
ABSTRACT
Defining the roadblocks responsible for cell cycle arrest in adult cardiomyocytes lies at the core of developing cardiac regenerative therapies. p53 and Mdm2 are crucial mediators of cell cycle arrest in proliferative cell types, however, little is known about their function in regulating homeostasis and proliferation in terminally differentiated cell types, like cardiomyocytes. To explore this, we generated a cardiac-specific conditional deletion of p53 and Mdm2 (DKO) in adult mice. Herein we describe the development of a dilated cardiomyopathy, in the absence of cardiac hypertrophy. In addition, DKO hearts exhibited a significant increase in cardiomyocyte proliferation. Further evaluation showed that proliferation was mediated by a significant increase in Cdk2 and cyclin E with downregulation of p21Cip1 and p27Kip1. Comparison of miRNA expression profiles from DKO mouse hearts and controls revealed 11 miRNAs that were downregulated in the DKO hearts and enriched for mRNA targets involved in cell cycle regulation. Knockdown of these miRNAs in neonatal rat cardiomyocytes significantly increased cytokinesis with an upregulation in the expression of crucial cell cycle regulators. These results illustrate the importance of the cooperative activities of p53 and Mdm2 in a network of miRNAs that function to impose a barrier against aberrant cardiomyocyte cell cycle re-entry to maintain cardiac homeostasis.
Introduction
Trp53 (p53) is a tumor suppressor gene encoding a transcription factor that is the most frequent target for mutations in tumors.Citation1 In addition to its tumor suppressive abilities, p53 acts as a cellular stress sensor,Citation2-4 functioning in regulating tissue homeostasisCitation5 and senescence.Citation6-8 Based on the observations that p53 knockout mice develop early onset spontaneous tumors with 100% penetrance,Citation9 that individuals with Li-Fraumeni syndrome who inherit a mutant p53 allele have heightened susceptibility for tumorigenesis,Citation10 and that cells with dysfunctional p53 exhibit aberrantly high levels of proliferation,Citation11 the importance of p53s role in growth control is unequivocal. Due to the diverse effects of p53 within a cell, its activity is tightly regulated through ubiquitination by the E3 ubiquitin ligase, murine double minute 2 (Mdm2), which targets p53 for proteasomal degradation.Citation12-15 Mdm2-null mice experience a very early embryonic lethality, due to high levels of p53, impairing organ development through extensive apoptosis and diminished cell proliferation.Citation16,Citation17
The most thoroughly characterized biochemical role of p53 within the cell is as a transcriptional activator, whereby it can mediate cell cycle arrest through activation of important cell cycle inhibitors such as cyclin-dependent kinase inhibitor 1A (p21Cip1)Citation1 and other genes that control the G2/M transition, as well as cytokinesis.Citation18 The upregulation of p21Cip1 expression by p53 leads to G1 phase cell cycle arrest, through p21Cip1-mediated inhibition of cyclin-dependent kinase 1 (Cdk1), cyclin-dependent kinase 2 (Cdk2) and proliferating cell nuclear antigen (PCNA).Citation18,Citation19
In contrast, p53-mediated transcriptional repression has been a contentious area of research, as the mechanism by which this occurs is largely unknown.Citation20 Recently microRNAs (miRNAs) have been suggested as the main mediators of p53-regulated gene repression.Citation21 miRNAs are a group of small non-coding RNAs with the capability of expansive genetic regulation through RNA silencing, mediated by base pairing to their complementary mRNA targets.Citation22-24 By targeting specific mRNAs for degradation or translational repression, miRNAs act as inhibitors of gene expression, and are involved in almost all developmental and pathological processes in animals, including the multiple pathways regulated by the tumor suppressor p53.Citation22
Many p53-regulated miRNAs have been shown to have robust effects on the cell cycle, and can themselves act as tumor suppressors.Citation25 Furthermore, cell cycle-related factors have bioinformatically been shown to be over-represented in the profiles of mRNAs targeted by p53-regulated miRNAs.Citation26-28 Additionally, p53-regulated miRNAs have been found to be involved in a myriad of signaling and biologic processes which regulate growth and regeneration, cardiac function, homeostasis, and the progression of cardiac remodeling, hypertrophy and heart failure.Citation29
Heart failure is a leading cause of morbidity and mortality in Canada.Citation30 As the quality of life and prognosis for patients with heart failure is poor, continued development of therapies to prevent and treat heart failure are urgently needed.Citation33 In the adult, cardiomyocytes are terminally differentiated cells that exist in a non-proliferative, post-mitotic state.Citation31,Citation32 Thus, the heart is particularly vulnerable to injury, as the regenerative capacity of this organ is poor. Therefore, determining the roadblocks responsible for maintaining cardiomyocyte cell cycle exit lies at an important crossroad in the development of potential regenerative therapies to promote cardiac repair following injury.
The p53 stress response in tumor prevention and tissue homeostasis is well characterized in murine models, however the normal physiologic role of the p53/Mdm2 circuitry in the heart has remained elusive. Notably, patients with end-stage heart failure exhibit an increase in p53 protein levels.Citation34 Given the crucial role of the p53/Mdm2 circuitry in promoting growth arrest, the aim of this study was to characterize the effect of p53 and Mdm2 in the maintenance of cardiac function and cell cycle arrest. Furthermore, we hypothesized that these effects may be in fact mediated by miRNAs.
Herein, we report that p53 and Mdm2 are integral to the continued monitoring and maintenance of cardiac performance. Furthermore, we demonstrate that differentiated cardiomyocytes regain proliferative capacity after the deletion of both p53 and Mdm2, through the downregulation of a group of miRNAs which act as inhibitors of genes involved in cell cycle regulation. The use of antagomirs targeting this subset of miRNAs in vitro further substantiated these observations. These data support the existence of a novel role for p53 and Mdm2 in the heart, to maintain cell cycle arrest through the regulation of a subset of anti-proliferative miRNAs.
Methods
Mdm2 and p53 conditional mutant mice
All animal usage in this study was in accordance with approved institutional animal care guidelines of the UHN (AUP 1379, Canadian Council in Animal Care). The mcm transgenic miceon a C57BL/6J background were obtained from Jackson (Bar Harbor, ME04609 USA; strain name: B6.FVB(129)-Tg(Myh6-cre/Esr1*)1Jmk/J; stock number: 005657). The Mdm2f/f mice (strain number 01XH9) and p53f/f mice (strain number 01XC2) were obtained from the mouse repository of the National Cancer Institute/National Institutes of Health at Frederick (Rockville, MD 20852 USA). The Mdm2f/f and p53f/f strainswere backcrossed onto C57BL/6J inbred mice (Jackson; stock number 000664) for at least 8 generations. Age-matched syngeneic adult male mice (12–13-week-old; 22–27 g body weight) were used in this study. All experiments used controls of matched age and sex. Genotyping was performed by PCR using alkaline hydrolysis of genomic DNA isolated from tail tips using the Terra PCR Direct Kit (mcm, Mdm2f/f, p53f/f; Clontech) or AccuStart II GelTrack PCR Supermix and the following oligonucleotides:
mcm (forward 5′-AGGTGGACCTGA TCATGGAG-3′; reverse 5′-ATACCGGAGATCATGCAAGC-3′)
Mdm2f/f (forward 5′-CTGTGTGAGCTGAGGGAGATGTG-3′; reverse 5′-CCTGGATTTAATCTGCAGCACTC-3′)
p53f/f (forward 5′-CACAAAAACAGGTTAAACCCAG-3′; reverse 5′-AGCACATAGGAGGC AGAGAC-3′).
To achieve inactivation of Mdm2 and p53 in the adult mouse heart, we used the Cre-loxP recombination system of bacteriophage P1. We used an inducible cardiomyocyte-specific transgenic mouse in which the cardiac muscle α-myosin heavy chain 6 (Myh6) promoter drives the expression of tamoxifen (Tam)-inducible Cre recombinase protein fused to 2 mutant estrogen receptor ligand-binding domains (mcm). In this strain, the mcm fusion protein is expressed only in cardiomyocytes and is retained in the cytoplasm. Administration of Tam induces mcm nuclear translocation thereby permitting Cre-mediated recombination. This system allowed us to explore conditional mutations of p53 and Mdm2 that might differ from the germ line absence of both of these factors in the adult heart. Next, we crossed mcm transgenic mice with mice carrying conditional Mdm2f/f and p53f/f alleles to obtain Mdm2f/f;mcm (Mdm2KO), p53f/f;mcm (p53KO) and Mdm2f/f;p53f/f;mcm (DKO), mice on a C57BL/6J background. In Mdm2f/f mice, Mdm2 exons 7 to 9 are flanked with loxP sites to facilitate inactivation of the RING finger domain which exerts ubiquitin ligase activity toward p53. p53f/f mice carry loxP sites in p53 introns 1 to 10 to ensure Cre-mediated elimination of the majority of the coding sequence.
To achieve conditional genetic ablation of Mdm2 and p53 in adult cardiomyocytes in vivo, we injected an ethanol/peanut oil (P2144, Sigma-Aldrich) emulsion of 4-Hydroxytamoxifen (H6278, Sigma-Aldrich) intraperitoneally once daily for 4 consecutive days (+Tam mice). The day of the fourth Tam injection was arbitrarily set as day 0. In the absence of Tam, adult Mdm2f/f, p53f/f, Mdm2f/f;p53f/f and mcm mice exhibited no evidence of fibrosis or cardiac dysfunction as analyzed by immunofluorescence and echocardiography. Mdm2f/f;p53f/f;mcm mice were also injected intraperitoneally once daily for 4 consecutive days with only peanut oil (-Tam mice, vehicle-injected control) and served as the control group for this study.
Echocardiography
Echocardiography in anesthetized mice (2% isoflurane, 98% oxygen) was performed using a 15-MHz linear ultrasound transducer (Vivid7; GE). Body temperature was maintained at 37 °C. M-mode measurements of the LV end-diastolic diameter (LVEDD) and LV end-systolic diameter (LVESD) were made in triplicate from short-axis views at the level of the papillary muscle and averaged over 3. LVEDD was measured at the time of the apparent maximal LV diastolic dimension, whereas LVESD was measured at the time of the most systolic excursion of the posterior wall. LV fractional shortening (FS) was calculated as follows: FS = (LVEDD -LVESD)/LVEDDx100%.
Electrocardiographic telemetry
Mice were anaesthetized (1% isoflurane inhalation) and implanted with radiofrequency electrocardiogram (ECG) transmitters (DSI PhysioTel Transmitter EA-F20; Data Sciences International, St. Paul, MN 55112 USA). Transmitters were aseptically inserted into a dorsal subcutaneous tissue pocket with the positive lead of the transmitter tunneled subcutaneously to the left anterior chest wall above the apex of the heart and the negative lead to the right shoulder. This configuration approximates lead II on the body surface ECG. ECG recordings commenced 7 d following implantation to allow for adequate recovery from surgery. Tam was injected as described above. ECG was acquired in 5 minute intervals every hour using Dataquest A.R.T. acquisition software (Version 4.1; DSI). ECG was recorded until the mice succumbed. All telemetry ECG data was analyzed offline using Dataquest A.R.T. analysis software (Version 4.1; DSI).
Nanostring nCounter miRNA assay
Total RNA was isolated by the Trizol method (15596–026; Life Technologies). 33 ng of total RNA from each heart was used as input for the nCounter Mouse v1.5 miRNA Expression Assay (Nanostring; GXA-MMIR15–12) and was processed according to the manufacturer's protocols. Raw array data was processed using the Nanostring nSolver Analysis Software 2.5. The mean of the negative controls plus one standard deviation was subtracted for background correction. Normalization of the data for sample/RNA content was performed using the geometric mean of the top 100 most highly expressed genes. Normalized miRNA expression levels were transformed from ratios (treatment vs. vehicle-injected control) to fold change values and analyzed using a Student's t-test to identify significantly differentially expressed miRNA between samples. Heatmaps were generated with the fold change values using Euclidean distance as the distance metric and the average as the linkage method. Pathway analysis was executed with the online platform miRSystem ver. 20150312 (http://mirsystem.cgm.ntu.edu.tw/index.php). The nCounter miRNA assay data are available in the ArrayExpress database (http://www.ebi.ac.uk/arrayexpress; accession number E-MTAB-5531)
miRNA and mRNA real-time qPCR
For miRNA real-time qPCR, total RNA isolated from mouse hearts, rat neonatal cardiomyocytes or rat neonatal fibroblasts with Trizol was reverse transcribed using the qScript microRNA cDNA Synthesis Kit (Quanta BioSciences, Inc.) using an initial input of 500ng of RNA. qPCR was performed using the Roche Light Cycler 480, a PerfeCTa miRNA assay unique for each individual miRNA along with the PerfeCTa Universal PCR Primer (specific to the unique sequence of the oligo-dT adaptor primer) and PerfeCTA SYBR Green SuperMix (5ng cDNA/25µl reaction). Relative miRNA expression and fold change was calculated by the ΔΔCt method, and was normalized to the endogenous control RNU6 (small RNA component of spliceosome) using the LC480 SW 1.5.1 analysis software.
For standard mRNA real-time qPCR, total RNA was isolated with Trizol, and purified using the RNeasy Plus Mini Kit (Qiagen). We used 1.0 pg total RNA/reaction for first-strand cDNA synthesis using random primers (Retroscript AM1710; Ambion). Real-time qPCR assays were performed using 50 ng DNA/20 µl reaction volume and the QuantiFast SYBR Green PCR kit (Qiagen) on a GeneAmp PCR System 9700 (Applied Biosystems). Relative quantification of gene expression levels was performed using the ΔΔCt method with normalization to Npm1.
Western Blot
Protein was isolated from mouse hearts using the Nuclear and Cytoplasmic Extraction Kit (ThermoFisher) according to the manufacturer's protocol. For Western Blot, we used SDS/PAGE gels (4–12% Bolt Bis-Tris Plus; Invitrogen), PVDF membranes (iBlot; Invitrogen), primary antibodies against p53 (BML-SA293; Enzo), Mdm2 (M8558; Sigma), Npm1 (B0556; Sigma), p21 (556431; BD), p27 (610241; BD), Cdk2 (sc-163; SantaCruz) and Cyclin E (4129; Cell Signaling), horseradishperoxidase (HRP) conjugated secondary antibodies (HRP-sheep anti-mouse IgG, RPN4201 and HRP-goat anti-rabbit IgG, RNP4301; GE Life Sciences), and the Luminata Crescendo HRP substrate for chemiluminescence detection (WBLUR0100; EMD Millipore). Blots were imaged with the BioRad ChemiDoc XRS+ and analyzed with Image Lab 5.0 software.
Immunocomplex kinase assay
Cellular extracts in supplemented RIPA Buffer were incubated with antibodies to Cdk2 covalently linked to protein A agarose beads (Seize X Protein A Immunoprecipitation Kit; Pierce) for 3 h at 4°C. Immunocomplexes were washed 3 times with ice-cold kinase buffer (Cell Signaling) composed of 25 mM Tris-HCl (pH 7.5), 5 mM E-glycerophosphate, 2 mM dithiothreitol (DTT), 0.1 mM Na3VO4, 10 mM MgCl2 supplemented with 1 mM ATP (Cell Signaling). We incubated 5 pg histone H1 (H4524; Sigma) with anti-Cdk2-immunocomplexes 50 µl kinase buffer for 30 min at 30°C. We stopped the reactions by adding 25 µl 2x SDS sample buffer (Cell Signaling). The amount of phosphorylation of histone H1 on threonine (Thr) residues was determined by immunoblotting with anti-Phospho-Thr Cdk substrate antibodies (Cell Signaling). Protein samples were resolved by SDS-PAGE using 4–12% and 3–8% NuPAGE pre-cast gels (Life Technologies), and PVDF membranes (iBlot; Life Technologies). The following secondary antibodies were used for chemiluminescence detection of proteins: horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (no. 7074; Cell Signaling), HRP-conjugated anti-mouse IgG (no. 7076; Cell Signaling), and Luminata Crescendo (WBLUR0100, Millipore)
Neonatal rat cardiomyocyte and fibroblast isolation and culture
Ventricular cardiomyocytes from 2 to 3 day old wild type Wistar rats (Charles River) were isolated using collagenase II (355 U/mL, Worthington) and pancreatin (0.8 mg/mL, Sigma), as described previously.Citation35 A purified population of cardiomyocytes was plated onto Collagen I-coated cell culture dishes (1 × 106 cells/well in 6 well plates, 150,000 cells/well in 48 well plates; or 150,000 cells/chamber in 8 chamber glass slides, Corning). Cardiomyocytes were cultured in DMEM/F12 medium containing 3 mM Na-pyruvate, 2 mM glutamine, antibiotics (Gibco), 10% fetal bovine serum (Invitrogen), 10% horse serum (Wisent) and 25 µM arabinosylcytosine (AraC; Sigma) to inhibit remaining non-cardiomyocyte proliferation for 48 hours. 500,000 fibroblasts were seeded/well in 6 well plates, and cultured in DMEM/F12 medium containing 10% fetal bovine serum (Invitrogen) and antibiotics. Cells were cultured to confluency before isolation of RNA for RT-qPCR.
TUNEL Assay
Detection of fragmented genomic DNA was performed by terminal deoxynucleotidyl transferase-mediated dUTP nick-end-labeling (TUNEL) according to the manufacturer's instruction (no. 1684795910; Roche). Samples were co-stained with cardiac-specific anti-sarcomeric α-actinin after permeabilization. Specimens were permeabilized in 1.0% Triton X-100 (X100; Sigma-Aldirch) in TBS (20mM Tris, 150 mM NaCl), pH 7.6 for 60 minutes at room temperature.
Immunofluorescence
Neonatal rat cardiomyocytes were fixed with 4% PBS-buffered formalin for 7 minutes at room temperature and permeabilized with 1% Triton-X-100 in 1X-Tris-buffered saline for 60 minutes. Whole mouse hearts were fixed with 4% PBS-buffered formalin for 30 minutes are room temperature. Cryosections (10 μm thickness) on positively charged micro slides (Superfrost Plus; VWR) were subsequently permeabilized in 1% Triton X-100 in 1X-Tris-buffered saline for 60 minutes. Cells or cryosections were stained for 2 hours with primary antibodies diluted in 1% Triton-X-100 in 1X-Tris-buffered saline: AuroraB (Sigma, cat. # A5102), Gata4 (Becton Dickinson, cat. # 560327), Mef2A (ab32866; Abcam), α-actinin, sarcomeric (A7811; Sigma), BrdU (MCA2060; Sigma), Phospho-Ser28-Histone H3 (H9908; Sigma), Birc5 (2810; Cell Signaling) and/or Alexa Fluor 488-conjugated wheat germ agglutinin (WGA, W7024; GE Life Sciences. Cells were then washed and incubated for 45 minutes in their respective secondary antibodies conjugated to Alexa Fluor-488 or Alexa Fluor-555 (Life Technologies) and Genomic DNA was stained with 4',6-diamidino-2-phenylindole dihydrochloride (Dapi, D1306; GE Life Sciences). Finally, cells or cryosections were fixed again with 4% PBS-buffered formalin for 7 minutes at room temperature. High resolution 3 dimensional confocal laser scanning microscopy was subsequently performed on a Zeiss LSM700 confocal microscope equipped with 4 solid laser lines (405 nm, 488 nm, 555 nm, 639 nm) and X-Cite fluorescence light source for visual mode as illumination device, Plan Apochromat 40x/1.4 Oil DIC M27 objective lens and LSM Zen 2009 data acquisition software (AOMF). Digital images were overlaid using ImageJ software 1.45 (U. S. National Institutes of Health, Bethesda, Maryland, USA, http://rsbweb.nih.gov/ij/). Numbers of α-actinin-positive, Dapi-positive cardiomyocytes per longitudinal section were determined by planimetry of immunofluorescence microphotographs using ImageJ software.
For the quantitative analyses of M-phase cardiomyocytes and proliferating cardiomyocytes in LV tissue sections, numbers of α-actinin-positive, Dapi-positive, Pi-H3 positive or Birc5 positive cardiomyocytes per 6 random fields (500 nuclei/field) were counted per longitudinal myocardial section comprising the LV, septum and right ventricle using Adobe Photoshop (version 6.0) software.
For the quantitative analysis of proliferating isolated neonatal rat ventricular cardiomyocytes, total AuroraB positive, α-actinin-positive, Dapi-positive cardiomyocytes were counted in an individual chamber of an 8 chamber Collagen I coated glass slide (Corning), n = 4 squares per antagomir treatment group.
For BrdU-labeling experiments, we intraperitoneally injected 5-Bromo-2’-deoxyuridine (BrdU; 160 mg/kg body weight cumulative dosage) once daily for 2 d (2d and 3d; 6d and 7d). Cryosections (10 μm thickness) on coverslips were incubated in 2 N HCl for 25 min at room temperature. Specimens were permeabilized in 1% Triton X-100 for 60 minutes at room temperature. Genomic DNA was co-stained with 4',6-diamidino-2-phenylindole dihydrochloride (Dapi; D1306, Life Sciences). Digital images were overlaid using ImageJ software 1.45 (U. S. National Institutes of Health, Bethesda, Maryland, USA, http://rsbweb.nih.gov/ij/). Numbers of α-actinin-positive, Dapi-positive, Brdu positive cardiomyocytes per longitudinal section were determined by planimetry of immunofluorescence microphotographs using ImageJ software.
For the quantitative analyses of S-phase cardiomyocytes in LV tissue sections, 6 random fields (500 nuclei/field) were counted per longitudinal myocardial section comprising the LV, septum and right ventricle using Adobe Photoshop (version 6.0) software.
3D image reconstruction from Z-stack images was performed with IMARIS software version 7.11 (Bitplane, Zurich, Switzerland) and ZEN (Carl Zeiss AG). Fluorescence tiling of whole longitudinal specimen was performed on an Olympus BX50 microscope equipped with EXFO fluorescent unit, UPlanSApo 10x/0.40 and UPlanSApo 20x/0.75 objective lenses, Semrock Quad Sedat Filter Set DA/FI/TR/Cy5–4 × 4M-B, Photometrics CoolSNAP HQ2 camera and Molecular Device Metamorph data acquisition software (AOMF – Advanced Optical Microcopy Facility UHN, Princess Margaret Hospital, Ontario Cancer Institute, Toronto, ON Canada).
miRNA antagomirs
miRIDIAN microRNA Hairpin Inhibitors were ordered from Dhamarcon (5 nmol). The Anti-cel-67 targeting a Caenorhabditis elegans miRNA not expressed in mammals was used as a negative control. Cardiomyocytes were transfected using the TransIT TKO Transfection Reagent (Mirus Bio) and 50 nM of BLOCK-iT Alexa Fluor 555 Red Fluorescent Oligo (Positive Control, Thermo Fisher) to determine transfection efficiency. Transfection efficiency was assessed by fluorescent microscopy, calculating the number of Mef2A positive, Alexa Fluor 555 positive cells/well. Transfection of antagomirs (25 nM each) was performed using the standard transfection protocol provided by Mirus Bio. in cardiomyocyte media (DMEM/F12) containing 1% fetal bovine serum, 1% horse serum, antibiotics and no AraC, 48 hours following cell plating. 24 hours after transfection, media was replaced, and 48 hours following transfection, cells were fixed for immunofluorescence or RNA was isolated.
Cell Cycle RT-qPCR Array
RNA was purified using the method described above from cardiomyocytes transfected with an antagomir cocktail (25 nM each) or a negative control (cel-67 100 nM). Concentrations were determined using a NanoDrop 2000 spectrophotometer (Thermo Scientific). Equal amounts of RNA (400ng/sample) were reverse transcribed into cDNA using the First Strand cDNA Synthesis (Qiagen) kit. qPCR was performed according to the manufacturer's protocol.
Transmission electron microscopy analysis
LV apexes from 13 week old DKO mice at 8d post-Tam or vehicle injected controls were rinsed in PBS (pH 7.4) and then fixed in 3.5% glutaraldehyde/Sorensen's phosphate buffer (pH 7.4) at room temperature for 24 h. After then being washed in phosphate buffer, samples were treated with 2% osmium tetroxide for 1 h. The samples were then dehydrated in a series of aqueous ethanol solutions 60, 75, 85, 95, and 100% and placed into propylene oxide for 30 min before being infiltrated with a 1:1 followed by 1:3 propylene–epon resin mixtures for 60 min each. The LV tissues were then transferred into pure epon resin and degassed under partial vacuum to ensure infiltration. Following overnight curing at 80°C, sections (100 nm thickness) were cut and placed on copper grids, stained, and examined with a Hitachi H-7000 TEM at HV = 75 kV with a digital image acquisition system.
Statistical analyses
Numerical data are expressed as either the ratio change, % change or fold change of the means ± the standard error of the mean (sem). The Student's t-test was used to determine significance between experiments with only 2 variables. One-way analysis of variance (ANOVA) with Tukey's Multiple Comparison Post-Test was used to analyze data between multiple independent (unrelated) groups using the GraphPad Prism 5.01 software. Level of significance was first determined by ANOVA analyses (P < 0.05), and subsequently significance between every pairwise group in the experiment was determined by Tukey's Multiple Comparison Test.
Results
Ablation of p53 and Mmd2 induces cardiomyopathy and early mortality
We crossed transgenic mice expressing Cre recombinase flanked by mutated estrogen receptors (MerCreMer; mcm) with mice carrying loxP flanked alleles (f/f) of p53andMdm2, to obtain p53f/f;mcm (p53KO), Mdm2f/f;mcm (Mdm2KO) and p53f/f;Mdm2f/f;mcm (DKO) animals. These strains developed normally without an obvious cardiac phenotype in the absence of tamoxifen (Tam). Four consecutive daily intraperitoneal Tam injections induced genetic ablation of p53 and/or Mdm2 with high recombination efficiency (Fig. S1). The last day of tamoxifen injection was arbitrarily set to 0. At 3 months post-Tam, p53KO mice were normal without significant differences in heart/body weight (HBW) ratios when compared with wild-type controls.Citation36 In contrast, at 14 d post-Tam, Mdm2KO mice developed concentric cardiac hypertrophy, with an average increase of 72% (P < 0.001) in HBW ratios in comparison to wild-type littermates (data not shown). Upon knockout of p53 and Mdm2 to generate double knockout (DKO) mice, heart weight:body weight ratios (HBW) were not significantly different between DKO and vehicle-injected control mice by 8 d after Tam injection (P > 0.05) (). However, on microscopic examination of longitudinal cardiac sections stained with Masson, the DKO mice exhibited left ventricular (LV) dilatation with thinning of the LV walls () and a concomitant reduction in fractional shortening (23 ± 4%; P < 0.001) () when compared with control mice. This was accompanied with a 46% reduction (P < 0.01) in cross-sectional area of cardiomyocytes in DKO hearts as compared with vehicle-injected control mice at 8 d post Tam ( and ). RT-qPCR of DKO cardiac RNA samples revealed that these mice also developed alterations in key markers of hypertrophy and heart failure, such as atrial natriuretic peptide (ANP), which became significantly upregulated, and brain natriuretic peptide (BNP), α-myosin heavy chain (α-MHC) and β-myosin heavy chain (β-MHC), all of which were significantly downregulated compared with controls (). Surprisingly, the co-deletion of p53 and Mdm2 in DKO was lethal with 100% penetrance by 8 d (P < 0.001), whereas such an effect on life span was not observed in p53KO and Mdm2KO mice post-Tam (). Furthermore, in p53KO animals, which have preserved cardiac function,Citation36 the cardiac-specific genes are expressed in a reciprocal manner (). Thus, following the ablation of both p53 and Mdm2 in the heart, there is the development of a dilated cardiomyopathy without hypertrophy, followed by sudden death.
Figure 1. Mice with acute genetic ablation of p53 and Mdm2 develop dilated cardiomyopathy. (A) Heart-weight corrected for body weight of DKO mice (8d post-Tam). Animals were 15 weeks old at the time of analysis. Data are means ± s.e.m. n = 14. (B) Representative Masson staining of longitudinal cardiac sections of DKO mice 8d post-Tam (right) and vehicle-injected control mice (left). Data are means ± s.e.m. n = 6. (C) Fractional shortening (FS) of DKO mice determined by M-mode echocardiography 8d post-Tam. Data are means ± s.e.m. n = 6. (D) Quantification of cross-sectional area of adult DKO cardiomyocytes 8d post-Tam shown in (F). Data are means ± s.e.m. n = 6. (E) Quantification of extracellular matrix area in left ventricular sections from DKO mice post-Tam shown in (F). Data are means ± s.e.m. n = 6. (F) Analysis of remodeling in the left ventricular free wall, confocal immunofluorescence microscopy was used using co-staining with wheat germ agglutinin (extracellular matrix, ECM; green), anti-α-actinin (cardiomyocytes; red) and DAPI (nuclear DNA; blue) in p53KO (3 months), Mdm2KO (14d) and DKO (8d) post-Tam. (G) 3D reconstitution of typical confocal micrographs further illustrating the extensive cardiac fibrosis (ECM; green) in DKO mice at 8d post-Tam.
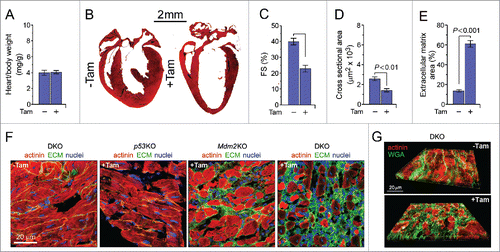
Figure 2. Premature death following acute genetic ablation of p53 and Mdm2 is elicited by drastic bradycardia followed by asystole. (A) Expression levels of hypertrophic and sarcomeric marker genes: atrial natriuretic factor (ANP), brain natriuretic factor (BNP), α-myosin heavy chain (α-MHC), β-myosin heavy chain (β-MHC), phospholamban (Pln), ryanodine receptor (Ryr2), troponin C (Tnnc1), tropomyosin (Tpm1), and titin (Ttn) as analyzed by RT-qPCR in DKO (8d), Mdm2KO (14d) and p53KO (3 months) mice post-Tam compared with vehicle-injected controls. Data are represented as the mRNA expression ratio change compared with vehicle-injected controls for each primer using the ΔΔCt method ± s.e.m, n = 4. A significant difference between the mean normalized delta Ct values versus vehicle-injected control is indicated by *P ≤ 0.05 and **P ≤ 0.01, determined by the Student's T-test. (B) Kaplan-Meier survival curves of conditional DKO, Mdm2KO and p53KO mice post-Tam. n = 10. (C) Transmission electron micrographs of left ventricular samples from vehicle-injected control (left) and DKO (right) mice. The absence of the M-line (white arrow) and the diffuse nature of Z-line (black arrow) indicate a potential change in the alignment of the actin-myosin filaments within the sarcomere. (D-G) Tam-injected DKO mice develop varying degrees of heart block and bradycardia as determined by telemetric electrocardiography. (D) DKO mice post-Tam develop sinus arrest, (E) bradycardia with loss of the circadian rhythm, (F) atrio-ventricular block, and (G) ventricular tachycardia. U, voltage. mV, millivolt. sec, seconds. One representative result of 4 independent experiments is shown. Averaged heart rate in beats per minute. One representative result of 4 independent experiments is shown.
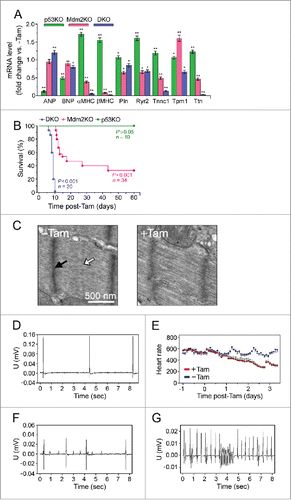
Structurally, DKO cardiomyocytes exhibit abnormalities within the sarcomeric architecture (). In addition, there is extensive interstitial fibrosis in the DKO mice, with evidence of fibrosis, although to a lesser extent, in the Mdm2KO mice (). The p53KO mice did not exhibit increased deposition of extracellular matrix, at the time points analyzed. With the early death and the degree of fibrosis observed in the DKO mice, it was important to determine if the presence of cardiac arrhythmias could be the precipitant for early death. As such, radio-telemetry units were implanted in DKO mice (+Tam) and in vehicle injected controls (-Tam). We noted irregularities involving all levels of the conduction system, including various degrees of atrio-ventricular block, ventricular tachycardia, as well as sinus arrest (). DKO succumbed as a result of profound bradycardia () followed by asystole. Consistent with this phenotype, RT-qPCR revealed a significant downregulation of phospholamban (Pln), ryanodine receptor 2 (Ryr2), troponin (Tnnc1), tropomyosin (Tpm1) and titin (Ttn), all of which are known to be mutated or deregulated in diverse arrhythmogenic conditionsCitation37 ().
Overlapping roles of p53 and Mdm2 inhibit cardiomyocyte proliferation
As we observed the presence of cardiomyocytes with reduced cross-sectional area but unchanged HBW ratios in DKO mice, we surmised that the p53/Mdm2 circuitry may not only mediate growth-related processes, but also may regulate cell cycle arrest in differentiated cells. Therefore, we investigated whether loss of p53 and Mdm2 induces DNA synthesis in DKO hearts post-Tam by intraperitoneally injecting 5-Bromo-2’-deoxyuridine (BrdU). Three dimensional confocal laser scanning microscopic analysis of cardiac sections using anti-sarcomeric α-actinin, a cytoplasmic cardiomyocyte-specific transcription marker, and BrdU antibodies showed that 78 ± 19 cardiomyocytes/mm2 in DKO hearts were undergoing DNA synthesis at 8 d post-Tam (P < 0.001) ( and ; Fig. S2). Interestingly, Mdm2KO hearts also exhibited evidence of DNA synthesis (17 ± 3 cardiomyocytes/mm2; P < 0.05), albeit to a significantly lower extent when compared with DKO post-Tam (P < 0.001) (). In contrast, deficiency of p53 alone was insufficient to induce DNA synthesis in cardiomyocytes ().
Figure 3. Co-deletion of p53 and Mdm2 triggers proliferation of adult cardiomyocytes in vivo. (A) Quantitative analysis of cardiomyocytes in S phase in p53KO, Mdm2KO and DKO mice at 4d and 8d post-Tam. The last day of Tam injection was arbitrarily set as 0 day. Data are means ± s.e.m. n = 6. (B) Confocal immunofluorescence microscopic analysis of S phase cardiomyocytes in DKO post-Tam. 5-Bromo-2’-deoxyuridine (BrdU) was intraperitoneally injected at 4d and 8d. Hearts were harvested at 4d and 8d post-Tam, and fixed tissue sections were subjected to immunostaining using anti-α-actinin (red) in conjunction with anti-BrdU to detect S phase nuclei, and DAPI to stain genomic DNA (blue). The same immunofluorescence analyses were performed for cardiac specimens from p53KO and Mdm2KO mice (data not shown). (C) Time course quantification of cardiomyocytes in M phase in p53KO, Mdm2KO and DKO mice post-Tam. Data are means ± s.e.m. n = 6. (D) Representative immunofluorescence micrograph of left ventricular sections in DKO mice at 8d post-Tam. Specimen were co-stained using antibodies recognizing Ser28 phosphorylated Histone H3 (Pi-H3) during M phase, and cardiomyocyte-specific sarcomeric actinin. The same immunofluorescence analyses were performed for p53KO and Mdm2KO LV sections (data not shown). (E) Induction of adult cardiomyocyte cytokinesis in the absence of p53KO and Mdm2KO in DKO mice at 8d post-Tam. Data are means ± s.e.m. n = 6. (F) Representative immunofluorescence micrograph of left ventricular tissue samples from DKO at 8d post-Tam using antibodies recognizing Birc5 (survivin) during cytokinesis. (G-J) 3D reconstitution of typical confocal micrographs show Birc5 (survivin) positive midbody structure (green) between 2 dividing daughter cardiomyocytes (red). Fixed DKO LV tissue sections were stained for indirect immunofluorescence microscopy analysis with antibody to α-actinin (red), antibody to Birc5 (green), and DAPI (blue) for nuclear DNA at 8d post-Tam. (K) Depiction of the distribution of Pi-H3 positive cardiomyocytes (red circles) throughout the myocardium of DKO mice at 8d post-Tam as analyzed by wide field immunofluorescence micrographs of a representative longitudinal cardiac-section using cardiomyocyte-specific antibodies to α-actinin (red) and Pi-H3 (green; middle panel). (L) Expression levels of cyclin/cyclin-dependent kinase inhibitors (Cdki) p21 and p27 in p53KO, Mdm2KO and DKO hearts in the presence and absence of Tam. Levels of endogenously expressed Cdki proteins in total LV heart tissue samples (60 mg/lane) were analyzed by immunoblotting using antibodies specific to p21 and p27. For normalization, Western blots were probed with anti-nucleophosmin (Npm1) antibody. One result of 3 independent experiments is shown. (M) Cdk2 activities in LV extracts of p53KO, Mdm2KO and DKO hearts were determined using histone H1 as substrate. Histone phosphorylation in the kinase reaction was analyzed by immunoblotting using anti-phospho-threonine antibodies. One result of 3 independent experiments is shown. (N) Quantitative analysis of Cdk2 kinase activities. Activation of Cdk2 through downregulation of inhibitory p21 and p27 is essential for cardiomyocyte proliferation. Data are means ± s.e.m, n = 3. (O) Quantitative analysis of non-cardiomyocytes (NCM) in S phase in p53KO, Mdm2KO and DKO hearts at 4d and 8d post-Tam. Left ventricular sections of p53KO, Mdm2KO and DKO mice exposed to Tam were analyzed for BrdU positive, DAPI positive and α-actinin negative S phase cells. Data are means ± s.e.m. n = 6. (P) Time course quantification of NCM in M phase. Left ventricular sections were co-stained for indirect immunofluorescence microscopy using antibodies recognizing Ser28 phosphorylated Histone H3 (Pi-H3) during M phase. Only Pi-H3 positive, DAPI positive and α-actinin negative cells were counted. Data are means ± s.e.m. n = 6. (Q) Acute deletion of Mdm2 triggers apoptosis. Analysis of apoptosis was performed by TUNEL immunofluorescence microscopy of fixed left ventricular sections derived from p53KO, Mdm2KO and DKO at 4d and 8d post-Tam. Data are means ± s.e.m. n = 4.
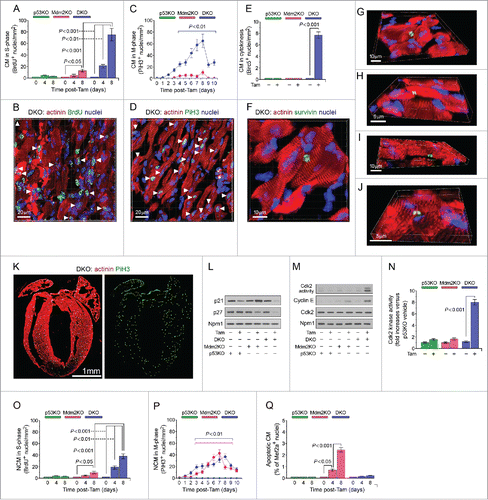
Next, we assessed cardiomyocyte mitosis by fluorescence microscopy of cardiac sections using anti-phospho-histone H3 (Pi-H3), an M-phase-specific nuclear marker with double immunostaining of anti-actinin. Microscopic inspection revealed a significantly increased number of Pi-H3 positive cardiomyocytes in DKO mice (65 ± 23 cells/mm2; P < 0.001) at 8 d after Tam administration, in comparison to control injected animals ( and , Fig. S3). Again, this effect was dramatically weaker in Mdm2KO (11 ± 4 cells/mm2; P < 0.05) and was not observed in p53KO post-Tam (P > 0.05) ().
It is well known that cardiomyocytes can be binucleated or polyploid, therefore, M-phase can occur independently of cell division. To investigate whether cardiomyocytes from DKO hearts completed mitosis by dividing into 2 cells (cytokinesis), we used anti-Birc5 (survivin) antibodies to detect midbody structures during daughter cell separation in fixed left ventricular specimens. We observed that ablation of p53/Mdm2 induced cytokinesis in 7.9 ± 1 cardiomyocytes/mm2 (P > 0.001) (; Fig. S3) post-Tam. This effect was not observed in either of the Mdm2KO and p53KO strains. Of interest, mitotic cardiomyocytes were distributed over all 4 heart chambers suggesting that the combined loss of p53 and Mdm2 endows, in principle, a widespread effect on adult cardiomyocytes to undergo DNA synthesis, progress through mitosis and ultimately, divide to form new daughter cells. ().
Based on these findings, we were curious to determine if non-cardiomyocytes (NCM) also exhibited increased proliferation in the hearts of these knockout mice. Indeed, we found that in the Mdm2KO and DKO mice, following Tam injection, there was a significant increase in non-cardiomyocytes in S and M phase ( and ). These results are consistent with the significant increase in extracellular matrix remodeling exhibited by these 2 strains ().
As p53 exerts its role as a tumor suppressor not only through cell cycle inhibition but also through induction of apoptosis,Citation1 we were interested to determine whether the deletion of p53 and Mdm2 in the heart could affect cardiomyocyte viability. We observed a marked increase in the number of TUNEL-positive cardiomyocyte nuclei in fixed left ventricular specimen from Tam-injected Mdm2KO mice (2.3 ± 0.46%; P < 0.001) as compared with controls (0.1 ± 0.02%; P < 0.001) ( and Fig. S4). Notably, significant numbers of TUNEL-positive CM nuclei were not observed in DKO and p53KO mice post-Tam. These results are consistent with the observation that loss of Mdm2 leads to upregulated levels of p53 in Mdm2KO mice (Fig. S1), thus promoting the increased occurrence of apoptosis.
Taken together, we surmise that the increased fibrosis exhibited in the Mdm2KO mice occurs as a result of the upregulated level of cardiac cell death in this strain. In the DKO mice, however, we speculate that the decreased size of the cardiomyocytes triggers non-cardiomyocyte proliferation and extracellular matrix deposition to prevent cardiac rupture, as an attempt to maintain cardiac function.
Loss of p53/Mdm2 downregulates p21Cip1 and p27Kip1 and induces Cyclin E
The lack of regenerative capacity of adult mammalian cardiomyocytes is thought to be caused in part by the unavailability of G1 cyclin-Cdks, crucial positive modulators of the cell cycle, and high levels of inhibitory cell cycle regulators, p21Cip1 and p27Kip1. To elucidate the effect of p53/Mdm2 gene ablation on these cell cycle regulators, their expression levels were examined. Immunoblot analysis revealed that p21Cip1 and p27Kip1 protein levels were downregulated in left ventricular extracts from DKO hearts treated with Tam (), with a concomitant upregulation of cyclin E levels (). Because progression through G1 and entry into S phase are tightly regulated by the enzymatic activity of Cdk2, its’ phosphotransferase activity was investigated via immune complex in vitro kinase assays. Injection of Tam did not lead to an increase in Cdk2-associated activity in p53KO and Mdm2KO mice post-Tam (). In contrast, Cdk2-dependent kinase activity was induced 6.7-fold exclusively in DKO mice post-Tam (). The gain of Cdk2 activity is consistent with the greatly reduced levels of Cdk2-inhibitory p21Cip1 and p27Kip1 and induction of cyclin E, observed in this strain. Thus, our results imply an important role for these Cdk inhibitors in the maintenance of cell cycle arrest by p53 and Mdm2 in differentiated cardiomyocytes.
The tumor suppressor p53/Mdm2 circuitry regulates expression of a unique subset of miRNAs within the heart
miRNAs are crucial modulators of cell growth and proliferation, 2 physiologic processes that we observed to be deregulated with the ablation of p53 and Mdm2. We therefore hypothesized that p53 and Mdm2 could maintain cardiomyocyte cell cycle arrest, in part, through the regulation of anti-proliferative miRNAs. To evaluate the role of p53 and Mdm2 as regulators of miRNA expression within the heart, total miRNA expression was profiled from the DKO mouse hearts, and compared with single knockouts (p53KO and Mdm2KO), and vehicle-injected controls, using the Nanostring nCounter miRNA platform. The nCounter miRNA array profiled over 600 mouse miRNAs. Heat map construction using the nSolver analysis software revealed that the Mdm2KO and DKO miRNA profiles were closely related, whereas the p53KO's miRNA profile was distinct ( and ). Setting the miRNA expression fold change cut off as greater than 1.2 or less than -1.2, a total of 89 significantly changed miRNA transcripts out of 600 (∼15%) were identified in the 3 mutant hearts compared with the vehicle-injected controls, with a P-value of ≤ 0.01 ().
Figure 4. Significant miRNA transcriptional changes in the absence of p53 and/or Mdm2 create unique miRNA profiles. (A) Heat map demonstrating miRNA fold change in the heart between the 3 different strains post-Tam injection vs. vehicle-injected controls. 600 miRNAs were profiled using the nCounter mouse miRNA panel (Nanostring). Fold change values are depicted by color intensity, where blue = repressed and red = induced. DKO (8d), Mdm2 (14d) and p53KO (3 months) mice were analyzed post-Tam, n = 3 mice per strain. (B) Heat map representing significant (P ≤ 0.01) miRNA fold changes between the 3 different strains post-Tam injection vs. vehicle-injected control with a fold change ≥ 1.2 ( ± ). 81 miRNA transcripts (rows) out of 600 (13.5%) were significantly changed in at least one of the 3 strains (columns). (C) Venn diagram analysis illustrating the overlap of significantly up and downregulated miRNAs between the 3 KOs vs. control. The 15 miRNAs uniquely downregulated in the DKO are the target miRNAs of interest. (D-F) Top 20 differentially regulated genes in (D) p53KO vs. vehicle-injected control, (E) Mdm2KO vs. vehicle-inject control and (F) DKO vs. vehicle-injected control. (P ≤ 0.01) (G) Scatter plot matrix representing normalized Log2 counts for each miRNA profiled of p53KO (green), Mdm2KO (pink) and DKO (blue) (Y axis) compared with Log2 counts of vehicle injected control (orange, X axis). Each point represents one miRNA. R2 values indicate that the p53KO miRNA profile is more closely related to that of the vehicle control than of the Mdm2KO or DKO profiles.
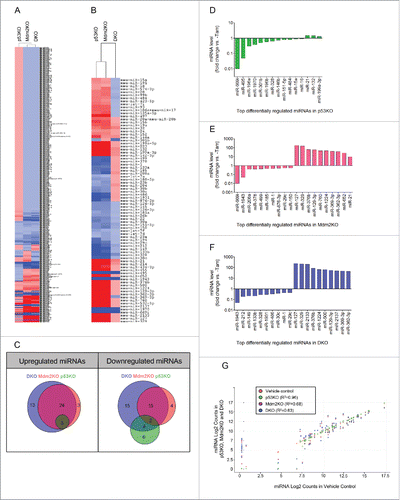
The top 20 differentially regulated miRNAs were determined for each strain. Members of the miR-15 family, well-characterized p53-regulated miRNAs,Citation38 were in the top 12 downregulated miRNAs in the p53KO hearts. These miRNAs were also significantly downregulated in the DKO, but were upregulated in the Mdm2KO, likely due to the known higher levels of p53. Interestingly, miR-1 and miR-133b, classic muscle specific miRNAs which play a critical role in myogenic differentiation,Citation39 were in the top 10 downregulated miRNAs of DKO mouse hearts (). Although the change in expression of many miRNAs overlapped between the 3 different strains, each knockout also had a subset of uniquely up and downregulated miRNAs (). We defined the subset of interest as the miRNAs uniquely downregulated in the DKO strain, as we hypothesized that inhibition of these miRNAs in wild type cardiomyocytes could promote proliferation, similarly to what is observed in the hearts of DKO mice. We identified 15 uniquely downregulated miRNAs with a significant fold change of less than −1.2 in the DKO mouse hearts (Table S1).
p53/Mdm2-regulated miRNAs are enriched for gene targets involved in cell cycle progression
To characterize the enriched target genes of the p53/Mdm2-regulated miRNAs, we used the miRSystem platform (http://mirsystem.cgm.ntu.edu.tw/index.php). In this analysis, miR-1931 was eliminated from the analysis since it did not yield any predicted target genes. Of the 14 remaining targets, 2 miRNAs (miR-133a and miR-133b) did not have target genes directly within the cell cycle pathway and were not analyzed further. To validate the miRNAs identified in the screen, RT-qPCR was performed using the same RNA sampled for the array. We found that 11/12 miRNAs were significantly downregulated in the DKO, compared with the vehicle-injected control (). In contrast, miR-212 showed an opposite expression pattern than what was determined by the array, and was eliminated from further study.
Figure 5. miRNAs downregulated in the DKO hearts are enriched for target genes within the cell cycle pathway. (A) Reverse transcription and quantitative PCR (RT-qPCR) was performed to validate miRNA expression values measured by the Nanostring nCounter mouse miRNA panel. Data are represented as the miRNA expression ratio change in DKO mouse hearts compared with controls for each primer using the ΔΔCt method ± s.e.m, n = 3. A significant difference between the mean normalized delta Ct values for the DKO vs. control is indicated by *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 determined by the Student's t-test. The 11 validated miRNAs correspond to the final miRNA hits extracted from the Nanostring microarray, whereas miR-212 was removed from further analysis. (B) Percent of each miRNA's total target genes found within the cell cycle pathway (*P ≤ 0.05, **P ≤ 0.01). (C) The number of cell cycle activating and inhibiting genes targeted by the 11 miRNA hits.
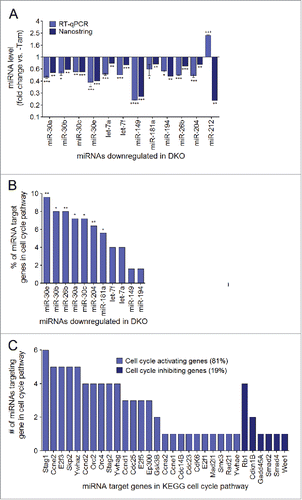
Cell cycle target genes of the remaining 11 miRNAs were subsequently investigated in depth. The most enriched biologic pathways that the target genes of these miRNAs belong to are illustrated in Figure S5. The cell cycle pathway was within the top 30th percentile of biologic pathways for this group of miRNAs, indicating that these miRNAs were enriched for target genes that function directly within this pathway (). More specifically, 26/32 miRNA target genes found within the cell cycle pathway were involved in cell cycle activation (81%), whereas only 6/32 target genes were involved in cell cycle inhibition (, Fig. S6A and B). This finding suggests that upon downregulation of these 11 miRNAs, the inhibition over a battery of key cell cycle activating genes may be alleviated, promoting their re-expression within the cell. In addition, a high degree of redundancy between the identified miRNAs and their targets within the cell cycle was observed (). Thus, we identified 11 miRNAs that were downregulated uniquely in DKO mouse hearts compared with vehicle-injected controls, and target at least one gene within the cell cycle pathway (Table S2). We defined these miRNAs as p53/Mdm2-regulated miRNAs, and surmised that they may function in cardiomyocytes as cell cycle inhibitors.
p53/Mdm2-regulated miRNAs are novel cell cycle inhibitors in cardiomyocytes
We next determined the expression levels of the 11 miRNAs in primary rat neonatal cardiomyocytes which do not proliferation appreciably, in vitro, and compared this to cardiac fibroblasts, which are known to proliferate extensively in response to cardiac injury.Citation40 RT-qPCR revealed that the expression of all 11 miRNAs was significantly decreased in fibroblasts compared with cardiomyocytes by up to 80% (Fig. S7) and therefore strengthened our hypothesis that inhibition of these miRNAs in wild type cardiomyocytes could promote cell cycle re-entry and proliferation.
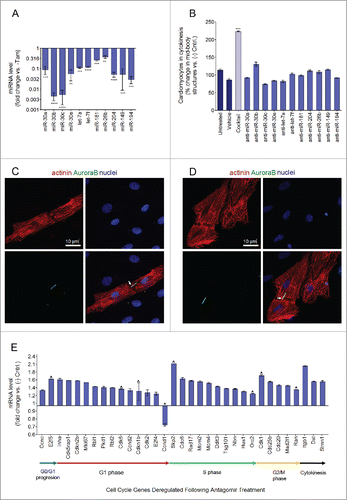
Antagomirs are chemically modified, cholesterol-conjugated, single-stranded RNA analogs complementary to miRNAs, that antagonize the function of miRNAs.Citation41,Citation42 To inhibit the 11 miRNAs identified, we transfected rat neonatal cardiomyocytes with a cocktail of 11 antagomirs targeting each individual miRNA (transfection efficiency of 93% ± 6%, Fig. S8). We were able to confirm that the use of an antagomir cocktail was highly effective in inhibiting the expression of the 11 target miRNAs (). In addition, inhibition of the 11 identified miRNAs led to greater than a 100% increase in the occurrence of cytokinetic events as compared with negative control transfected cardiomyocytes (P ≤ 0.01, n = 3) (). To quantify the degree of cardiomyocyte proliferation, we used immunofluorescence staining for AuroraB kinase (AuroraB, cytokinesis marker), co-stained with α-sarcomeric-actinin (cardiomyocyte-specific sarcomere marker), and DAPI (nuclear DNA marker) ( and ). Conversely, inhibition of any of the 11 miRNAs individually using a single antagomir was not sufficient to induce cardiomyocyte proliferation. These results highlight that the identified miRNAs likely function synergistically to maintain cell cycle arrest in cardiomyocytes, as opposed to in isolation.
Figure 6. Inhibition of p53/Mdm2-regulated miRNAs promotes cardiomyocyte cytokinesis. (A) Transfection of cardiomyocytes with the 11 antagomir cocktail efficiently knocks down all 11 target miRNAs. RT-qPCR performed for cardiomyocytes transfected with either a cocktail of all 11 antagomirs (25 nM each) or anti cel-67 (negative control, 100 nM). Data are represented as represented as miRNA expression ratio change of antagomir treated group compared with anti-cel-67 negative control group for each primer ± s.e.m, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001, n = 3 and analyzed by Student's T-test. (B) Cardiomyocytes were either not transfected, treated with only transfection reagent (MirusBio TransIT TKO), or transfected with anti-cel-67 (negative control antagomir, 100 nM) a cocktail of all 11 antagomirs (25 nM each, red bar), or each antagomir individually (25 nM, gray bars). Data represented as % change of AuroraB positive mid-body structures in treated cells compared with negative control, ± s.e.m. Data analysis by one-way ANOVA (P = 0.0035) and Tukey's Multiple Comparison Test to identify differences between groups, P ≤ 0.001 for antagomir treatment compared with negative control treatment, n = 4. (C-D) 40x confocal immunofluorescence images of cardiomyocytes transfected with a cocktail of all 11 antagomirs (25 nM each) undergoing cytokinesis. Cells were fixed and stained for α-actinin (red, cardiomyocyte specific sarcomere marker) DAPI (blue, nuclei), and AuroraB (green, cytokinesis marker). White arrows indicate AuroraB positive mid-body structures between 2 cells undergoing cytokinesis (E) Significantly changed cell cycle related genes (P ≤ 0.05; ratio change of > 1.2 or < 0.5) following treatment of cardiomyocytes with a cocktail of 11 antagomirs to inhibit identified p53/Mdm2-regulated miRNAs compared with treatment with a negative control antagomir (anti-cel-67). The Qiagen Cell Cycle RT2 Profiler PCR Array was used to profile 86 cell cycle related genes following antagomir treatment compared with anti-cel-67 treatment by RT-qPCR. Values are represented as the ratio change of treatment vs. control ± s.e.m, n = 4 and analyzed by the Student's t-test. Red chevrons indicate genes that the miRSystem software predicted to be target genes for the 11 inhibited miRNAs.
p53/Mdm2-regulated miRNAs inhibit cardiomyocyte proliferation by targeting cell cycle genes
To elucidate the mechanism by which inhibition of the 11 identified miRNAs promotes cell cycle reactivation, a cell cycle pathway specific RT-qPCR array (Qiagen) was used. This array profiles the expression of 86 cell cycle related genes (), and was used to determine which specific cell cycle targets genes were affected by the antagomir treatment (listed in , Fig. S6A and B). Of the 32 genes within the cell cycle pathway predicted to be targeted by the 11 identified miRNAs, 20 were contained in the RT-qPCR cell cycle array. This array would therefore allow us to confirm certain predicted miRNA target genes, and also provide the opportunity to identify novel, previously unidentified, gene targets. The result of this analysis revealed a widespread upregulation in factors involved in cell cycle re-entry and progression. More specifically, inhibition of the 11 miRNAs caused a significant (P≥ 0.05) upregulation of 32 genes involved in cell cycle regulation (∼36% of genes on the array) () that could be found in all 4 stages of the cell cycle. For example, we found an upregulation of cyclin D2, which promotes G1 phase progression, minichromosome maintenance proteins 2/4 (Mcm2/4) and origin of replication complex subunit 2 (Orc2) which are necessary for DNA replication in S phase, cell division cycle 25 (Cdc25) which controls the rate limiting step of Cdk1 activation in late G2 phase, and importantly, Cdk1, the essential kinase that induces M phase initiation and progression ().Citation43 Only one gene from this array, cyclin D1, was found to be significantly downregulated by the antagomir treatment. This supports the canonical role of miRNAs as inhibitors of gene expression, such that their inhibition by antagomirs allowed for a widespread upregulation of their target genes. Notably, 40% (8/20) of the miRNAs’ predicted cell cycle target genes profiled by the array were upregulated following miRNA inhibition (). The extensive effects that inhibition of p53/Mdm2-regulated miRNAs has on the expression of a plethora of crucial cell cycle genes supports their role as strategic mediators of cell cycle arrest in cardiomyocytes.
Discussion
In this paper, we present evidence for a non-canonical role of p53 in maintaining normal cardiac function, in cooperation with its inhibitor Mdm2. As demonstrated in germ line p53 knockout mice and embryonic in situ hybridization studies, p53 plays a role in the regulation of normal embryonic development, including neural tube formationCitation44 and cardiogenesis.Citation45 In our study, p53 and Mdm2 were acutely inactivated in the physiologic context of the adult heart. This genetic manipulation led to the development of heart failure and sudden death within 10 d. Intriguingly, we have previously demonstrated that acute deletion of p53 was itself insufficient to evoke gross cardiac abnormalities.Citation36 This observation led us to realize that, in addition to p53, basal Mdm2 activity is indispensable for the control of proper heart function. Together, the p53/Mdm2 circuitry is a major regulator of cardiac differentiation and function in experimental murine models.
As an essential function of p53 is to induce cell-cycle arrest under conditions of stress, combined with the observation of smaller cardiomyocytes in DKO mice, we investigated whether p53/Mdm2 also regulated the cell cycle in cardiomyocytes under basal conditions. Ablation of p53 and Mdm2 in the adult mouse heart was sufficient to induce cardiomyocyte cell cycle re-entry in all 4 chambers of the heart. Nnot only did cell cycle re-entry occur, but it was completed to its entirety, resulting in the formation of the midbody just before cytokinesis. Importantly, knockout of either gene individually was insufficient to promote cardiomyocyte proliferation.
In wild type mice, cardiomyocyte cell cycle arrest is characterized by the inactivity of Cdk2 because of undetectable cyclin E and the presence of high levels of the Cdk inhibitors p21Cip1 and p27Kip1. Mechanistically, we show that p53/Mdm2 inactivation suffices to downregulate these critical Cdk inhibitors, and activate Cdk2 through cyclin E induction. Thus, these data suggest that p53, together with Mdm2 maintain multiple roadblocks that inhibit cardiomyocyte cell cycle entry and cell division, making them novel inhibitors of cardiomyocyte proliferation.
It is well known that p53 represses gene transcription of many genes via direct transactivation of various miRNAs, some of which inhibit the translation of crucial cell cycle activators. As our findings demonstrate that the presence of either p53 or Mdm2 is necessary and sufficient to block cardiomyocyte cell cycle re-entry, this supports the importance of Mdm2 in the context of possible miRNA regulation within the p53 pathway. We thus sought to determine the role of p53 and Mdm2 on miRNA regulation in the heart. Following knockout of p53 and Mdm2, we were able to identify distinct populations of miRNAs that are regulated by each gene individually, as well as in tandem. Loss of p53 led to the downregulation of 12 miRNAs whereas 19 and 29 miRNAs were downregulated in the Mdm2KO and DKO, respectively. Furthermore, knockout of p53 produced an upregulation of only 3 miRNAs, whereas the Mdm2KO had 29 upregulated genes, and DKO had 36, many of which overlapped between the 2 strains, indicating that the over-activation of p53 in the Mdm2KO is not the sole cause of the gene expression changes observed. This highlights a potentially new role for Mdm2, in regulating the stability of other miRNA transcriptional activators yet to be identified.
The cell cycle is a molecular cascade that involves numerous feedback loops, with an expansive number of regulators with overlapping functions. The robustness of this network highlights the evolutionary importance of this pathway, as the cell utilizes the tight interplay of a large number of proteins with degrees of functional redundancy, such that the loss of a single factor does not result in a mitotic catastrophe.Citation46 For example, knockout of Cdk2, Cdk4 or Cdk6 alone does not produce a significant effect on cell proliferation.Citation47-50 Cdk1 has been shown to compensate for the loss of Cdk2 and is capable to bind cyclin E in S phase,Citation50 and Cdk2 was shown to assume the functions of Cdk4/6 upon their loss.Citation49 It therefore follows that miRNAs could play an important role in maintaining quiescence in differentiated cells, as their capacity to regulate the expression of an expansive number of genes gives them the ability to simultaneously modify the expression of a multitude of cell cycle regulators, overcoming the internal redundancy of this pathway.
We found that cardiac ablation of p53 and Mdm2 led to the downregulation of a unique subset of 11 miRNAs which were not significantly changed in the hearts of either single knockout. Through bioinformatic target gene analysis, we found that these miRNAs possess multiple target genes within the cell cycle, and that a large majority of these target genes are involved in cell cycle activation. Upon inhibition of these miRNAs in wild type cardiomyocytes through the use of antagomirs, over a 100% increase in cytokinesis was elicited. This indicates that these miRNAs have a broad effect over cell cycle control, and that their inhibition not only induces re-entry, but the completion of this pathway to its entirety, resulting in the formation of new cardiomyocytes. Furthermore, a degree of redundancy in miRNA target genes was an important realization, as inhibiting each of the identified miRNAs individually proved to be insufficient to promote cardiomyocyte proliferation. This suggests that the expression of multiple miRNAs with overlapping targets can promote synergy, increasing their potency within the cell to shut off biologic pathways, while enhancing others.Citation51
To determine a mechanism by which cardiomyocyte proliferation was enhanced, we investigated the expression of key cell cycle regulators following transfection of the antagomir cocktail inhibiting the 11 identified miRNAs. This treatment upregulated the expression of a significant percentage cell cycle regulating genes, including multiple genes predicted by the target gene prediction software that we used. Conversely, out of the 86 cell cycle genes profiled, only cyclin D1 was significantly downregulated by the treatment. This demonstrates that the knockdown of the identified miRNAs alleviates their inhibition over a large number of genes essential for cell cycle entry (early G1 phase), progression (G1/S transition, S phase, G2/M transition, M phase) and completion (cytokinesis). The majority of the genes upregulated were cell cycle activators, but key cell cycle inhibitors such as retinoblastoma (Rb), retinoblastoma-like protein 1 (Rbl1/p107) and retinoblastoma-like protein 2 (Rbl2/p130) were also induced. This finding indicates that a large majority of the cardiomyocytes were likely in G1 phase, a phase of the cell cycle in which the expression of these cell cycle inhibitors is important for the timely coordination of E2f-mediated gene expression.Citation52 Factors that promote G1/S phase (Cdk6, Skp2), S phase (Mcm2, Mcm4) and G2/M transition (Cdc25b) were also upregulated. Importantly, this would be redundant if Cdk1 was not also expressed.Citation53 We were able to demonstrate a significant upregulation of the expression of Cdk1 in cardiomyocytes, indicating that not only can the cardiomyocytes enter into the cell cycle, but can also proceed through mitosis. In addition, factors involved in M phase (Mad2l1) and progression through cytokinesis (Stmn1) were significantly upregulated following antagomir treatment.Citation54 Collectively, we have been able to show that many of the factors controlling the intricate orchestration of cell cycle re-entry and progression are controlled by a group of “anti-proliferative” miRNAs regulated by p53/Mdm2. Therefore, the inhibition of these miRNAs permits the re-entry and controlled cycling of cardiomyocytes through each phase of the cell cycle.
Numerous other studies have also attempted to induce cardiomyocyte proliferation with the expectation of promoting cardiac regeneration or repair. Overexpression of cell cycle activators such as cyclin D1, D2 or D3 in cardiac-specific transgenic mice resulted in upregulated DNA synthesis.Citation55 More specifically, transgenic overexpression of cyclin D2 in mice was the only model where persistent DNA synthesis was observed, albeit, at a low efficiency.Citation55 In a different model system, cyclin D1 overexpression in cardiomyocytes upregulated DNA synthesis more efficiently. However, no progression into M phase was observed due to a lack of Cdk1 activation.Citation56 In contrast, constitutive overexpression of cyclin A2 induced an increased number of cardiomyocytes in M phase as detected by phosphorylated histone H3.Citation57 While the loss of cell cycle inhibitors p27Citation58 or Meis1Citation59 has yielded promise through the indirect upregulation of multiple positive cell cycle regulators that become suppressed during terminal differentiation, the exact mechanism remains to be elucidated.
The demonstration of cardiomyocytes entering the cell cycle and undergoing cytokinesis is problematic. Surrogate markers of cell division, such as phosphorylated histone H3, a marker of M phase, or the antigen Ki-67, are more commonly used, as opposed to detecting the actual event of cytokinesis. One of the major strengths of our study is the detection of the midbody structure, in vivo. This structure is formed at the end of M phase and is present throughout cytokinesis.Citation60 The detailed analysis requiring 3-dimensional reconstruction to ascertain the cellular origin of the signal, provides the unequivocal evidence of cardiomyocytes undergoing cell division.
Induction of cardiomyocyte proliferation likely requires targeting multiple cell cycle checkpoints, as opposed to a single cell cycle regulating factor.Citation61 In our study, we have shown that targeting multiple miRNAs to affect the cell cycle at all stages leads to induction of cardiomyocyte proliferation. In other studies, the Hippo pathway, a highly evolutionarily conserved signaling pathway that regulates proliferation, organ size, and cell survival, has been shown to regulate cardiomyocyte proliferation.Citation62-64 Specifically, it has been shown that knockdown of the upstream effector proteins in the Hippo pathway, or overexpression of the downstream target protein Yes associated protein 1 (Yap1), can promote cardiomyocyte proliferation and ventricular wall thickening.Citation62 Conversely, inhibition of Yap1 during the course of cardiac development leads to cardiomyocyte hypoplasia and lethality.Citation63 The overexpression of phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit β (Pik3cb), a downstream target of Yap1, can upregulate cardiomyocyte proliferationCitation64 at a frequency of less than 0.01%, indicating how inefficient this approach is.Citation62 For comparison, the rate of cytokinesis observed in our study is 1.7%.
Administration of signaling molecules to the myocardium has also promoted cardiomyocyte cell cycle re-entry. For example, treatment of cardiomyocytes with fibroblast growth factor-1, along with an inhibitor to p38 MAPK, promotes cardiomyocyte proliferation, through a mechanism that promoted initial sarcomere dedifferentiation, followed by proliferation.Citation65 In another approach, infusion of the molecule neuregulin which signals through the Erbb2 receptor tyrosine kinase 2/4 pathway, was also shown to promote fetal and adult cardiomyocyte proliferation.Citation66 Neuregulin was subsequently infused into patients with heart failure and promoted an acute improvement of cardiac function that lasted for up to 3 months. However, the mechanism of this functional improvement remains to be elucidated.Citation67
miRNAs also have emerged as important regulators of cardiac development and the mediation of cell cycle arrest. Overexpression of miR-590 and miR-199a was capable of promoting cardiomyocyte proliferation.Citation68 Others have demonstrated that downregulating the expression of miR-15Citation69 and miR-34Citation70 family or the miR-17–92Citation71 cluster, can promote cardiomyocyte cell cycle re-entry and upregulate cytokinesis. miRNAs are attractive therapeutic targets because the miRNA mimic and antagomir technologies that have been developed can be delivered as synthetic small molecules that have the capability of entering into cells and exerting their function in vivo transiently and without the use of viral vecotrs or other genome editing techniques such as CRISPR.Citation72 This is important because our work clearly demonstrates that uncontrolled levels of proliferation can have detrimental effects on the structural integrity of the heart.
In all, the technique of stimulating endogenous cardiomyocyte cell cycle re-entry and proliferation is attractive in the field of cardiac regeneration, because it is through this mechanism that regeneration occurs in lower vertebrates, as well as in neonatal mammals.Citation73 This method promotes the generation of new, autologous cardiomyocytes, which implies that they will be mechanically, electrically and vascularly integrated into the myocardium, because they arise from cells that already have these properties, as opposed to exogenous cells that have to surpass the hurdles posed by engraftment. Future work remains to determine whether the level of cardiomyocyte proliferation that has been successfully induced in study and in others, is sufficient to expand the cardiomyocyte population in a clinically meaningful manner. Importantly, the optimal method for cardiomyocyte cell cycle reactivation is still up for debate. It is possible that taking a multi-pronged approach by combining multiple methods such as targeting miRNAs, in tandem with the administration of certain cytokines such as fibroblast growth factor-1, might be more effective than any one method on their own.
In conclusion, this present study provides strong genetic evidence for the role of the p53/Mdm2 pathway in the maintenance of adult cardiac homeostasis and the post-mitotic state. Furthermore, we have shown that cardiomyocyte proliferation is inhibited by the synergistic function of a group of miRNAs under the regulation of this critical tumor suppressor circuitry. These data provide a new level of understanding as to how cell cycle arrest is regulated in the heart, and how p53 and Mdm2 participate in the mediation of this process. Therefore, manipulation of the p53/Mdm2 pathway, through the downstream miRNA targets to regenerate cardiomyocytes, could potentially be a novel approach to be harnessed in future clinical strategies to improve cardiac function after injury.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KCCY_S_1346758.docx
Download MS Word (8.9 MB)Funding
Funding provided by Government of Canada | CIHR | Institute of Circulatory and Respiratory Health (ICRH) (130288).
References
- Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer 2014; 14:359-70; PMID:24739573; https://doi.org/10.1038/nrc3711
- Daniely Y, Dimitrova DD, Borowiec JA. Stress-Dependent Nucleolin Mobilization Mediated by p53-Nucleolin Complex Formation. Mol. Cell. Biol 22, 6014-22 (2002); PMID:12138209; https://doi.org/10.1128/MCB.22.16.6014-6022.2002
- Olson MOJ. Sensing cellular stress: another new function for the nucleolus? Sci. STKE 2004; 2004:pe10.
- Bouafia A, Corre S, Gilot D, Mouchet N, Prince S, Galibert MD. p53 Requires the Stress Sensor USF1 to Direct Appropriate Cell Fate Decision. PLoS Genet 2014; 10:e1004309; PMID:24831529; https://doi.org/10.1371/journal.pgen.1004309
- Olovnikov I, Kravchenko J, Chumakov P. Homeostatic functions of the p53 tumor suppressor: regulation of energy metabolism and antioxidant defense. Semin Cancer Biol 2009; 19:32-41; PMID:19101635; https://doi.org/10.1016/j.semcancer.2008.11.005
- Itahana K, Dimri G, Campisi J. Regulation of cellular senescence by p53. Eur. J. Biochem 2001; 268:2784-91; PMID:11358493; https://doi.org/10.1046/j.1432-1327.2001.02228.x
- Qian Y, Chen X. Senescence regulation by the p53 protein family. Methods Mol. Biol 2013; 965:37-61; PMID:23296650
- Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005; 436:725-30; PMID:16079851; https://doi.org/10.1038/nature03918
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Curr Biol 1994; 4:1-7; PMID:7922305; https://doi.org/10.1016/S0960-9822(00)00002-6
- Malkin D. Li-fraumeni syndrome. Genes Cancer 2011; 2:475-84; PMID:21779515; https://doi.org/10.1177/1947601911413466
- Zhao Z, Zuber J, Diaz-Flores E, Lintault L, Kogan SC, Shannon K, Lowe SW. p53 loss promotes acute myeloid leukemia by enabling aberrant self-renewal. Genes Dev 2010; 24:1389-402; PMID:20595231; https://doi.org/10.1101/gad.1940710
- Hu W, Feng Z, Levine AJ. The Regulation of Multiple p53 Stress Responses is Mediated through MDM2. Genes Cancer 2012; 3:199-208; PMID:23150753; https://doi.org/10.1177/1947601912454734
- Shi D, Gu W. Dual Roles of MDM2 in the Regulation of p53: Ubiquitination Dependent and Ubiquitination Independent Mechanisms of MDM2 Repression of p53 Activity. Genes Cancer 2012; 3:240-8; PMID:23150757; https://doi.org/10.1177/1947601912455199
- Iwakuma T, Lozano G. MDM2, An Introduction 2003;. 1:993-1000.
- Moll UM, Petrenko O. The MDM2-p53 interaction. Mol Cancer Res 2003; 1:1001-8; PMID:14707283
- de Oca Luna RM, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 1995; 378:203-6; PMID:7477326; https://doi.org/10.1038/378203a0
- Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 1995; 378:206-8; PMID:7477327; https://doi.org/10.1038/378206a0
- Agarwal ML, Agarwal A, Taylor WR, Stark GR. p53 controls both the G2/M and the G1 cell cycle checkpoints and mediates reversible growth arrest in human fibroblasts (p21/WAF1/Li-Fraumeni cells/tetracycline/mimosine/cyclin-cyclin-dependent kinase). Cell Biol 1995; 92:8493-97.
- El-Deiry W, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993; 75:817-25; PMID:8242752; https://doi.org/10.1016/0092-8674(93)90500-P
- Fischer M, Steiner L, Engeland K. The transcription factor p53: not a repressor, solely an activator. Cell Cycle 2014; 13:3037-58; PMID:25486564; https://doi.org/10.4161/15384101.2014.949083
- Hünten S, Kaller M, Drepper F, Oeljeklaus S, Bonfert T, Erhard F, Dueck A, Eichner N, Friedel CC, Meister G, et al. p53-Regulated Networks of Protein, mRNA, miRNA, and lncRNA Expression Revealed by Integrated Pulsed Stable Isotope Labeling With Amino Acids in Cell Culture (pSILAC) and Next Generation Sequencing (NGS) Analyses. Mol. Cell. Proteomics 2015; 14:2609-29; PMID:26183718; https://doi.org/10.1074/mcp.M115.050237
- He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet 2004; 5:522-31; PMID:15211354; https://doi.org/10.1038/nrg1379
- Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 2014; 15:509-24; PMID:25027649; https://doi.org/10.1038/nrm3838
- Wahid F, Shehzad A, Khan T, Kim YY. MicroRNAs: Synthesis, mechanism, function, and recent clinical trials. Biochim Biophys Acta - Mol Cell Res 2010; 1803:1231-43; https://doi.org/10.1016/j.bbamcr.2010.06.013
- Hermeking H. MicroRNAs in the p53 network: micromanagement of tumour suppression. Nat Rev Cancer 2012; 12:613-26; PMID:22898542; https://doi.org/10.1038/nrc3318
- Raver-Shapira N, Marciano E, Meiri E, Spector Y, Rosenfeld N, Moskovits N, Bentwich Z, Oren M. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell 2007; 26:731-43; PMID:17540598; https://doi.org/10.1016/j.molcel.2007.05.017
- Chang T-C, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH, Feldmann G, Yamakuchi M, Ferlito M, Lowenstein CJ, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol. Cell 2007; 26:745-52; PMID:17540599; https://doi.org/10.1016/j.molcel.2007.05.010
- Bommer GT, Gerin I, Feng Y, Kaczorowski AJ, Kuick R, Love RE, Zhai Y, Giordano TJ, Qin ZS, Moore BB, et al. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr. Biol 2007; 17:1298-307; PMID:17656095; https://doi.org/10.1016/j.cub.2007.06.068
- Hinkel R, Ng JKM, Kupatt C. Targeting microRNAs for cardiovascular therapeutics in coronary artery disease. Curr Opin Cardiol 2014; 29:586-94; PMID:25159281
- Hunt SA, Abraham WT, Chin MH, Feldman AM, Francis GS, Ganiats TG, Jessup M, Konstam MA, Mancini DM, Michl K, Oates JA, Rahko PS, Silver MA, Stevenson LW. Y. C. ACC/AHA 2005 guideline update for the diagnosis and management of chronic heart failure in the adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guideli. Am. Coll. Cardiol. Web Site. Circulation 2006; 113(7):e166-286; available at: http://www.acc.org/clinical/guidelines/failure//index.pdf
- Bolli R, Chugh AR, D'Amario D, Loughran JH, Stoddard MF, Ikram S, Beache GM, Wagner SG, Leri A, Hosoda T, et al. Cardiac stem cells in patients with ischaemic cardiomyopathy (SCIPIO): Initial results of a randomised phase 1 trial. Lancet 2011; 378:1847-57; PMID:22088800
- Senyo SE, Lee RT, Kühn B. Cardiac regeneration based on mechanisms of cardiomyocyte proliferation and differentiation. Stem Cell Res 2014; 13:532-41; PMID:25306390
- Krum H, Abraham WT. Heart failure. Lancet 2009; 373:941-55; PMID:19286093
- Song H, Conte JV, Foster AH, McLaughlin JS, Wei C. Increased p53 protein expression in human failing myocardium.e. J Hear Lung Transplant 1999; 18:744-9.
- Ehler E, Moore-Morris T, Lange S. Isolation and Culture of Neonatal Mouse Cardiomyocytes. J Vis Exp 2013; e50154-e50154; https://doi.org/10.3791/50154
- Mak TW, Hauck L, Grothe D, Billia F. p53 regulates the cardiac transcriptome. Proc Natl Acad Sci U S A 2017; 114:2331-36; PMID:28193895
- van Opbergen CJM, Delmar M, van Veen TAB. Potential new mechanisms of pro-arrhythmia in arrhythmogenic cardiomyopathy: focus on calcium sensitive pathways. Netherlands Hear J 2017; 25(3):157-69; https://doi.org/10.1007/s12471-017-0946-7
- Shi L, Jackstadt R, Siemens H, Li H, Kirchner T, Hermeking H. p53-induced miR-15a/16-1 and AP4 form a double-negative feedback loop to regulate epithelial-mesenchymal transition and metastasis in colorectal cancer. Cancer Res 2014; 74:532-42; PMID:24285725; https://doi.org/10.1158/0008-5472.CAN-13-2203
- Koutsoulidou A, Mastroyiannopoulos NP, Furling D, Uney JB, Phylactou LA. Expression of miR-1, miR-133a, miR-133b and miR-206 increases during development of human skeletal muscle. BMC Dev Biol 2011; 11:34; PMID:21645416; https://doi.org/10.1186/1471-213X-11-34
- Chen W, Frangogiannis NG. Fibroblasts in post-infarction inflammation and cardiac repair. Biochim Biophys Acta 2013; 1833:945-53; https://doi.org/10.1016/j.bbamcr.2012.08.023
- Krützfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature 2005; 438:685-89; PMID:16258535; https://doi.org/10.1038/nature04303
- Krützfeldt J, Kuwajima S, Braich R, Rajeev KG, Pena J, Tuschl T, Manoharan M, Stoffel M. Specificity, duplex degradation and subcellular localization of antagomirs. Nucleic Acids Res 2007; 35:2885-92; PMID:17439965; https://doi.org/10.1093/nar/gkm024
- Rhind N, Russell P. Signaling pathways that regulate cell division. Cold Spring Harb. Perspect Biol 2012; 4:a005942-; PMID:23028116; https://doi.org/10.1101/cshperspect.a005942
- Sah VP, Attardi LD, Mulligan GJ, Williams BO, Bronson RT, Jacks T. A subset of p53-deficient embryos exhibit exencephaly. Nat Genet 1995; 10:175-80; PMID:7663512; https://doi.org/10.1038/ng0695-175
- Krinka D, Raid R, Pata I, Kärner J, Maimets T. In situ hybridisation of chick embryos with p53-specific probe and their immunostaining with anti-p53 antibodies. Anat. Embryol. (Berl) 2001; 204:207-15; PMID:11681800; https://doi.org/10.1007/s004290100195
- Bueno MJ, Malumbres M. MicroRNAs and the cell cycle. Biochim. Biophys. Acta - Mol. Basis Dis 2011; 1812:592-601; https://doi.org/10.1016/j.bbadis.2011.02.002
- Berthet C, Kaldis P. Cdk2 and Cdk4 cooperatively control the expression of Cdc2. Cell Div 2006; 1:10; PMID:16759374; https://doi.org/10.1186/1747-1028-1-10
- Malumbres M, Sotillo R, Santamaría D, Galán J, Cerezo A, Ortega S, Dubus P, Barbacid M. Mammalian Cells Cycle without the D-Type Cyclin-Dependent Kinases Cdk4 and Cdk6. Cell 2004; 118:493-504; PMID:15315761; https://doi.org/10.1016/j.cell.2004.08.002
- Berthet C, Aleem E, Coppola V, Tessarollo L, Kaldis P. Cdk2 Knockout Mice Are Viable. Curr Biol 2003; 13:1775-85; PMID:14561402; https://doi.org/10.1016/j.cub.2003.09.024
- Aleem E, Kiyokawa H, Kaldis P. Cdc2–cyclin E complexes regulate the G1/S phase transition. Nat. Cell Biol 2005; 7:831-6; PMID:16007079; https://doi.org/10.1038/ncb1284
- Huang W, Tian SS, Hang PZ, Sun C, Guo J, Du ZM. Combination of microRNA-21 and microRNA-146a Attenuates Cardiac Dysfunction and Apoptosis During Acute Myocardial Infarction in Mice. Mol Ther Acids 2016; 5:e296; https://doi.org/10.1038/mtna.2016.12
- Hurford RK, Cobrinik D, Lee M-H, Dyson N. pRB and p107/p130 are required for the regulated expression of different sets of E2F responsive genes. Genes Dev 1997; 11(11):1447-63.
- Abe S, Nagasaka K, Hirayama Y, Kozuka-Hata H, Oyama M, Aoyagi Y, Obuse C, Hirota T. The initial phase of chromosome condensation requires Cdk1-mediated phosphorylation of the CAP-D3 subunit of condensin II. Genes Dev 2011; 25:863-74; PMID:21498573; https://doi.org/10.1101/gad.2016411
- Baquero MT, Hanna JA, Neumeister V, Cheng H, Molinaro AM, Harris LN, Rimm DL. Stathmin expression and its relationship to microtubule-associated protein tau and outcome in breast cancer. Cancer 2012; 118:4660-9; PMID:22359235; https://doi.org/10.1002/cncr.27453
- Pasumarthi KBS, Nakajima H, Nakajima HO, Soonpaa MH, Field LJ. Targeted Expression of Cyclin D2 Results in Cardiomyocyte DNA Synthesis and Infarct Regression in Transgenic Mice. Circ Res 2004; 96:110-118; PMID:15576649; https://doi.org/10.1161/01.RES.0000152326.91223.4F
- Tane S, Okayama H, Ikenishi A, Amemiya Y, Nakayama KI, Takeuchi T. Two inhibitory systems and CKIs regulate cell cycle exit of mammalian cardiomyocytes after birth. Biochem Biophys Res Commun 2015; 466:147-154; PMID:26363457; https://doi.org/10.1016/j.bbrc.2015.08.102
- Cheng RK, Asai T, Tang H, Dashoush NH, Kara RJ, Costa KD, Naka Y, Wu EX, Wolgemuth DJ, Chaudhry HW. Cyclin A2 induces cardiac regeneration after myocardial infarction and prevents heart failure. Circ Res. 2007; 100(12):1741-1748; PMID:17495221; https://doi.org/10.1161/CIRCRESAHA.107.153544
- Tamamori-Adachi M, Hayashida K, Nobori K, Omizu C, Yamada K, Sakamoto N, Kamura T, Fukuda K, Ogawa S, Nakayama KI, et al. Down-regulation of p27Kip1 promotes cell proliferation of rat neonatal cardiomyocytes induced by nuclear expression of cyclin D1 and CDK4. Evidence for impaired Skp2-dependent degradation of p27 in terminal differentiation. J Biol Chem. 2004; 279(48):50429-50436. doi:10.1074/jbc.M403084200; PMID:15371458; https://doi.org/10.1074/jbc.M403084200
- Muralidhar SA, Sadek HA. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Etiol Morphog Congenit Hear Dis From Gene Funct Cell Interact to Morphol. 2016; 497(7448):93-101. https://doi.org/10.1007/978-4-431-54628-3_11
- Murata-Hori M, Tatsuka M, Wang Y-L. Probing the dynamics and functions of aurora B kinase in living cells during mitosis and cytokinesis. Mol Biol Cell 2002; 13:1099-108; PMID:11950924; https://doi.org/10.1091/mbc.01-09-0467
- Lin Z, Pu WT. Strategies for cardiac regeneration and repair. Sci Transl Med 2014; 6:239rv1; PMID:24898748; https://doi.org/10.1126/scitranslmed.3006681
- Lin Z, Zhou P, von Gise A, Gu F, Ma Q, Chen J, Guo H, van Gorp PR, Wang DZ, Pu WT. Pi3kcb links Hippo-YAP and PI3K-AKT signaling pathways to promote cardiomyocyte proliferation and survival. Circ Res 2015; 116:35-45; PMID:25249570; https://doi.org/10.1161/CIRCRESAHA.115.304457
- von Gise A, Lin Z, Schlegelmilch K, Honor LB, Pan GM, Buck JN, Ma Q, Ishiwata T, Zhou B, Camargo FD, et al. YAP1, the nuclear target of Hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proc Natl Acad Sci 2012; 109:2394-99; https://doi.org/10.1073/pnas.1116136109
- Zhou J. An emerging role for Hippo-YAP signaling in cardiovascular development. J Biomed Res 2014; 28:251-4.
- Engel FB, Schebesta M, Duong MT, Lu G, Ren S, Madwed JB, Jiang H, Wang Y, Keating MT. p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes Dev 2005; 19:1175-87; https://doi.org/10.1101/gad.1306705.chaemic
- Bersell K, Arab S, Haring B, Kühn B. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell 2009; 138:257-70; PMID:19632177; https://doi.org/10.1016/j.cell.2009.04.060
- Jabbour A, Hayward CS, Keogh AM, Kotlyar E, McCrohon JA, England JF, Amor R, Liu X, Li XY, Zhou MD, et al. Parenteral administration of recombinant human neuregulin-1 to patients with stable chronic heart failure produces favourable acute and chronic haemodynamic responses. Eur J Heart Fail 2011; 13:83-92; PMID:20810473; https://doi.org/10.1093/eurjhf/hfq152
- Eulalio A, Mano M, Dal Ferro M, Zentilin L, Sinagra G, Zacchigna S, Giacca M. Functional screening identifies miRNAs inducing cardiac regeneration. Nature 2012; 492:376-81; PMID:23222520; https://doi.org/10.1038/nature11739
- Porrello ER, Johnson BA, Aurora AB, Simpson E, Nam YJ, Matkovich SJ, Dorn GW 2nd, van Rooij E, Olson EN. MiR-15 family regulates postnatal mitotic arrest of cardiomyocytes. Circ Res 2011; 109:670-9; PMID:21778430; https://doi.org/10.1161/CIRCRESAHA.111.248880
- Yang Y, Cheng HW, Qiu Y, Dupee D, Noonan M, Lin YD, Fisch S, Unno K, Sereti KI, Liao R. MicroRNA-34a Plays a Key Role in Cardiac Repair and Regeneration Following Myocardial Infarction. Circ Res 2015; 117(5):450-59; PMID:26082557; https://doi.org/10.1161/CIRCRESAHA.117.305962
- Chen J, Huang ZP, Seok HY, Ding J, Kataoka M, Zhang Z, Hu X, Wang G, Lin Z, Wang S, et al. Mir-17-92 cluster is required for and sufficient to induce cardiomyocyte proliferation in postnatal and adult hearts. Circ Res 2013; 112(12):1557-1566; PMID:23575307; https://doi.org/10.1161/CIRCRESAHA.112.300658
- Wahlquist C, Jeong D, Rojas-Muñoz A, Kho C, Lee A, Mitsuyama S, van Mil A, Park WJ, Sluijter JP, Doevendans PA, et al. Inhibition of miR-25 improves cardiac contractility in the failing heart. Nature 2014; 508:531-5; PMID:24670661; https://doi.org/10.1038/nature13073
- Graham RM, Harvey RP. The Ontogeny of Cardiac Regeneration. Circ Res 2011; 108:1304-05; PMID:21617133; https://doi.org/10.1161/RES.0b013e318222ba1a