
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
The purpose of this research has been deciphering the Warburg paradox, the biochemical enigma unsolved since 1923. We solved it by demonstrating that its specific character, i.e. the forced aerobic lactate exportation, represents a crucial metabolic device to counteract the cytotoxic effect produced by an excess of pyruvate at the connection of glycolysis with the Krebs cycle. This solution was verified by exposing cancer cells of different histogenesis to pyruvate concentrations higher than the physiological ones, after showing that these concentrations are totally innocuous when injected into mice. The mechanism of the pyruvate cytotoxicity relies on the saturation of the respiratory chain, leading to a negative shift of the cytosolic NADP/NADPH ratio and the consequent restriction of the purine synthesis and the related cell apoptosis. The reducing equivalents generated by glycolysis and by cytosolic metabolism compete each other for their disposal trough the respiratory chain; this makes it that the cytotoxicity of pyruvate is inversely related to the mitochondrial number and efficiency of various cell types. Thus, the cytotoxicity is high in anaplastic cancer stem cells, whose mitochondria are extremely few and immature (cristae-poor); on the contrary, no inhibition is brought about in adult differentiated cells, physiologically rich of mature mitochondria. All this generates the pyruvate anticancer selectivity, together with the lack of a general toxicity, making pyruvate represent an ideal candidate for a radical non toxical anticancer treatment.
Introduction
The modern oncology converges to the idea that the cancer stem cells (CSCs) pool may be selected target to eradicate the neoplastic disease [Citation1]. This perspective highly incites investigations on origin, fate and evolution of the cell populations confluent in the neoplastic stem cell compartment. Innumerable studies are aimed at finding aggressive treatments selective for the CSCs rather than the stem cells normally living in differentiated tissues. The limelight is nowadays occupied by the search of metabolic features distinguishing normal from cancer stem compartments, to be employed for selective anticancer treatments. Up to date, the exhausting exploration of this field suggests that the unique pathognonomic character sufficient to identify the neoplastic malignant transformation is the so called Warburg effect.
The aerobic glycolysis and the Warburg's effect: Dfinition and oncological implications
So long as 1923, Otto Warburg [Citation2] distinguished two glycolytic pathways on the basis of the final exportation of lactate in the cellular environment. He demonstrated that one pathway produces lactate only in the absence of O2 (the anaerobic glycolysis), whereas the other produces lactate despite the presence of O2 (the aerobic glycolysis); the anaerobic glycolysis was brought about by any type of cells, whereas the aerobic glycolysis was carried out only by embryonic tissues and by whatsoever anaplastic cancer. This finding is nowadays the basis, evidencing lactate in aerobic tissues by the PET methodology, that is regarded as a clinical clue of malignancy development [Citation3].
Since the first description, the aerobic glycolysis appeared a paradoxical energetic waste (the Warburg effect, or paradox). Indeed, the arrest of the aerobic glucose metabolism at the pyruvate level, with lactate exportation, reduces the energetic yield to 2 ATP per glucose against the 36 obtainable from the whole molecule.
The Warburg effect and the connected metabolic crossways have been object of intensive investigations in our laboratory throughout the last forty years, initially employing the ascites hepatoma AH130 [Citation4–7]. This highly anaplastic tumor was generated by treating Wistar rats with the carcinogen o-aminoazotoluene and has been widely used as in vivo model of experimental cancer [Citation8,9]. After serial transplantations in rat peritoneal cavity, this tumor became composed of isolated spheroidal cells, fed by the ascites fluid extruded from the peritoneal vessels. Recently, we showed that these cells display a Pluripotent-like cell phenotype, which expresses fundamental Embryonic Transcription Factors (ETFs), such as Nanog, Klf4 and c-Myc [Citation10]: these factors operate a transcriptional circuitry that controls the cell cycle as a function of pO2. At advances stages of tumor development in vivo, the intraperitoneal pO2 is near zero, making the bulk survive as an anaerobic population arrested in G0/G1.
Experiments performed with this model provided the following fundamental information reported below. Anaplastic cancer possess a huge capacity of aerobic glycolysis, converting glucose to lactate at percentages increasing with glucose external concentrations. This is shown in , which reports the glycolytic balance of AH130 hepatoma cells at their recruitment into the S phase from the anaerobic resting state. This synchronized cell entering into the S phase is measured by the rate of radioactive 14C-thymidine incorporation into DNA.
Table 1. The tumor cell recruitment into the S phase and the aerobic glycolysis as function of glucose external concentration. Data refer to H130 Hepatoma, adapted from the experiments described in 6 and 11.
When incubated in 15 mM glucose, this G1/S transition consumed 11 μmol/106 cells in 18 hours, exporting 18 μmol of lactate: this means that 80% of imported glucose was converted to lactate. Incubating the same cells in 0.5 mM glucose continuously renewed, the sugar uptake fell down to 1 μmol (95% decrease), and lactate exportation decreased to 0.7 μmol (97% decrease). Thus, cancer can uptake enormous amounts of glucose, exporting lactate proportionally to the sugar uptake. Noteworthy, cell recruitment into S phase turned out to be even higher in 0.5 mM in environmental glucose than in 15 mM.
On the whole, the amount of glucose necessary for the optimal cell growth is far less than that the cell can uptake, provided that the amount exceeding the optimum is totally converted to lactate. With 10 mM glucose, a concentration frequently used in cell culture, this conversion produces an energy waste that can be calculated as (34 × 11 = ) 374 ATP.
shows that cell recruitment into the mitotic cycle is abolished in anaerobiosis (nitrogen incubation), as well as by blocking the mitochondrial respiratory chain with a specific inhibitor of the cytochrome-oxidase, antimycin A [Citation7,11]; on the other hand, no inhibition of cell recruitment was produced blocking the mitochondrial ATP generation, uncoupling the oxidative phosphorylation with 2,4 dinitrophenol (DNP). In addition to that, the total intracellular ATP content turned out to be the same with either antimycin A or DNP treatment. Equally, the same was the rate of an energy-dependent process, such as protein synthesis estimated by 3H – Lysine incorporation into proteins.
Table 2. Effects of various treatments on recruitment of cell H130 Hepatoma into S phase. Data refer to time interval 0/18hrs as µmoles or µatoms/min/106 viable cells. Various are means ± standard error, in parenthesis number of experiments. n.d. not determined; n.s. not significant; ## = P<0.05; ### = P<0.01. Data were adapted from 11.
These data appear even more relevant considering the rate of O2 consumption (ΔQ O2/min) in the presence of various inhibitors. The block of this parameter with nitrogen incubation produced an 80% inhibition of cell recruitment, whereas no significant effect was obtained by uncoupling the oxidative phosphorylation with DNP, despite the doubling of O2 consumption brought about by this inhibitor [Citation7,11]. These data demonstrate that the energy for cancer growth is entirely supplied by respiration in that it affords the electron transport to oxygen through the respiratory chain, without significant contribution by the oxidative phosphorylation. These bioenergetic features reflect precisely those of the cristae-poor mitochondria of stem cells adapted to the physiological hypoxia [Citation12]. This suggested that the metabolic network controlling the cancer stem recruitment resides in the intimate mechanisms of this adaptation.
The genesis and maintenance of stem cells in the physiological hypoxia: The hypoxic niche
Recent studies showed that gradients of pO2 below 1% is the selective environment for stem cells, in line with the concept of physiological hypoxia, introduced to describe the naturally low oxygen tension (pO2 > 2) experienced by cells during the normal development [Citation13]. Oxygen concentrations range from 1% to 5% in the uterine environment [Citation14], and in the mouse embryo cells in low oxygen (<2%) are widespread until maternal and foetal blood interfaces around midgestion [Citation13,15]. The physiological hypoxia transcriptionally activates genes that are involved in energy metabolism, promoting tolerance to hypoxia by decreasing the cellular requirement and increasing the supply of oxygen [Citation13,16,17].
In a series of papers [Citation18–22], it has been postulated that such physiological hypoxia is localized also in restricted areas of adult differentiated tissues, allowing stem cells survival in the resting state. This localization was indeed sustained by the demonstration that bone marrow microareas were hypoxic relative to other tissues and that this localization was a crucial factor in the determination of stem cell fate and maintenance. These studies introduced the concept of Hypoxic Niche [Citation19], in keeping with the observation that hemopoietic stem cells (HSCs) are confined to regions of the bone marrow blood where the blood pO2 is lower than in other tissues and is equivalent to that of blood in jugular vein. These cells are in strict contact with several stromal and progenitor cells, physically residing between the HSCs and cells close to blood vessels. This arrangement represents a selective locus apt to preserve the cellular at pO2 below 1%, as compared to that of 6% in sinusoidal cavity. Actually, HSCs are selectively maintained in these niches, whereas the fast cycling early hemopoietic progenitors, with limited capacity of cell renewal, reside in areas far from the vasculature [Citation23].
The major advantage of residing in hypoxic niches is that stem cells can maintain indefinitely a slow-cycling proliferation rate, avoiding the oxidative stress associated with well oxygenated tissues [Citation19].
The CSCs evolution and reprogramming
CSCs represent the oncological equivalent of the physiological stem cell compartment. This view derives from a widely accepted model [Citation24], which proposes a cell hierarchical organization of stem cells originating from Embryonic Stem Cells (ESCs). The latter are physiologically generated from the inner mass of the embryo in the hypoxic gastrula environment (pO2 ≤ 2%), and are endowed with unlimited self renewal, together with the capacity of generating all types of tissues (Pluripotent Stem Cells = PSCs). These properties are maintained until the cells remain in hypoxic environments, expressing the ETFs, which operate the silencing of the differentiation genes. The occurrence of ESCs-like expressing ETFs in adult differentiated tissues, or in anaplastic tumors, represents an anomaly menacing an irreversible perturbation of growth. Thus, the identification and elimination of reprogrammed stem like cells in adult tissues is a crucial objective to pursue the eradication of the neoplastic disease.
In this light, we decided to focus our interest in anaplastic tumors expressing fundamental ETFs such as Nanog, Klf4, cMyc and Sox2. [Citation10,25–27].
The ectopic presence of ESCs raises the question of how these cells get into such anomalous locations. Nowadays, two speculative explanations have been proposed; a) ESCs migrate from gastrula to cancer, surviving in some ischemic areas of the tumor bulk; b) ESCs can be de novo generated in the context of adult tissues in the course of intense genetic and epigenetic alterations leading to the chromatin plasticity, which transforms differentiated tissues into immature cell populations. This plasticity is produced by the convergence of the two following processes:
| a) | Neoplastic Progression. This term was introduced by 28 to synthesise the fact that “Tumors change from bad to worse”. It consists of a series of gradual genetic and epigenetic mutations involving distinct, independently matched characters, which progressively drive the anaplastic dedifferentiation of cancer [Citation10,11,25]. | ||||
| b) | Cell reprogramming. This is the process inducing differentiated cells to invert their developmental potency down to obtain the pluripotent state typical of the induced PSCs (iPSCs) [Citation27]. This state is strongly facilitated by the neoplastic progression. | ||||
Mitochondrial reprogramming
A crucial information obtained in the course of the iPSCs generation is their glycolytic-oriented metabolism, in the absence of substantial energetic contribution by a rudimental cristae poor mitochondrial apparatus [Citation12]. Noteworthy, the differentiation promotes an intense mitochondrial biogenesis to form networks of elongated and cristae rich mitochondria entitled to support a competent oxidative metabolism [Citation12]. Moreover, the conversion of differentiated cells to iPSCs induces a profound mitochondrial regression from elongated and cristae-rich elements to small spherical and cristae-poor remnant structures. This, together with the up regulation of the glycolytic enzymes [Citation12,27]. These findings explain the fact that, in the dedifferentiated cancer cells, the respiration does not generate ATP, in keeping with the data reported in . This metabolic trim is typical of hypoxia adaptation, accounting up to 90% failure of chemotherapeutic treatments [Citation28].
The unveiling of Warburg paradox
Information so far led us to approach the solution of the Warburg paradox conceiving that its specific character, i.e. the forced aerobic exportation of lactate (recall ), represents a crucial metabolic device to counteract some cytotoxic effect brought about by an excess of pyruvate at the connection of glycolysis with the Krebs cycle. This hypothesis was verified exposing cancer cells to pyruvate at concentrations much higher than the physiological ones, after showing that these concentrations are totally innocuous when injected i.p. into mice (see below).
The anticancer cytotoxic activity of pyruvate
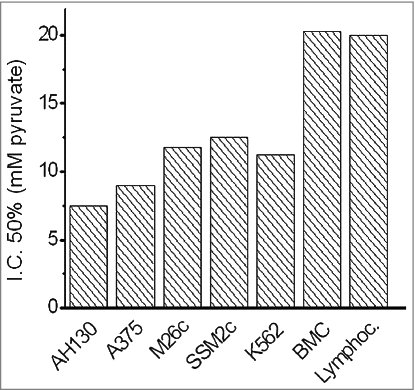
As shown in , Pyruvate determined an I.C.50 growth inhibition between 7.8 and 12.5 mM (reaching 100% inhibition around 20 mM) in different highly anaplastic tumors of different phenotypes, including AH130 Hepatoma, A375, M26C and SSM2C melanoma cell lines and K562 leukemia cell line. Substantially unaffected resulted bone marrow cells (BMC) and normal clonogenic lymphocytes.
Figure 1. The sensitivity of various cancer and normal stem cell populations to the cytotoxic activity of pyruvate. Values are expressed as IC50 of cell growth in the presence of varying concentrations of the substrate. AH130: Hepatoma; A375, M26C and SSM2C melanoma cell lines; K562 leukemia cell line; BMC: bone marrow cells; Lymphoc.: normal clonogenic lymphocytes. Values were determined after 18 hrs treatment, seeding 105 viable cells/well. Adapted from 25.
The pyruvate cancer cytotoxicity was then explored in vivo, injecting an anaplastic melanoma cell line (SSM2c, 25) in nude mice (nu/nu), (104 cells/mouse). Pyruvate was injected daily intraperitoneal (ip), at dosages sufficient to make all the organism contain the same concentration of pyruvate used in vitro (20 mM) and after checking that a fivefold higher dose (100 mM) resulted totally innocuous in control mice, throughout one year. The anticancer activity of pyruvate was definitely confirmed by i): the delay of metastatic nodules development in pyruvate treated mice as compared to controls; ii) the massive apoptosis characterizing the nodules developed in the pyruvate treated mice. As shown , the melanoma injection, followed throughout three months from the day of cancer cell inoculation, produced metastatic nodules developing with the two kinetics, representing, respectively, the nodules developed in controls (ctr, black kinetics), and the nodules developed in mice daily treated with 5.5 mg of pyruvate (pyr, red kinetics).
Figure 2. In vivo efficacy of a long-term pyruvate treatment. A) Time course of tumor masses growth in control (ctr) and daily treated mice (pyr). Tumor volumes were measured by caliper, following the formula (Width# Length# 0,54). B) Hematoxylin and eosin staining (panels a-c); Terminal dUTP Nick End-Labeling (TUNEL) assay (R&D systems) (panels d-f). Column I panel (a-d): picture of early necrosis, where single apoptotic cells are present; column II panel (b-e): picture of late apoptosis associated to an initial necrotic process; column III, panel c-f: massive cellular death to extensive apoptotic and necrotic processes.
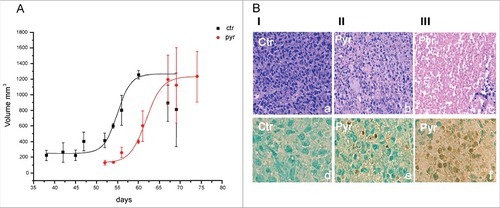
, column I, shows the vigorous melanoma cell population developing in the absence of pyruvate in control mice, stained with hematoxilin, panel (a) and negative for the apoptotic specific TUNEL reaction panel (d). Note the lack of evidence of any flogistic infiltrate.
column II and III shows the progressive cell apoptosis developing in the metastatic nodules, characterizing pyruvate treated mice. Column II (panel b) shows the initial evidence of necrosis and its apoptotic nature, revealed by TUNEL positive cells (panel e). The massive evolution of these two types of cell death is clearly shown in column III, panels (c) and (f).
The explanation of this Figure requires recalling some features of apoptosis. This is a complex, ATP-dependent process, due to multiple DNA mutations, which manifest only after latencies since the application of the initiating event [Citation29]. We showed that the impairment of the respiratory chain by pyruvate or antimycin A leads to a severe restriction of purine pool, without affecting the pyrimidine pool [Citation26]. Since DNA synthesis and turn-over are tightly coupled with the balance among the various deoxynucleotides, alteration of this balance produces high ratios of mis-incorporation of this precursors into DNA, and hence genetic mutations [Citation30]. Up to a certain limit, these mutations can be repaired by an efficient p53; on the contrary, they trigger cell apoptosis when p53 is deleted or altered as usually happens in anaplastic tumors. This explains why the pyruvate cytotoxicity produces the apoptotic type of cell death. [Citation30].
Altogether, this sequence explains the apoptotic delay with respect to the trigger stimulus and mirrors the apoptotic effect produced by pyruvate. Note the total absence of signs of inflammation (leukocytes, hyperhaemia), that always accompany the instantaneous cell death produced by highly toxic insults.
On the whole, data in are consistent with a temporal sequence like that represented by the kinetics reported in panel A.Data so far indicate that pyruvate fulfils the requisites we conceived proper to act as the cytotoxic agent, active at the connection between glycolysis and Krebs cycle. In other words, the elimination of pyruvate as lactate indeed represents an indispensable metabolic device to allow an unrestricted growth of cells with an immature mitochondrial apparatus. In anaplastic cancer stem cells, this in turn allows the use of the combined energy supply by the glycolytic pathway, without saturating the respiratory chain. This device provides the decisive selective advantage inscribed in the Warburg paradox. As simple as that.
The metabolic network underlying the pyruvate cytotoxicity
As a first step to explore the general impact of pyruvate, we compared its cytotoxicity with the other fundamental substrates of the Krebs cycle.
As shown in , this effect is strictly proportional to the rate of oxygen consumption (ΔQO2) measured as described in 4 and 32, while pyruvate turned out to be by far the most active. This is in keeping with the fact that pyruvate is the only glycolytic substrate directly connecting glycolysis to the Krebs cycle before entering any prior metabolization.
Figure 3. The role of cellular redox-state in the cytotoxic activity of physiological agents: the involvement of purine and folate metabolism. (A) The inhibition of cell recruitment into the S phase of AH130 cells by the Krebs cycle substrates, as a function of the corresponding rate of O2 consumption. O2 consumption (ΔQO2) is expressed as μl of O2 per mg of dry weight per hour. Ketog, α-ketoglutarate; Succ, Succinate; Mal, Malate; Fum, Fumarate; Citr, Citrate; OxalAc, Oxalacetate; Pyr, pyruvate. All substrates are used at [10 mM]. R = rate of 14C-Thymidine incorporation into DNA per 90 min per 106 viable cells at 18 hours of incubation in air. Values are expressed as % of control. (B) Effects of purine and pyrimidines on pyruvate inhibition of AH130 cell recruitment into the S phase. Abbreviations: pyr, pyruvate; ade, adenine; guan, guanine; thym, thymidine; cyt, cytosine; urac, uracile. Pyruvate was added at 10 mM; all bases are used at 0.5 mM. All additions were performed at time 0. Values are means ± SEM of 3 separate experiments. (C) antimycin A (ant.A) inhibition of adenine pool and its removal by folate in AH130 cells. Values are expressed as nmol/5 × 107 viable cells at 18 hours of incubation in air and are means ± SEM of 6 separate experiments. Ant.A (6 × 10−6M), F (100 μmol), gln, glutamine (5 mM).were added at t = 0. (D) The effects of various treatments on the intracellular NADP/NADPH ratio in AH130 cells. NADP and NADPH were estimated by HPLC technique and are expressed as percentage of control at t = 18 hours of incubation in air, and are means ± SEM of 5 separate experiments. ##P < 0.02; ###P < 0.001. Adapted from 25.
![Figure 3. The role of cellular redox-state in the cytotoxic activity of physiological agents: the involvement of purine and folate metabolism. (A) The inhibition of cell recruitment into the S phase of AH130 cells by the Krebs cycle substrates, as a function of the corresponding rate of O2 consumption. O2 consumption (ΔQO2) is expressed as μl of O2 per mg of dry weight per hour. Ketog, α-ketoglutarate; Succ, Succinate; Mal, Malate; Fum, Fumarate; Citr, Citrate; OxalAc, Oxalacetate; Pyr, pyruvate. All substrates are used at [10 mM]. R = rate of 14C-Thymidine incorporation into DNA per 90 min per 106 viable cells at 18 hours of incubation in air. Values are expressed as % of control. (B) Effects of purine and pyrimidines on pyruvate inhibition of AH130 cell recruitment into the S phase. Abbreviations: pyr, pyruvate; ade, adenine; guan, guanine; thym, thymidine; cyt, cytosine; urac, uracile. Pyruvate was added at 10 mM; all bases are used at 0.5 mM. All additions were performed at time 0. Values are means ± SEM of 3 separate experiments. (C) antimycin A (ant.A) inhibition of adenine pool and its removal by folate in AH130 cells. Values are expressed as nmol/5 × 107 viable cells at 18 hours of incubation in air and are means ± SEM of 6 separate experiments. Ant.A (6 × 10−6M), F (100 μmol), gln, glutamine (5 mM).were added at t = 0. (D) The effects of various treatments on the intracellular NADP/NADPH ratio in AH130 cells. NADP and NADPH were estimated by HPLC technique and are expressed as percentage of control at t = 18 hours of incubation in air, and are means ± SEM of 5 separate experiments. ##P < 0.02; ###P < 0.001. Adapted from 25.](/cms/asset/3e9cf28a-d665-4906-8777-f71cdf6cc531/kccy_a_1403679_f0003_b.gif)
This study was continued by recalling the data reported in , showing that the pyruvate inhibition of tumor growth depends on the rudimental mitochondrial apparatus of cancer cells, which support the electron flux to oxygen, without operating the oxidative phosphorylation. It ensues that the pyruvate impairs some mitochondrial function other than the ATP generation. This function can only be the regulation of the cellular redox state expressed by the cytosolic NADP/NADPH ratio, according to the reaction(1)
(1) The reoxidation of this NADPH is brought about through the shuttles systems, which transport the reducing equivalents (H) across the mitochondrial external membrane.
This mechanism operates in one crucial step of the synthesis of the purine ring summarized in the following equation:(2)
(2) catalyzed by the methylenedehyrogenase, where CH-FH4 represents the group 2 of the adenine ring (see also ). This reaction accounts for the dependence of purine synthesis on the NADP/NADPH ratio and, hence, on reoxidation of the reducing equivalent through the respiratory chain. All this produces that the inhibition or saturation of this chain restricts the purine pools with the arrest of the cell cycle.
Such involvement of purine synthesis in the pyruvate cytotoxicity was confirmed by showing that this inhibition is 100% removed by adding preformed purines (50% by guanine), while is totally unaffected by adding the pyrimidine base cytosine and uracil (). The dependence of the purine pools on the respiratory chain was explored by testing its response to antimycin A. As shown in , this inhibitor determined a 70% restriction of the Adenine pool, totally removed by folate (F). Moreover, antimycin A produced a 90% reduction of the NADP/NADPH ratio ().
The definite evidence that NADP/NADPH ratio gears the purine synthesis was obtained by modifying this ratio according to the classical reaction:(3)
(3) catalyzed by the Dihydrofolic reductase (DHIFR), selectively inhibited by the antineoplastic Metotrexate (MTX).
As predicted by this equation, the addition of high doses of FH4 produces an increment of NADPH and the equivalent decrease of NADP, with the consequent reduction of the NADP/NADPH ratio, and hence the arrest of cell cycle. A substantial reduction of the NADP/NADPH ratio is also produced by adding 5 mM glutamine (gln), as an effect of glutaminolysis pathway.
The addition of the reduced form of FH4 strongly diminishes the cell content of NADP and enhanced NADPH, while, the addition of F tends to diminish NADPH and enhances NADP.
A strong increment of NADPH is also produced by the antineoplastic agent Metotrexate (MTX), which is the selective inhibitor of the enzyme catalyzing the above reaction, Dihydrofolic reductase (DHFR).
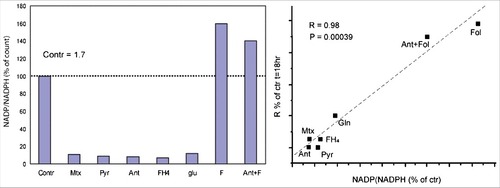
The NADP/NADPH ratios measured in all the conditions experimented in this study, expressed of percentages of control, are condensed in . When these data are compared with the corresponding rates of cancer cell recruitment into the cycle, R, (as in ), it emerged that this recruitment is a linear function of the NADP/NADPH ratio ().
Figure 4. The central role of the NADP/NADPH ratio in the control of ascites cell recruitment into S. A. Effects of various factors on the NADP/NADPH ratio. F clearly shows an opposite effect respect to all the others substrates, resulting much higher even than the control. The stimulating effect of F removes almost completely the antimycin inhibition. The ratio was determined at 18 hours of incubation in air. Additions were performed at t = 0. Contr: control; Mtx. methotrexate; Pyr: pyruvate; Ant: antimycin; FH4 tetrahydrofolate.; glu: glutamine; F: folate; B. The correlation between AH130 cell recruitment into S and the NADP/NADPH ratio under various conditions. Correlation index, R = 0.98. #P < 0.05; ##P < 0.02; ###P < 0.001. Adapted from 25.
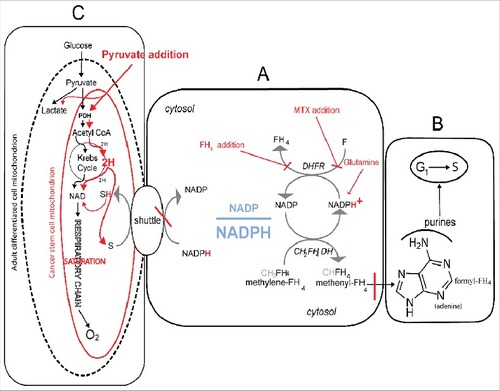
This complex metabolic network is outlined in the , where it is summarized the metabolic network described above.
Figure 5. The metabolic network which regulates the cell cycle activation at the O2 dependent G1/S transition and its modification by pyruvate addition. The core of the network is the cellular redox-state expressed by the cytosolic NADP/NADPH ratio. This ratio (A) regulates the transfer of reducing equivalents (H) from the methylene-tetrahydrofolate (CH2-FH4) to methenyl-trahydrofolate (CH-FH4), a crucial NADP-dependent reaction generating NADPH. This is a limiting step of the synthesis of purine ring (B), required for the amplification of purine pools indispensable for the G1/S transition. A fundamental role in the regulation of the NADP/NADPH ratio is played by folate, whose reduction to FH4 by the dehydrofolic reductase (DHFR) generates NADP (A). The addition of an excess of FH4 impairs the DHFR activity, leading to an increment of NADPH, with the consequent reductive shift of the NADP/NADPH ratio and the inhibition of the G1/S transition. Consistently, FH4 mimics the effects of MTX, a powerful inhibitor of DHFR (A), incrementing the NADPH levels and inhibiting cell recruitment into S. Whatever the mechanism incrementing the cytosolic NADPH, including an excess of glutamine through the glutaminolytic pathway (A), has an inhibitory effect, unless the NADPH is removed through the shuttle mechanisms, which discharge the cytosolic reducing equivalents onto the mitochondrial respiratory chain. The frame above is regulated by the size and shape of the mitochondrial apparatus, see the oval sketches (C) in the Figure. Actually, the respiratory chain, which is the mitochondrial core, deeply differs in adult differentiated cells as compared to stem cell and cancer undifferentiated cells; the adult cells normally contain a high number of differentiated cristae-rich mitochondria (dashed black line), whereas stem cancer cells contain very few cristae-poor mithocondria (red line). This difference implies that the two cell types can exploit two different amount of disposal of reducing equivalents (H) produced both by the glycolytic pathway as well as by the metabolic oxidative reduction carried out in the cytosol. In fact, these sources compete each other for the respiratory chain with the following consequences. Up to the point that no one of the two sources saturate the respiratory chain, glycolysis will be performed down to the final oxidation of glucose down to CO2 and H2O, without arresting glucose metabolism at the level of the pyruvate exportation through the lactate conversion. This is the reason why adult differentiated cell do not produce lactate but in anaerobiosis, or upon inhibition of respiratory chain (e.g. by antimycin A). This is what Warburg called the anaerobic glycolysis, which is the pathway that each cell must activate in severe hypoxia. On the other hand, whenever in aerobic conditions, the cytosolic sources tend to saturate the respiratory chain (e.g. in the presence of an excess of pyruvate) the cell brings about the total oxidation of glucose without lactate conversion, proportionally to the unsaturation of respiratory chain. Under this conditions, the cell will produce lactate despite the presence of O2, which is what Warburg defined aerobic glycolysis, the enigma remains so far the Warburg paradox, with the inscribed anticancer therapeutic suggestion explore in this study. The extreme pyruvate anticancer selectivity derives from the easy saturation of the respiratory chain of the immature cancer mitochondria, as compare to the vast number of the mature mithocondria of adult differentiated cells. Indeed the saturation of differentiated cells is not obtainable at concentrations thousand time higher than the cancer cytotoxic ones. Updated from 25.
The ultimate target of pyruvate: The cancer stem cell
As an experimental insight into this direction, we explored the pyruvate inhibition of the stem self renewal in highly anaplastic metastatic melanoma cell lines in vitro and in vivo.
The in vitro experiments were carried out using the human melanoma cell line A375 [Citation25]. In the in vitro procedures performed on these highly metastatic melanoma, Pyruvate drastically abolished sphere formation at the same extent as the conventional chemotherapic drug methotrexate ( panel B and C, respectively) [Citation25]. As shown in panel D, the addition of folate do not significantly alter the sphere formation with respect to the control (panel A), while FH4 (FH4, panel E) abolished the sphere generation, at the same extent as pyruvate (panel B) and MTX (panel C).
Figure 6. The cytotoxic effects of physiological agents on melanoma stem cells. Effects of various treatments on sphere formation derived from A375 melanoma cells (A–E). (F) Percentage of Cleaved caspase 3-positive cells after treatment with cytotoxic physiological agents. ctr: control; pyr: pyruvate; Mtx: methotrexate; F: folate; FH4: tetrahydrofolate. F = 100 μM, FH4 = 100 μM, pyr = 20 mM, MTX = 10−6M. (F) Updated from 25.
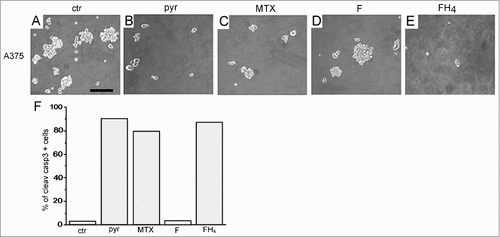
The sphere abolition was accompanied by an extensive apoptotic cell death, as demonstrated in by the strong positivity to the cleaved caspase 3, reported in the A375 melanoma cells upon the addition of pyruvate or FH4.
These data demonstrate that the physiological factors, such as FH4 and pyruvate, exert a strong cytotoxic effect on melanoma cells, similar to that of MTX, drastically interfering with stem cell renewal. Moreover, it is worth noting that, even in absence of these compounds, their inhibitory effect is evident also in secondary and tertiary spheres derived from the dissociation of the cells previously treated with FH4 and pyruvate, respectively [Citation25]. These results indicate that the cytotoxicity of the tested physiological agents not only reduces the melanoma stem cell generation, but it is also memorized, influencing the self-renewal ability by the further sphere generation after the agent removal.
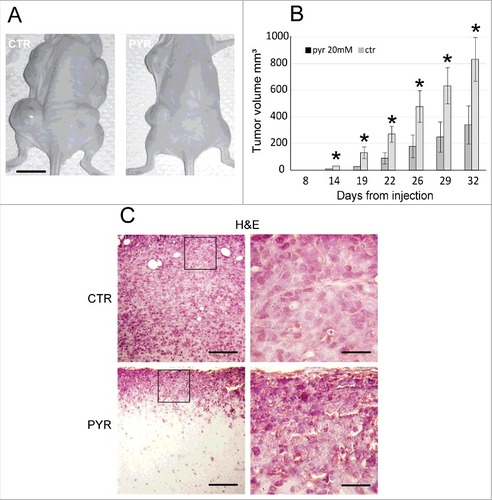
This “memorization process” was confirmed by the in vivo experiments reported in [Citation25]. Briefly, A375 melanoma cells with high stemness potential, obtained by dissociation from primary spheres, was split in 2 cultures, which were incubated for 1 week either in the presence of 20 mM pyruvate or PBS. At the end of this incubation, 104 viable pyruvate or PBS-treated cells were injected subcutaneously into nude mice to test their tumorigenicity. As shown in , pyruvate pre-treatment caused an inhibition of tumor growth, determining a remarkable reduction of metastasis number and size (). Histological examination of pyruvate-treated nodules shown a mass of apoptotic cells, in contrast with the metastatic nodules developed in PBS injected mice.
Figure 7. Pyruvate pretreatment of viable A375 melanoma cells strongly reduces the tumor development in vivo. (A) The inhibitory effect of pyruvate pre-treatment on melanoma growth in vivo. Scale bar = 10 mm. (B) Quantification of tumor volume. Values represent the averages of tumors developed at various times and expressed as mean ± SEM (n = 12, each). (C) Histological sections of tumors derived from PBS- (upper pictures) and pyruvate-treated melanoma stem cells (lower pictures) counterstained with Hematoxylin and Eosin (H&E). Scale bar left pictures = 100 μm, right pictures = 25 μm. Adapted from 25.
The milestones of the Radical anticancer therapy inscribed in the Warburg effect
On the whole, this study produced the following milestones to orient a radical therapy of anaplastic cancer:
| 1) | to be successful, such therapy must be selectively targeted to the stem compartment, which is resistant to chemo and radiochemical treatments. | ||||
| 2) | The stem compartment is segregated in and, adapted to, hypoxic districts (the hypoxic niches), where they survive in a resting state. This adaptation implies a remnant mitochondrial apparatus, whose respiratory chain is substantially devoid of the cristae supporting the electron transport and the correlated oxidative phosphorylation. | ||||
| 3) | As a consequence of point 2, stem cells surviving in hypoxic niches derive their energetic supply exclusively by the metabolic process that Warburg named anaerobic glycolysis, in which pyruvate is actively converted to lactate by the lactate dehydrogenase (LDH), in the reaction | ||||
All this generate an overall metabolic trim where glucose utilization is in part stopped at the pyruvate/lactate conversion, despite the aerobic environment. This competition underlines the aerobic glycolysis, at a rate inversely proportional to the maturated mitochondrial respiration. In embryonic tissues as well as anaplastic cancer, this equipment is poor, implying the necessity of this metabolic competition.
In the adult differentiated cells, the mitochondrial apparatus is normally so well developed to allow the total glucose oxidation through the Krebs cycle and the respiratory chain, avoiding any lactate production through the aerobic glycolysis.
The definition of Cancer cytotoxic physiological factors
Data so far lead to conclude that pyruvate is the prototype of physiogical agents, including also FH4 and Glutamine, that can be regarded as a category of Cancer Cytotoxic Physiological Factors (CCPFs), whose potential use in therapy deserves to be tested as to their clinical use. Indeed, all these factors are currently used in therapy at dosages that, in the case of pyruvate proved innocuous at dosages hundreds times higher than those used in this study to eliminate the cancer stem compartment. Since 1990s, pyruvate is employed as diet integrator at 5g/day for man or woman on a liquid diet [Citation31,32].
Clinical perspectives
CCPFs emerged from this study as anticancer factors targeted to the hypoxia selected cancer stem cells migrating out of their hypoxic niches, before they accomplish the intense mitochondriogenesis, that characterizes this stage. Two peculiarities make them represent an ideal agents to approach a radical therapy of the neoplastic disease; a) they are highly effective on stem compartments, the more the higher the tumor immaturity; b) they are totally innocuous to adult differentiated cell; c) their current commercial and extremely cheap availability.
On these bases, the CCPFs should be regarded as suitable to attempt a radical anticancer therapy, abolishing the cancer renewal with avoiding toxic and ineffectual chemotherapies.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Special thanks to Anna Marchi Mazzini for her generous support and encouragement, that helped us to complete this work. This work was supported by pluriennal grants financed by Ente Cassa di Risparmio di Firenze, Istituto Toscano Tumori (ITT), Associazione Italiana per la Ricerca sul Cancro (AIRC), Regione Toscana, Consiglio Nazionale delle Ricerche (CNR), Ministero dell'Università e della Ricerca Scientifica e Tecnologica (MURST), changed in Ministero dell'Istruzione, dell'Università e della Ricerca (MIUR).
Additional information
Funding
References
- Clarke MF, Dick JE, Dirks PB, et al. Cancer stem cells-perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339–9344. doi:10.1158/0008-5472.CAN-06-3126. PMID:16990346
- Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–270. PMID: 13351639.
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi:10.1126/science.1160809. PMID:19460998
- Del Monte U, Olivotto M. Research on the nature of the substrates of endogenous respiration of Yoshida's ascites hepatoma. Sperimentale. 1965;115:405–422. PMID: 4164352.
- Olivotto M, Paoletti F. Studies on the kinetic of initial cycle progression in vitro of ascites tumor cells subsequent to isolation from ascites fluid. Cell Tissue Kinet. 1980;13:605–612. doi: 10.1111/j.1365-2184.1980.tb00499.x. PMID:7417977
- Olivotto M, Paoletti F. The role of respiration in tumor cell transition from the non cycling to the cycling state. J Cell Physiol. 1981;107:243–249. doi:10.1002/jcp.1041070210. PMID:7251682
- Olivotto M, Caldini R, Chevanne M, et al. The respiration-linked limiting step of tumor cell transition from the non-cycling to the cycling state: its inhibition by oxidizable substrates and its relationships to purine metabolism. J Cell Physiol. 1983;116:149–158. PMID: 6863398. doi:10.1002/jcp.1041160205. PMID:6863398
- Arcangeli A, Del Bene MR, Ricupero L, et al. Fibronectin hyperpolarizes the plasma membrane potential of murine erythroleukemia cells. Ann NY Acad Sci. 1988;551:242–244. PMID: 3245662. doi:10.1111/j.1749-6632.1988.tb22341.x. PMID:3245662
- Arcangeli A, Del Bene MR, Poli R, et al. Mutual contact of murine erythroleukemia cells activates depolarizing cation channels, whereas contact with extracellular substrata activates hyperpolarizing Ca2+-dependent K+ channels. J Cell Physiol. 1989;139:1–8. doi:10.1002/jcp.11041390102. PMID:2468677
- Marzi I, Cipolleschi MG, D'Amico M, et al. The involvement of a Nanog, Klf4 and c-Myc transcriptional circuitry in the intertwining between neoplastic progression and reprogramming. Cell Cycle. 2013;12:353–364. doi:10.4161/cc.23200. PMID:23287475
- Olivotto M, Dello Sbarba P. Environmental restrictions within tumor ecosystems select for a convergent, hypoxia-resistant phenotype of cancer stem cells. Cell Cycle. 2008;7:176–187. doi:10.4161/cc.7.2.5315. PMID:18256528
- Folmes CDL, Nelson TJ. Somatic oxidative bioenergetics transition into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metabolism. 2011:14;264–271. doi:10.1016/j.cmet.2011.06.011. PMID:21803296
- Dunwoodie SL. The role of hypoxia in development of the mammalian embryo. Dev Cell. 2009;17:755–773. doi:10.1016/j.devcel.2009.11.008. PMID:20059947
- Maltepe E, Saugstad OD. Oxygen in health and disease: regulation of oxygen homeostasis-clinical implications. Pediatr Res. 2009;65:261–268. doi:10.1203/PDR.0b013e31818fc83f.
- Okazaki K, Maltepe E. Oxygen, epigenetics and stem cell fate. Regen Med. 2006;1:71–83. doi:10.2217/17460751.1.1.71. PMID:17465821
- Anderson JC, Blake AJ, Mills M, et al. A general one-step synthesis of beta-nitronitriles. Org Lett. 2008;10:4141–4143. doi:10.1021/ol801691c. PMID:18722454
- Seidel S, Garvalov BK, Wirta V, et al. 2010. A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2 alpha. Brain. 133:983–995. doi:10.1093/brain/awq042. PMID:20375133
- Borovski T, De Sousa E, Melo F, et al. Cancer stem cell niche: the place to be. Cancer Res. 2011;71:634–639. doi:10.1158/0008-5472.CAN-10-3220. PMID:21266356
- Cipolleschi MG, Dello Sbarba P, Olivotto M. The role of hypoxia in the maintenance of hematopoietic stem cells. Blood. 1993;82:2031–2037. PMID: 8104535. PMID:8104535
- Cipolleschi MG, D'Ippolito G, Bernabei PA, et al. Severe hypoxia enhances the formation of erythroid bursts from human cord blood cells and the maintenance of BFU-E in vitro. Exp Hematol. 1997;25:1187–1194. PMID: 9328456. PMID:9328456
- Dello Sbarba P, Cipolleschi MG, Olivotto M. Hemopoietic progenitor cells are sensitive to the cytostatic effect of pyruvate. Exp Hematol. 1987;15:137–142. PMID: 3817047. PMID:3817047
- Mohyeldin A, Garzón-Muvdi T, Quiñones-Hinojosa A. Oxygen in stem cell biology: a critical component of the stem cell niche. Cell Stem Cell. 2010;7:150–161. doi:10.1016/j.stem.2010.07.007. PMID:20682444
- Kubota K, Soeda J, Misawa R, et al. Bone marrow-derived cells fuse with hepatic oval cells but are not involved in hepatic tumorigenesis in the choline-deficient ethionine-supplemented diet rat model. Carcinogenesis. 2008;29:448–454. doi:10.1093/carcin/bgm279. PMID:18174248
- Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755–768. doi:10.1038/nrc2499. PMID:18784658
- Cipolleschi MG, Marzi I, Santini R, et al. Hypoxia-resistant profile implies vulnerability of cancer stem cells to physiological agents, which suggests new therapeutic targets. Cell Cycle. 2014:13:268–278. doi:10.4161/cc.27031. PMID:24200964
- Stivarou T, Cipolleschi MG, D'Amico M, et al. The complex metabolic network gearing the G1/S transition in leukemic stem cells: Hints to a rational use of antineoplastic agents. Oncotarget. 2015;6:31985–31996. doi:10.18632/oncotarget.5155. PMID:26396171
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi:10.1016/j.cell.2006.07.024 doi:10.1016/j.cell.2006.07.024. PMID:16904174
- Abdullah LN, Chow EK. Mechanisms of chemoresistance in cancer stem cells. Clin Transl Med. 2013;23:1–9. doi:10.1186/2001-1326-2-3.
- Majno G, Joris I. Cells, tissues and diseases. Oxford (UK): Oxford University Press; 2004.
- Hastak K, Paul RK, Agarwal MK, et al. DNA synthesis from unbalanced nucleotide pools causes limited DNA damage that triggers ATR-CHK1-dependent p53 activation. Proc Natl Acad Sci USA. 2008;105:6314–6319. doi:10.1073/pnas.0802080105. PMID:18434539
- Koh-Banerjee PK, Ferreira MP, Greenwood M, et al. Effects of calcium pyruvate supplementation during training on body composition, exercise capacity, and metabolic responses to exercise. Nutrition. 2005;21:312–319. doi:10.1016/j.nut.2004.06.026. PMID:15797672
- Onakpoya I, Hunt K, Wider B, et al. Pyruvate supplementation for weight loss: a systematic review and meta-analysis of randomized clinical trials. Crit Rev Food Sci Nutr. 2014;54:17–23. doi:10.1080/10408398.2011.565890. PMID:24188231