was removed to apply the membrane to the cell surface and cells were irradiated. UV irradiation was performed using a UV lamp (SUV-16, As One, Japan) with a dose rate of 0.33 J/m2/s, which was monitored using a UV radiometer (UVX Radiometer, UVP) at 254 nm. We then added 2 ml of PBS, removed the filter, and cultured the cells in fresh medium for the indicated times. Cells were then fixed with 4% paraformaldehyde for 10 min, treated with 0.15% Triton-X100/PBS, and processed for immunofluorescence.
Cross-linking and immunoprecipitation
Cells were harvested by centrifugation at 3,000 rpm (900×g) for 3 min, washed twice with PBS, and fixed for 10 min at room temperature with 1 mL PBS containing 0.1% paraformaldehyde. After washing twice with 1 mL PBS, the cells were lysed with 400 μL 0.1% Triton X-100 containing CSK buffer (10 mM Pipes, pH 7.9, 100 mM NaCl, 300 mM sucrose, 0.1% Triton X-100, 1 mM phenyl-methyl-sulfonyl-fluoride, 10 mM ß-glycerophosphate, 1 mM Na3VO4, 10 mM NaF, and 1× protease inhibitor cocktail [Roche]) on ice. Cell lysates were sonicated (output 3, duty cycle 20%, 3 sets of 5 beats and 1 min interval) on ice. Cells lysates were adjusted to 400 μL and 2 mg/mL protein, and clarified by centrifugation 13,500 rpm (17,700×g) for 15 min at 4˚C. The supernatants (320 μL) were mixed with 10 μL bed volume of anti-FLAG M2 Affinity gel (SIGMA), and rotated for 90 min at 4˚C. The affinity gel beads were washed 3 times with 200 µL CSK buffer and mixed with 1×SDS sample buffer and boiled to make the bound fraction sample.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
1295179_Supplemental_Material.zip
Download Zip (8.1 MB)Acknowledgements
We thank Dr. Y. Nakatsu for valuable comments and discussion.
Funding
This work was financially supported by Grant-in-Aid for Scientific Research on Innovative Area (25131718) and Grants-in-Aid for Basic Scientific Research (B) (26291025) and for Challenging Exploratory Research (26650064) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan to HN, and by Grants-in-Aid for Basic Scientific Research (C) (25430171) to YS.
Related Research Data
References
- Ciccia A, Elledge SJ. The DNA damage response: Making it safe to play with knives. Mol Cell 2010; 40:179-204; PMID:20965415; http://dx.doi.org/10.1016/j.molcel.2010.09.019
- Masai H, Matsumoto S, You Z, Yoshizawa-Sugata N, Oda M. Eukaryotic chromosome DNA replication: Where, when, and how? Annu Rev Biochem 2010; 79:89-130; PMID:20373915; http://dx.doi.org/10.1146/annurev.biochem.052308.103205
- Havens CG, Walter JC. Mechanism of CRL4Cdt2, a PCNA-dependent E3 ubiquitin ligase. Genes Dev 2011; 25:1568-82; PMID:21828267; http://dx.doi.org/10.1101/gad.2068611
- Jin J, Arias EE, Chen J, Harper JW, Walter JC. A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol Cell 2006; 23:709-21; PMID:16949367; http://dx.doi.org/10.1016/j.molcel.2006.08.010
- Sansam CL, Shepard JL, Lai K, Ianari A, Danielian PS, Amsterdam A, Hopkins N, Lees JA. DTL/CDT2 is essential for both CDT1 regulation and the early G2/M checkpoint. Genes Dev 2006; 20:3117-29; PMID:17085480; http://dx.doi.org/10.1101/gad.1482106
- Higa LA, Wu M, Ye T, Kobayashi R, Sun H, Zhang H. CUL4-DDB1 ubiquitin ligase interacts with multiple WD40-repeat proteins and regulates histone methylation. Nat Cell Biol 2006; 8:1277-83; PMID:17041588; http://dx.doi.org/10.1038/ncb1490
- Nishitani H, Lygerou Z, Nishimoto T, Nurse P. The Cdt1 protein is required to license DNA for replication in fission yeast. Nature 2000; 404:625-8; PMID:10766248; http://dx.doi.org/10.1038/35007110
- Maiorano D, Moreau J, Mechali M. XCDT1 is required for the assembly of pre-replicative complexes in Xenopus laevis. Nature 2000; 404:622-5; PMID:10766247; http://dx.doi.org/10.1038/35007104
- O'Donnell M, Langston L, Stillman B. Principles and concepts of DNA replication in bacteria, archaea, and eukarya. Cold Spring Harb Perspect Biol 2013; 5:a010108; PMID:23818497; http://dx.doi.org/10.1101/cshperspect.a010108
- Moldovan GL, Pfander B, Jentsch S. PCNA, the maestro of the replication fork. Cell 2007; 129:665-79; PMID:17512402; http://dx.doi.org/10.1016/j.cell.2007.05.003
- Arias EE, Walter JC. PCNA functions as a molecular platform to trigger Cdt1 destruction and prevent re-replication. Nat Cell Biol 2006; 8:84-90; PMID:16362051; http://dx.doi.org/10.1038/ncb1346
- Nishitani H, Sugimoto N, Roukos V, Nakanishi Y, Saijo M, Obuse C, Tsurimoto T, Nakayama KI, Nakayama K, Fujita M, et al. Two E3 ubiquitin ligases, SCF-Skp2 and DDB1-Cul4, target human Cdt1 for proteolysis. EMBO J 2006; 25:1126-36; PMID:16482215; http://dx.doi.org/10.1038/sj.emboj.7601002
- Senga T, Sivaprasad U, Zhu W, Park JH, Arias EE, Walter JC, Dutta A. PCNA is a cofactor for Cdt1 degradation by CUL4/DDB1-mediated N-terminal ubiquitination. J Biol Chem 2006; 281:6246-52; PMID:16407252; http://dx.doi.org/10.1074/jbc.M512705200
- Slenn TJ, Morris B, Havens CG, Freeman RM, Jr, Takahashi TS, Walter JC. Thymine DNA glycosylase is a CRL4Cdt2 substrate. J Biol Chem 2014; 289:23043-55; PMID:24947512; http://dx.doi.org/10.1074/jbc.M114.574194
- Kim Y, Starostina NG, Kipreos ET. The CRL4Cdt2 ubiquitin ligase targets the degradation of p21Cip1 to control replication licensing. Genes Dev 2008; 22:2507-19; PMID:18794348; http://dx.doi.org/10.1101/gad.1703708
- Abbas T, Shibata E, Park J, Jha S, Karnani N, Dutta A. CRL4(Cdt2) regulates cell proliferation and histone gene expression by targeting PR-Set7/Set8 for degradation. Mol Cell 2010; 40:9-21; PMID:20932471; http://dx.doi.org/10.1016/j.molcel.2010.09.014
- Nishitani H, Shiomi Y, Iida H, Michishita M, Takami T, Tsurimoto T. CDK inhibitor p21 is degraded by a proliferating cell nuclear antigen-coupled Cul4-DDB1Cdt2 pathway during S phase and after UV irradiation. J Biol Chem 2008; 283:29045-52; PMID:18703516; http://dx.doi.org/10.1074/jbc.M806045200
- Oda H, Hubner MR, Beck DB, Vermeulen M, Hurwitz J, Spector DL, Reinberg D. Regulation of the histone H4 monomethylase PR-Set7 by CRL4(Cdt2)-mediated PCNA-dependent degradation during DNA damage. Mol Cell 2010; 40:364-76; PMID:21035370; http://dx.doi.org/10.1016/j.molcel.2010.10.011
- Tardat M, Brustel J, Kirsh O, Lefevbre C, Callanan M, Sardet C, Julien E. The histone H4 Lys 20 methyltransferase PR-Set7 regulates replication origins in mammalian cells. Nat Cell Biol 2010; 12:1086-93; PMID:20953199; http://dx.doi.org/10.1038/ncb2113
- Centore RC, Havens CG, Manning AL, Li JM, Flynn RL, Tse A, Jin J, Dyson NJ, Walter JC, Zou L. CRL4(Cdt2)-mediated destruction of the histone methyltransferase Set8 prevents premature chromatin compaction in S phase. Mol Cell 2010; 40:22-33; PMID:20932472; http://dx.doi.org/10.1016/j.molcel.2010.09.015
- Shibata E, Dar A, Dutta A. CRL4Cdt2 E3 ubiquitin ligase and proliferating cell nuclear antigen (PCNA) cooperate to degrade thymine DNA glycosylase in S phase. J Biol Chem 2014; 289:23056-64; PMID:24962565; http://dx.doi.org/10.1074/jbc.M114.574210
- Havens CG, Walter JC. Docking of a specialized PIP Box onto chromatin-bound PCNA creates a degron for the ubiquitin ligase CRL4Cdt2. Mol Cell 2009; 35:93-104; PMID:19595719; http://dx.doi.org/10.1016/j.molcel.2009.05.012
- Havens CG, Shobnam N, Guarino E, Centore RC, Zou L, Kearsey SE, Walter JC. Direct Role for proliferating cell nuclear antigen (PCNA) in substrate recognition by the E3 Ubiquitin ligase CRL4-Cdt2. J Biol Chem 2012; 287:11410-21; PMID:22303007; http://dx.doi.org/10.1074/jbc.M111.337683.
- Michishita M, Morimoto A, Ishii T, Komori H, Shiomi Y, Higuchi Y, Nishitani H. Positively charged residues located downstream of PIP box, together with TD amino acids within PIP box, are important for CRL4(Cdt2) -mediated proteolysis. Genes Cells 2011; 16:12-22; PMID:21143559; http://dx.doi.org/10.1111/j.1365-2443.2010.01464.x
- Ralph E, Boye E, Kearsey SE. DNA damage induces Cdt1 proteolysis in fission yeast through a pathway dependent on Cdt2 and Ddb1. EMBO Rep 2006; 7:1134-9; PMID:17039252; http://dx.doi.org/10.1038/sj.embor.7400827
- Stathopoulou A, Roukos V, Petropoulou C, Kotsantis P, Karantzelis N, Nishitani H, Lygerou Z, Taraviras S. Cdt1 is differentially targeted for degradation by anticancer chemotherapeutic drugs. PLoS One 2012; 7:e34621; PMID:22479651; http://dx.doi.org/10.1371/journal.pone.0034621
- Sakaguchi H, Takami T, Yasutani Y, Maeda T, Morino M, Ishii T, Shiomi Y, Nishitani H. Checkpoint kinase ATR phosphorylates Cdt2, a substrate receptor of CRL4 ubiquitin ligase, and promotes the degradation of Cdt1 following UV irradiation. PLoS One 2012; 7:e46480; PMID:23029527; http://dx.doi.org/10.1371/journal.pone.0046480
- Higa LA, Mihaylov IS, Banks DP, Zheng J, Zhang H. Radiation-mediated proteolysis of CDT1 by CUL4-ROC1 and CSN complexes constitutes a new checkpoint. Nat Cell Biol 2003; 5:1008-15; PMID:14578910; http://dx.doi.org/10.1038/ncb1061
- Ishii T, Shiomi Y, Takami T, Murakami Y, Ohnishi N, Nishitani H. Proliferating cell nuclear antigen-dependent rapid recruitment of Cdt1 and CRL4Cdt2 at DNA-damaged sites after UV irradiation in HeLa cells. J Biol Chem 2010; 285:41993-2000; PMID:20929861; http://dx.doi.org/10.1074/jbc.M110.161661
- Roukos V, Kinkhabwala A, Colombelli J, Kotsantis P, Taraviras S, Nishitani H, Stelzer E, Bastiaens P, Lygerou Z. Dynamic recruitment of licensing factor Cdt1 to sites of DNA damage. J Cell Sci 2011; 124:422-34; PMID:21224399; http://dx.doi.org/10.1242/jcs.074229
- Raman M, Havens CG, Walter JC, Harper JW. A genome-wide screen identifies p97 as an essential regulator of DNA damage-dependent CDT1 destruction. Mol Cell 2011; 44:72-84; PMID:21981919; http://dx.doi.org/10.1016/j.molcel.2011.06.036
- Shiomi Y, Hayashi A, Ishii T, Shinmyozu K, Nakayama J, Sugasawa K, Nishitani H. Two different replication factor C proteins, Ctf18 and RFC1, separately control PCNA-CRL4Cdt2-mediated Cdt1 proteolysis during S phase and following UV irradiation. Mol Cell Biol 2012; 32:2279-88; PMID:22493068; http://dx.doi.org/10.1128/MCB.06506-11
- Friedberg EC, Aguilera A, Gellert M, Hanawalt PC, Hays JB, Lehmann AR, Lindahl T, Lowndes N, Sarasin A, Wood RD. DNA repair: From molecular mechanism to human disease. DNA Repair 2006; 5:986-96; PMID:16955546; http://dx.doi.org/10.1016/j.dnarep.2006.05.005
- Gillet LC, Scharer OD. Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem Rev 2006; 106:253-76; PMID:16464005; http://dx.doi.org/10.1021/cr040483f
- Sugasawa K. Multiple DNA damage recognition factors involved in mammalian nucleotide excision repair. Biochemistry (Mosc) 2011; 76:16-23; PMID:21568836; http://dx.doi.org/10.1134/S0006297911010044
- Scrima A, Konickova R, Czyzewski BK, Kawasaki Y, Jeffrey PD, Groisman R, Nakatani Y, Iwai S, Pavletich NP, Thoma NH. Structural basis of UV DNA-damage recognition by the DDB1-DDB2 complex. Cell 2008; 135:1213-23; PMID:19109893; http://dx.doi.org/10.1016/j.cell.2008.10.045
- Sugasawa K, Okuda Y, Saijo M, Nishi R, Matsuda N, Chu G, Mori T, Iwai S, Tanaka K, Hanaoka F. UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell 2005; 121:387-400; PMID:15882621; http://dx.doi.org/10.1016/j.cell.2005.02.035
- Aboussekhra A, Biggerstaff M, Shivji MK, Vilpo JA, Moncollin V, Podust VN, Protic M, Hubscher U, Egly JM, Wood RD. Mammalian DNA nucleotide excision repair reconstituted with purified protein components. Cell 1995; 80:859-68; PMID:7697716; http://dx.doi.org/10.1016/0092-8674(95)90289-9
- Shivji MK, Podust VN, Hubscher U, Wood RD. Nucleotide excision repair DNA synthesis by DNA polymerase epsilon in the presence of PCNA, RFC, and RPA. Biochemistry 1995; 34:5011-7; PMID:7711023; http://dx.doi.org/10.1021/bi00015a012
- Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem 2005; 74:681-710; PMID:15952900; http://dx.doi.org/10.1146/annurev.biochem.74.082803.133243
- Iyer RR, Pluciennik A, Burdett V, Modrich PL. DNA mismatch repair: Functions and mechanisms. Chem Rev 2006; 106:302-23; PMID:16464007; http://dx.doi.org/10.1021/cr0404794
- Zlatanou A, Despras E, Braz-Petta T, Boubakour-Azzouz I, Pouvelle C, Stewart GS, Nakajima S, Yasui A, Ishchenko AA, Kannouche PL. The hMsh2-hMsh6 complex acts in concert with monoubiquitinated PCNA and Pol eta in response to oxidative DNA damage in human cells. Mol Cell 2011; 43:649-62; PMID:21855803; http://dx.doi.org/10.1016/j.molcel.2011.06.023
- Pena-Diaz J, Bregenhorn S, Ghodgaonkar M, Follonier C, Artola-Boran M, Castor D, Lopes M, Sartori AA, Jiricny J. Noncanonical mismatch repair as a source of genomic instability in human cells. Mol Cell 2012; 47:669-80; PMID:22864113; http://dx.doi.org/10.1016/j.molcel.2012.07.006
- Ogi T, Limsirichaikul S, Overmeer RM, Volker M, Takenaka K, Cloney R, Nakazawa Y, Niimi A, Miki Y, Jaspers NG, et al. Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Mol Cell 2010; 37:714-27; PMID:20227374; http://dx.doi.org/10.1016/j.molcel.2010.02.009
- Soria G, Belluscio L, van Cappellen WA, Kanaar R, Essers J, Gottifredi V. DNA damage induced Pol eta recruitment takes place independently of the cell cycle phase. Cell Cycle (Georgetown, Tex) 2009; 8:3340-8; PMID:19806028; http://dx.doi.org/10.4161/cc.8.20.9836
- Tsanov N, Kermi C, Coulombe P, Van der Laan S, Hodroj D, Maiorano D. PIP degron proteins, substrates of CRL4Cdt2, and not PIP boxes, interfere with DNA polymerase eta and kappa focus formation on UV damage. Nucleic Acids Res 2014; 42:3692-706; PMID:24423875; http://dx.doi.org/10.1093/nar/gkt1400
- Bacquin A, Pouvelle C, Siaud N, Perderiset M, Salome-Desnoulez S, Tellier-Lebegue C, Lopez B, Charbonnier JB, Kannouche PL. The helicase FBH1 is tightly regulated by PCNA via CRL4(Cdt2)-mediated proteolysis in human cells. Nucleic Acids Res 2013; 41:6501-13; PMID:23677613; http://dx.doi.org/10.1093/nar/gkt397
- Morino M, Tanaka M, Shiomi Y, Nishitani H. Imaging analysis to determine chromatin binding of the licensing factor MCM2-7 in mammalian cells. Methods Mol Biol 2014; 1170:529-37; PMID:24906334; http://dx.doi.org/10.1007/978-1-4939-0888-2_29
- Almeida KH, Sobol RW. A unified view of base excision repair: Lesion-dependent protein complexes regulated by post-translational modification. DNA Repair 2007; 6:695-711; PMID:17337257; http://dx.doi.org/10.1016/j.dnarep.2007.01.009
- Hong Z, Jiang J, Hashiguchi K, Hoshi M, Lan L, Yasui A. Recruitment of mismatch repair proteins to the site of DNA damage in human cells. J Cell Sci 2008; 121:3146-54; PMID:18765568; http://dx.doi.org/10.1242/jcs.026393
- Barone F, McCulloch SD, Macpherson P, Maga G, Yamada M, Nohmi T, Minoprio A, Mazzei F, Kunkel TA, Karran P, et al. Replication of 2-hydroxyadenine-containing DNA and recognition by human MutSalpha. DNA Repair 2007; 6:355-66; PMID:17188944; http://dx.doi.org/10.1016/j.dnarep.2006.11.002
- Duckett DR, Drummond JT, Murchie AI, Reardon JT, Sancar A, Lilley DM, Modrich P. Human MutSalpha recognizes damaged DNA base pairs containing O6-methylguanine, O4-methylthymine, or the cisplatin-d(GpG) adduct. Proc Natl Acad Sci USA 1996; 93:6443-7; PMID:8692834; http://dx.doi.org/10.1073/pnas.93.13.6443
- Mazurek A, Berardini M, Fishel R. Activation of human MutS homologs by 8-oxo-guanine DNA damage. J Biol Chem 2002; 277:8260-6; PMID:11756455; http://dx.doi.org/10.1074/jbc.M111269200
- Jascur T, Fotedar R, Greene S, Hotchkiss E, Boland CR. N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) triggers MSH2 and Cdt2 protein-dependent degradation of the cell cycle and mismatch repair (MMR) inhibitor protein p21Waf1/Cip1. J Biol Chem 2011; 286:29531-9; PMID:21725088; http://dx.doi.org/10.1074/jbc.M111.221341
- Kielbassa C, Roza L, Epe B. Wavelength dependence of oxidative DNA damage induced by UV and visible light. Carcinogenesis 1997; 18:811-6; PMID:9111219; http://dx.doi.org/10.1093/carcin/18.4.811
- Mu D, Tursun M, Duckett DR, Drummond JT, Modrich P, Sancar A. Recognition and repair of compound DNA lesions (base damage and mismatch) by human mismatch repair and excision repair systems. Mol Cell Biol 1997; 17:760-9; PMID:9001230; http://dx.doi.org/10.1128/MCB.17.2.760
- Hoffman PD, Wang H, Lawrence CW, Iwai S, Hanaoka F, Hays JB. Binding of MutS mismatch repair protein to DNA containing UV photoproducts, “mismatched” opposite Watson-Crick and novel nucleotides, in different DNA sequence contexts. DNA Repair 2005; 4:983-93; PMID:15996534; http://dx.doi.org/10.1016/j.dnarep.2005.04.018
- Morino M, Nukina K, Sakaguchi H, Maeda T, Takahara M, Shiomi Y, Nishitani H. Mitotic UV irradiation induces a DNA replication-licensing defect that potentiates G1 arrest response. PLoS One 2015; 10:e0120553; PMID:25798850; http://dx.doi.org/10.1371/journal.pone.0120553
- Bendjennat M, Boulaire J, Jascur T, Brickner H, Barbier V, Sarasin A, Fotedar A, Fotedar R. UV irradiation triggers ubiquitin-dependent degradation of p21(WAF1) to promote DNA repair. Cell 2003; 114:599-610; PMID:13678583; http://dx.doi.org/10.1016/j.cell.2003.08.001
- Mansilla SF, Soria G, Vallerga MB, Habif M, Martinez-Lopez W, Prives C, Gottifredi V. UV-triggered p21 degradation facilitates damaged-DNA replication and preserves genomic stability. Nucleic Acids Res 2013; 41:6942-51; PMID:23723248; http://dx.doi.org/10.1093/nar/gkt475
- Ichikawa M, Nakane H, Marra G, Corti C, Jiricny J, Fitch M, Ford JM, Ikejima M, Shimada T, Yoshino M, et al. Decreased UV sensitivity, mismatch repair activity and abnormal cell cycle checkpoints in skin cancer cell lines derived from UVB-irradiated XPA-deficient mice. Mutat Res 2000; 459:285-98; PMID:10844242; http://dx.doi.org/10.1016/S0921-8777(00)00005-7
- Yoshino M, Nakatsu Y, te Riele H, Hirota S, Kitamura Y, Tanaka K. Additive roles of XPA and MSH2 genes in UVB-induced skin tumorigenesis in mice. DNA Repair 2002; 1:935-40; PMID:12531021; http://dx.doi.org/10.1016/S1568-7864(02)00144-1
- Nishitani H, Taraviras S, Lygerou Z, Nishimoto T. The human licensing factor for DNA replication Cdt1 accumulates in G1 and is destabilized after initiation of S-phase. J Biol Chem 2001; 276:44905-11; PMID:11555648; http://dx.doi.org/10.1074/jbc.M105406200