
ABSTRACT
Tumor-associated macrophages (TAM) are prominent components of tumor microenvironment (TME) and capable of promoting cancer progression. However, the mechanisms for the formation of M2-like TAMs remain enigmatic. Here, we show that lactate is a pivotal oncometabolite in the TME that drives macrophage M2-polarization to promote breast cancer proliferation, migration, and angiogenesis. In addition, we identified that the activation of ERK/STAT3, major signaling molecules in the lactate signaling pathway, deepens our molecular understanding of how lactate educates TAMs. Moreover, suppression of ERK/STAT3 signaling diminished tumor growth and angiogenesis by abolishing lactate-induced M2 macrophage polarization. Finally, research data of the natural compound withanolide D provide evidence for ERK/STAT3 signaling as a potential therapeutic strategy for the prevention and treatment of breast cancer. These findings suggest that the lactate-ERK/STAT3 signaling pathway is a driver of breast cancer progression by stimulating macrophage M2-like polarization and reveal potential new therapeutic targets for breast cancer treatment.
Introduction
Breast cancer is the most commonly diagnosed malignant tumor in women worldwide and contributes significantly, as the primary cause, to female cancer-related mortality [Citation1]. Tumor-associated macrophages (TAM) are prominent components of the breast tumor microenvironment (TME). Moreover, TAM accumulation in tumors correlates with a poor clinical outcome [Citation2]. TAMs show typical M2 macrophage properties and can secrete IL-6 and VEGF to promote tumor angiogenesis, matrix metalloproteinase to promote tumor metastasis and local immunosuppressive factors to mediate immune escape [Citation3]. Thus, TAM-targeted therapeutics are promising novel anti-cancer strategies.
Macrophages exhibit high plasticity in response to various external signals [Citation4]. Depending on signals present in the microenvironment, macrophages are polarized into two distinct phenotypes: the classically activated (M1) or the alternative activated (M2) macrophages. M1-like macrophages play a role in killing invading pathogens and tumor cells, secrete pro-inflammatory cytokines, and express nitric oxide synthase (iNOS). M2-like macrophages, important in tissue remodeling, wound healing and tumor progression, secrete anti-inflammatory cytokines. M2-like macrophages exhibit high expression of mannose receptor (CD206 or MRC-1) and arginase 1 (Arg-1). Some signaling molecules participate in macrophage M2-polarization, such as PI3K/Akt–ERK signaling [Citation5], STAT3 [Citation6], HIF1α [Citation7], STAT6 [Citation8], etc. Tumor-associated macrophages (TAMs) closely resemble M2-polarized macrophages [Citation9]. However, the mechanisms of M2-like TAM formation remain elusive.
We previously demonstrated that the abnormal oncometabolites such as succinate caused nonmetabolic dysregulation and promoted cancer angiogenesis [Citation10,Citation11]. In tumors, where oxygen availability is limited and fluctuating, cells undergo metabolic adaptations, including a switch to aerobic glycolysis, termed the “Warburg effect,” which results in higher rates of glycolysis, reduced pyruvate oxidation, and increased lactate production [Citation12]. Elevated lactate concentrations in the tumor microenvironment, ranging from 5 to 40 mmol/L, may, under some circumstances, be used as metabolic fuel [Citation13]. In addition to predominate carbon source of the TCA cycle for cancer [Citation14], previous studies have shown that lactate is correlated with cell re-education in the TME [Citation13]. However, the signals involved in the communication between lactate and macrophages are poorly defined. Here, we show that lactate drives macrophage differentiation toward an alternatively activated (M2) phenotype via ERK/STAT3 signaling activation, which then facilitates angiogenesis, cancer cell migration, and invasion. Inhibition of ERK/STAT3 signaling with an inhibitor or natural product reduces lactate-induced M2 macrophage polarization and obstructs breast cancer progression in vitro and in vivo. These findings uncover the lactate signaling cascade as a potential new therapeutic target for breast cancer treatment.
Results
Tumor-derived lactate induced M2 Macrophage polarization
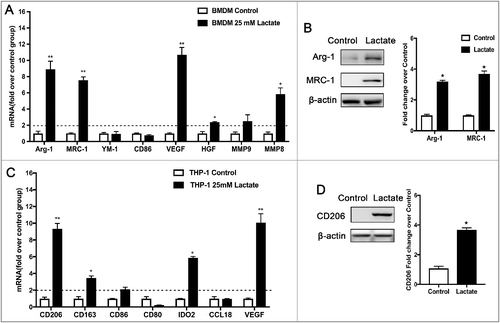
Macrophages in the tumor microenvironment can be educated by cancer cells [Citation15]. To examine whether cancer cell-derived lactate can modulate macrophage M2 activation, Western blot and QPCR analyses of bone marrow-derived macrophages (BMDMs) and the THP-1 cell line showed that lactate enhanced the expression of M2 macrophage markers. As shown in , the expression of Arg-1 and MRC-1 mRNA were increased 8.9-fold and 7.6-fold, respectively, by lactate in BMDM. The expression of Arg-1 and MRC-1 protein were also increased. Moreover, similar results were also achieved in THP-1 polarization model after treatment with lactate. Consistent with this observation, the expression of CD206 and VEGF were enhanced by lactate in THP-1 cells ( and ). Together, these results suggest that cancer cell-derived lactate may stimulate macrophage M2 polarization.
Figure 1. Lactate promotes M2-like macrophage polarization. (A) The relative mRNA expression of M2-like macrophages was analyzed by QPCR in BMDMs with or without lactate treatment for 12 h (n = 3). *P < 0.05. (B) Western blot for Arg-1 and MRC-1 in BMDMs after treatment with lactate for 24 h. β-actin was used as a loading control. The Arg-1/β-actin or MRC-1/β-actin ratio was quantified and is shown as fold changes compared with control (n = 3). *P < 0.05, **P < 0.01 compared with control. (C) The relative mRNA expression of M2-like macrophages was analyzed by QPCR in THP-1 cells with or without lactate treatment for 12 h (n = 3). *P < 0.05. (D) Western blot for CD206 in THP-1 cells after treatment with lactate for 24 h. β-actin was used as a loading control. The CD206/β-actin ratio was quantified and is shown as fold changes compared with control (n = 3). *P < 0.05, **P < 0.01 compared with control group.
Lactate-stimulated M2-like macrophage polarization promotes breast cancer cell proliferation, migration, and angiogenesis in Vitro
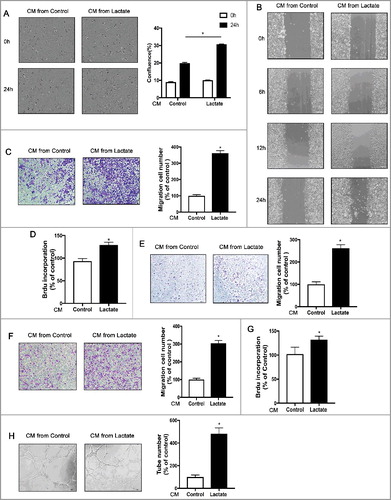
M2 macrophages have been shown to promote tumor growth, metastasis, and angiogenesis [Citation16]. Thus, we investigated whether lactate-induced M2 macrophages promote breast cancer cell proliferation, migration, and angiogenesis in vitro. First, the effects of the conditioned medium (CM) from various pretreated macrophages were examined on breast cancer cell proliferation. Compared with CM from untreated control macrophages, CM from lactate-activated macrophages significantly increased the proliferation of breast cancer cells ( and ). Second, wound-healing migration assays and Boyden chamber assays were used to examine the effects of lactate-induced M2 macrophages on the motility of breast cancer cells. Compared with untreated control macrophages, the lactate-stimulated macrophages significantly enhanced the number of migrating cancer cells (, and ). Proliferation assays, Boyden chamber assays, and capillary-like tube formation assays were used to examine the pro-angiogenesis ability of lactate-stimulated macrophages. As shown in , compared with the control group, the CM from lactate-stimulated macrophages significantly enhanced the proliferation, migration and capillary-like tube formation of pHUVECs. Together, these data suggest that macrophage activation by lactate promotes breast cancer cell proliferation, migration, and angiogenesis in vitro.
Figure 2. Lactate-activated macrophages promote cancer cell migration and angiogenesis in vitro. (A) MCF7 cells were treated with the conditioned medium (CM) from THP-1 cells treated with or without lactate and monitored by live imaging; Scale bar represents 50 μm. Confluence was quantified and compared with control (B) wound-healing migration assay of MCF7 cells migration. (C,E and F) Boyden chamber assay of MDA-MB-231 cells(C), MCF7 cells (E), and pHUVECs(F) migration. Cells were plated on the upper chamber inserts with serum-free medium, and CM from control THP-1 cells or lactate-activated THP-1 cells were put in the lower chambers. After migration for 6 h (F) or 24 h (C, E), the migrated cells were stained with crystal violet and counted as cells per field of view under the microscope (n = 5). (D,G) Lactate-activated THP-1 cells induced the proliferation of MCF7 cells(D) and pHUVECs(G). (H) Lactate-activated macrophage induced capillary-structure formation of endothelial cells. pHUVECs were placed in 96-well plates coated with Matrigel (2.0 × 104 cells/well) and stimulated with CM from THP-1 cells that were treated with or without lactate. After 6 h, tubular structures were photographed (magnification, × 100).*P < 0.05, **P < 0.01 compared with control group.
Lactate promotes M2 macrophage polarization by activating ERK/STAT3 signaling pathway
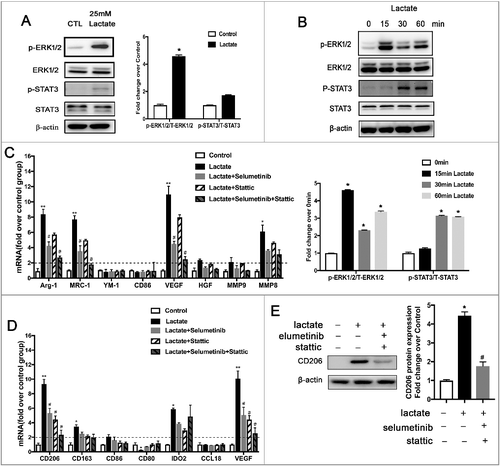
Both STAT3 and ERK1/2 signaling activation can drive M2 macrophage polarization via different mechanisms in the TME [Citation5,Citation17]. Therefore, in the following experiments, we detected the expression of STAT3 and ERK1/2 by Western blot analysis to illustrate the underlying mechanisms. As shown in and , the expression of phosphorylated STAT3 and phosphorylated ERK1/2 (p-ERK1/2) was significantly increased in lactate-stimulated macrophages. Additionally, we utilized the selective inhibitors, selumetinib and stattic (well-known MEK and STAT3 inhibitors, respectively), to explore whether STAT3 and ERK1/2 signaling are of importance in M2 macrophage polarization stimulated by lactate. Western blot and quantitative PCR (QPCR) analyses showed that the ERK1/2 inhibitor in combination with the STAT3 inhibitor significantly decreased the expression of M2 macrophage markers, including CD206 and Arg-1, induced by lactate, (). Collectively, these data suggest that lactate promotes M2 macrophage polarization by activating the ERK/STAT3 signaling pathway.
Figure 3. Macrophage phenotypic switch induced by lactate is associated with increased ERK/STAT3 signaling. (A and B) THP-1 cells were stimulated with lactate or PBS for 24 h (A) or a different time (B). Expression and phosphorylation levels of ERK1/2 and STAT3 in THP-1 cells were determined by Western blots. (C and D) The relative mRNA expression of M2-like macrophages was analyzed by QPCR in BMDM(C) and THP-1 cells(D) which were pretreated with Selumetinib (10 mM) or/and Stattic (10 mM) for 30 min and then exposed to lactate or PBS for 12 h (n = 3). *P < 0.05 compared with control. (E) THP-1 cells were pretreated with Selumetinib (10 mM) and Stattic (10 mM) for 30 min and then exposed to lactate or PBS for 24 h. Phosphorylation levels of ERK1/2 and STAT3 in macrophages were determined by Western blot. Data are representative of results obtained in three independent experiments. *P < 0.05, **P < 0.01 compared with control group; #P < 0.05 compared with lactate group.
The functions of M2 macrophages stimulated by lactate were abolished by inhibition of ERK/STAT3 activation
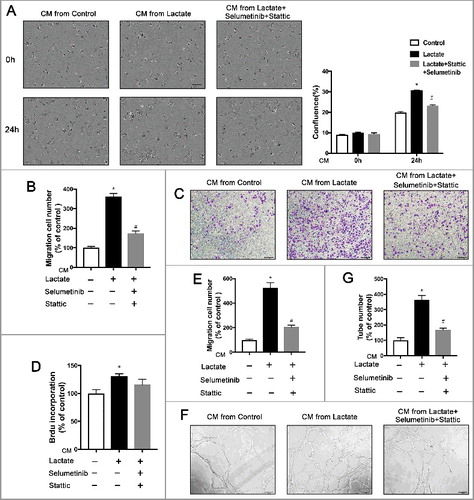
To confirm the role of ERK/STAT3 signaling in M2 macrophage polarization activated by lactate, we next examined the effect of the protumor and pro-angiogenic functions of lactate-activated M2 macrophages after exposure to selumetinib and stattic, an MEK and a STAT3 inhibitor, respectively, in vitro. As shown in and , inhibition of ERK/STAT3 signaling in macrophages diminished not only breast cancer cell proliferation but also breast cancer cell migration induced by lactate-activated macrophages. Moreover, inhibition of ERK/STAT3 signaling decreased angiogenesis induced by lactate-activated macrophages in vitro (). These results indicate a crucial role of ERK/STAT3 signaling in macrophage M2 activation upon education by lactate.
Figure 4. Inhibition of the ERK/STAT3 signaling impedes breast cancer migration and angiogenesis by suppressing Lactate-induced M2 macrophage polarization. (A) MCF7 cells were treated with the conditioned medium (CM) from macrophages which were pretreated with Selumetinib (10 mM) and Stattic (10 mM) for 30 min and then treated with lactate or PBS and monitored by live imaging, Scale bar represents 50 μm. Confluence was quantified and compared with control. (B, C and E) Boyden chamber assay of the MDA-MB-231 cell (B), and pHUVEC (C,E) migration. Cells were plated on the upper chamber inserts with serum-free medium, and CM from control macrophages or lactate-activated macrophages which were pretreated with Selumetinib (10 mM) and Stattic (10 mM) for 30 min was put in the lower chambers. After migration for 6 h(C) or 24 h(B), the migrated cells were stained with crystal violet and counted as cells per field of view under the microscope (n = 5). (D) The proliferation of pHUVECs induced by lactate-activated macrophages was suppressed by inhibition of the ERK/STAT3 signaling. pHUVECs cells were treated with cultured medium(CM) from macrophages which were pretreated with Selumetinib (10 mM) and Stattic (10 mM) for 30 min and then treated with lactate or PBS. (F and G) Capillary-structure formation of endothelial cells induced by lactate-activated macrophages was suppressed by inhibition of ERK/STAT3 signaling. pHUVECs were placed in 96-well plates coated with Matrigel (2.0 × 104 cells/well) and stimulated with CM from macrophages which were pretreated with Selumetinib (10 mM) and Stattic (10 mM) for 30 min and then treated with lactate or PBS. After 6 h, tubular structures were photographed (magnification, × 100). *P < 0.05, **P < 0.01 compared with control group; #P < 0.05 compared with lactate group.
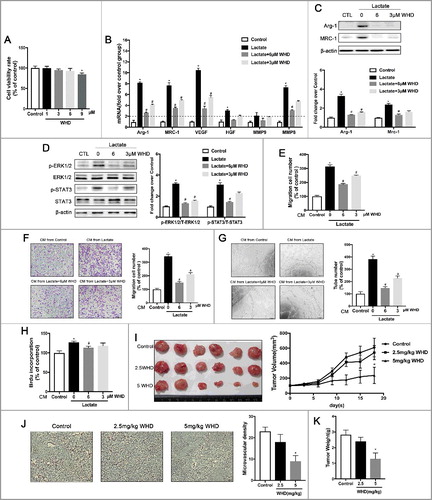
Withanolide D decreased cancer growth and angiogenesis by attenuation of lactate-induced M2 macrophage polarization through ERK/STAT3 signaling inhibition
Our present data demonstrated that suppression of the lactate signaling pathway that induces M2-like macrophages should change macrophage functions to prevent cancer proliferation, migration, and angiogenesis. It is believed that natural compounds are a good source of new drugs [Citation18]. Withanolide D (C4b-C5b, C6b-epoxy-1-oxo-,20b, dihydroxy- 20S, 22R-witha-2,24-dienolide; C28H38O6, MW 470.6; WithaD) is a steroidal lactone isolated from the leaves and roots of Withania Somnifera [Citation19], which is one of the most reputed Indian medicinal plants and forms the essential constituent of more than 100 traditional medicine formulations [Citation20]. In an attempt to explore the effects of WithaD on macrophage activation, the expression of surface M2-characteristic markers was measured using Western blots and QPCR. As shown in , inducible expression of MRC-1 and Arg-1 in lactate-activated macrophages was inhibited by WithaD treatment. Moreover, WithaD abrogated the expression of phosphorylated STAT and phosphorylated ERK1/2 in macrophages stimulated by lactate (). To establish whether WithaD altered macrophage function as well as cytokine production and surface marker expression, we evaluated the ability of WithaD -treated M2 macrophages activated by lactate to promote breast cancer proliferation, migration, and angiogenesis in vitro and in vivo (). Compared with the control group, macrophages pretreated with WithaD not only failed to promote cancer cell proliferation and migration ( and ) but also abrogated angiogenesis in vitro and in vivo. We found that tumor volume () was significantly reduced in the 5 mg/kg WithaD-treated group. The mean tumor weight of the 5 mg/kg WithaD-treated group was also much smaller than that of the control group (). At the same time, there was no significant difference between control and WithaD-treated mouse body weight (Data were not shown), implying few side-effects of WithaD at the therapeutic dosage. Tumor sections stained with anti-vWF antibodies revealed that WithaD inhibited new blood vessels as well as pruned preexisting tumor vessels (). Thus, WithaD modulation of lactate-activated M2 macrophages activation suppressed breast cancer growth and angiogenesis via inhibition of ERK/STAT3 signaling.
Figure 5. WithaD suppresses breast cancer growth and angiogenesis by impeding lactate-induced M2 macrophage polarization via the ERK/STAT3 signaling pathway. (A) The cell viability was detected by the CCK8 assay. BMDMs were treated with different concentrations of WithaD for 24 h. (B) WithaD suppresses M2-like macrophage polarization induced by lactate. The relative mRNA expression of M2-like macrophages was analyzed by QPCR in BMDMs pre-treated with WithaD for 30 min and then with or without lactate treatment for 12 h (n = 3). *P < 0.05. (C) Western blot for Arg-1 and MRC-1 in BMDMs stimulated by lactate for 24 h after pretreated with WithaD for 30 min. β-actin was used as a loading control. The Arg-1/β-actin or MRC-1/β-actin ratio was quantified and is shown as fold changes compared with control (n = 3). *P < 0.05, **P < 0.01 compared with control. (D) BMDMs were pretreated with WithaD for 30 min and then were stimulated with lactate or PBS for 24 h. Expression and phosphorylation levels of ERK1/2 and STAT3 in BMDMs were determined by Western blot. (E and F) Boyden chamber assay of the MDA-MB-231 cell (E), and pHUVEC (F) migration. Cells were plated on the upper chamber inserts with untreated (Control) media, and culture medium(CM) from lactate-activated macrophages which were pretreated with different concentration of WithaD for 30 min was put in the lower chambers. After migration for 6 h(F) or 24 h(E), the migrated cells were stained with crystal violet and counted as cells per field of view under the microscope (n = 5). (G) Capillary-structure formation of endothelial cells induced by lactate-activated macrophages was suppressed by WithaD. pHUVECs were placed in 96-well plates coated with Matrigel (2.0 × 104 cells/well) and stimulated with CM from macrophages which were pretreated with WithaD for 30 min and then treated with lactate or PBS. After 6 h, tubular structures were photographed (magnification, × 100). (H) The proliferation of pHUVECs induced by lactate-activated macrophages was suppressed by WithaD. pHUVECs cells were treated with CM from macrophages which were pretreated with WithaD for 30 min and then treated with lactate or PBS. (I-K) WithaD suppressed tumor growth and angiogenesis by suppressing lactate-induced M2 macrophage polarization. (I) WithaD suppressed tumor growth. Photographs of different treatment group tumors, along with the graph of tumor volume and tumor weight (K). (J) Immunohistochemistry of tumor slides stained with antibodies against vWF; arrows show new blood vessels in the tumor, with the statistical results of microvessels on the right. *P < 0.05, **P < 0.01 compared with control group; #P < 0.05 compared with lactate group.
Discussion
The Warburg effect is a well-known characteristic of breast cancer [Citation21] and the Reverse Warburg effect is specifically associated with poor overall survival of breast cancer [Citation22]. The activation of oncogenic signaling pathways induces the reprogramming of glucose metabolism in tumor cells, and aerobic glycolysis may also occur in the stromal compartment that surrounds the tumor [Citation23]. As a consequence of the Warburg effect and reverse Warburg effect, cancer cells and stromal cells secrete large amounts of lactate to the extracellular microenvironment [Citation24]. Lactate is a 3-carbon hydroxycarboxylic acid that has two stereoisomers, d-lactate, and l-lactate. L-lactate is the predominant physiological−enantiomer. Under healthy conditions, Lactate produced by most tissues in the human body is cleared by the liver and, to a lesser extent, by the kidneys. On a systemic level, lactate can serve as a source of energy by being carried to the liver and reconverted into glucose via the Cori cycle [Citation25]. Metabolic reprogramming of tumor cells modifies the metabolic fluxes, restructuring the Krebs cycle and enhancing glycolysis. The enhanced glycolytic carbon flux, in turn, leads to the production of high amounts of lactate. It is estimated that tumor cells produce up to 40 times more lactate than normal cells [Citation26]. Meanwhile, innate immune cells present in the tumor microenvironment also produce lactate at some points [Citation25]. Lactate is not only the predominate carbon source in the TCA cycle for cancer but is also of particular importance given its molecular effects on signaling within cancer and stromal cells [Citation14,Citation27]. Lactate suppresses antitumor immunity, increases tumor angiogenesis through endothelial cells [Citation28] and modulates the tumor microenvironment to promote tumor progression. Thus, anti-tumor therapies targeting lactate production and the lactate signaling pathway are of increasing interest. In this study, we found that intratumoral lactate-induced M2-like macrophage polarization, which was confirmed by increased mannose receptor and ARG1 expression in the macrophages. M2-like macrophage polarization induced by lactate is responsible for pro-angiogenesis and cancer progression. It is important to determine the details of this signaling pathway.
The breast tumor microenvironment (TME) comprised of recruited host stromal cells and tumor cells, has recently emerged as an important player in tumor progression, with the potential as a target for future treatment [Citation2]. The TME includes immune system elements (such as macrophages and lymphocytes), cells composing blood vessels, fibroblasts, myofibroblasts, mesenchymal stem cells, adipocytes and the extracellular matrix (ECM). Among these cells, tumor-associated macrophages (TAM) are a prominent component of the breast tumor microenvironment. Clinicopathological studies have suggested that a high TAM density correlates with poor prognosis in human breast carcinomas [Citation29]. Over the years, studies of the role of TAMs in breast cancer progression have identified TAMs to be capable of inducing angiogenesis, remodeling the tumor extracellular matrix to aid invasion, enabling breast cancer cells to evade host immune system and recruiting immunosuppressive leukocytes to the tumor microenvironment, similar to M2 macrophages [Citation30]. However, precisely how TAMs are educated by TME is still poorly defined.
Consistent with our findings, recent studies show that lactate is a pivotal factor driving macrophage M2 polarization [Citation31]. The signals from lactate that elicit M2-like functional polarization of TAMs are still not fully understood. Many signals are involved in M2-like macrophage polarization, for instance, ERK1/2, STAT3, HIF1α, and STAT6 and so forth, the activation of which can upregulate M2 macrophage polarization via different ways in the TME [Citation5,Citation15]. In this study, we found that lactate activated ERK1/2 and STAT3 signaling in macrophages. The M2-like macrophage markers were decreased by ERK/STAT3 inhibitors even if lactate existed. STAT3 signaling pathway is an important mechanism for inducing M2-type polarization of TAMs or THP-1 cells [Citation32]. The essential role of ERK was proved in M2 polarization of various types of IL-4-stimulated BM-derived macrophages [Citation33]. Therefore, suppression of both STAT3 and ERK signaling is a good strategy for treatment of cancer [Citation34], spared nerve injury (or neuropathic pain) [Citation35] and so forth. Here, we have shown the additive effects of the combination of selumetinib and stattic on macrophage polarization. Our results suggest that the combination of STAT3 and ERK inhibitors is more suitable to reverse M2 phenotype. Interestingly, selumetinib (ERK inhibitor) seems to be more active in mouse BMDM models and stattic (STAT3 inhibitor) in human THP-1 model, which may be associated with the different species. Moreover, the promoted functions of angiogenesis, migration, and proliferation of lactate-activated M2-like macrophage were abrogated by inhibition of ERK1/2 and STAT3 signaling. These data suggest that activation of the ERK 1/2 and STAT3 signaling pathways is of importance in macrophage M2-polarization induced by lactate.
In the present study, no significant change of YM-1 expression was observed in mouse BMDM exposed to lactate. Alternatively activated macrophages (M2 macrophages) are usually associated with anti-inflammatory and homeostatic functions linked to wound healing, fibrosis and tissue repair [Citation36]. Recently, five of the main M2 stimuli have been characterized including IL-4, IgG, IL-10, glucocorticoids and M-CSF [Citation37]. The M2 stimuli are complex and include transient and stable gene signatures which are involved in different transcription factors and signaling. Although Ym1 was considered as M2 makers, in some circumstance, its expression didn't change upon M2 stimuli. For example, when macrophage was polarized to the M2 phenotype, aryl hydrocarbon receptor-null cells exhibited lower levels of Fizz1, Ym1, and IL-10. In contrast, arginase activity was increased when compared to wild-type macrophages [Citation38]. Our data is consistent with the previous finding in which lactate induced M2 polarization of macrophages by increasing the expression of Arg1, Mrc1, but not higher levels of Ym1 [Citation13]. Our study not only confirms that lactate is an M2 macrophage driving signal but also identifies activation of ERK1/2 and STAT3 signaling as a key mechanism. Thus, our data suggest targeting ERK/STAT3 signaling is a powerful way to modulate macrophage phenotypes, and could serve as a promising cancer therapeutic strategy.
We have demonstrated that lactate secreted by tumors induces protumoral M2-like macrophages. In addition, blocking of the lactate signaling pathway directly improves the function of macrophages, even if the intratumoral concentration of lactate is not decreased. Therefore, we predict that suppression of the lactate signaling pathway that induces M2-like macrophages should improve the macrophage phenotype to prevent cancer. Natural compounds are a major source of current chemotherapeutic agents, and the chemotherapeutic and chemopreventive effects of such compounds have been demonstrated both in preclinical and clinical studies [Citation18]. Therefore, identification of candidate compounds with antitumor activity from the natural compound library has an important role in the research and development of new drugs. Withania somnifera formulation inhibited the pSTAT-3 expression in Ehrlich ascites tumor tissues [Citation39]. Accumulating evidence have shown that WithaD elicited apoptosis in malignant cells [Citation40], and reduced the levels of p-ERK in K562 and MOLT-4 cells [Citation41]. However, the exact mechanism of these anti-cancer properties is not clear. In this study, we investigated the impact of WithaD on M2 macrophage polarization induced by lactate. Our results provide the first evidence that WithaD effectively prevents macrophage M2-polarization by decreasing ERK/STAT3 activation, leading to suppression of breast cancer progression and tumor angiogenesis in vitro and in vivo.
Collectively, these data show that lactate is a pivotal factor in the TME that drives macrophage M2-polarization to promote breast cancer progression. In addition, we identified that activation of ERK/STAT3 is essential signaling in lactate signaling pathway, thereby deepening our molecular understanding of how lactate educates TAMs. Moreover, suppression of ERK/STAT3 signaling diminished tumor growth and angiogenesis by abolishing lactate-induced M2 macrophage polarization. Finally, research data of the natural compound WHD provides evidence that ERK/STAT3 signaling is a therapeutic strategy for the prevention and treatment of breast cancer.
Methods
Cell culture
THP-1, pHUVCEs, MDA-MB231, and MCF7 cell lines were cultured in culture media containing 10% FBS (Sigma) and 1:100 antibiotic-antimycotic (Gibco, Grand Island, NY, USA). The THP-1, MDA-MB231, and MCF7 cell lines were purchased from the American Type Culture Collection. The conditioned medium (CM) was obtained from serum-free media of cultured THP-1 or BMDM cells containing 0.25% FBS for 24 h after phosphate-buffered saline or lactate acid stimulation for 12 h. Primary human umbilical vascular endothelial cells (pHUVECs) were obtained from human umbilical cord veins by collagenase treatment, as described previously, and used at early passages 1–6. The pHUVECs were cultured in endothelial cell medium (ECM) (1001, Sciencell, San Diego, CA, USA) [Citation10]. All cells were cultured at 37°C under a humidified 95%:5% (v/v) mixture of air and CO2.
Proliferation and Live cell Imaging
For live cell imaging experiments, MCF7 cells were plated at single-cell density (1 × 105 cells/well) on four-well chambered coverslips (Thermo Fisher Scientific, San Jose, CA, USA). Individual MCF7 cells were allowed to settle and adhere to 12 h before the media was exchanged to phenol red-free CM media. Two-dimensional (2D) live cell imaging of MCF7 cells was performed on a Zeiss microscope with a 37°C incubated live imaging chamber, filled with humidified air supplemented with 5% CO2. Multiple positions were performed to capture multiple division events during any given experiment. Images were captured at 0 h and 24 h.
Wound-healing migration assay
Primary (p)HUVECs or breast cancer cells were plated to full confluence in 24-well plates pre-coated with 0.1% gelatin (Sigma, St. Louis, MO, USA) and were subsequently starved with serum-free medium containing 0.25% FBS for 6 h (pHUVECs) or 12 h (breast cancer cells) to inactivate cell proliferation. Subsequently, the monolayer of cells was wounded by a 200 µl sterile pipette tip and washed with phosphate-buffered saline to remove the detached cells. Different CMs were added to the wells. Images were then photographed (Olympus IX5, Olympus America Inc., Center Valley, PA USA; magnification, × 100) [Citation42]. The distance between the wound gap was measured in micrometers. Three independent experiments were performed.
Boyden chamber assay
Migration of breast cancer cells or pHUVECs was examined using 24-well Boyden chambers (Corning, NY, USA) with transwell inserts of 8 μM pore size, as previously reported. CM from lactate-activated macrophages which were pretreated with 10 mM Selumetinib (s1008, Selleck), 10 mM Stattic (s7024, Selleck) or withanolide D (JJL55240202020202, Shanghai jinjinle Industry Co., Ltd) for 30 min was put in the lower chambers. pHUVECs (1.8 × 104 cells per well) or breast cancer cells (105 cells per well) suspended in serum-free ECM were seeded in the upper compartment of the Boyden chamber and cultured at 37°C for 6 h (pHUVECs) or 24 h (breast cancer cells). The migrated cells were fixed with 4% PFA and stained with crystal violet (0.05%; Sigma), and then counted as cells per field of view under a microscope (Olympus IX5, Olympus America Inc., Center Valley, PA USA; magnification, × 100). Three independent experiments were performed.
Capillary-like tube formation assay
Briefly, Growth factor–reduced Matrigel (356231, BD Biosciences) was put into pre-chilled 96-well plates (60 μL Matrigel/well). Matrigel was polymerized for 60 min at 37°C. pHUVECs were incubated in serum-free ECM containing 0.25% FBS for 6 h, subsequently pretreated with various dilutions of WithaD and inhibitors for 30 min, and then pipetted onto the Matrigel in 96-well plates (2.0 × 104 cells per well) suspended in different CM. After 6 h of incubation at 37°C, the tubular structures of the endothelial cells were then photographed (Olympus IX5, Olympus America Inc., Center Valley, PA USA; magnification, × 100). The tube number of the capillary-like structure was measured. Inhibition percentage was expressed using serum-free ECM; wells containing 0.25% FBS -treated were control, 100%.
RNA isolation and QPCR
Total RNA was isolated from cells or mouse tissues using Trizol reagent (Invitrogen, Carlsbad, CA, USA), following the manufacturer's instructions. RNA (200–500 ng) was removed from all genomic DNA with RNase-free DNase I (Sigma, St. Louis, MO, USA), and then reverse-transcribed into cDNA using the QPCR RT kit (Invitrogen, Carlsbad, CA, USA). The cDNA was analyzed using real-time QPCR (SYBR Green PCR Master mix; Roche Applied Science, Mannheim, Germany) with a StepOnePlusTM Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). Each reaction was performed in triplicate in a 96-well plate. The expression of each gene was normalized by the expression of GAPDH.
Primers
The following primers were used to determine the relative gene expression using quantitative PCR. Mouse primers: Arg1: FP, 5′- CAT TGG CTT GCG AGA CGT AGA C-3′; RP, 5′- GCT GAA GGT CTC TTC CAT CAC C-3′. MRC1: FP, 5′- GTT CAC CTG GAG TGA TGG TTC TC-3′; RP, 5′- AGG ACA TGC CAG GGT CAC CTT T-3′. Ym1: FP, 5′- TAC TCA CTT CCA CAG GAG CAG G-3′; RP, 5′- CTC CAG TGT AGC CAT CCT TAG G-3′. CD86: FP, 5′- ACG TAT TGG AAG GAG ATT ACA GCT-3′; RP, 5′- TCT GTC AGC GTT ACT ATC CCG C-3′. VEGFA: FP, 5′- CTG CTG TAA CGA TGA AGC CCT G-3′; RP, 5′- GCT GTA GGA AGC TCA TCT CTC C-3′. Hgf: FP, 5′- GTC CTG AAG GCT CAG ACT TGG T-3′; RP, 5′- CCA GCC GTA AAT ACT GCA AGT GG-3′. Mmp9: FP, 5′- GCT GAC TAC GAT AAG GAC GGC A-3′; RP, 5′- TAG TGG TGC AGG CAG AGT AGG A-3′. Mmp8: FP, 5′- GAT GCT ACT ACC ACA CTC CGT G-3′; RP, 5′- TAA GCA GCC TGA AGA CCG TTG G-3′. GAPDH: FP, 5′- CAT CAC TGC CAC CCA GAA GAC TG -3′; RP, 5′- ATG CCA GTG AGC TTC CCG TTC AG -3′. Human primers: CD206: FP, 5′- AGC CAA CAC CAG CTC CTC AAG A-3′; RP, 5′- CAA AAC GCT CGC GCA TTG TCC A-3′. CD163: FP, 5′- CCA GAA GGA ACT TGT AGC CAC AG-3′; RP, 5′- CAG GCA CCA AGC GTT TTG AGC TA-3′. CD86: FP, 5′- CCA TCA GCT TGT CTG TTT CAT TCC-3′; RP, 5′- GCT GTA ATC CAA GGA ATG TGG TC-3′. CD80: FP, 5′- CTC TTG GTG CTG GCT GGT CTT T-3′; RP, 5′- GCC AGT AGA TGC GAG TTT GTG C-3′. IDO2: FP, 5′- GTT ATG TCT GGC AGG AAG GAG AG-3′; RP, 5′- GTC CAG TTC GTC AGC ACC AAG T-3′. CCL18: FP, 5′- GTT GAC TAT TCT GAA ACC AGC CC-3′; RP, 5′- GTC GCT GAT GTA TTT CTG GAC CC-3′. VEGFA: FP, 5′- TTG CCT TGC TGC TCT ACC TCC A-3′; RP, 5′- GAT GGC AGT AGC TGC GCT GAT A-3′. GAPDH: FP, 5′- GTC TCC TCT GAC TTC AAC AGC G-3′; RP, 5′- ACC ACC CTG TTG CTG TAG CCA A -3′.
Western blot
After receiving the indicated treatments, macrophages were harvested for protein extraction. Protein concentration was examined by a BCA assay (Thermo Fisher Scientific, San Jose, CA, USA). Samples of 20 μg of protein were applied to SDS polyacrylamide gels. After protein transfer, membranes were incubated with antibodies (1: 1,000) for Arg-1 (Rabbit monoclonal; #93668, CST, Danvers, MA, USA.), MRC-1/CD206 (Rabbit polyclonal; ab64693, Abcam, Cambridge, MA, U.K.), p/t-ERK, p/t-STAT3, or β-actin (Rabbit monoclonal: #4695, #4370, #9145, #4970; Mouse monoclonal: #9139, CST, Danvers, MA, USA) overnight at 4°C. Membranes were washed and then incubated with HRP-conjugated secondary antibodies (1: 1,000) for 1 h at room temperature. Reactive bands were detected with an ECL Western Blot Detection Kit (35050, Thermo, USA). The relative intensities of the bands were quantified by densitometry and normalized to β-actin signal levels. Densitometric quantification was performed by Image-Pro Plus 6.0 software (Media Cybernetics, Inc. Rockville, MD, USA).
Human breast tumor xenograft mouse model
The experimental protocol was established according to institutional ethical guidelines for Animal Research and Care and approved by the Ethics Committee of the Second Affiliated Hospital of Nanjing Medical Universi [Citation42]. Six-week-old female BALB/c nude mice (National Rodent Laboratory Animal Resources, Shanghai, China) weighing approximately 22 g were randomly divided into three groups of six each. MDA-MB-231 cells were s.c. injected into the mice (8 × 106 per mouse). After the tumors grew to approximately 100 mm3, the mice were injected intraperitoneally with either vehicle or WithaD (5 mg/kg or 2.5 mg/kg) every day for 18 days. The tumor size was measured with a caliper (calculated volume = shortest diameter2 × longest diameter/2) at three-day intervals.
Histology and immunohistochemistry
The procedure and protocol of staining have been reported in detail in our previous report [Citation42]. Solid tumors were removed and fixed with 10% formaldehyde and embedded in paraffin. Specific blood vessel staining was performed on 5-μm deparaffinized sections with a specific blood vessel staining kit (von Willebrand Factor; Millipore, Billerica, MA, USA). Section images were obtained with using a Leica DM 4000B photomicroscope (Leica, Wetzlar, Germany) at a magnification of 200 ×. Blood vessels were counted (n = 5).
Statistical analysis
All data reported herein represent means ± SD from at least three independent experiments. Statistical comparisons between the treated groups and the control group were performed by one-way analysis of variance (ANOVA) followed by Dunnett's test, and the difference between two groups was analyzed by Student's t-test. A P value of <0.05 was considered statistically significant.
Disclosure of potential conflicts of interest
The authors declare no conflict of interest.
Acknowledgments
The authors thank the National Natural Science Foundation of China and the Jiangsu Provincial Talent Work Leading Group Office for supporting this work.
Additional information
Funding
Related Research Data
References
- Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012 CA Cancer J Clin 2015.65:87–108. doi:10.3322/caac.21262.
- Choi J, Gyamfi J, Jang H, et al. The role of tumor-associated macrophage in breast cancer biology. Histol Histopathol. 2017;11916;
- Sossey-Alaoui K, Pluskota E, Bialkowska K, et al. Kindlin-2 regulates the growth of breast cancer tumors by activating CSF-1-Mediated macrophage infiltration. Cancer Res. 2017;77:5129–5141. PMID:28687620
- Boniakowski AE, Kimball AS, Jacobs BN, et al. Macrophage-mediated inflammation in normal and diabetic wound healing. J Immunol. 2017;199:17–24. doi:10.4049/jimmunol.1700223. PMID:28630109
- Zhang W, Xu W, Xiong S. Macrophage differentiation and polarization via phosphatidylinositol 3-kinase/Akt-ERK signaling pathway conferred by serum amyloid P component. J Immunol. 2011;187:1764–1777. doi:10.4049/jimmunol.1002315. PMID:21753147
- Sun L, Chen B, Jiang R, et al. Resveratrol inhibits lung cancer growth by suppressing M2-like polarization of tumor associated macrophages. Cell Immunol. 2017;311:86–93. doi:10.1016/j.cellimm.2016.11.002. PMID:27825563
- Raggi F, Pelassa S, Pierobon D, et al. Regulation of human macrophage M1-M2 polarization balance by hypoxia and the triggering receptor expressed on myeloid cells-1. Front Immunol. 2017;8:1097. doi:10.3389/fimmu.2017.01097. PMID:28936211
- Rahman K, Vengrenyuk Y, Ramsey SA, et al. Inflammatory Ly6Chi monocytes and their conversion to M2 macrophages drive atherosclerosis regression. J Clin Invest. 2017;127:2904–2915. doi:10.1172/JCI75005. PMID:28650342
- Shinohara H, Kuranaga Y, Kumazaki M, et al. Regulated polarization of tumor-associated macrophages by miR-145 via colorectal cancer-derived extracellular vesicles. J Immunol. 2017;199:1505–1515. doi:10.4049/jimmunol.1700167. PMID:28696255
- Mu X, Zhao T, Xu C, et al. Oncometabolite succinate promotes angiogenesis by upregulating VEGF expression through GPR91-mediated STAT3 and ERK activation. Oncotarget. 2017;8:13174–13185. doi:10.18632/oncotarget.14485. PMID:28061458
- Zhao T, Mu X, You Q. Succinate: An initiator in tumorigenesis and progression. Oncotarget. 2017;8:53819–53828. PMID:28881853
- Vaitheesvaran B, Xu J, Yee J, et al. The Warburg effect: a balance of flux analysis. Metabolomics. 2015;11:787–796. doi:10.1007/s11306-014-0760-9. PMID:26207106
- Colegio OR, Chu NQ, Szabo AL, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513:559–563. doi:10.1038/nature13490. PMID:25043024
- Faubert B, Li KY, Cai L, et al. Lactate metabolism in human lung tumors. Cell. 2017;171:358–371.e9. doi:10.1016/j.cell.2017.09.019. PMID:28985563
- Peda JD, Salah SM, Wallace DP, et al. Autocrine IL-10 activation of the STAT3 pathway is required for pathological macrophage differentiation in polycystic kidney disease. Dis Model Mech. 2016;9:1051–1061. doi:10.1242/dmm.024745. PMID:27491076
- Li R, Hebert JD, Lee TA, et al. Macrophage-Secreted TNFalpha and TGFbeta1 influence migration speed and persistence of cancer cells in 3D tissue culture via independent pathways. Cancer Res. 2017;77:279–290. doi:10.1158/0008-5472.CAN-16-0442. PMID:27872091
- Zhang J, Cao J, Ma S, et al. Tumor hypoxia enhances non-small cell lung cancer metastasis by selectively promoting macrophage M2 polarization through the activation of ERK signaling. Oncotarget. 2014;5:9664–9677. doi:10.18632/oncotarget.1856. PMID:25313135
- Suta S, Maggi F, Nicoletti M, et al. New drugs from old natural compounds: scarcely investigated sesquiterpenes as new possible therapeutic agents. Curr Med Chem. 2018;25:1–18. doi:10.2174/0929867324666170404150351. PMID: 28393689
- Issa ME, Wijeratne EMK, Gunatilaka AAL, et al. Withanolide D exhibits similar cytostatic effect in drug-resistant and drug-sensitive multiple myeloma cells. Front Pharmacol. 2017;8:610. doi:10.3389/fphar.2017.00610. PMID: 28943850
- Batista PH, de Lima KS, Pinto F, et al. Withanolides from leaves of cultivated Acnistus arborescens. Phytochemistry. 2016;130:321–327. doi:10.1016/j.phytochem.2016.07.003. PMID: 27498045
- Hart PC, Mao M, de Abreu AL, et al. MnSOD upregulation sustains the Warburg effect via mitochondrial ROS and AMPK-dependent signalling in cancer. Nat Commun. 2015;6:6053. doi:10.1038/ncomms7053. PMID: 25651975
- Witkiewicz AK, Whitaker-Menezes D, Dasgupta A, et al. Using the “reverse Warburg effect” to identify high-risk breast cancer patients: stromal MCT4 predicts poor clinical outcome in triple-negative breast cancers. Cell Cycle. 2012;11:1108–1117. doi:10.4161/cc.11.6.19530. PMID: 22313602
- Liberti MV, Dai Z, Wardell SE, et al. A predictive model for selective targeting of the warburg effect through GAPDH inhibition with a natural product. Cell Metab. 2017;26:648–659.e8. doi:10.1016/j.cmet.2017.08.017. PMID: 28918937
- Tekade RK, Sun X. The Warburg effect and glucose-derived cancer theranostics. Drug Discov Today. 2017.; doi:10.1016/j.drudis.2017.08.003.
- Romero-Garcia S, Moreno-Altamirano MM, Prado-Garcia H, et al. Lactate contribution to the tumor microenvironment: mechanisms, effects on immune cells and therapeutic relevance. Front Immunol. 2016;7:52. doi:10.3389/fimmu.2016.00052. PMID: 26909082
- Holm E, Hagmuller E, Staedt U, et al. Substrate balances across colonic carcinomas in humans. Cancer Res. 1995;55:1373–1378. PMID: 7882338
- Feng J, Yang H, Zhang Y, et al. Tumor cell-derived lactate induces TAZ-dependent upregulation of PD-L1 through GPR81 in human lung cancer cells. Oncogene. 2017;36:5829–5839. doi:10.1038/onc.2017.188. PMID: 28604752
- Vegran F, Boidot R, Michiels C, et al. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-kappaB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011;71:2550–2560. doi:10.1158/0008-5472.CAN-10-2828. PMID: 21300765
- Chen XW, Yu TJ, Zhang J, et al. CYP4A in tumor-associated macrophages promotes pre-metastatic niche formation and metastasis. Oncogene. 2017;36:5045–5057. doi:10.1038/onc.2017.118. PMID: 28481877
- Niu Z, Shi Q, Zhang W, et al. Caspase-1 cleaves PPARgamma for potentiating the pro-tumor action of TAMs. Nat Commun. 2017;8:766. doi:10.1038/s41467-017-00523-6. PMID: 28974683
- Chen P, Zuo H, Xiong H, et al. Gpr132 sensing of lactate mediates tumor-macrophage interplay to promote breast cancer metastasis. Proc Natl Acad Sci U S A. 2017;114:580–585. doi:10.1073/pnas.1614035114. PMID: 28049847
- He W, Zhu Y, Mu R, et al. A Jak2-selective inhibitor potently reverses the immune suppression by modulating the tumor microenvironment for cancer immunotherapy. Biochem Pharmacol. 2017;145:132–146. doi:10.1016/j.bcp.2017.08.019. PMID: 28859967
- Veremeyko T, Yung AWY, Dukhinova M, et al. Cyclic AMP pathway suppress autoimmune neuroinflammation by inhibiting functions of encephalitogenic CD4 T cells and enhancing M2 macrophage polarization at the site of inflammation. Front Immunol. 2018;9:50. doi:10.3389/fimmu.2018.00050. PMID: 29422898
- Zhou J, Zhao T, Ma L, et al. Cucurbitacin B and SCH772984 exhibit synergistic anti-pancreatic cancer activities by suppressing EGFR, PI3K/Akt/mTOR, STAT3 and ERK signaling. Oncotarget. 2017;8:103167–103181. doi:10.18632/oncotarget.21704. PMID: 29262554
- Ding CP, Guo YJ, Li HN, et al. Red nucleus interleukin-6 participates in the maintenance of neuropathic pain through JAK/STAT3 and ERK signaling pathways. Exp Neurol. 2018;300:212–221. doi:10.1016/j.expneurol.2017.11.012. PMID: 29183675
- Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. 2011;11:750–761. doi:10.1038/nri3088. PMID: 22025054
- Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000prime Rep. 2014;6:13. doi:10.12703/P6-13. PMID: 24669294
- Climaco-Arvizu S, Dominguez-Acosta O, Cabanas-Cortes MA, et al. Aryl hydrocarbon receptor influences nitric oxide and arginine production and alters M1/M2 macrophage polarization. Life Sci. 2016;155:76–84. doi:10.1016/j.lfs.2016.05.001. PMID: 27153778
- Malik F, Kumar A, Bhushan S, et al. Immune modulation and apoptosis induction: two sides of antitumoural activity of a standardised herbal formulation of Withania somnifera. Eur J Cancer. 2009;45:1494–1509. doi:10.1016/j.ejca.2009.01.034. PMID: 19269163
- Mondal S, Bhattacharya K, Mallick A, et al. Bak compensated for Bax in p53-null cells to release cytochrome c for the initiation of mitochondrial signaling during Withanolide D-induced apoptosis. PLoS One. 2012;7:e34277. doi:10.1371/journal.pone.0034277. PMID: 22479585
- Mondal S, Mandal C, Sangwan R, et al. Withanolide D induces apoptosis in leukemia by targeting the activation of neutral sphingomyelinase-ceramide cascade mediated by synergistic activation of c-Jun N-terminal kinase and p38 mitogen-activated protein kinase. Mol Cancer. 2010;9:239. PMID: 20836852
- Mu X, Shi W, Sun L, et al. Pristimerin, a triterpenoid, inhibits tumor angiogenesis by targeting VEGFR2 activation. Molecules. 2012;17:6854–6868. doi:10.3390/molecules17066854. PMID: 22669041