
ABSTRACT
Recovery from DNA damage is critical for cell survival. However, serious damage cannot be repaired, leading to cell death for prevention of abnormal cell growth. Previously, we demonstrated that 4N-DNA accumulates via the initiation of an abnormal interphase without cytokinesis and that re-replication occurs during a prolonged recovery period in the presence of severe DNA damage in mitotic cells. Mitotic phosphorylated Plk1 is typically degraded during mitotic exit. However, Plk1 has unusually found to be dephosphorylated in mitotic slippage without cytokinesis during recovery from mitotic DNA damage. Here, we investigated how Plk1 dephosphorylation is established during recovery from mitotic DNA damage. Mitotic DNA damage activated ATM and Chk1/2 and repressed Cdk1 and Greatwall protein kinase, followed by PP2A activation through the dissociation of ENSA and PP2A-B55. Interaction between Plk1 and PP2A-B55α or PP2A-B55δ was strongly induced during recovery from mitotic DNA damage. Moreover, the depletion of PP2A-B55α and/or PP2A-B55δ by siRNA transfection led to the recovery of Plk1 phosphorylation and progression of the cell cycle into the G1 phase. Therefore, to adapt to severe DNA damage, the activated Greatwall/ENSA signaling pathway was repressed by ATM/Chk1/2, even in mitotic cells. Activation of the PP2A-B55 holoenzyme complex induced the dephosphorylation of Plk1 and Cdk1, and finally, mitotic slippage occurred without normal chromosome segregation and cytokinesis.
Introduction
Activation of DNA damage response halts cell cycle progression and provides time for the repair of lesions; this process is called the DNA damage checkpoint. The molecular network involved in this checkpoint is well established: two phosphatidylinositol 3-kinase-related kinases, ATM and ATR as sensor kinases are activated, which then phosphorylate a large number of substrate proteins to regulate the checkpoint process, DNA repair, and cell removal [Citation1]. Checkpoint activation is mediated by two checkpoint kinases, Chk1 and Chk2, which are the targets of ATM and ATR. Checkpoint kinases phosphorylate various cell cycle regulators to halt cell division [Citation2]. Cdc25 phosphatase is a key regulator of the cell cycle that promotes the activation of cyclin-dependent protein kinases (Cdks) by dephosphorylating Cdks at inhibitory phosphorylation sites [Citation3]. Chk1 and Chk2 play critical roles in preventing Cdk activation through the inhibition of Cdc25 [Citation4,Citation5]. When DNA damage occurs in the G2 phase, Cdk1 is inactivated through the inhibition of Cdc25C followed by ATM/Chk activation, and mitotic entry is effectively blocked until the damage is repaired. In addition to Cdk1, recent studies have revealed that the Greatwall kinase is critical for regulating DNA damage responses in the G2 phase and controlling the timing of mitotic entry after DNA damage [Citation6,Citation7]. The Greatwall kinase phosphorylates ENSA and Arpp-19, which specifically interact with and inhibit the PP2A-B55 complex. The inhibition of PP2A maintains the phosphorylation state of Cdk1 substrates, allowing the progression of mitotic entry [Citation8,Citation9]. In previous studies, we investigated DNA damage response during mitosis, but not during the G2 phase. DNA damage during early mitosis induces the inactivation of mitosis-specific protein kinases such as Cdk1 and Plk1 in an ATM/Chk-dependent manner. Under these conditions, cells skip the entire late mitotic process, such as anaphase-promoting complex/cyclosome (APC/C) activation and cytokinesis, and instead enter the G1 phase with 4N-DNA content [Citation10,Citation11]. During the long-term response to mitotic DNA damage, 8N-DNA content is generated through re-replication. Depending on p53 activity, cells adapt with multiploidy or die through apoptosis [Citation12]. Plk1 plays key roles in centrosome maturation, bipolar spindle establishment, and APC/C activation from the beginning to the end of the mitotic phase [Citation13,Citation14]. Plk1 is phosphorylated and activated during mitosis and degraded by APC/C at the end of mitosis. In addition to its mitotic functions, Plk1 is a regulator of recovery from cell cycle arrest associated with G2-DNA damage by directly targeting multiple DNA damage checkpoint factors, including Claspin, 53BP1, and Chk2 [Citation15,Citation16]. Downregulation of Plk1 has been shown to delay mitotic entry after the G2-DNA damage checkpoint [Citation17,Citation18]. We previously reported that DNA damage stress can occur in mitotic cells in which Plk1 has already been activated [Citation10] and that for recovery from severe mitotic DNA damage, Plk1 is inactivated by dephosphorylation but not by degradation. Dephosphorylation of Plk1 is dependent on ATM/Chk activation and PP2A activity [Citation19]. In this study, we demonstrate how dephosphorylation of Plk1 occurs during the mitotic DNA damage response and how ATM/Chk activation is linked to the functionality of PP2A, which causes Plk1 dephosphorylation.
Materials and methods
Cell culture and treatments
Wild-type HCT116 cells were cultured in Dulbecco’s modified Eagle’s medium containing 10% FBS (Corning). To synchronize cells during prometaphase, they were treated with 100 ng/ml nocodazole (Sigma) for 16 h and collected by shaking. To induce DNA damage in mitotic and interphase cells, cells were treated with 5 μM doxorubicin (Sigma) for 1 h and 6 h, respectively. For recovery from the damage, cells were incubated in a fresh medium for an indicated time following wash. Finally, for the inhibition of ATM kinase and PP2A, cells were treated with 100 μM KU55933 (Sigma) and 100 nM okadaic acid (Alexis), respectively.
DNA constructs and ectopic expression
In cloning vectors, human cDNAs of PP2A-B55α, -B56γ (Sino Biological Inc.), -B55δ (Origene), -B56α, -B56β (GenScipt), and a catalytic subunit (by cDNA construction) were amplified by PCR using oligonucleotide primers (). The PCR products were cloned into pCMV-Tag2B, pEGFP-C3, and pGEX4T-2 vectors to obtain FLAG-, GFP-, and GST-fusion protein constructs, respectively. For expression in mammalian cells, DNA constructs were transfected into cells using CalPhos Mammalian Transfection Kit (Takara-Clontech) following the procedure recommended by the manufacturer with modification. In brief, 20 μg of DNA in 500 μl calcium solution was precipitated by adding 500 μl 2x HBS buffer and dropped into cells cultured until 50% confluent in a 100-mm dish. After 24 h of DNA removal, cells were harvested and used for further study.
Table 1. Oligonucleotide primers used for PCR.
Flow cytometry
Cells were harvested, fixed in 80% cold ethanol for more than 16 h, and incubated with 100 μg/ml RNase A at 37°C for 2 h. Fixed cells were stained with 40 μg/ml propidium iodide (Sigma), and DNA contents were analyzed by flow cytometry using FACScan Flow Cytometer (Becton Dickinson). The data were analyzed using the Cell Quest program (Becton Dickinson).
Immunological assays and antibodies
Cells were lysed in a lysis buffer (0.5% NP-40, 150 mM NaCl, 20 mM Tris-HCl, pH 8.0, 2 mM EDTA, pH 8.0, 2 mM EGTA, pH 8.0) containing protease and phosphatase inhibitors. For western blotting, cell lysate was separated on SDS-PAGE and proteins were transferred on a PVDF membrane (Millipore). The proteins on the membrane were incubated with primary and secondary antibodies diluted in TBS containing 0.1% Tween-20 and 5% skim milk or 5% BSA, and protein signals were visualized using the ECLTM system (Amersham Biosciences). For immunoprecipitation, cell lysate was incubated with the indicated antibody and protein A/G agarose bead. Beads were subjected to SDS-PAGE for immunoblotting. For immunofluorescence analysis, cells were grown on fibronectin-coated glass coverslips and fixed in 4% paraformaldehyde for 2 h at 4°C, followed by treatment with cold methanol for dehydration for 2 min. After blocking, proper primary and FITC- or Cy3-labeled secondary antibodies were added for staining. Fluorescence signals were detected using a confocal microscopy (LSM510, Zeiss). Antibodies against ATM, phospho-ATM (S1981), Chk1, phospho-Chk1 (S345), Chk2, phospho-Chk2 (T68), phospho-Plk1 (T210), phospho-Cdc25 (S216), phospho-Cdc25 (T198), phospho-Cdc25 (T48), phospho-Cdk1 (T161), Greatwall, and ENSA were purchased from Cell Signaling. Antibodies against the PP2A catalytic subunit and Cdk1 were purchased from Millipore. Antibodies against Cdc25, phospho-Cdk1 (Y14/T15), PP2A-B55, PP2A-B56, and actin were purchased from Santa Cruz Biotechnology. Further, anti-Plk1 and anti-FLAG antibodies were obtained from Invitrogen and Sigma, respectively.
In vitro phosphatase assays
PP2A activity was performed using PP2A Immunoprecipitation Phosphatase Assay Kit (Millipore) according to the procedure recommended by the manufacturer with modification. Briefly, endogenous PP2A immunoprecipitated using an anti-PP2A antibody incubated with a synthetic phosphopeptide (K-R-pT-I-R-R) at 30°C for 10 min with shaking. Free phosphate from the phosphopeptide by PP2A activity was estimated based on color using the Malachite Green phosphate detection solution and quantified as optical density measured at a wavelength of 650 nm. For in vitro phosphatase assay, we followed the procedure reported previously with modification [Citation20]. FLAG-PP2A-B55α and B55δ-transfected HeLa cell lysates were subjected to immunoprecipitation using an anti-FLAG antibody. Immunoprecipitates were preincubated at 4°C for 10 min with PP2A assay buffer (20 mM Tris-HCl, pH 7.4, 1% Triton X-100, 250 mM sucrose, 1 mM MnCl2, and 0.1% β-mercaptoethanol) and 2 µg GST-PP2AC. Then, 0.5 µg of active His-Plk1 (Thermo Fisher Scientific) was added to the reaction solution to a final volume of 20 µl. Phosphatase reactions were performed at 30°C for 30 min. Phosphorylation of Plk1 was detected by the anti-phospho-Plk1(T210) band intensity.
RNA interference
SiRNAs targeting ATM (UCUGUACUUGAUAGACACU), Chk1 (GUGGAUUUUCUAAGCACAU), Chk2 (CCUGUGGAGAGGUAAAGCU), PP2A-B55α (GACAGCGUGCCAUUCGACA), and PP2A-B55δ (GUGUUCGUCUACAGCAGUA) were generated by Bioneer (Daejeon, Korea). Cells were grown in a 6-well culture dish at a density of 2 × 105 cells/well and were transfected with siRNA using LipofectamineTM RNAiMAX (Invitrogen) following the manufacturer’s instructions. To confirm gene silencing, genes were ectopically overexpressed to rescue the silencing effect. DNA constructs were transfected 6 h after siRNA transfection.
Results
The ATM/Chk pathway is activated during mitotic DNA damage response
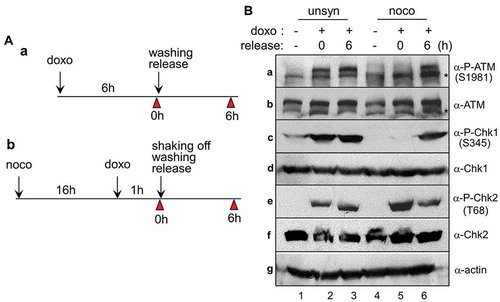
In cells with DNA damage, the checkpoint machinery generally operates correctly, with an attempt to achieve recovery from lesions. For the induction of interphase DNA damage, wild-type HCT116 cells cultured in an unsynchronized manner were treated with 5 μM doxorubicin for 6 h and released into a fresh medium for recovery for indicated times (). In addition, for the induction of mitotic DNA damage, prometaphase cells prepared by nocodazole treatment for 16 h were treated with doxorubicin for 1 h and released for recovery (). It has long been known that autophosphorylation of ATM kinase at serine-1981 and phosphorylation of Chk1 at serine-345 and of Chk2 at threonine-68 are required for an appropriate DNA damage response [Citation21–Citation23]. Indeed, doxorubicin treatment in unsynchronized cells induced the phosphorylation of ATM, Chk1, and Chk2 immediately after recovery from mitotic DNA damage (, lanes 2 and 3 in a, c, and e). In the case of mitotic DNA damage response, phosphorylation of ATM kinase and Chk1 was observed during a recovery period of 6 h after doxorubicin treatment (, lanes 5 and 6 in a and c). Compared with the phosphorylation of ATM and Chk1, phosphorylation of Chk2 at threonine-68 rapidly increased immediately after doxorubicin treatment and continued to decrease during recovery (, lanes 5 and 6 in e). These results suggest that mitotic DNA damage induces a DNA damage checkpoint pathway in an ATM/Chk-dependent manner.
Figure 1. ATM and Chk1/2 activate during recovery from DNA damage. (A) Experimental design for the induction of DNA damage and recovery in unsynchronous (a) and mitotic cells (b). Cells were harvested for indicated times (arrowhead). doxo and noco indicate doxorubicin and nocodazole, respectively. (B) Activation of DNA damage indicators following the doxorubicin treatment of cells that were subsequently regrown in fresh media. Unsynchronous (unsyn) and mitotic cells (mitotic) were treated with doxorubicin and released into fresh media for indicated time. The indicated proteins and their phosphorylated epitopes were detected by immunoblotting. 1 and 4, no treatment with doxorubicin; 2 and 5, immediately after treatment with doxorubicin; 3 and 6, washing and regrown for 6 h. Asterisk (*) indicates nonspecific signals recognized by ATM antibodies.
Greatwall and other mitosis-specific kinases were dephosphorylated during recovery from mitotic DNA damage
We next investigated whether any modification of key mitotic regulators and DNA damage-related proteins could occur during recovery from mitotic DNA damage. For this, mitotic cells treated with doxorubicin were released into a fresh medium for recovery from mitotic DNA damage and collected at a tighter time interval (). After mitotic arrest, cell division normally occurred within 100 min of release and the cells entered G1 phase with 2N-DNA content (, noco in a and b). However, cells containing mitotic DNA damage by doxorubicin treatment did not divide even after 2 h of release, and the cells persisted with 4N-DNA content thereafter (, noco/doxo in a and b). Similarly, when mitotic cells were damaged by gamma-radiation, cells persisted with 4N-DNA content during recovery (Supplementary Figure 1) [Citation10]. Although these cells contained 4N-DNA, the mitosis-specific phosphorylation signals of mitotic proteins totally decreased (, lane 4 in c) and the nuclear membrane was regenerated during recovery for 6 h (Supplementary Figure 2) [Citation10]. These results indicate that mitotic cells containing DNA damage enter interphase during recovery without cell division.
Figure 2. Inactivation of mitotic kinases through dephosphorylation during recovery from mitotic DNA damage. (A) Experimental design for the induction of DNA damage and recovery in mitotic cells. Cells were harvested for indicated times (arrowhead). doxo and noco indicate doxorubicin and nocodazole, respectively. (B) Cellular changes in mitotic cells with DNA damage during recovery. Cells treated with nocodazole to facilitate prometaphase arrest (noco) or followed by doxorubicin treatment to induce DNA damage (noco/doxo) were released into fresh media for damage recovery for indicated time. The changes in cell division, DNA content, and mitotic index were detected by time-lapse microscopy (a), FACS analysis (b), and immunostaining with anti-MPM2 antibody (c), respectively. DNA contents in cells are indicated as 2N and 4N. (C) The indicated proteins and phosphorylated forms were analyzed by immunoblotting from cell extracts. Cells were treated with nocodazole to facilitate prometaphase arrest (noco) or followed by doxorubicin treatment to induce DNA damage (noco/doxo). 1, mitotic cells; 2, immediately after doxorubicin treatment in mitotic cells; 3–5, cells regrown in fresh media for recovery from mitotic DNA damage for 3, 6, and 9 h, respectively.
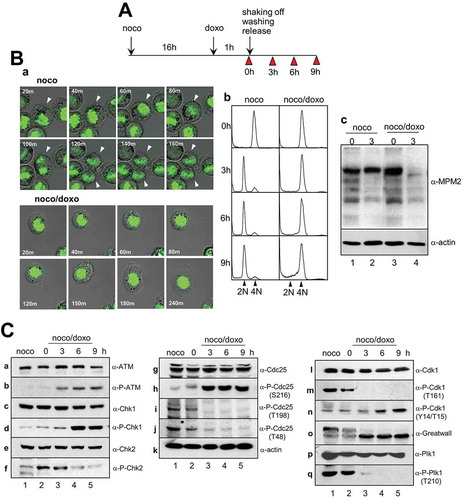
The active phosphorylation of ATM kinase and Chk1 again gradually increased from 3 h of recovery (). Remarkably, unlike ATM and Chk1, phosphorylation of Chk2 occurred immediately during DNA damage stress and gradually disappeared during recovery (, lanes 2 and 3 in f). Cdc25C, one of the key mitotic regulators, is involved in the activation of Cdk1. Inhibitory phosphorylation of Cdc25 at serine-216, which is known to occur due to Chk1 and Chk2, was highly induced after 3 h of recovery from mitotic DNA damage (, lanes 3 and 5 in h). Phosphorylation of Cdc25C by Cdk1 or Plk1 was detected in both undamaged mitotic cells and cells immediately after DNA damage (, lanes 1 and 2). Previously, we reported that mitotic cells with severe DNA damage failed to undergo cell division and entered interphase with 4N-DNA content [Citation10]. Therefore, Cdc25C phosphorylation by mitotic kinases completely disappeared upon entry into a G2-like G1 phase (, lanes 3–5 in i and j). The major downstream target of Cdc25C phosphatase in mitosis is Cdk1, and inhibitory phosphorylation of Cdk1 at tyrosin-14 and threonine-15 should be dephosphorylated by Cdc25C for mitotic initiation and progression. According to Cdc25C inhibition by Chk1 and Chk2 during damage recovery, inhibitory phosphorylations of Cdk1 at tyrosine-14 and threonine-15 gradually accumulated over time (), indicating that Cdk1 becomes inactivated during recovery from mitotic DNA damage. One of the downstream targets of Cdk1 known to contribute to timely mitotic entry is the Greatwall kinase, which plays a pivotal role in maintaining the mitotic phosphorylation of various Cdk1 substrates [Citation24–Citation26]. Owing to the accumulation of inhibitory phosphorylation of Cdk1 by Cdc25C inactivation, phosphorylation of the Greatwall kinase as seen in mitosis gradually decreased during recovery from mitotic DNA damage (, lanes 3–5 in o). Moreover, under these conditions, the active phosphorylation of two major mitotic kinases, Cdk1 and Plk1 completely disappeared after 3 h of recovery (, lanes 3–5 in m and q). These findings suggest that the activation of ATM/Chk by the mitotic DNA damage response induces the dephosphorylation of Plk1, possibly through the repression of the Cdc25/Cdk1/Greatwall signaling pathway during recovery from mitotic DNA damage.
PP2A activity during recovery from mitotic DNA damage is dependent on the ATM/Chk pathway
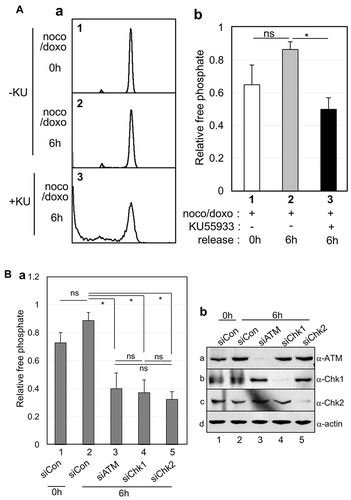
PP2A plays a critical role in the regulation of almost all major pathways of the cell cycle, such as the G1/S transition, DNA synthesis, and mitotic initiation [Citation27,Citation28]. In addition to cell cycle functions, to investigate whether PP2A activity is modulated in ATM activation during the mitotic DNA damage response, mitotic cells were treated with KU55933 to inhibit the ATM kinase prior to doxorubicin treatment. As reported in our previous papers, while normal mitotic cells entered the G1 phase within 6 h (, panel 1 in a), mitotic cells with DNA damage could not complete cytokinesis and retained 4N-DNA content after 6 h of recovery in a fresh medium (, panel 2 in a) [Citation10,Citation19]. On treatment with KU55933, even damaged mitotic cells did not divide and retained 4N-DNA, and the cells were highly defective after 6 h of recovery (, panel 3 in a). When KU55933 was applied, PP2A activity decreased by approximately 50% during the 6-h recovery from mitotic DNA damage (, gray and black bars in b). The depletion of DNA damage regulators showed a similar effect as that following treatment with KU55933. When siRNA constructs of ATM, Chk1, and Chk2 were transfected, the expression of these proteins was blocked in cells () and PP2A activities decreased in compared with the activities of control cells (). These results suggest that PP2A activity during recovery from mitotic DNA damage is maintained by the activation of the ATM/Chk pathway.
Figure 3. PP2A activity is maintained depending on ATM/Chk activation during recovery from mitotic DNA damage. (A) Effects of ATM inhibitors during mitotic DNA damage. The ATM inhibition effect by KU55933 treatment on cell division and PP2A activity were detected by FACS analysis (a) and phosphatase assay (b) during mitotic damage recovery. Cells were treated with nocodazole to arrest them in prometaphase (noco) or followed by doxorubicin treatment to induce DNA damage (noco/doxo). 1, mitotic cells; 2, doxorubicin treatment administered to mitotic cells; and 3, doxorubicin and KU55933 treatment administered to mitotic cells. Cells were regrown in fresh media for 6 h (6h). (B) PP2A activity was impaired by depletions of ATM, Chk1, and Chk2 during recovery from mitotic DNA damage. PP2A activities in the depleted cells significantly decreased compared with those in control cells (*P < 0.001) (a), and protein expressions were clearly blocked using the indicated siRNA constructs (b). 1, mitotic cells with control siRNA immediately after DNA damage (0h); 2–5, mitotic cells with damage transfected with the indicated siRNAs were regrown in fresh media for 6 h (6h). Proteins were detected by immunoblot analysis using the indicated antibodies.
Phosphorylation of ENSA at serine-67 is essential for its interaction with PP2A-B55 regulatory subunits
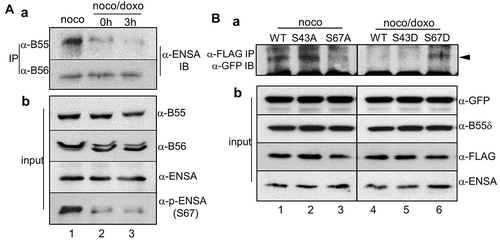
Previously, we reported that PP2A is involved in Plk1 dephosphorylation via PP2A activity inhibition using low concentration (~100 nM) of okadaic acid and depletion of the PP2A catalytic subunit [Citation10]. Generally, the B-type subunits of PP2A are responsible for the regulatory complexity and substrate specificity of PP2A via various distinct isoforms [Citation28,Citation29]. In normal mitotic cells, the Greatwall kinase phosphorylates and activates ENSA, which is one of the PP2A-B55 inhibitors [Citation30–Citation32]. In previous experiments, the Greatwall kinase already activated in mitotic cells was dephosphorylated and possibly inactivated during recovery from mitotic DNA damage (, lanes 3–5 in o). Therefore, the inhibition of the Greatwall kinase was considered to maintain PP2A in an active state through the regulation of ENSA. While endogenous ENSA was strongly associated with PP2A-B55 regulatory subunits in mitotic cells (, lane 1 in the upper panel in a), the interaction dramatically decreased during recovery from mitotic DNA damage (, lanes 2 and 3 in the upper panel in a). On the other hand, PP2A-B56 regulatory subunits and ENSA interacted consistently and very weakly during mitosis regardless of DNA damage (, lanes 1–3 in the lower panel in a). In previous reports, phosphorylation of ENSA at serine-67 by the Greatwall kinase makes ENSA for potent inhibitor of PP2A-B55 [Citation30,Citation33]. As expected, endogenous ENSA in mitotic cells was phosphorylated at serine-67, and this phosphorylation signal dramatically disappeared following mitotic DNA damage and during mitotic recovery (, input in b). Unlike wild-type and S43A control mutant ENSA, ectopic ENSA-S67A phosphorylation-defective mutant did not interact with PP2A-B55δ in mitotic cells (, lane 3 in panel a). However, ENSA-S67D phospho-mimic mutant clearly interacted with B55δ under the mitotic DNA damage condition, even when wild-type ENSA did not (, lanes 4 and 6). These data suggest that PP2A activity was inhibited in normal mitotic cells by strong interaction between the PP2A regulatory subunit and phospho-ENSA (particularly phosphorylated at serine-67), and that B55, not B56, mainly functions in PP2A activity during recovery from mitotic DNA damage by releasing from ENSA.
Figure 4. PP2A-B55 regulatory subunits and ENSA are dissociated during mitotic DNA damage recovery. (A) In vivo protein interaction. Endogenous PP2A-B55 or PP2A-B56 was immunoprecipitated, and the bound ENSA (a) and amount of input proteins (b) were detected by immunoblot analysis using the indicated antibodies. (B) Phosphorylation of ENSA at serine-67 is essential for its interaction with PP2A-B55δ. GFP-B55δ and FLAG-ENSA mutants were ectopically co-expressed in cells, and FLAG-ENSA proteins were immunoprecipitated with an anti-FLAG antibody. Bound B55δ was detected by immunoblot analysis using an anti-GFP antibody. Lanes 1 and 4, wild-type FLAG-ENSA; lane 2, FLAG-ENSA S43A; lane 3, FLAG-ENSA S67A; lane 5, FLAG-ENSA S43D; and lane 6, FLAG-ENSA S67D. Prometaphase cells were prepared by 16-h nocodazole treatment (noco) and mitotic cells with DNA damage were released in fresh media for 3 h for recovery from mitotic DNA damage (noco/doxo). Bound B55δ is indicated with arrowhead.
PP2A-B55 regulatory subunits are involved in Plk1 dephosphorylation during recovery from mitotic DNA damage
To determine whether Plk1 dephosphorylation during recovery from mitotic DNA damage is caused by PP2A activation through B55 regulation, association between B55 subunits and Plk1 was examined. While endogenous B55 subunits and Plk1 did not interact in mitotic cells, their interaction gradually increased during recovery from mitotic DNA damage (, arrowhead in a). Further, to investigate the types of PP2A regulatory subunits involved in Plk1 dephosphorylation during recovery from mitotic DNA damage, we created expression constructs of five isoforms of B55 and B56 subunits, which were tagged by FLAG fragment, and subjected them to protein-protein interaction analysis. Ectopic B-type subunits were immunoprecipitated using an anti-FLAG antibody, and bound Plk1 was detected by immunoblotting. The results showed that Plk1 was detected in the immunoprecipitates of B55α and B55δ during recovery from mitotic DNA damage (, arrowheads in panels a and b). Although Plk1/B55α interaction was detected immediately after DNA damage and maintained during recovery, Plk1/B55δ interaction appeared later in the recovery process (, arrowheads in lanes 2 and 3 in panels a and b). As expected, Plk1 did not significantly bind to B56α, B56β, or B56γ (, arrowheads in panels c-f). For in vitro protein binding analysis with exogenous B55 proteins, we constructed GST-B55α and GST-B55δ and expressed them in an Escherichia coli system. GST-B55 proteins were purified using glutathione agarose beads, and the beads were incubated with mitotic DNA damaged cell extracts. As expected, GST-B55α and GST-B55δ interacted with endogenous Plk1 in the mitotic DNA damaged cells stronger than in normal mitotic cells (, lanes 2–3 in a). Moreover, when PP2A-B55α and/or PP2A-B55δ was depleted, the phosphorylation of Plk1 restored during 6-h release from mitotic DNA damage (, lanes 2–4 in α-P-Plk1 in a). Restoration of Plk1 phosphorylation by PP2A-B55δ silencing was rescued by overexpression of exogenous PP2A-B55δ (, lanes 2 and 3 in α-P-Plk1 in b). Under these conditions, 4N-DNA cells were re-divided and entered the G1 phase, and the concentration of 2N cells increased by approximately by 10% after 6-h recovery from mitotic DNA damage (, panel b).
Figure 5. Plk1 phosphorylation is restored by the depletion of PP2A-B55α/δ during recovery after mitotic DNA damage. To examine in vivo interaction between PP2A-B55α/δ and Plk1 during DNA damage recovery, endogenous PP2A-B55 was immunoprecipitated with an anti-PP2A-B55 antibody and the bound Plk1 were detected by immunoblot analysis using the indicated antibodies (A). The bound Plk1 and IgG heavy chain of antibody used for IP are indicated with an arrowhead and asterisk, respectively. (B) FLAG-tagged PP2A regulatory subunits were ectopically expressed and immunoprecipitated with an anti-FLAG antibody. Bound endogenous Plk1 was detected by immunoblot analysis (α-FLAG IP/α-Plk IB). The bound Plk1 and IgG heavy chain of antibody used for IP are indicated with an arrowhead and asterisk, respectively. Amount of proteins used were detected by immunoblot analysis using the indicated antibodies (input). a, FLAG-PP2A-B55α; b, FLAG-PP2A-B55δ; c, FLAG-PP2A-B56α; d, FLAG-PP2A-B56β; e, FLAG-PP2A-B56γ. 1, mitotic cells; 2, immediately after doxorubicin treatment in mitotic cells (0h); and 3, cells regrown in fresh media for recovery from mitotic DNA damage for 3 h (3h). (C) In vitro protein-binding analysis of GST-tagged PP2A-B55α/δ and Plk1. Bound Plk1 was detected by an anti-Plk1 antibody (a), and the amount of proteins used was detected by the indicated antibodies (b). 1, GST only; 2, GST-B55α; and 3, GST-B55δ. (D) The effect of depletion of PP2A-B55α/δ on Plk1 modification. Plk1 phosphorylation was restored by depletion of PP2A-B55α/δ during 6 h of recovery from mitotic DNA damage (a), and the restoration of phosphorylation was rescued by ectopic expression of PP2A-B55δ (b). In part b, closed and open arrowheads indicate exogenous FLAG-B55δ protein and endogenous B55δ, respectively. (E) Cell division was recovered and G1 population increased during recovery from mitotic DNA damage for 6 h by simultaneous depletion of B55α and B55δ. DNA contents are indicated as 2N and 4N. (f) In vitro phosphatase assay. HeLa cells were transfected with FLAG-B55α and/or FLAG-B55δ. Proteins were immunoprecipitated by the anti-FLAG antibody and were subjected to an in vitro phosphatase assay as described in the Materials and methods.
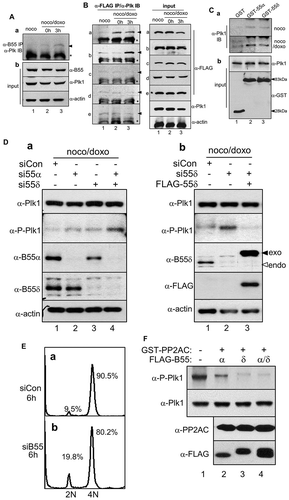
To confirm the Plk1 can be dephosphorylated by PP2A-B55 complex directly, in vitro phosphatase assays were performed. FLAG-PP2A-B55α and/or -B55δ were overexpressed in mitotic cells with DNA damage and subsequently subjected to immunoprecipitation. Immunopurified B55s and recombinant GST-PP2A catalytic subunit were pre-incubated to reconstitute the PP2A holoenzyme and then used in a phosphatase assay incorporating an active His-fused full-length Plk1 as a substrate. PP2A activity toward Plk1 was analyzed by immunoblotting with an anti-phospho-Plk1(T210) antibody. As shown in , GST-PP2AC with B55α and/or δ showed significantly reduced levels of Plk1 phosphorylation (, lanes 2–4). These findings indicate that PP2A activity is restored by the dissociation of PP2A-B55 from ENSA during recovery from mitotic DNA damage and that Plk1 dephosphorylation occurs in a B55α- and B55δ-dependent manner.
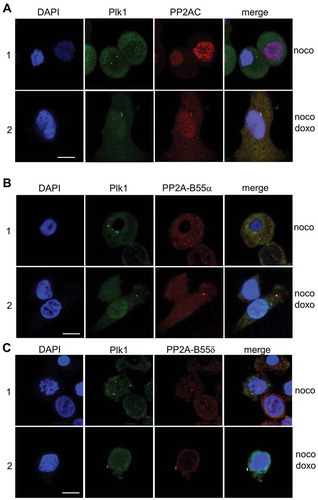
The connection between PP2A activity and Plk1 dephosphorylation could also be demonstrated by the localization of PP2A and Plk1. The co-localization of PP2A catalytic subunit and Plk1 on centrosome was found in approximately 70% of the cells after 6 h of recovery from mitotic DNA damage (, panel 2). In addition, endogenous Plk1 was co-localized with PP2A-B55α and PP2A-B55δ, which were ectopically expressed, in 60 and 67% of the cells with mitotic DNA damage, respectively (, panels 2 in B and C). However, in mitotic cells without mitotic DNA damage, co-localization of these proteins was not detected (, panels 1 in A-C).
Figure 6. PP2A is co-localized with Plk1 in cells during mitotic DNA damage response. Subcellular co-localization of the endogenous PP2A catalytic subunit and Plk1 (A) and PP2A-B55 regulatory subunits and Plk1 (B) were detected by immunofluorescence analysis. 1, immediately after doxorubicin treatment in mitotic cells (0h); 2, cells regrown in fresh media for recovery from mitotic DNA damage for 6 h (6h). Nuclear detection was performed by DAPI staining. Plk1 and PP2A-C/PP2A-B55 were indicated as FITC and Cy3 signals, respectively. Scale bar indicates 50 μm.
Discussion
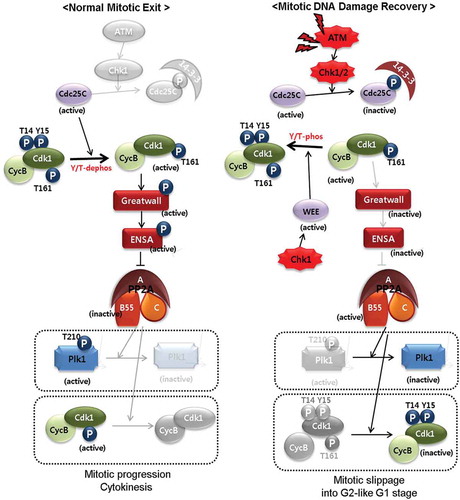
Generally, cell cycle checkpoints that respond to DNA damage are encountered by interphase cells. If DNA damage is induced in the G2 phase, mitotic entry cannot begin until repairs have been completed. Activation of the DNA damage checkpoint is induced by damage-sensing kinases such as ATM/ATR and the effector kinases Chk1/Chk2. Regarding cascade signaling, Chk1/Chk2 phosphorylates and inactivates Cdc25 before mitotic entry, and this inactivation causes the accumulation of inhibitory phosphorylation of Cdk1, resulting in G2 arrest. We have been studying mitotic DNA response and its recovery for many years. Unlike during DNA damage in the G2 phase, unique features observed during recovery from mitotic DNA damage are the dephosphorylation and inactivation of mitosis-specific kinases already activated during mitosis, progression of the cell cycle into the G1 phase without cytokinesis, and establishment of multiploidy in relation to p53 [Citation10–Citation12,Citation19]. Plk1 is phosphorylated and activated during G2/M transition, plays various roles in mitotic progression, and is generally degraded by APC/C at the point of mitotic exit. However, unusual Plk1 dephosphorylation has been detected during recovery from mitotic DNA damage. Although we previously clarified that Plk1 dephosphorylation is induced by the activation of the ATM/Chk pathway and occurs in a PP2A-dependent manner [Citation10,Citation19], it was unclear as to how the ATM pathway and PP2A function are related to Plk1 regulation during recovery from mitotic DNA damage. During mitotic entry, one of the Cdk1 substrates is the Greatwall kinase, which is one of the essential mitotic protein kinases that phosphorylates and activates PP2A inhibitors such as ENSA and Arpp19 [Citation26,Citation33,Citation34]. PP2A inhibition by Greatwall/ENSA during mitotic entry results in the maintenance of the mitotic phosphorylation of Cdk1 substrates and stable mitotic initiation. Conversely, the dephosphorylation of Cdk1 substrates by PP2A activation would be necessary for mitotic exit. For this, Cdk1 and the Greatwall kinase are inactivated by cyclin B degradation and by PP1 and PP2A, respectively [Citation35]. Apart from its functions in normal mitotic entry and exit, the Greatwall kinase promotes recovery from DNA damage in Xenopus egg extracts. Depletion of the Greatwall kinase activates DNA damage response, whereas the addition of wild-type but not kinase-dead mutant inhibits damage-related molecules [Citation7]. In this study, we hypothesized the role of the Greatwall kinase during recovery from mitotic DNA damage. Plk1 dephosphorylation during recovery from mitotic DNA damage possibly occurs via PP2A activation followed by Greatwall kinase inactivation and further ENSA inactivation from 3 h of recovery from mitotic DNA damage (, panel o and q).
The Greatwall kinase activated by hyperphosphorylation phosphorylates ENSA at serine-67 during mitosis, and this is essential for its interaction with PP2A-B55δ and subsequent inactivation of PP2A, facilitating complete phosphorylation of mitotic substrates. When the Greatwall kinase is inactivated, PP2A-B55 reactivates, which leads to dephosphorylation of cyclin B1-cdc2 and mitotic exit [Citation31,Citation32,Citation36]. Indeed, in this study, phosphorylation at serine-67 was essential for interaction between ENSA and PP2A-B55δ using ENSA phosphorylation site mutants, and dephosphorylated ENSA dissociated from PP2A-B55δ (). These results suggested that the inactivation of the Greatwall kinase during mitotic DNA damage response leads to dephosphorylation of ENSA and the dissociation of PP2AB55δ from ENSA.
Degradation of Plk1 does not occur because APC/C is not activated due to the slippage of any subsequent mitotic event immediately after DNA damage during prometaphase. Indeed, in our previous studies, one of the APC/C substrates, cyclin B, was not degraded during recovery from mitotic DNA damage, although cells entered the G1 phase with 4N-DNA content [Citation10,Citation19]. Since PP2A activity and substrate specificity are determined by regulatory subunits, we investigated the types of subunit involved in PP2A activity for Plk1 dephosphorylation. Based on the association between ENSA and Plk1 (), a depletion and rescue experiment, and in vitro phosphatase assay (), PP2A-B55α and PP2A-B55δ are possibly involved in Plk1 dephosphorylation and mitotic exit without cell division during recovery from mitotic DNA damage. These results are consistent with those of previous reports on the functions of PP2A in mitotic exit [Citation24,Citation25,Citation35,Citation37–Citation39]. Based on the above results of our study and previous reports, we suggest a pathway model for the recovery from mitotic DNA damage and unusual regulation of Plk1 (). Briefly, by DNA damage in mitotic cells, a common mechanism of DNA damage regulators, ATM and Chks are activated under this condition. As known, Chk1 and Chk2 regulate mitotic Cdc25C negatively and Wee1 positively, followed by the inhibition of Cdk1 [Citation40–Citation42]. The active phosphorylation of the Greatwall kinase by Cdk1 in mitosis is difficult to maintain and then PP2A inhibitors such as ENSA are inactivated. Interaction between ENSA and PP2A-B55α or PP2A-B55δ was detected in mitotic cells, but their association disappeared during mitotic DNA damage response. B55 subunits then associated with Plk1 during recovery (), indicating that PP2A-B55α and PP2A-B55δ are the possible regulators during recovery for hours from mitotic DNA damage and have functions that specify PP2A substrate under these conditions. In addition to Plk1 dephosphorylation, the activatory phosphorylation of Cdk1 at threonine-161 could be a target for PP2A (). Owing to Plk1 and Cdk1 inactivation, many processes during anaphase and cytokinesis could fail, and cells enter the G1 phase with 4N-DNA content. In this report, we showed a checkpoint pathway for recovery from mitotic DNA damage. In conclusion, Greatwall/ENSA repression is induced by the ATM/Chk pathway, and the PP2A-B55 holoenzyme complex plays an important role in recovery from mitotic DNA damage by abolishing the mitotic phosphorylation of key mitotic kinases. Subsequently, if DNA damage cannot be repaired owing to its severity, cells die through apoptosis or adapt via multiploidy depending on p53 activity [Citation11,Citation12].
Figure 7. The ATM/Chk/Greatwall/PP2A signaling model for normal mitotic exit (A) and for abnormal mitotic exit during recovery from mitotic DNA damage (B). Blurred parts in the cartoon are events that were inactive or did not occur.
ACKNOWLEDGMEMTS
We thank Min-Yi Han for technical support in preparing DNA constructs. This research was supported by Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Science and ICT (NRF-2016R1A1A3A04005189).
Supplemental Material
Download MS Word (718.1 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer. 2003;3:155–168.
- Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439.
- Aressy B, Ducommun B. Cell cycle control by the CDC25 phosphatases. Anticancer Agents Med Chem. 2008;8:818–824.
- Boutros R, Dozier C, Ducommun B. The when and wheres of CDC25 phosphatases. Curr Opin Cell Biol. 2006;18:185–191.
- Shechter D, Costanzo V, Gautier J. Regulation of DNA replication by ATR: signaling in response to DNA intermediates. DNA Repair (Amst). 2004;3:901–908.
- Wong PY, Ma HT, Lee HJ, et al. MASTL(Greatwall) regulates DNA damage responses by coordinating mitotic entry after checkpoint recovery and APC/C activation. Sci Rep. 2016;6:22230.
- Peng A, Yamamoto TM, Goldberg ML, et al. A novel role for greatwall kinase in recovery from DNA damage. Cell Cycle. 2010;9:4364–4369.
- Glover DM. The overlooked greatwall: a new perspective on mitotic control. Open Biol. 2012;2:120023.
- Virshup DM, Kaldis P. Cell biology. Enforcing the Greatwall in mitosis. Science. 2010;330:1638–1639.
- Jang YJ, Ji JH, Choi YC, et al. Regulation of Polo-like kinase 1 by DNA damage in mitosis. Inhibition of mitotic PLK-1 by protein phosphatase 2A. J Biol Chem. 2007;282:2473–2482.
- Hyun SY, Rosen EM, Jang YJ. Novel DNA damage checkpoint in mitosis: mitotic DNA damage induces re-replication without cell division in various cancer cells. Biochem Biophys Res Commun. 2012;423:593–599.
- Hyun SY, Jang YJ. p53 activates G(1) checkpoint following DNA damage by doxorubicin during transient mitotic arrest. Oncotarget. 2015;6:4804–4815.
- Combes G, Alharbi I, Braga LG, et al. Playing polo during mitosis: PLK1 takes the lead. Oncogene. 2017;36:4819–4827.
- Petronczki M, Lenart P, Peters JM. Polo on the Rise-from Mitotic Entry to Cytokinesis with Plk1. Dev Cell. 2008;14:646–659.
- van Vugt MA, Gardino AK, Linding R, et al. A mitotic phosphorylation feedback network connects Cdk1, Plk1, 53BP1, and Chk2 to inactivate the G(2)/M DNA damage checkpoint. PLoS Biol. 2010;8:e1000287.
- Yoo HY, Kumagai A, Shevchenko A, et al. Adaptation of a DNA replication checkpoint response depends upon inactivation of Claspin by the Polo-like kinase. Cell. 2004;117:575–588.
- van Vugt MA, Bras A, Medema RH. Polo-like kinase-1 controls recovery from a G2 DNA damage-induced arrest in mammalian cells. Mol Cell. 2004;15:799–811.
- van Vugt MA, Medema RH. Checkpoint adaptation and recovery: back with Polo after the break. Cell Cycle. 2004;3:1383–1386.
- Lee HJ, Hwang HI, Jang YJ. Mitotic DNA damage response: polo-like kinase-1 is dephosphorylated through ATM-Chk1 pathway. Cell Cycle. 2010;9:2389–2398.
- Chiyoda T, Sugiyama N, Shimizu T, et al. LATS1/WARTS phosphorylates MYPT1 to counteract PLK1 and regulate mammalian mitotic progression. J Cell Biol. 2012;197:625–641.
- So S, Davis AJ, Chen DJ. Autophosphorylation at serine 1981 stabilizes ATM at DNA damage sites. J Cell Biol. 2009;187:977–990.
- Kastan MB, Lim DS. The many substrates and functions of ATM. Nat Rev Mol Cell Biol. 2000;1:179–186.
- Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21:4129–4139.
- Burgess A, Vigneron S, Brioudes E, et al. Loss of human Greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin B-Cdc2/PP2A balance. Proc Natl Acad Sci U S A. 2010;107:12564–12569.
- Vigneron S, Brioudes E, Burgess A, et al. Greatwall maintains mitosis through regulation of PP2A. Embo J. 2009;28:2786–2793.
- Yu J, Zhao Y, Li Z, et al. Greatwall kinase participates in the Cdc2 autoregulatory loop in Xenopus egg extracts. Mol Cell. 2006;22:83–91.
- Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J. 2001;353:417–439.
- Wlodarchak N, Xing Y. PP2A as a master regulator of the cell cycle. Crit Rev Biochem Mol Biol. 2016;51:162–184.
- Wang F, Zhu S, Fisher LA, et al. Protein interactomes of protein phosphatase 2A B55 regulatory subunits reveal B55-mediated regulation of replication protein A under replication stress. Sci Rep. 2018;8:2683.
- Mochida S, Maslen SL, Skehel M, et al. Greatwall phosphorylates an inhibitor of protein phosphatase 2A that is essential for mitosis. Science. 2010;330:1670–1673.
- Blake-Hodek KA, Williams BC, Zhao Y, et al. Determinants for activation of the atypical AGC kinase Greatwall during M phase entry. Mol Cell Biol. 2012;32:1337–1353.
- Lorca T, Castro A. The Greatwall kinase: a new pathway in the control of the cell cycle. Oncogene. 2013;32:537–543.
- Gharbi-Ayachi A, Labbe JC, Burgess A, et al. The substrate of Greatwall kinase, Arpp19, controls mitosis by inhibiting protein phosphatase 2A. Science. 2010;330:1673–1677.
- Rangone H, Wegel E, Gatt MK, et al. Suppression of scant identifies Endos as a substrate of greatwall kinase and a negative regulator of protein phosphatase 2A in mitosis. PLoS Genet. 2011;7:e1002225.
- Ma S, Vigneron S, Robert P, et al. Greatwall dephosphorylation and inactivation upon mitotic exit is triggered by PP1. J Cell Sci. 2016;129:1329–1339.
- Vigneron S, Gharbi-Ayachi A, Raymond AA, et al. Characterization of the mechanisms controlling Greatwall activity. Mol Cell Biol. 2011;31:2262–2275.
- Manchado E, Guillamot M, de Carcer G, et al. Targeting mitotic exit leads to tumor regression in vivo: modulation by Cdk1, Mastl, and the PP2A/B55alpha,delta phosphatase. Cancer Cell. 2010;18:641–654.
- Hegarat N, Vesely C, Vinod PK, et al. PP2A/B55 and Fcp1 regulate Greatwall and Ensa dephosphorylation during mitotic exit. PLoS Genet. 2014;10:e1004004.
- Lorca T, Bernis C, Vigneron S, et al. Constant regulation of both the MPF amplification loop and the Greatwall-PP2A pathway is required for metaphase II arrest and correct entry into the first embryonic cell cycle. J Cell Sci. 2010;123:2281–2291.
- Furnari B, Rhind N, Russell P. Cdc25 mitotic inducer targeted by chk1 DNA damage checkpoint kinase. Science. 1997;277:1495–1497.
- Chen MS, Ryan CE, Piwnica-Worms H. Chk1 kinase negatively regulates mitotic function of Cdc25A phosphatase through 14-3-3 binding. Mol Cell Biol. 2003;23:7488–7497.
- Lee J, Kumagai A, Dunphy WG. Positive regulation of Wee1 by Chk1 and 14-3-3 proteins. Mol Biol Cell. 2001;12:551–563.