
ABSTRACT
Recently, we demonstrated that sterile α motif and HD domain containing protein 1 (SAMHD1) is a major barrier in acute myelogenous leukemia (AML) cells to the cytotoxicity of cytarabine (ara-C), the most important drug in AML treatment. Ara-C is intracellularly converted by the canonical dNTP synthesis pathway to ara-CTP, which serves as a substrate but not an allosteric activator of SAMHD1. Using an AML mouse model, we show here that wild type but not catalytically inactive SAMHD1 reduces ara-C treatment efficacy in vivo. Expanding the clinically relevant substrates of SAMHD1, we demonstrate that THP-1 CRISPR/Cas9 cells lacking a functional SAMHD1 gene showed increased sensitivity to the antimetabolites nelarabine, fludarabine, decitabine, vidarabine, clofarabine, and trifluridine. Within this Extra View, we discuss and build upon both these and our previously reported findings, and propose SAMHD1 is likely active against a variety of nucleoside analog antimetabolites present in anti-cancer chemotherapies. Thus, SAMHD1 may constitute a promising target to improve a wide range of therapies for both hematological and non-haematological malignancies.
Introduction
Sterile α motif and histidine/aspartic acid domain containing protein 1 (SAMHD1) was identified in 2011 to restrict human immunodeficiency virus type 1 (HIV-1) in myeloid cells, a breakthrough in unravelling the, up to then, enigmatic reasons underlying the low permissivity of macrophages or dendritic cells to HIV-1 infection.Citation1,2 Soon after this discovery, the restriction mechanism of SAMHD1 was suggested to involve its enzymatic triphosphohydrolase activity, which was proposed to deplete intracellular deoxynucleoside triphosphate (dNTP) substrates for HIV-1 reverse transcription.Citation3,4 Subsequent studies showed that this activity of SAMHD1 is subjected to allosteric regulation by nucleotides, with the first allosteric site (A1) requiring a guanine nucleotide (GTP or dGTP) and the second allosteric site (A2) requiring any dNTP, which together allow formation of the catalytically active SAMHD1 tetramer (). Owing to its activity toward all canonical dNTPs and the elegant allosteric regulatory mechanism, SAMHD1 has been proposed to oppose ribonucleotide reductase (RNR) as a major regulator of DNA precursor pools, and a cell-cycle dependent differential activation of RNR (S-phase) and SAMHD1 (G1/0-phase) has been suggested.Citation5 Underscoring a fundamental biologic role for SAMHD1, germ-line mutations in the gene encoding SAMHD1 are associated with human diseases, such as the neurodegenerative and hyperinflammatory Aicardi–Goutières syndrome (AGS).Citation6 In addition, somatic aberrations of SAMHD1 have been found in chronic lymphocytic leukemia,Citation7 lung cancer,Citation8 and colorectal cancer.Citation9 More recently, we and others have identified SAMHD1 as an obstacle toward antimetabolite-based cancer therapies,Citation10-12 which will be the focus of this Extra View.
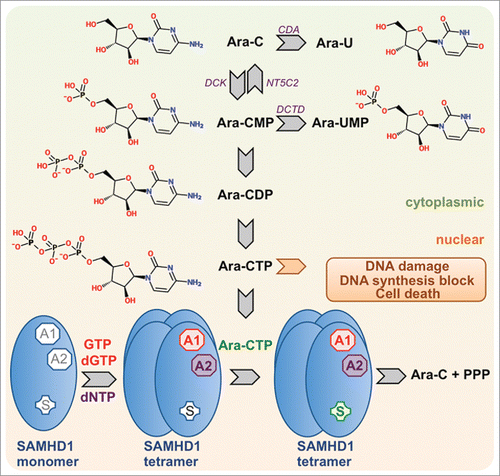
Figure 1. Intracellular conversion of cytarabine (ara-C) to ara-CTP and detoxification by SAMHD1. The schematic depicts canonical pathways for the intracellular synthesis of ara-CTP, the active metabolite of ara-C, which exerts DNA-damage and antiproliferative downstream effects by interfering with DNA synthesis. SAMHD1 is activated by binding of GTP or dGTP to allosteric site 1 (AS1), binding of a dNTP to allosteric site 2 (AS2) and binding of its substrate in the catalytic site. Ara-CTP is a substrate for SAMHD1 but not an allosteric activator. Abbreviations: CDA, cytidine deaminase; DCK, deoxyctidine kinase; NT5C2, cytosolic nucleotidase-II; DCTD, deoxycytidylate deaminase.
SAMHD1 is a mediator of ara-C toxicity in AML cell models
The global annual incidence of leukemia is about 350,000 cases, predominantly consisting of acute myelogenous leukemia (AML).Citation13 With the exception of pediatric patients,Citation14 only about one fifth of AML patients survive a 5-year period after diagnosis, and survival rates decrease substantially with the age at diagnosis.Citation15 Standard treatment of AML involves combination therapy of an anthracycline (most commonly doxorubicin or daunorubicin) and cytarabine (ara-C).Citation15 The latter is a nucleoside analog that was first used for AML-induction therapy in 1968Citation16 and was later shown to also be very effective in post-remission therapy.Citation17 However, most of patients will eventually succumb to resistant diseases and relapses,Citation15-17 and while it has been accepted that a worse clinical outcome directly correlates with a decrease in intracellular levels of the active metabolite ara-CTP,Citation18-26 the underlying molecular reasons for this remained enigmatic. Shedding light on this, we and others have recently shown that SAMHD1 is a major determinant in dictating sensitivity of AML blasts to ara-C treatment, as the cell-active triphosphate metabolite of ara-C, ara-CTP, is a SAMHD1 substrate.Citation10-12 In particular, RNA interference (RNAi)-mediated SAMHD1 knockdown, CRISPR/Cas9-induced SAMHD1 knockout or SAMHD1 degradation with Vpx-containing virus-like particles (VLPs) from simian immunodeficiency virus (SIV) increased sensitivity of AML cells to ara-C cytotoxicity.Citation10-12 Confirming the importance of the catalytic activity of SAMHD1, overexpression of wild type SAMHD1, but not allosteric site mutant D137N or the catalytic site mutants D311A or H233A, significantly reduced ara-C cytotoxicity.Citation10,11 Interestingly, despite a proposed role of SAMHD1 phosphorylation in the restriction of retroviruses,Citation27 we did not find any evidence for a role of the SAMHD1 phosho-site T592 in ara-CTP turn over,Citation11 which is consistent with experimental evidence that dNTPase activity might be dispensable for HIV-1 restriction.Citation28 SAMHD1 phosphorylation ablates tetramer-formation as well as HIV-1 restriction, however the dNTPase activity of phospho-SAMHD1 is only affected in conditions of low nucleotide levels.Citation29 Our study also investigated the mechanism underlying ara-C cytotoxicity and showed that activation of the intra-S-phase and DNA-damage response pathways are substantially elevated in ara-C treated leukemic cells lacking SAMHD111, consistent with the established mechanism of action of ara-C.Citation30-33 While Schneider et al. specifically looked at AML tumor cell lines, our study provides evidence that other haematological malignancies may involve SAMHD1 as a barrier to treatment efficacy and could possibly be antagonised to improve therapy.Citation11 Similar to monocytic THP-1 cells, the cutaneous T-cell lymphoma line Hut-78, derived from a patient with Sézary syndrome,Citation34 was also sensitized to ara-C treatment when SAMHD1 was depleted, and reconstitution of dNTPase-proficient SAMHD1 reduced ara-C cytotoxicity.Citation11 This is also in support of a negative correlation of SAMHD1 mRNA expression and ara-C cytotoxicity in a panel of cell lines containing both myeloid and lymphoid neoplasms;Citation11 hence SAMHD1s role as a modifier of ara-C toxicity is not restricted to myeloid neoplasms.
Mouse models confirm in vivo role of SAMHD1 dNTPase activity in reducing ara-C treatment efficacy
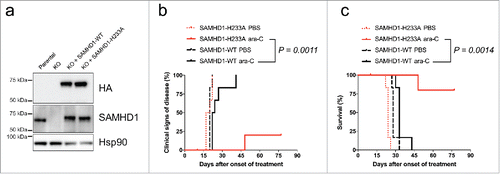
To address whether human AML tumor cells with differential SAMHD1 expression would respond differently to ara-C treatment, we used both a heterotopic as well as an orthotopic AML mouse model. Firstly, nude mice were subcutaneously transplanted with CRISPR/Cas9 THP-1 cell clones expressing SAMHD1 or not.Citation11 Secondly, we injected CRISPR/Cas9 HL-60/iva cell clones containing or lacking a functional SAMHD1 gene, respectively, into the tail-vein of NOD/SCID mice.Citation11 Lack of SAMHD1 expression dramatically increased the sensitivity of AML xenotransplants to ara-C induced toxicity, resulting in pronounced survival improvements.Citation11 As mentioned above, it has been reported that restriction of retroviral infection by SAMHD1 can be uncoupled from its dNTPase activity,Citation28 and thus we wanted to confirm that modulation of ara-C efficacy in vivo is dependent on the enzymatic activity of SAMHD1 and not mediated by other functions of SAMHD1. To perform in vivo structure-function analyses, we reconstituted SAMHD1 expression by lentiviral transduction and ectopically expressed either wild type or the catalytically inactive H233A mutant of SAMHD1 in HL-60/iva CRISPR/Cas9 SAMHD1−/− cells. We confirmed equal expression of the SAMHD1 variants by immunoblotting () and then xenotransplanted NOD/SCID mice i.v. Subsequently, these mice were treated with 50 mg•kg−1 ara-C for 5 consecutive days from day 6 post xenotransplantation, and signs of disease of these mice were monitored by veterinarian examination as described previously.Citation11 Mice transplanted with cells expressing wild type SAMHD1 were substantially more resistant to ara-C treatment and developed signs of disease after a median time of 23 days, while 5 out of 6 mice in which cells were engrafted expressing the H233A mutant of SAMHD1 were still without signs of disease at the time of sacrifice ( and ). This demonstrates that in vivo detoxification of ara-C requires catalytically competent SAMHD1.
Figure 2. Overexpression of wild type but not catalytic-inactive SAMHD1 confers resistance to ara-C treatment in vivo. HL-60/iva CRISPR/Cas9 cells lacking endogenous SAMHD1 expression were transduced with a lentiviral vector encoding for HA-tagged wild type (black) or the catalytically-inactive H233A mutant (red) SAMHD1. Equal expression levels of ectopic SAMHD1 were confirmed by western blotting (a). Cells were xenotransplanted into NOD/SCID IL2R−/− female mice; (n = 12 for each cell line), which were subsequently treated with either PBS or ara-C. Clinical signs of disease (b) and percentage of survival (c) were determined over time. For details see Methods.
SAMHD1 controls the therapeutic response of AML to ara-C
Targeting SAMHD1 with RNAi or Vpx-VLP treatment in patient-derived AML blasts sensitized those to ara-C-induced toxicity, although there was some donor-to-donor variability in the magnitude of sensitization.Citation10,11 A retrospective analysis of the adult AML cohort from The Cancer Genome Atlas (TCGA),Citation10,11 as well as the Therapeutically Applicable Research To Generate Effective Treatments (TARGET) cohort of children with AML,Citation11 demonstrated that patients with lower SAMHD1 expression levels in AML blasts had better clinical outcome. Interestingly, our analyses did not show a significant difference in SAMHD1 expression in patients that achieved complete remission as compared with patients that did not.Citation11 The survival advantage only became apparent later, i.e. during consolidation courses. This seemingly is a contradiction to the data presented by Schneider et al. that based their conclusions for the clinical significance of SAMHD1 upon immunohisto- and immunocytochemistry. As the therapy regimens did not differ substantially between the patient cohorts analyzed in both studies, this discrepancy might be explained by methodological differences. We assessed mRNA expression of SAMHD1 as a continuous measure, whereas Schneider et al. scored the staining intensity by eye categorically. It can be reasoned that protein levels do not necessarily correlate with mRNA abundance, while another possible confounder may be staining of patient-AML specific non-SAMHD1 epitopes due to the polyclonal nature of the antibody used by Schneider et al., however there is no reason to assume that unspecific staining would be more prevalent in AML patients that fail induction therapy. Ultimately, only the analysis of an independent patient cohort by immunohistochemistry, preferably using a more specific antibody, may resolve this apparent contradiction. This is not merely an academic discussion, as this will have large implications when considering SAMHD1 as a possible biomarker for treatment stratification. If the mRNA levels analyzed in 2 independent patient cohorts in our study are representative, dose modification (i.e., dose reduction for low SAMHD1 expressers and/or dose escalation for high SAMHD1 expressers) would only be justified in the post-remission phase. We feel that this is in line with the notion that anthracyclines contribute most to remission induction,Citation35 and in line with the study by Mayer et al., which showed improved survival in AML with escalating doses of ara-C during the post-remission phase.Citation17 In any case, reliable cut-off values of SAMHD1 expression applicable for dose-adjustments would have to be determined by prospective studies specifically designed for this purpose and cannot simply be inferred from retrospective analyses. Furthermore, the therapeutic phase with most negative impact of SAMHD1 will also inform the study design of implementing putative SAMHD1 inhibitors into AML treatment protocols. In our manuscript, we suggested to explore Vpx as a biologic SAMHD1 inhibitor.Citation11 As this protein ultimately has to be delivered to the cytoplasm/nucleoplasm, we made use of non-replicating VLPs as vehicle, whose pseudotyping with VSV-G guarantees delivery to the cytoplasmic compartment.Citation36 In fact, Vpx-VLPs have been shown to increase transduction efficiency as a direct effect of Vpx,Citation37,38 and Vpx has been suggested to improve the efficacy of lentiviral gene therapy.Citation39,40 Lentiviral vectors are being tested in clinical trials, mainly for ex vivo transduction of hematopoetic stem cells to correct hemoglobinopathies and primary immunodeficiencies,Citation41,42 for instance. To treat the systemic disease AML, in vivo delivery of Vpx would be required. Pre-clinical safety and toxicity data are available for lentiviral vectors, but studies for in vivo transduction to correct non-haematological genetic disorders are only in preparation.Citation43 One major obstacle for Vpx therapy in AML is the efficiency of bone marrow targeting, however in vivo mouse experiments show that this barrier can be overcome.Citation44,45 Alternative strategies to deliver Vpx to leukemic blasts are mRNA-based therapeutics,Citation46 the use of small DNA viruses,Citation47 liposomal packaging of Vpx proteinCitation48,49 or coupling of Vpx to antibodies targeting AML cells.Citation50
SAMHD1 potentially bears some tumor suppressor functionality,Citation7-9,51-53 and thus transient inhibition of SAMHD1 only for the time of co-administration of ara-C or other antimetabolites would be preferable from a toxicity and safety perspective. Hence, viral transduction strategies with potential off-target effectsCitation54 might conceptually be the least favorable approach, and small molecule inhibitors would be ideal for controllable, safe, transient and cost-effective SAMHD1 inhibition. As germline mutations of SAMHD1 are causing autoimmune-like symptoms in AGS patients,Citation6 it is also important to evaluate inflammatory reactions in putative SAMHD1 inhibitor trials. If existent, these might even enhance anti-tumor effects e.g. due to interferon secretion. Although to date no cell-active SAMHD1 inhibitors have been reported, methodologies have been developed that can be used to screen small molecule libraries in vitro.Citation55,56 From these and other studies, several nucleoside triphosphate-based small molecules have been reported to successfully inhibit SAMHD1 in vitro,Citation12,55-57 and although these compounds currently lack potency, they provide starting points for rational design of SAMHD1 inhibitors.
Following the line of discussion in our recent manuscript regarding targeting SAMHD1 to improve therapy,Citation11 one could speculate that AML patients with high SAMHD1 levels in leukemic blasts would eventually have a better prognosis than low SAMHD1 expressers due to the tumor suppressive activity of SAMHD1 in putative post-treatment residual AML cells. In any case, further studies on the role of SAMHD1 in maintaining genome stability and cell homeostasis are needed. In addition to general dNTP pool homeostasis, potential further explanations for tumor suppressor functions of SAMHD1 could be sanitation of endogenously occurring or stress-induced modified nucleotides, like dUTPCitation58 or oxidised dNTPs,Citation59-61 or perhaps the potential role of SAMHD1 in the DNA damage response,Citation7 where it could be involved in fine-tuning dNTP requirements for repair synthesis.
SAMHD1 neutralizes the cytotoxic effects of diverse nucleoside-based antimetabolites
SAMHD1 is a promiscuous dNTPase that is regulated allosterically by dNTPs, and thus it is likely that several dNTP analogs, whether endogenously occurring or as part of antimetabolite-based therapies, will be SAMHD1 substrates and/or activators. Among the first modified nucleotides to be tested as substrates were several nucleoside reverse transcriptase inhibitors (NRTIs), and overall these were restrictive to SAMHD1-induced hydrolysis. Interestingly, SAMHD1 was reported to increase activity of these nucleoside analogs in cells by depleting the competing endogenous nucleoside triphosphates.Citation59,62 Despite lack of activity toward NRTIs, several base-modified nucleotides were reported as SAMHD1 substrates, such as O6-methyl-dGTP and 5-methyl-2′-dCTP,Citation59 however the biological relevance of this has yet to be examined. The development of a continuous enzyme-coupled assay for measuring the triphosphohydrolase activity of SAMHD1 enabled the investigation of potential nucleotide substrates, activators and inhibitors.Citation55 It was in this report that activity of SAMHD1 toward the nucleoside triphosphate metabolite of clofarabine, an anti-cancer drug used to treat hematological malignancies, was demonstrated.Citation55 Clofarabine-triphosphate was also suggested to be an allosteric activator.Citation55 This was confirmed in our study, and we also demonstrated that SAMHD1 modulates clofarabine-induced toxicity in THP-1 and Hut-78 cells,Citation11 however to a lesser extent than ara-C (see and for comparison). Supportive of a wider role of SAMHD1 in controlling clofarabine cytotoxicity, a significant negative correlation was observed between SAMHD1 expression and clofarabine-induced cytotoxicity in a panel of 133 haematological and lymphoid tissue-derived cell lines ().
Figure 3. Diverse anti-neoplastic nucleoside analogs are more toxic in the absence of SAMHD1. THP-1 CRISPR/Cas9 control cells (black) or cells lacking a functional SAMHD1 gene (red) were treated in parallel with the indicated concentrations of cytarabine (a), vidarabine (b), nelarabine (c), fludarabine (d), clofarabine (e), decitabine (f) or trifluridine (g). Cells were treated for 3 days, or 6 d in the case of decitabine to obtain maximal cytotoxicity as described previously,Citation87 and cell viability was determined using a colorimetric proliferation inhibition assay. Representative experiments from a total of at least 2 independent experiments performed in triplicate are shown. EC50 values (for ctrl SAMHD1+/+ vs. g2–2 SAMHD1−/−) were calculated using a non-linear regression curve fit (for details see Methods): a: 53.6 µM vs. 0.4 µM, b: 657.6 µM vs. 87.8 µM; c: 2114 µM vs. 65.2 µM, d: 8.3 µM vs. 1.5 µM; e: 135.8 nM vs. 57.7 nM; f: 521 µM vs. 0.5 µM; g: 20.7 µM vs. 3.0 µM. Curves were compared by means of Extra-sum-of-squares F tests (**: P ≤ 0.01; ****: P ≤ 0.0001).
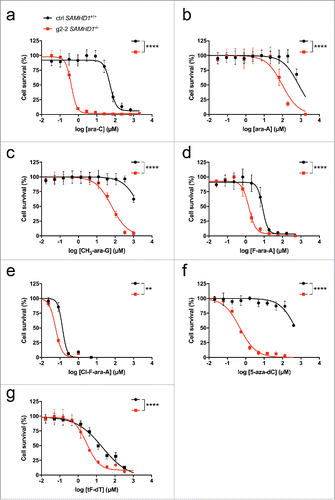
Figure 4. Correlation of SAMHD1 mRNA expression and sensitivity of cells to specific nucleoside analogs. Pearson correlations of SAMHD1 mRNA expression with clofarabine (a), nelarabine (b) or decitabine (c) sensitivity are shown in a panel of haematopoietic and lymphoid tissue-derived cell lines. mRNA expression data was obtained from the Cancer Cell Line Encyclopaedia (http://www.broadinstitute.org/ccle)Citation88 and area under curve (AUC) measurements from the Cancer Therapeutic Response Portal (http://www.broadinstitute.org/ctrp).Citation89,90 Pearson correlations were calculated using Prism 6 (GraphPad Software), number of XY pairs: clofarabine = 133, nelarabine = 117, decitabine = 133.
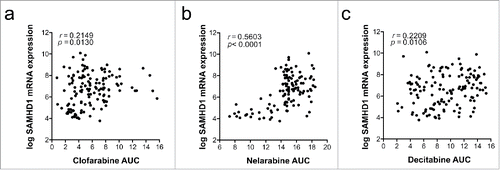
A recent study used in silico predictions to identify additional substrates of SAMHD1, and verified several of these predictions using an in vitro HPLC-based assay.Citation12 To determine whether SAMHD1 modulates cytotoxicity of these and other compounds, we treated parental THP-1 or THP-1 CRISPR/Cas9 SAMHD1 knockout cells with a panel of these nucleoside analogs and measured cytotoxicity as described previously.Citation11 As anticipated, THP-1 cells lacking SAMHD1 were 2 orders of magnitude more sensitive to cytarabine treatment (). Similarly, SAMHD1−/− cells displayed increased sensitivity to vidarabine (), nelarabine (), fludarabine (), decitabine () and trifluridine (), indicating that triphosphate variants of these drugs could be substrates for SAMHD1. Although vidarabine is mainly used as an antiviral, it has been suggested as an antineoplastic agent,Citation63 and trifluridine is another example of an antiviral drug that recently has been repurposed for the use in cancer treatment.Citation64-66 In addition to a possible role for future cancer therapies, this may become interesting when considering SAMHD1 as a possible intrinsic resistance gene toward treatment of herpes viral infections.Citation67 In further support of a key role for SAMHD1 in controlling cytotoxicity of nelarabine and decitabine, significant negative correlations of SAMHD1 mRNA expression and toxicity of these drugs were observed in a panel of myeloid and lymphoid cells lines ( and ). We also reported that gemcitabine triphosphate and 6-thioguanine triphosphate are not substrates of SAMHD1, and accordingly, the absence of SAMHD1 did not affect toxicity of these drugs.Citation11 The data presented here, together with the recent reports,Citation10,12,55 suggests that SAMHD1 is a key player in reducing the efficacy of antimetabolite-based cancer therapies; further investigations will determine whether this is the case and to which extent this is relevant in clinical settings. The complex allosteric regulation of SAMHD1 in combination with the enzymatic activity creates a scenario in which the net effect of a SAMHD1-interacting nucleoside toward SAMHD1 activity is composed to varying degrees of (i) being a substrate and/or competitive inhibitor at the catalytic site as well as (ii) an allosteric activator or inhibitor at either one or both of the allosteric sites.Citation55 Eventually, a very simple strategy for choosing the right antimetabolite for a SAMHD1-positive cancer might be to select one that is not a SAMHD1-substrate.
Future perspectives–SAMHD1 and beyond
Our study suggests that targeting SAMHD1 bears the potential to improve outcome in AML therapy when combined with high-dose ara-C treatment.Citation11 As shown by Hollenbaugh et al. and the present work, SAMHD1 may also have activity toward triphosphate metabolites of other drugs used against AML (in particular fludarabine and clofarabine) and drugs used against other types of cancers like nelarabine for T-lymphoblastic lymphoma and leukemia, decitabine for myelodysplastic syndrome (MDS), and trifluridine that in combination with the thymidine phosphorylase (TP) inhibitor tipiracil, is used against metastasised colorectal cancer and has been approved by the FDA and EMA.Citation68 Tipiracil is a prime example of how the understanding of both anabolic and catabolic antimetabolite metabolism is driving the development of combination therapies to improve the efficacy of anti-cancer drugs. TP is a glycosyltransferase that removes the deoxyribose monophosphate from thymidine leaving thymine as second reaction product.Citation69 It has been recognized that trifluridine monophosphate (tF-dTMP) is also a substrate for TP, counteracting the intracellular toxification of trifluridine.Citation70 Catabolic enzymes that reduce the effective concentration of antimetabolites have, however, been recognized long before. The deaminsases cytosine deaminase (CDA) and deoxycytidylate deaminase (DCTD) convert dC or dCMP, respectively, and its analogs ara-C or ara-CMP as well as 5-aza-dC or 5-aza-dCMP,Citation71-73 to their uracil derivatives that have no or much lower antineoplastic activityCitation74 (). Indeed, CDA inhibitors like tetrahydrouridine have been developed to increase both plasma (inhibiting plasma and liver CDA) and intracellular concentrations of ara-C.Citation72,75,76 Another example of augmenting the efficacy of antimetabolites is the use of small molecule inhibitors targeting dUTPase, which hydrolyses dUTP and 5-FdUTP, an active metabolite of 5-FU, to their monophosphate forms, thereby limiting their toxic incorporation into nascent DNA.Citation58,77,78 Hence, inhibitors of dUTPase can increase the efficacy of 5-FU.Citation79,80 A more recent target to improve cytotoxic therapies with thiopurines has emerged by discovering that the nudix enzyme family member NUDT15 can hydrolyse triphosphates of 6-thioguanine and mercaptopurine.Citation81,82 Hence, also in this case, combining these drugs with a NUDT15 inhibitor might improve therapy outcomes. It should, however, be added that these drugs are particularly suited for maintenance therapy strategies over longer periods of time. Hence, a careful pre-selection of patients that have a selective overexpression of NUDT15 in tumor cells should be aimed at when studying NUDT15 inhibitors clinically to omit drastic increases in toxicity – that even today are reasons to pause or cancel a treatment with thiopurines.Citation83
Based on substitutions of the stereoselective 2′ sugar moiety, Hollenbaugh et al. predicted nelarabine triphosphate, fludarabine triphosphate, BV-ara-UTP, floxuridine triphosphate, trifluridine triphosphate, sorivudine triphosphate and cladribine triphosphate as SAMHD1 substrates. It will be of interest to simulate interactions of other nucleotide drugs with the SAMHD1 catalytic site, possibly in an effort for rational design of competitive SAMHD1 inhibitors.
Several SAMHD1 inactivating single nucleotide polymorphisms (SNPs) have been described in AGS, some of them located close to the active site.Citation6,84 Certain SAMHD1 SNPs were also reported for chronic lymphoblastic leukemia.Citation7 In addition, a recent study associated SAMHD1 SNPs with colon cancer.Citation9 While some of these SNPs have been shown to affect dNTPase function, possibly even in a dNTP-specific manner, there are currently hundreds of SNPs published in public databases for which the effects on SAMHD1 dNTPase activity and stability are unknown. Future studies will have to investigate their role in cancer, their antagonisation of antimetabolite-based chemotherapies and their potential to resist small molecule inhibitors of SAMHD1. Considering compounds that bind to the active site in a competitive manner, development of resistance to these inhibitors while retaining full ara-CTPase activity seems rather unlikely, but not impossible. Allosteric inhibitors, however, are conceptually more prone for resistance development as evidenced by allosteric inhibitors of HIV-1 reverse transcriptase.Citation85
The fact that trifluridine triphosphate is a potential substrate of SAMHD1 could have important implications for SAMHD1-directed therapies that might be relevant for non-hematological solid tumors.Citation86 It is important to gain more insight into why certain nucleoside analogs such as 5-FU, gemcitabine, and trifluridine are clinically most useful against solid tumors, whereas other analogs like ara-C, clofarabine, nelarabine, fludarabine and decitabine are almost exclusively reserved for myeloid and lymphoid neoplasms. Our preliminary analyses indicate that this discrepancy cannot be solely explained by differential SAMHD1 expression (data not shown). Whether general differences in tumor biology, e.g., differences in proliferation rates, or tumor-type specific transporters, metabolic enzymes or differentially activated damage-response pathways constitute the main reason for a relative resistance of solid tumors to various antimetabolites, remains to be investigated. We are hopeful that these kinds of studies will ultimately increase the arsenal of drugs that can be used to more efficiently target solid tumors.
Concluding remarks
In conclusion, our study along with the reports by Schneider et al. and Hollenbaugh et al. demonstrate that SAMHD1 is a key barrier to ara-C efficacy during AML consolidation therapy and that SAMHD1 may be a suitable target to enhance chemotherapies against other malignancies.Citation10-12 The search for drugs to target and counteract SAMHD1 to improve chemotherapeutic treatments will hopefully lead to the discovery of compounds that, in combination with existing drugs, would reduce side effects and simultaneously make them more effective. A better understanding of their physiologic metabolism will hopefully pave the way for a less empirical and more rational design of future cancer treatments with antimetabolites. Personalised cytotoxic therapy of the future could select the best combinations from a plethora of antimetabolites in clinical use and tune their efficacy by selecting the right combination of accompanying inhibitors.
Methods
Plasmids and viral vectors
pCSxW encoding hemagglutinin-tagged wild type or catalytically-inactive H233A mutant SAMHD1, HIV-1 GagPol expression vector pCMV-ΔR8.91 and pMD.G (kind gift from D.Trono, School of Life Sciences, Lausanne, Switzerland) were described before.Citation11 To generate guideRNA-resistant versions of these SAMHD1 expression plasmids we PCR-amplified the SAMHD1 cDNAs using forward primer (EcoRI) OTS1478 5′-ATC gaa ttc ATG CAG CGA GCC GAT TCC GAG CAG CCG TCA GCA CGA CCC AGA TGT GAC GAT TCA CCA AGA ACC CCC TCA AAC AC-3′ which contains non-coding nucleotide changes (underlined) and reverse primer (NotI) OTS1393 5′-GCA Tgc ggc cgc TCA CAT TGG GTC ATC TTT AAA AAG C-3′ and cloned the amplicon back into pCSxW-HA using EcoRI/NotI. pLentiCRISPRv2 (Addgene plasmid #52961) encoding SAMHD1 specific gRNAs was described before.Citation11 For viral vector production 293T cells were transfected according to described previously protocols.Citation11
Generation of THP-1 SAMHD1 CRISPR/Cas9 cell lines and antibodies
The construction of THP-1 and HL-60/iva CRISPR/Cas9 single cell clones with disrupted SAMHD1 gene has been described before.Citation11 We reconstituted expression of HA-tagged wild type or H233A mutant SAMHD1 by transduction with pCSxW derived lentiviral vectors encoding guide RNA-resistant SAMHD1 at a multiplicity of infection (MOI) of ∼100. Due to this high MOI there was no need to drug-select for ectopic expression. We used a monoclonal rat anti-HA antibody 3F10 (Roche) directly coupled to horse-reddish peroxidase to detect ectopically expressed SAMHD1, mouse monoclonal anti-SAMHD1 antibody 1F9 (Abcam) or rabbit polyclonal anti-Hsp90 antibody H-114 (Santa Cruz) as loading control.
Mouse experiments
Ectopic SAMHD1 expression was achieved by lentiviral transduction of HL-60/iva CRISPR/Cas9 SAMHD1−/− cells. Ectopically expressed wild type or the catalytically-inactive H233A mutant SAMHD1 contained synonymous nucleotide substitutions to escape the guide RNAs expressed in the cell. Transduced cell bulks were xenotransplanted intravenously into NOD/SCID mice (each n = 6). Mice were treated with 50 mg•kg−1 ara-C for 5 consecutive days from day 6 post xenotransplantation. Signs of disease of these mice were monitored by veterinarian examination with endpoints defined as described previously.Citation11
Cytotoxicity assay
To measure cytotoxicity we used the colorimetric CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) (Promega) as described previously.Citation11 Absorbance was measured using a Glomax Multi+ plate reader (Promega), and EC50 values were calculated using non-linear logistic regression analyses in Prism 6 (GraphPad Software), and statistics were performed by means of Extra-sum-of-squares F tests (**: P≤0.01; ****: P≤0.0001).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank M. Nordenskjöld and S. Eriksson for laboratory assistance, D. Gavhed and K. Edfeldt for administrative assistance, T. Lundbäck and H. Axelsson for discussion. We would like to express our gratitude toward NCI's Office of Cancer Genomics (OCG), The Cancer Genome Atlas (TCGA) and Therapeutically Applicable Research To Generate Effective Treatments (TARGET) initiative for granting public access to their AML databases and the Broad Institute for access to the Cancer Therapeutics Response Portal (CTRP).
Funding
This work was funded by an EMBO Long-Term Fellowship (ALTF-605–2014 to S.G.R.), the German Research Foundation (DFG) (SCHA1950/1–1 to T.S.) and partially through the Bundesministerium für Bildung und Forschung (BMBF) supported Immunoquant project (0316170 C) and HIVERA: EURECA project (01KI1307B), the Swedish Children's Cancer Foundation (PR2016–0044 and TJ2016–0040 to N.H.); PR2013–0002 and PR2014–0048 to T.H.; PR2015–0005 to J.I.H), the Swedish Cancer Society (CAN 2013/396 to J.I.H; CAN 2012/770 and CAN 2015/255 to T.H.,), the Swedish Research Council (2012–5935 and 2013–3791 to T.H.), and the Stockholm County Council (ALF project) (20150016 to J.I.H.).
Related Research Data
References
- Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Segeral E, Yatim A, Emiliani S, Schwartz O, Benkirane M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 2011; 474:654-7; PMID:21613998; https://doi.org/10.1038/nature10117
- Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S, Florens L, Washburn MP, Skowronski J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 2011; 474:658-61; PMID:21720370; https://doi.org/10.1038/nature10195
- Lahouassa H, Daddacha W, Hofmann H, Ayinde D, Logue EC, Dragin L, Bloch N, Maudet C, Bertrand M, Gramberg T, et al. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat Immunol 2013; 14:877; https://doi.org/10.1038/ni0813-877a
- Goldstone DC, Ennis-Adeniran V, Hedden JJ, Groom HC, Rice GI, Christodoulou E, Walker PA, Kelly G, Haire LF, Yap MW, et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 2011; 480:379-82; PMID:22056990; https://doi.org/10.1038/nature10623
- Franzolin E, Pontarin G, Rampazzo C, Miazzi C, Ferraro P, Palumbo E, Reichard P, Bianchi V. The deoxynucleotide triphosphohydrolase SAMHD1 is a major regulator of DNA precursor pools in mammalian cells. Proc Natl Acad Sci U S A 2013; 110:14272-7; PMID:23858451; https://doi.org/10.1073/pnas.1312033110
- Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW, Carr IM, Fuller JC, Jackson RM, Lamb T, Briggs TA, et al. Mutations involved in Aicardi-Goutieres syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet 2009; 41:829-32; PMID:19525956; https://doi.org/10.1038/ng.373
- Clifford R, Louis T, Robbe P, Ackroyd S, Burns A, Timbs AT, Wright Colopy G, Dreau H, Sigaux F, Judde JG, et al. SAMHD1 is mutated recurrently in chronic lymphocytic leukemia and is involved in response to DNA damage. Blood 2014; 123:1021-31; PMID:24335234; https://doi.org/10.1182/blood-2013-04-490847
- Wang JL, Lu FZ, Shen XY, Wu Y, Zhao LT. SAMHD1 is down regulated in lung cancer by methylation and inhibits tumor cell proliferation. Biochem Biophys Res Commun 2014; 455:229-33; PMID:25449277; https://doi.org/10.1016/j.bbrc.2014.10.153
- Rentoft M, Lindell K, Tran P, Chabes AL, Buckland RJ, Watt DL, Marjavaara L, Nilsson AK, Melin B, Trygg J, et al. Heterozygous colon cancer-associated mutations of SAMHD1 have functional significance. Proc Natl Acad Sci U S A 2016; 113:4723-8; PMID:27071091; https://doi.org/10.1073/pnas.1519128113
- Schneider C, Oellerich T, Baldauf HM, Schwarz SM, Thomas D, Flick R, Bohnenberger H, Kaderali L, Stegmann L, Cremer A, et al. SAMHD1 is a biomarker for cytarabine response and a therapeutic target in acute myeloid leukemia. Nat Med 2017; 23(2):250-255
- Herold N, Rudd SG, Ljungblad L, Sanjiv K, Myrberg IH, Paulin CB, Heshmati Y, Hagenkort A, Kutzner J, Page BD, et al. Targeting SAMHD1 with the Vpx protein to improve cytarabine therapy for hematological malignancies. Nat Med 2017; 23:256-63; PMID:28067901; https://doi.org/10.1038/nm.4265
- Hollenbaugh JA, Shelton J, Tao S, Amiralaei S, Liu P, Lu X, Goetze RW, Zhou L, Nettles JH, Schinazi RF, et al. Substrates and Inhibitors of SAMHD1. PLoS One 2017; 12:e0169052; PMID:28046007; https://doi.org/10.1371/journal.pone.0169052
- Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 2015; 136:E359-86; PMID:25220842; https://doi.org/10.1002/ijc.29210
- Taga T, Tomizawa D, Takahashi H, Adachi S. Acute myeloid leukemia in children: Current status and future directions. Pediatr Int 2016; 58:71-80; PMID:26645706; https://doi.org/10.1111/ped.12865
- Ossenkoppele G, Lowenberg B. How I treat the older patient with acute myeloid leukemia. Blood 2015; 125:767-74; PMID:25515963; https://doi.org/10.1182/blood-2014-08-551499
- Ellison RR, Holland JF, Weil M, Jacquillat C, Boiron M, Bernard J, Sawitsky A, Rosner F, Gussoff B, Silver RT, et al. Arabinosyl cytosine: a useful agent in the treatment of acute leukemia in adults. Blood 1968; 32:507-23; PMID:4879053
- Mayer RJ, Davis RB, Schiffer CA, Berg DT, Powell BL, Schulman P, Omura GA, Moore JO, McIntyre OR, Frei E, 3rd. Intensive postremission chemotherapy in adults with acute myeloid leukemia. Cancer and Leukemia Group B. N Engl J Med 1994; 331:896-903; PMID:8078551; https://doi.org/10.1056/NEJM199410063311402
- Zittoun R, Marie JP, Delanian S, Suberville AM, Thevenin D. Prognostic value of in vitro uptake and retention of cytosine arabinoside in acute myelogenous leukemia. Semin Oncol 1987; 14:269-75; PMID:3473680
- Early AP, Preisler HD, Slocum H, Rustum YM. A pilot study of high-dose 1-beta-D-arabinofuranosylcytosine for acute leukemia and refractory lymphoma: Clinical response and pharmacology. Cancer Res 1982; 42:1587-94; PMID:6949642
- Estey E, Plunkett W, Dixon D, Keating M, McCredie K, Freireich EJ. Variables predicting response to high dose cytosine arabinoside therapy in patients with refractory acute leukemia. Leukemia 1987; 1:580-3; PMID:3669771
- Heinemann V, Jehn U. Rationales for a pharmacologically optimized treatment of acute nonlymphocytic leukemia with cytosine arabinoside. Leukemia 1990; 4:790-6; PMID:2232893
- Hiddemann W, Schleyer E, Unterhalt M, Kern W, Buchner T. Optimizing therapy for acute myeloid leukemia based on differences in intracellular metabolism of cytosine arabinoside between leukemic blasts and normal mononuclear blood cells. Ther Drug Monit 1996; 18:341-9; PMID:8857548; https://doi.org/10.1097/00007691-199608000-00005
- Kessel D, Hall TC, Rosenthal D. Uptake and phosphorylation of cytosine arabinoside by normal and leukemic human blood cells in vitro. Cancer Res 1969; 29:459-63; PMID:5250069
- Rustum YM, Riva C, Preisler HD. Pharmacokinetic parameters of 1-beta-D-arabinofuranosylcytosine (ara-C) and their relationship to intracellular metabolism of ara-C, toxicity, and response of patients with acute nonlymphocytic leukemia treated with conventional and high-dose ara-C. Semin Oncol 1987; 14:141-8; PMID:3589689
- Smyth JF, Robins AB, Leese CL. The metabolism of cytosine arabinoside as a predictive test for clinical response to the drug in acute myeloid leukaemia. Eur J Cancer 1976; 12:567-73; PMID:1066282; https://doi.org/10.1016/0014-2964(76)90164-X
- Yamauchi T, Kawai Y, Goto N, Kishi S, Imamura S, Yoshida A, Urasaki Y, Fukushima T, Iwasaki H, Tsutani H, et al. Close correlation of 1-beta-D-arabinofuranosylcytosine 5′-triphosphate, an intracellular active metabolite, to the therapeutic efficacy of N(4)-behenoyl-1-beta-D-arabinofuranosylcytosine therapy for acute myelogenous leukemia. Jpn J Cancer Res 2001; 92:975-82; PMID:11572766; https://doi.org/10.1111/j.1349-7006.2001.tb01188.x
- Cribier A, Descours B, Valadao AL, Laguette N, Benkirane M. Phosphorylation of SAMHD1 by cyclin A2/CDK1 regulates its restriction activity toward HIV-1. Cell Rep 2013; 3:1036-43; PMID:23602554; https://doi.org/10.1016/j.celrep.2013.03.017
- Welbourn S, Dutta SM, Semmes OJ, Strebel K. Restriction of virus infection but not catalytic dNTPase activity is regulated by phosphorylation of SAMHD1. J Virol 2013; 87:11516-24; PMID:23966382; https://doi.org/10.1128/JVI.01642-13
- Arnold LH, Groom HC, Kunzelmann S, Schwefel D, Caswell SJ, Ordonez P, Mann MC, Rueschenbaum S, Goldstone DC, Pennell S, et al. Phospho-dependent regulation of SAMHD1 oligomerisation couples catalysis and restriction. PLoS Pathog 2015; 11:e1005194; PMID:26431200; https://doi.org/10.1371/journal.ppat.1005194
- Kufe D, Spriggs D, Egan EM, Munroe D. Relationships among Ara-CTP pools, formation of (Ara-C)DNA, and cytotoxicity of human leukemic cells. Blood 1984; 64:54-8; PMID:6587917
- Major PP, Egan EM, Herrick DJ, Kufe DW. Effect of ARA-C incorporation on deoxyribonucleic acid synthesis in cells. Biochem Pharmacol 1982; 31:2937-40; PMID:7138584; https://doi.org/10.1016/0006-2952(82)90266-0
- Plunkett W, Begleiter A, Liliemark JO, Reed JC. Why do drugs work in CLL? Leuk Lymphoma 1996; 22 Suppl 2:1-11; PMID:9021705; https://doi.org/10.3109/10428199609102699
- Robak P, Robak T. Older and new purine nucleoside analogs for patients with acute leukemias. Cancer Treat Rev 2013; 39:851-61; PMID:23566572; https://doi.org/10.1016/j.ctrv.2013.03.006
- Kaltoft K, Bisballe S, Rasmussen HF, Thestrup-Pedersen K, Thomsen K, Sterry W. A continuous T-cell line from a patient with Sezary syndrome. Arch Dermatol Res 1987; 279:293-8; PMID:3498444; https://doi.org/10.1007/BF00431220
- Fernandez HF, Sun Z, Yao X, Litzow MR, Luger SM, Paietta EM, Racevskis J, Dewald GW, Ketterling RP, Bennett JM, et al. Anthracycline dose intensification in acute myeloid leukemia. N Engl J Med 2009; 361:1249-59; PMID:19776406; https://doi.org/10.1056/NEJMoa0904544
- Matos PM, Marin M, Ahn B, Lam W, Santos NC, Melikyan GB. Anionic lipids are required for vesicular stomatitis virus G protein-mediated single particle fusion with supported lipid bilayers. J Biol Chem 2013; 288:12416-25; PMID:23493401; https://doi.org/10.1074/jbc.M113.462028
- Goujon C, Jarrosson-Wuilleme L, Bernaud J, Rigal D, Darlix JL, Cimarelli A. With a little help from a friend: increasing HIV transduction of monocyte-derived dendritic cells with virion-like particles of SIV(MAC). Gene Ther 2006; 13:991-4; PMID:16525481; https://doi.org/10.1038/sj.gt.3302753
- Berger G, Goujon C, Darlix JL, Cimarelli A. SIVMAC Vpx improves the transduction of dendritic cells with nonintegrative HIV-1-derived vectors. Gene Ther 2009; 16:159-63; PMID:18668143; https://doi.org/10.1038/gt.2008.128
- Berger G, Durand S, Goujon C, Nguyen XN, Cordeil S, Darlix JL, Cimarelli A. A simple, versatile and efficient method to genetically modify human monocyte-derived dendritic cells with HIV-1-derived lentiviral vectors. Nat Protoc 2011; 6:806-16; PMID:21637200; https://doi.org/10.1038/nprot.2011.327
- Durand S, Nguyen XN, Turpin J, Cordeil S, Nazaret N, Croze S, Mahieux R, Lachuer J, Legras-Lachuer C, Cimarelli A. Tailored HIV-1 vectors for genetic modification of primary human dendritic cells and monocytes. J Virol 2013; 87:234-42; PMID:23077304; https://doi.org/10.1128/JVI.01459-12
- Booth C, Gaspar HB, Thrasher AJ. Treating immunodeficiency through HSC gene therapy. Trends Mol Med 2016; 22:317-27; PMID:26993219; https://doi.org/10.1016/j.molmed.2016.02.002
- Mansilla-Soto J, Riviere I, Boulad F, Sadelain M. Cell and gene therapy for the beta-thalassemias: Advances and prospects. Hum Gene Ther 2016; 27:295-304; PMID:27021486; https://doi.org/10.1089/hum.2016.037
- Alton EW, Beekman JM, Boyd AC, Brand J, Carlon MS, Connolly MM, Chan M, Conlon S, Davidson HE, Davies JC, et al. Preparation for a first-in-man lentivirus trial in patients with cystic fibrosis. Thorax 2017; 72:137-47; PMID:27852956; https://doi.org/10.1136/thoraxjnl-2016-208406
- Frecha C, Costa C, Negre D, Amirache F, Trono D, Rio P, Bueren J, Cosset FL, Verhoeyen E. A novel lentiviral vector targets gene transfer into human hematopoietic stem cells in marrow from patients with bone marrow failure syndrome and in vivo in humanized mice. Blood 2012; 119:1139-50; PMID:22117040; https://doi.org/10.1182/blood-2011-04-346619
- Frecha C, Fusil F, Cosset FL, Verhoeyen E. In vivo gene delivery into hCD34+ cells in a humanized mouse model. Methods Mol Biol 2011; 737:367-90; PMID:21590405
- Sahin U, Kariko K, Tureci O. mRNA-based therapeutics–developing a new class of drugs. Nat Rev Drug Discov 2014; 13:759-80; PMID:25233993; https://doi.org/10.1038/nrd4278
- Kienle E, Senis E, Borner K, Niopek D, Wiedtke E, Grosse S, Grimm D. Engineering and evolution of synthetic adeno-associated virus (AAV) gene therapy vectors via DNA family shuffling. J Vis Exp 2012; 3819; PMID:22491297; https://doi.org/10.3791/3819
- Chen JJ, Jin MX, Zhu SL, Li F, Xing Y. The Synthesis and characteristic study of transferrin-conjugated liposomes carrying brain-derived neurotrophic factor. Biomed Mater Eng 2014; 24:2089-99; PMID:25226906
- Morrell NT, Leucht P, Zhao L, Kim JB, ten Berge D, Ponnusamy K, Carre AL, Dudek H, Zachlederova M, McElhaney M, et al. Liposomal packaging generates Wnt protein with in vivo biological activity. PLoS One 2008; 3:e2930; PMID:18698373; https://doi.org/10.1371/journal.pone.0002930
- Gamis AS, Alonzo TA, Meshinchi S, Sung L, Gerbing RB, Raimondi SC, Hirsch BA, Kahwash SB, Heerema-McKenney A, Winter L, et al. Gemtuzumab ozogamicin in children and adolescents with de novo acute myeloid leukemia improves event-free survival by reducing relapse risk: Results from the randomized phase III Children's Oncology Group trial AAML0531. J Clin Oncol 2014; 32:3021-32; PMID:25092781; https://doi.org/10.1200/JCO.2014.55.3628
- Merati M, Buethe DJ, Cooper KD, Honda KS, Wang H, Gerstenblith MR. Aggressive CD8(+) epidermotropic cutaneous T-cell lymphoma associated with homozygous mutation in SAMHD1. JAAD Case Rep 2015; 1:227-9; PMID:27051737; https://doi.org/10.1016/j.jdcr.2015.05.003
- Kodigepalli KM, Li M, Liu SL, Wu L. Exogenous expression of SAMHD1 inhibits proliferation and induces apoptosis in cutaneous T-cell lymphoma-derived HuT78 cells. Cell Cycle 2017; 16:179-88; PMID:27929746; https://doi.org/10.1080/15384101.2016.1261226
- Bonifati S, Daly MB, St Gelais C, Kim SH, Hollenbaugh JA, Shepard C, Kennedy EM, Kim DH, Schinazi RF, Kim B, et al. SAMHD1 controls cell cycle status, apoptosis and HIV-1 infection in monocytic THP-1 cells. Virology 2016; 495:92-100; PMID:27183329; https://doi.org/10.1016/j.virol.2016.05.002
- Frecha C, Szecsi J, Cosset FL, Verhoeyen E. Strategies for targeting lentiviral vectors. Curr Gene Ther 2008; 8:449-60; PMID:19075628; https://doi.org/10.2174/156652308786848003
- Arnold LH, Kunzelmann S, Webb MR, Taylor IA. A continuous enzyme-coupled assay for triphosphohydrolase activity of HIV-1 restriction factor SAMHD1. Antimicrob Agents Chemother 2015; 59:186-92; PMID:25331707; https://doi.org/10.1128/AAC.03903-14
- Seamon KJ, Stivers JT. A high-throughput enzyme-coupled assay for SAMHD1 dNTPase. J Biomol Screen 2015; 20:801-9; PMID:25755265; https://doi.org/10.1177/1087057115575150
- Seamon KJ, Hansen EC, Kadina AP, Kashemirov BA, McKenna CE, Bumpus NN, Stivers JT. Small molecule inhibition of SAMHD1 dNTPase by tetramer destabilization. J Am Chem Soc 2014; 136:9822-5; PMID:24983818; https://doi.org/10.1021/ja5035717
- Ladner RD. The role of dUTPase and uracil-DNA repair in cancer chemotherapy. Curr Protein Pept Sci 2001; 2:361-70; PMID:12374095; https://doi.org/10.2174/1389203013380991
- Amie SM, Bambara RA, Kim B. GTP is the primary activator of the anti-HIV restriction factor SAMHD1. J Biol Chem 2013; 288:25001-6; PMID:23880768; https://doi.org/10.1074/jbc.C113.493619
- Gad H, Koolmeister T, Jemth AS, Eshtad S, Jacques SA, Strom CE, Svensson LM, Schultz N, Lundback T, Einarsdottir BO, et al. MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature 2014; 508:215-21; PMID:24695224; https://doi.org/10.1038/nature13181
- Rudd SG, Valerie NC, Helleday T. Pathways controlling dNTP pools to maintain genome stability. DNA Repair (Amst) 2016; 44:193-204; PMID:27311542; https://doi.org/10.1016/j.dnarep.2016.05.032
- Huber AD, Michailidis E, Schultz ML, Ong YT, Bloch N, Puray-Chavez MN, Leslie MD, Ji J, Lucas AD, Kirby KA, et al. SAMHD1 has differential impact on the efficacies of HIV nucleoside reverse transcriptase inhibitors. Antimicrob Agents Chemother 2014; 58:4915-9; PMID:24867973; https://doi.org/10.1128/AAC.02745-14
- Honma Y, Niitsu N. Vidarabine and 2-deoxycoformycin as antileukemic agents against monocytic leukemia. Leuk Lymphoma 2000; 39:57-66; PMID:10975384; https://doi.org/10.3109/10428190009053539
- Sueda T, Sakai D, Kudo T, Sugiura T, Takahashi H, Haraguchi N, Nishimura J, Hata T, Hayashi T, Mizushima T, et al. Efficacy and safety of regorafenib or TAS-102 in patients with metastatic colorectal cancer refractory to standard therapies. Anticancer Res 2016; 36:4299-306; PMID:27466548
- Scagliotti G, Nishio M, Satouchi M, Valmadre G, Niho S, Galetta D, Cortinovis D, Benedetti F, Yoshihara E, Makris L, et al. A phase 2 randomized study of TAS-102 versus topotecan or amrubicin in patients requiring second-line chemotherapy for small cell lung cancer refractory or sensitive to frontline platinum-based chemotherapy. Lung Cancer 2016; 100:20-3; PMID:27597276; https://doi.org/10.1016/j.lungcan.2016.06.023
- Bando H, Doi T, Muro K, Yasui H, Nishina T, Yamaguchi K, Takahashi S, Nomura S, Kuno H, Shitara K, et al. A multicenter phase II study of TAS-102 monotherapy in patients with pre-treated advanced gastric cancer (EPOC1201). Eur J Cancer 2016; 62:46-53; PMID:27208903; https://doi.org/10.1016/j.ejca.2016.04.009
- Griffith JF, Fitzwilliam JF, Casagrande S, Butler SR. Experimental herpes simplex virus encephalitis: Comparative effects of treatment with cytosine arabinoside and adenine arabinoside. J Infect Dis 1975; 132:506-10; PMID:171318; https://doi.org/10.1093/infdis/132.5.506
- Miyamoto Y, Lenz HJ, Baba H. A novel antimetabolite: TAS-102 for metastatic colorectal cancer. Expert Rev Clin Pharmacol 2016; 9:355-65; PMID:26677869; https://doi.org/10.1586/17512433.2016.1133285
- Elamin YY, Rafee S, Osman N, KJ OB, Gately K. Thymidine phosphorylase in cancer; Enemy or friend? Cancer Microenviron 2016; 9:33-43; PMID:26298314; https://doi.org/10.1007/s12307-015-0173-y
- Lenz HJ, Stintzing S, Loupakis F. TAS-102, a novel antitumor agent: a review of the mechanism of action. Cancer Treat Rev 2015; 41:777-83; PMID:26428513; https://doi.org/10.1016/j.ctrv.2015.06.001
- Momparler RL, Rossi M, Bouchard J, Vaccaro C, Momparler LF, Bartolucci S. Kinetic interaction of 5-AZA-2′-deoxycytidine-5′-monophosphate and its 5′-triphosphate with deoxycytidylate deaminase. Mol Pharmacol 1984; 25:436-40; PMID:6203026
- Serdjebi C, Milano G, Ciccolini J. Role of cytidine deaminase in toxicity and efficacy of nucleosidic analogs. Expert Opin Drug Metab Toxicol 2015; 11:665-72; PMID:25495470; https://doi.org/10.1517/17425255.2015.985648
- Heinemann V, Plunkett W. Modulation of deoxynucleotide metabolism by the deoxycytidylate deaminase inhibitor 3,4,5,6-tetrahydrodeoxyuridine. Biochem Pharmacol 1989; 38:4115-21; PMID:2688654; https://doi.org/10.1016/0006-2952(89)90693-X
- Jansen RS, Rosing H, Schellens JH, Beijnen JH. Deoxyuridine analog nucleotides in deoxycytidine analog treatment: Secondary active metabolites? Fundam Clin Pharmacol 2011; 25:172-85; PMID:20199587; https://doi.org/10.1111/j.1472-8206.2010.00823.x
- Riva C, Barra Y, Carcassonne Y, Cano JP, Rustum Y. Effect of tetrahydrouridine on metabolism and transport of 1-beta-D-arabinofuranosylcytosine in human cells. Chemotherapy 1992; 38:358-66; PMID:1286578; https://doi.org/10.1159/000239026
- Ferraris D, Duvall B, Delahanty G, Mistry B, Alt J, Rojas C, Rowbottom C, Sanders K, Schuck E, Huang KC, et al. Design, synthesis, and pharmacological evaluation of fluorinated tetrahydrouridine derivatives as inhibitors of cytidine deaminase. J Med Chem 2014; 57:2582-8; PMID:24520856; https://doi.org/10.1021/jm401856k
- McIntosh EM, Haynes RH. dUTP pyrophosphatase as a potential target for chemotherapeutic drug development. Acta Biochim Pol 1997; 44:159-71; PMID:9360704
- Grasser FA, Romeike BF, Niedobitek G, Nicholls J, Kremmer E. dUTPase in human neoplastic cells as a potential target for therapeutic intervention. Curr Protein Pept Sci 2001; 2:349-60; PMID:12369931; https://doi.org/10.2174/1389203013381053
- Miyakoshi H, Miyahara S, Yokogawa T, Endoh K, Muto T, Yano W, Wakasa T, Ueno H, Chong KT, Taguchi J, et al. 1,2,3-Triazole-containing uracil derivatives with excellent pharmacokinetics as a novel class of potent human deoxyuridine triphosphatase inhibitors. J Med Chem 2012; 55:6427-37; PMID:22715973; https://doi.org/10.1021/jm3004174
- Saito K, Nagashima H, Noguchi K, Yoshisue K, Yokogawa T, Matsushima E, Tahara T, Takagi S. First-in-human, phase I dose-escalation study of single and multiple doses of a first-in-class enhancer of fluoropyrimidines, a dUTPase inhibitor (TAS-114) in healthy male volunteers. Cancer Chemother Pharmacol 2014; 73:577-83; PMID:24452393; https://doi.org/10.1007/s00280-014-2383-2
- Valerie NC, Hagenkort A, Page BD, Masuyer G, Rehling D, Carter M, Bevc L, Herr P, Homan E, Sheppard NG, et al. NUDT15 Hydrolyzes 6-Thio-DeoxyGTP to Mediate the Anticancer Efficacy of 6-Thioguanine. Cancer Res 2016; 76:5501-11; PMID:27530327; https://doi.org/10.1158/0008-5472.CAN-16-0584
- Moriyama T, Nishii R, Perez-Andreu V, Yang W, Klussmann FA, Zhao X, Lin TN, Hoshitsuki K, Nersting J, Kihira K, et al. NUDT15 polymorphisms alter thiopurine metabolism and hematopoietic toxicity. Nat Genet 2016; 48:367-73; PMID:26878724; https://doi.org/10.1038/ng.3508
- Schmiegelow K, Nersting J, Nielsen SN, Heyman M, Wesenberg F, Kristinsson J, Vettenranta K, Schroeder H, Weinshilboum R, Jensen KL, et al. Maintenance therapy of childhood acute lymphoblastic leukemia revisited-Should drug doses be adjusted by white blood cell, neutrophil, or lymphocyte counts? Pediatr Blood Cancer 2016; 63:2104-11; PMID:27447547; https://doi.org/10.1002/pbc.26139
- Goncalves A, Karayel E, Rice GI, Bennett KL, Crow YJ, Superti-Furga G, Burckstummer T. SAMHD1 is a nucleic-acid binding protein that is mislocalized due to aicardi-goutieres syndrome-associated mutations. Hum Mutat 2012; 33:1116-22; PMID:22461318; https://doi.org/10.1002/humu.22087
- Menendez-Arias L, Betancor G, Matamoros T. HIV-1 reverse transcriptase connection subdomain mutations involved in resistance to approved non-nucleoside inhibitors. Antiviral Res 2011; 92:139-49; PMID:21896288; https://doi.org/10.1016/j.antiviral.2011.08.020
- Emura T, Murakami Y, Nakagawa F, Fukushima M, Kitazato K. A novel antimetabolite, TAS-102 retains its effect on FU-related resistant cancer cells. Int J Mol Med 2004; 13:545-9; PMID:15010854
- Hollenbach PW, Nguyen AN, Brady H, Williams M, Ning Y, Richard N, Krushel L, Aukerman SL, Heise C, MacBeth KJ. A comparison of azacitidine and decitabine activities in acute myeloid leukemia cell lines. PLoS One 2010; 5:e9001; PMID:20126405; https://doi.org/10.1371/journal.pone.0009001
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012; 483:603-7; PMID:22460905; https://doi.org/10.1038/nature11003
- Seashore-Ludlow B, Rees MG, Cheah JH, Cokol M, Price EV, Coletti ME, Jones V, Bodycombe NE, Soule CK, Gould J, et al. Harnessing connectivity in a large-scale small-molecule sensitivity dataset. Cancer Discov 2015; 5:1210-23; PMID:26482930; https://doi.org/10.1158/2159-8290.CD-15-0235
- Rees MG, Seashore-Ludlow B, Cheah JH, Adams DJ, Price EV, Gill S, Javaid S, Coletti ME, Jones VL, Bodycombe NE, et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat Chem Biol 2016; 12:109-16; PMID:26656090; https://doi.org/10.1038/nchembio.1986