
ABSTRACT
Nasopharyngeal carcinoma (NPC) is the most prevailing malignancy of the head and neck with unique geographic distribution. Southern China has one of the highest incidence rates of NPC in the world. Although radiotherapy and chemotherapy are the most important treatment modalities for NPC, recurrence, and metastasis severely interfere with the survival quality of patients. It is much-needed to find an effective method of NPC treatment with a good prognosis such as gene therapy. PFK1, a key regulatory enzyme of glycolysis, is frequently shown to be amplified and overexpressed in a variety of human cancers. However, the function of PFK1 and molecular mechanism in NPC is elusive. Here, we knockdown PFK1 expression by utilizing DNA vector-based RNA Interference. Western blotting and real-time PCR show that the expression of PFK1 is efficiently down-regulated in both protein and mRNA levels by stable transfection with PFK1 siRNA expression vector. In addition, stable knockdown of PFK1 expression inhibits cell growth, induces apoptosis, decreases the invasive capability and metastasis in the CNE2 human NPC cell line. This present study finds the importance of PFK1 which can be worked as a novel target in NPC treatment and holds great potential to be extended to other malignant cancers.
Introduction
Nasopharyngeal carcinoma (NPC), the most prevailing malignancy of the head and neck, is particularly high in Southeast Asia and China, with an estimated incidence of 20–80 cases per 100,000 annually in endemic regions [Citation1–4]. Due to a lack of specific biomarkers and effective treatments, the 5-year survival rate of NPC patients is only 50% [Citation5,Citation6]. Therefore, revealing the molecular mechanisms of the tumorigenesis, invasion, and metastasis in NPC is important for developing new effective therapeutic approaches. Recently, the glycolytic enzymes phosphofructokinase (PFK) family has been approved to be potential molecular targets for the development and survival of human malignant tumors [Citation7].
In oncology, the Warburg effect is a form of modified cellular metabolism based on aerobic fermentation found in cancer cells, which tend to favor anabolic glycolysis rather than the oxidative phosphorylation pathway which is the preference of most other cells of the body [Citation8]. PFK1, a kinase that catalyzes the phosphorylation of fructose-6-phosphate to fructose-1,6-bisphosphate, is essential for glycolysis, and plays a significant role in the deviant energetic metabolism of cancer cells [Citation9]. Amplification and overexpression of the PFK1 occur in a variety of cancers, including astroblastomas, oligodendrogliomas [Citation10], ovarian cancer [Citation11], cholangiocarcinoma [Citation12], bladder [Citation7], colorectal cancer [Citation13], as well as for glioblastoma [Citation14], which potentially promoting the carcinogenesis and maintenance of malignant tumors. Furthermore, aberrant expression of PFK1 is shown to be more frequently associated with multiple tumor progressions including development, differentiation, migration, invasion, and metastasis, as well as the resistance to chemotherapy and radiation treatment [Citation15]. Smerc et al. [Citation16] recently demonstrated that blocking up-regulation and aberrant activation of PFK1 dramatically reduced cancer cell proliferation in vitro and impaired metastatic tumor formation in vivo.
Gene expression between normal cells and cancer cells often has a large difference, which provides potential targets. Thus, a specific antagonist PFK1, such as siRNA, may become a novel strategy of molecular-targeted antitumor therapy. In our preliminary experiments, aberrant expression levels of PFK1 were significantly associated with the pathological differentiation, clinic stage, and lymph node metastases in obtained NPC tissues (data not have shown), implying PFK1 have an oncogenic function in tumorigenesis. In the present study, we further detected the expression of PFK1 in NPC tissues and found that up-regulated expression of PFK1 correlated with the progression of nasopharyngeal carcinoma. The antitumor activity of PFK1 in CNE2 NPC cell line was studied via knocking down expression by RNA interference (RNAi). We demonstrated that PFK1 inhibited cell growth and induced apoptosis as well as retarded cell migration and invasion in NPC cancer cells. PFK1 is not only important for the NPC diagnosis and therapeutic advancement as a reliable biomarker but also for in-depth understanding of their effects on cellar processes.
Methods
Tissue samples and cell culture
Nasopharyngeal tissues were obtained from 61 patients who were revealed to have a nasopharyngeal neoplasm through nasal endoscopic examination between January 2011 and December 2012 at the Department of Otorhinolaryngology, Huazhong University of Science and Technology Union Shenzhen Hospital (Previously known as Nanshan People’s Hospital), under an approved protocol of our University. The patients had not received any cancer therapy before admission. Tissues obtained from patients were preserved in liquid nitrogen within 5 min and stored at −80°C. Human NPC cell line CNE2 was obtained from Cell Lines Bank, Shanghai Institute of Biological Sciences. The cells were cultured in RPMI-1640 medium (Gibico, USA) supplemented with 1% Penn/Strep and 10% fetal bovine serum and incubated at 37°C in a humidified atmosphere of 5% CO2/95% air.
siRNA preparation and transfection
PFK1 siRNA was synthesized by Wuhan Google Bioengineering Co., Ltd, Hubei, China, and their sequences are listed in . All the oligonucleotides were purified by PAGE. The cells were incubated with a mixture of Lipofectamine 2000 (Invitrogen, USA) and PFK1 shRNAs following the instructions.
Table 1. The sequence of the oligonucleotides in this work
Western blotting analysis
Total protein from tissues and cell lines were obtained using RIPA buffer containing protease inhibitors followed by centrifugation at 12,000 × g for 10 min. For western blot analysis, 30 μg of proteins were resolved on SDS-PAGE, transferred onto PVDF membranes, and probed by anti-PFK1 antibodies (diluted 1:200, Abcam, USA) anti- active caspase-3, 9 antibodies (diluted 1:200, Santa Cruz Biotechnology, USA), anti-Bcl-2 antibody (diluted 1:200, Santa Cruz Biotechnology, USA), anti-Bax antibody (diluted 1:200, Santa Cruz Biotechnology, USA), or anti-PCNA antibody (diluted 1:200, Santa Cruz Biotechnology, USA) at 4°C overnight. The membranes were incubated with horseradish peroxidase-conjugated antibodies (Santa Cruz Biotechnology, USA) for 2 h. Bands on blots were visualized using chemiluminescence with Pierce ECL kits (Millipore, USA). β-Actin was used as an internal control.
Quantitative real-time reverse transcription PCR
Total RNA was extracted with Trizol (Invitrogen, CA, USA) agent. After obtaining RNAs, reverse transcription was carried on to produce cDNA by using the PrimeScript™ RT-PCR Kit (Takara, Shiga, Japan). Real time-PCR was performed as described [Citation17]. The primers for PFK1 were as follows: forward primer: 5'- CTACAGTCTCCAACAATGTCCC-3'; reverse primer: 5'- CCACCCATAGTCTCAATGATAAAC −3'; Primers for β-Actin: forward primer: 5'- GGGAAACTGTGGCGTGAT-3'; reverse primer: 5'- GGGTGTCGCTGTTGAAGT- 3'.
Enzyme assays
Activity assays for PFK1 were performed via an auxiliary enzyme assay. PFK1 activity was determined by a coupled reaction system by spectrophotometer at 340 nm (Lambda25 UV/VIS spectrophotometer, Perkin Elmer) [Citation18].
MTT assay
CNE2 cells were seeded in 96-well plates at a density of 5 × 103 cells/well for overnight. To evaluate the potentiation of cytotoxicity by RNA oligonucleotides or plasmids for different time periods. About 10 mL MTT solution (0.5 mg/ml) was added to medium and incubated with cells for 4 h in a 5% CO2 atmosphere at 37°C. The supernatant was removed followed by added 100 mL DMSO and then the OD at 490 nm was measured using a Quant Universal Microplate Spectrophotometer (Bio-Tek Instruments, Winooski, USA).
Flow cytometry analysis of apoptosis
Annexin V and PI fluorescein staining kit (Bender MedSystems, Austria) was utilized to measure CNE2 cell apoptosis. Briefly, 5 × 105 cells were suspended in 500 μL 1 × binding buffer (10 mM HEPES pH 7.4, 140 mM NaCl, 2.5 mM CaCl2). Cells were incubated with Annexin V (1:20) for 5 min followed by incubation with propidium iodide (PI, 1 mg/mL) for 15 min at room temperature in the dark. Apoptosis rate was evaluated by a FACScalibur flow cytometer (BD, Franklin Lakes, NJ, USA).
Wound healing assay
CNE2 cells were transfected with sh-PFK1 or scrambled negative control. The cells were grown to 90% confluency for 24 h after transfection, created wounds with a sterile 100 μL pipette tip. The cells were rinsed with a medium to remove any free-floating cells and debris and then incubated with fresh medium at 37°C. Wound healing was observed at different time points (0, 12, and 24 h) within the scrape line-and representative scrape lines were imaged. Results are presented as the percentage of wound healing, which was calculated as follows: [Wound area (initial) – Wound area (final)]/Wound area (initial) × 100%.
Transwell assay
Cell invasion assay was performed in a Transwell (Corning, NY, USA) with a pore size of 8.0 μm. About 1 × 105 cells were seeded in serum-free medium in the upper chamber, while medium containing 10% FBS in the lower chamber. After incubating for 48 h at 37°C, CNE2 cells in the upper chamber were carefully removed with a cotton swab. The cells traversed to the reverse face of the membrane were fixed with methanol and stained with crystal violet. Labeled invasive cells imaging was carried out on an inverted microscope.
Statistical analysis
All statistical analysis was performed by SPSS version 19.0 (IBM, Chicago, IL, USA) statistical software package. The data were described as mean± standard deviation (SD). The Student’s t-test was used to compare the means of two groups. Analysis of variance (ANOVA) and Tukey’s post hoc test method was used to perform multiple comparisons of numerical variables. Results with p < 0.05 were considered statistically significant.
Results
PFK1 is overexpressed in NPC tissues
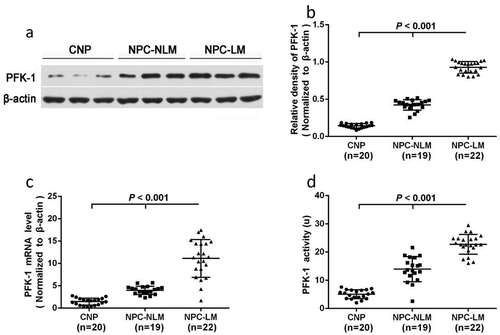
To investigate PFK1 expressions among nasopharyngeal tissue samples with different clinical stages, we explored three groups: chronic inflammatory tissue of nasopharyngeal (CNP), nasopharyngeal carcinoma without lymph node metastasis (CPN-NLM) and nasopharyngeal carcinoma lymph node metastasis (NPC-LM). As expected, the PFK1 expression level was significantly higher in CNP-LM tissues than that in other groups (–b)). Consistent with WB results, as shown in ), a significant increase in expression levels of the PFK1 mRNA was observed in CNP-LM and CNP-NLM group as compared to the control (P < 0.001). Both WB and qPCR analysis indicated that tumors with advanced clinical stages have a higher expression levels of PFK1. Moreover, the activity of the PFK1 enzyme and clinical-stage progression has a notable positive correlation, which suggested that PFK1 has effect on malignant biological behaviors as well ()).
Figure 1. Expression and activity of PFK1 in nasopharyngeal tissues. (a–b) Western blot of PFK1 in different tissues where β-actin protein was used as reference shows significant difference in PFK1 expression level. (c) Quantification of PFK1 mRNA expression in different nasopharyngeal tissues detected by qPCR. The expression of β-actin protein and mRNA was served as the controls for sample loading. (d) Analysis activity of PFK1 enzyme in different groups. All experiments were performed at least three times with consistent and repeatable results
PFK1 is efficiently knocked-down by DNA vector-based RNAi
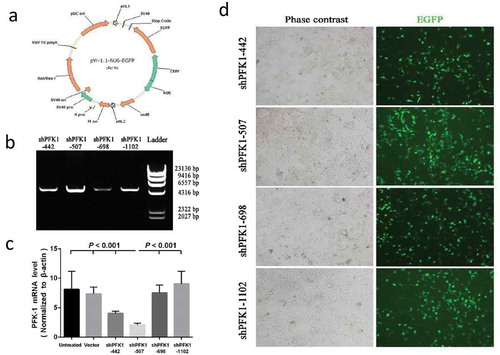
After comprehending the relative of PFK1 expression level and NPC stage progression, knockdown of PFK1 in CNE2 cells were carried on by DNA vector-based RNAi (Schematic illustration of the recombinant pYr-1.1-hU6-EGFP vector is shown in )). Here we tested four different PFK1 si-H-PFK1-442, sh-H-PFK1-507, si-H-PFK1-698, si-H-PFK1-1102, and RT-PCR analysis were carried on to evaluate the knockdown efficiencies. We demonstrated that vector-based RNAi could specifically and efficiently down-regulate the expression of the PFK1 gene, with knockdown efficiencies that might differ greatly between shRNAs ()). As shown in ), compared with that in vector control or untreated transfectants, the PFK1 mRNA levels in cells transduced with sh-H-PFK1-507 significantly reduced by nearly 4.0-fold (p < 0.001), however, the CNE2-si-H-PFK1-698 and CNE2-si-H-PFK1-1102 cells showed no marked reduction in the mRNA transcription (p > 0.05). Additionally, labeled cell imaging was recorded on an immunofluorescence microscope. As shown in ), more green fluorescence dots were observed in CNE2 cells treated with sh-H-PFK1-507, which were in agreement with the luciferase expression experiments.
Figure 2. Construction, identification and expression of PFK-1 shRNA recombinant plasmids. (a) Schematic diagram of the recombinant pYr-1.1-hU6-EGFP vector. (b) RT-PCR analysis identified the PFK-1 shRNA recombinant plasmids after digestion with restriction enzyme XhoI. The level of sh-PFK1-507 mRNA expression was higher compared with other RNA interference fragment. (c) RT-PCR analysis of PFK1 mRNA expression. The β-actin mRNA and protein expression served as controls for sample loading. (d) Immunofluorescence microscopy images (left: bright field, right: green fluorescence EGFP) of intracellular trafficking of different PFK1 in CNE2 cells at optimal ratios
Stable knockdown of PFK1 using sh-PFK1-507 in CNE2 cells
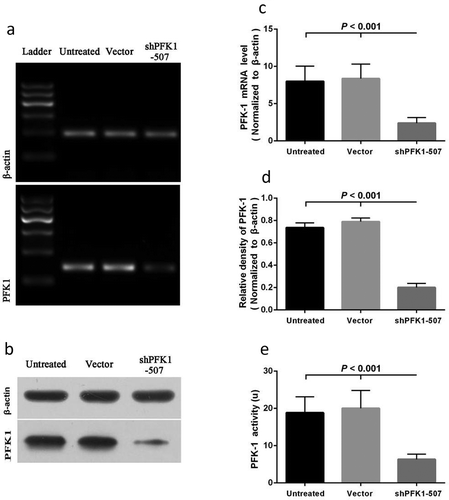
To further validate the roles of PFK1 in cancer development and the potentials of PFK1 down-regulation for NPC therapy, PFK1 stable knockdown experiments were performed in the CNE2 human NPC cell line. As shown in , stable infectious clone sh-PFK1-507 was established from the CNE2 cells, with the greatest suppression by sh-PFK1-507 infected CNE2 cells in mRNA levels (P < 0.001). No obvious effects on PFK1 expression were found between the empty vector group and the untreated group ()). Protein quantification demonstrated that levels of PFK1 expression were down-regulated by nearly 4.0-fold in sh-PFK1-507 cells; however, no significant difference was found between empty vector-transduced clone and untreated group. (p > 0.05, )). In line with the above results, significant decreases in expression levels of the PFK1 mRNA were observed in sh-PFK1-507 cells as compared to the control (p < 0.001, )). Furthermore, we also evaluated the effect of stable knockdown of PFK1 on its enzyme activity in NPC CNE2 cells. Moreover, the PFK1 activity was significantly reduced by approximately 3.0-fold with sh-PFK1-507 transduced (p < 0.001, )), speculating that the overexpression of PFK1 could increase its enzyme activity through activating some canonical pathway in NPC cancer cells.
Figure 3. Effects on shRNA-mediated silencing of PFK1 at the mRNA and protein levels. The CNE2 cells were stably transduced with a sh-PFK1-507 plasmid or a control vector. The expression of PFK1 mRNA (a) and protein (b) in stable clones was examined by RT-PCR and Western blot analysis. β-actin served as an internal control. mRNA or Protein quantification was obtained by densitometric analysis of the mRNA (c) or protein (d). (e) Effects of PFK1 knockdown by sh-PFK1-507 on enzyme activity. All experiments were performed at least three times with consistent and repeatable results
PFK1 knockdown induces apoptosis and inhibits proliferation of CNE2 cells
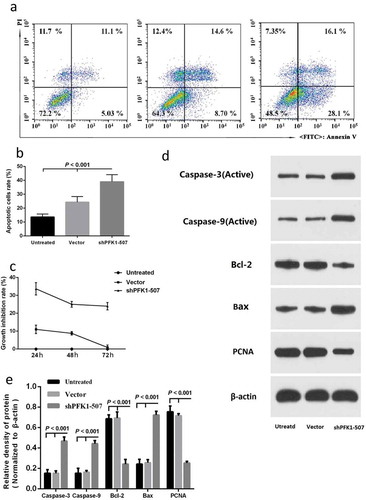
ShRNA-mediated down-regulation of PFK1 expression on CNE2 cell apoptosis was detected in order to research on the phenotypical changes. After obtained the most effective and specific sh-PFK1-507 transfectant, we following conducted Annexin V-FITC/PI assay to analyze cell apoptosis in CNE2 transfectant cells. As shown in and b), cell apoptotic rate of sh-PFK1-507-transduced treatments induced cell increased from 13.69 ± 2.06% to 39.01 ± 5.08% compared with the control group (p < 0.01). To further explore whether PFK1 knockdown by sh-PFK1-507 also has an inhibitory effect on CNE2 cell proliferation, we accomplished determination of cell growth via MTT assay for 24 h, 48 h, and 72 h, respectively. The growths of sh-PFK1-507 and control vector-transduced cells were obviously inhibited with increased incubation time from the cell growth curve ()). Accordingly, PFK1 promoted cell proliferation and suppressed apoptosis in NPC CNE2 cells. Furthermore, the sh-PFK1-507-transfected cells can significantly increase the expression of the active caspase-3, active caspase-9 compared with other groups (–e)). We also found an increased trend for Bax, and a reduced trend for Bcl-2 and PCNA in CNE2 cells transfected with sh-PFK1-507.
Figure 4. Effect of sh-PFK1-507 on apoptosis and proliferation of CNE2 cells. (a) Flow cytometry analysis of cell apoptosis. Cells were incubated in culture medium for 24 h after transfected with sh-PFK1-507. (b) The mean± SD (Standard deviation) percentage of apoptosis cells measured from Flow cytometry. Compared with control cells, the apoptotic rate of sh-PFK1-507 cells significantly increased (39.01 ± 5.08%; p < 0.001). (c) Detection of cell viability via MTT assay. Proliferation of CNE2 cells stably transfected with sh-PFK1-507, vector or untransfected CNE2 cells were evaluated. The highest inhibitory rate of sh-PFK1-507 transfected cells was 33.69 ± 3.47% (p < 0.05). (d–e) Apoptosis proteins (active caspase-3, active caspase-9, and Bax) were increased, and antiapoptosis (Bcl-2 and PCNA) were reduced in CNE2 cells transfected with sh-PFK1-507. All experiments were performed three times with consistent and repeatable results
PFK1 Knockdown inhibits the migration of CNE2 Cells
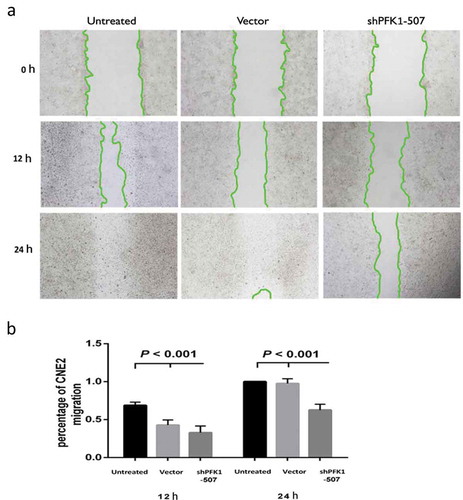
A wound healing assay was performed to confirm the PFK1 knockdown effect on migration of CNE2 cells. As shown in and b), the wound healing ratio at 12 h of untreated, vector, and shPFK1-507 cells was 0.69 ± 0.04, 0.43 ± 0.07, 0.33 ± 0.09, respectively. And 1 ± 0, 0.97 ± 0.06, 0.63 ± 0.08, respectively, at 24 h, ShPFK1-507 transduced CNE2 cells migration into the scratch wound area was significantly decelerated compared with control groups (P < 0.001), while there were no obvious differences between untreated and control vector cells (P > 0.05). Thus, PFK1 expression knockdown blocked the migratory ability of CNE2 cells.
Figure 5. Stable knockdown of PFK1 inhibited migration of CNE2 cells. (a) Migration of CNE2 cells into the scratch wound after 0, 12 and 24 h of culture with ShPFK1-507 in the Wound healing assay. (b) Migration rate of CNE2 after incubation in 12 and 24 h
PFK1 knockdown inhibits the invasion of CNE2 cells
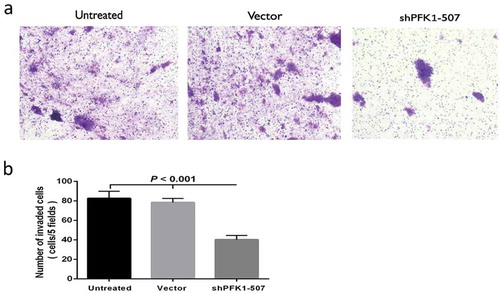
CNE2 cells were transferred to Matrigel-coated transwell chambers for 48 h in order to explore the effect on invasion potential of PFK1 by vector-based shRNA-mediated suppression. The cells invaded through the membrane coated with Matrigel-and the amount of shPFK1-507 cells was significantly less than the untreated or control Vector cells ()). The analysis of quantification implied that invading cells of ShPFK1-507 were 2-folds decreased comparing with untreated and Vector cells (p < 0.001, )). It is easy to say that stable silence for PFK1 evidently reduced the invasive capability of CNE2 cells.
Figure 6. Stable knockdown of PFK1 inhibited invasion of CNE2 cells. (a) Representative cells invade through Matrigel and membrane. (b) Amount of invaded cells in different groups. All experiments were performed at least three times with consistent and repeatable results
Discussion
Nasopharyngeal carcinoma which arises from squamous epithelial cells is frequently diagnosed in advanced stages or after metastases due to the anatomical position and lack of specific prodromal symptomatology [Citation5,Citation19,Citation20]. The prognosis of patients with NPC is relatively poor with a median survival period of about 31.30 months [Citation21]. With the development of molecular inhibitors such as nivolumab, pembrolizumab, and Bevacizumab, the prognosis of patients with recurrent and/or metastatic NPC has improved tremendously [Citation22]. It is needed to realize the function of a certain gene and the molecular mechanism of NPC.
The 6-phosphofructo-1-kinase (PFK1) is a key regulatory enzyme of glycolysis. The function of PFK1 is catalyzing the Mg-ATP-dependent phosphorylation of fructose-6-phosphate to converse into fructose 1, 6-bisphosphate, and the release of Mg-ADP as a byproduct [Citation23]. Glycosylation inhibits PFK1 activity and redirects the flux of glucose from glycolysis through the pentose phosphate pathway (PPP) which provides a selective growth advantage to cancer cells. Blocking glycosylation of PFK1 at Ser529 could reduce cancer cell proliferation in vitro and impair tumor formation in vivo [Citation23]. Moreover, Wang et al. has found that PITA interacts with p53 and specifically suppresses transcription of the glycolysis regulator TIGAR exhibited increased PFK1 activity and an elevated glycolytic rate [Citation24]. It has also showed that miR-135 accumulates specifically in response to glutamine deprivation and requires ROS-dependent activation of mutant p53, which directly promotes miR-135 expression to suppress aerobic glycolysis by targeting PFK1 [Citation25]. PFKFB3 is one of the allosteric activators of PFK1 as well as the most potent stimulator of glycolysis which is believed to contribute to tumor progression of different types of cancer [Citation15,Citation26]. Thus, PFK1 has been confirmed to be strikingly overexpressed in several human cancers including Glioblastoma [Citation14], cervical cancer [Citation10], oligodendrogliomas [Citation10], bladder cancer [Citation18], and cholangiocarcinoma [Citation12]. Altenberg et al. has shown that the expression of glycolysis genes were ubiquitously upregulated in 24 cancers [Citation26], which including rate-limiting enzymes PFK1, concentration-dependent enzymes HK2, GAPDH, TPI, PGK1, and ENO1 [Citation27]. Furthermore, other researchers [Citation7,Citation16] and our study (unpublished data) have revealed that PFK1 expression has a positive correlation with tumor stage, differential degree, tumor proliferation, and invasive ability. NPC patients with an extremely strong PFK1 overexpression have relatively shorter survival rates than those with lower PFK1 expression, indicating that elevated expression level of PFK1 could be an independent factor of bad prognosis (unpublished data). Additionally, the activity of PFK1 was remarkably increased in the primary sites of breast cancer patients by comparing para-cancer tissues or patients with breast benign lesions. Metastatic tumor tissues presented a two-fold increase in this particulate activity when compared to nonmetastatic tumor samples [Citation28]. In the current study, we confirmed that the expression and activity of PFK1 were remarkably increasing both in NPC tissue samples and CNE2 cell lines. Thus, the mechanistic function and expression type of PFK1 suggests that PFK1 may play a role in promoting carcinogenesis and malignant progression of NPC under certain pathological conditions. The correlation between the high expression level of PFK1 and the advanced stages of NPC or other cancers suggests that PFK1 expression regulation could provide a new idea in gene therapy.
RNAi, a posttranscriptional gene-silencing mechanism in most eukaryotic cells that modulate a wide range of genetic regulatory pathways, was first discovered in Caenorhabditis elegans [Citation29]. Recently, RNAi technology has been proven to be a newly advanced effective tool for target gene silencing which can therefore be used widely for analyzing the function of genes and as a therapeutic target in cancer in vivo and in vitro [Citation30–34]. Currently, knockdown of PFK1 by vector-based RNAi as a therapeutic model for CNE2 treatment is not yet established. In the present study, we found that the expression level of PFK1 mRNA and protein was remarkably suppressed by RNAi according to RT-PCR and Western bolt analysis.
Next, we used RNAi as a strategy to specifically knockdown of PFK1 to explore that whether inhibition of PFK1 could regulate the biologic functions of CNE2 NPC cells. Based on our results, knockdown of PFK1 expression significantly inhibits cell proliferation of the CNE2 cell line via MTT assay. As expected, it can also induce apoptosis by flow cytometry analysis, comparing to the rare in CNE2 cells transfected with mock siRNA. As it is well known, apoptosis is a physiological process controlled by multiple genes, in which various apoptosis-related proteins are implicated. Caspase-3 is known to be play an executioner role in cell apoptosis, which can be activated by both extrinsic (caspase-8 activation) and intrinsic (caspase-9 activation) pathways to induce cell shrinkage, nuclear condensation, and DNA fragmentation [Citation35]. Moreover, the Bcl-2 family also has a great influence on tumor cell apoptosis. It is comprised of antiapoptotic members such as Bcl-2, Bcl-xL, and Mcl-1 and pro-apoptotic members such as Bax, Bad, and Bak [Citation36]. As shown in –e), the expression level of active caspase-3, active caspase-9, and Bax was increased, while the expression levels of Bcl-2 and PCNA were steadily reduced in CNE2 cells transfected with sh-PFK1-507. These data demonstrating that PFK1 plays an important role in promoting antiapoptotic proteins and inhibiting pro-apoptotic proteins, which are consistent with those in a study [Citation7] of PFK1 knockdown promotes apoptosis and inhibits cell proliferation in muscle-invasive bladder cancer cell.
In addition, some clinical studies indicate that PFK1 levels are closely associated with the degree of tumor cell invasion and PFK1 plays key roles in tumor progression and metastasis [Citation7,Citation16,Citation28,Citation37]. These clinical results prompted us to investigate whether stable silencing of PFK1 expression could inhibit CNE2 progression by altering tumor cell migration and invasion. Our cell wound healing experiments revealed that a long-term RNAi knockdown of PFK1 could result in significant suppression of migration in CNE2 cells. The invasive capabilities of in vitro CNE2 cells, a hallmark of cancer progression and metastasis, also significantly reduced by vector-based PFK1-specific siRNA.
In summary, silencing the PFK1 gene with vector-based PFK1 siRNA can significantly promote apoptotic effects and inhibit proliferation, migration, invasion in CNE2 cells. These, therefore, raise the possibility that PFK1 is a reliable biomarker for the molecular diagnosis of NPC and a promising candidate target for NPC therapy at least in CNE2 cells. Animal studies using vector-based PFK1-specific siRNA are currently on the way to evaluate their in vivo usefulness.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Data availability statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Disclosure statement
The authors declare no potential conflicts of interest.
Additional information
Funding
References
- Chen YP, Chan ATC, Le QT, et al. Nasopharyngeal carcinoma. Lancet. 2019;394:64–80.
- Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
- Chen M, Tang LL, Sun Y, et al. Treatment outcomes and feasibility of partial neck irradiation for patients with nasopharyngeal carcinoma with only retropharyngeal lymph node metastasis after intensity-modulated radiotherapy. Head Neck. 2014;36:468–473.
- M F J, Sheng W, Cheng W, et al. Incidence and mortality of nasopharyngeal carcinoma: interim analysis of a cluster randomized controlled screening trial (PRO-NPC-001) in southern China. Ann Oncol. 2019;30:1630–1637.
- Wang JY, Li XF, Li PZ, et al. MicroRNA-23b regulates nasopharyngeal carcinoma cell proliferation and metastasis by targeting E-cadherin. Mol Med Rep. 2016;14:537–543.
- He HL, Lee YE, Liang PI, et al. Overexpression of JAK2: a predictor of unfavorable prognosis for nasopharyngeal carcinoma. Future Oncol. 2016;12:1887–1896.
- Sun CM, Xiong DB, Yan Y, et al. Genetic alteration in phospho fructokinase family promotes growth of muscle-invasive bladder cancer. Int J Biol Markers. 2016;31:286–293.
- Alfarouk KO, Verduzco D, Rauch C, et al. Glycolysis, tumor metabolism, cancer growth and dissemination. A new pH-based etiopathogenic perspective and therapeutic approach to an old cancer question. Oncoscience. 2014;1:777–802.
- Amano Y, Mandai M, Yamaguchi K, et al. Metabolic alterations caused by HNF1β expression in ovarian clear cell carcinoma contribute to cell survival. Oncotarget. 2015;6:26002–26017.
- MorenoSánchez R, RodríguezEnríquez S, MarínHerná- ndez A, et al. Energy metabolism in tumor cells. Febs J. 2010;274:1393–1418.
- Xiang JD, Zhou LN, Zhuang Y, et al. Lactate dehydrogenase is correlated with clinical stage and grade and is downregulated by si-STAB1 in ovarian cancer. Oncol Rep. 2018;40:2788–2797.
- Fujiwara H, Tateishi K, Misumi K, et al. Effect of high glucose on expression of glycolytic enzymes in cholangiocarcinoma cells. Sci Rep. 2019;9:18859.
- Cui SS, Yang X, Zhang LH, et al. LncRNA MAFG-AS1 promotes the progression of colorectal cancer by sponging miR-147b and activation of NDUFA4. Biochem Biophys Res Commun. 2018;506:251–258.
- Lee JH, Liu R, Li J, et al. EGFR-phosphorylated platelet isoform of phosphofructokinase 1 promotes PI3K activation. Mol Cell. 2018;70:197–210.
- Yalcin A, Clem BF, Imbert-Fernandez Y, et al. 6-Phosphofructo-2-kinase (PFKFB3) promotes cell cycle progression and suppresses apoptosis via Cdk1-mediated phosphorylation of p27. Cell Death Dis. 2014;5:e1337.
- Smerc A, Sodja E, Legisa M. Posttranslational modification of 6-phosphofructo-1-kinase as an important feature of cancer metabolism. PloS One. 2011;6:e19645.
- Donato L, D’Angelo R, Alibrandi S, et al. Effects of A2E-induced oxidative stress on retinal epithelial cells: new insights on differential gene response and retinal dystrophies. Antioxidants. 2020;9:307.
- Jia W, Ren C, Wang L, et al. CD109 is identified as a potential nasopharyngeal carcinoma biomarker using aptamer selected by cell-SELEX. Oncotarget. 2016;7:55328–55342.
- Li Y, Tang X, He Q, et al. Overexpression of mitochondria mediator gene TRIAP1 by miR-320b loss is associated with progression in nasopharyngeal carcinoma. PLoS Genet. 2016;12:e1006183.
- Siti-Azrin AH, Norsa’adah B, Naing NN. Five-year survival and median survival time of nasopharyngeal carcinoma in hospital universiti Sains Malaysia. Asian Pac J Cancer Prev. 2014;15:6455–6459.
- Szturz P, Vermorken JB. Immunotherapy in head and neck cancer: aiming at EXTREME precision. BMC Med. 2017;15:110.
- Lee N, Harris J, Pfister DG, et al. Long-term update of a phase II Study of concurrent chemoradiotherapy using radiation + bevacizumab (BV) for locally or regionally advanced nasopharyngeal cancer (NPC): RTOG 0615. Int J Radiat Oncol Biol Phys. 2020;106:1119.
- Yi W, Clark PM, Mason DE, et al. PFK1 glycosylation is a key regulator of cancer cell growth and central metabolic pathways. Science. 2012;337:975–980.
- Wang S, Peng Z, Wang S, et al. Author Correction: KRAB-type zinc-finger proteins PITA and PISA specifically regulate p53-dependent glycolysis and mitochondrial respiration. Cell Res Cell Res. 2018;28:605.
- Yang Y, Ishak Gabra MB, Hanse EA, et al. MiR-135 suppresses glycolysis and promotes pancreatic cancer cell adaptation to metabolic stress by targeting phosphofructokinase-1. Nat Commun. 2019;10:809.
- Altenberg B, Greulich KO. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics. 2004;84:1014–1020.
- Glunde K, Jiang L, Moestue SA, et al. Magnetic resonance spectroscopy and imaging guidance in molecular medicine: targeting and monitoring of choline and glucose metabolism in cancer. NMR Biomed. 2011;24:673–690.
- Tatiana EB, Marta SF, Mauro SP. Cellular distribution of phosphofructokinase activity and implications to metabolic regulation in human breast cancer. Mol Genet Metab. 2003;79:294–299.
- Ambros V. A hierarchy of regulatory genes controls a larva-to-adult developmental switch in C. elegans. Cell. 1989;57:49–57.
- Gui SJ, Ding RL, Wan YP, et al. Knockdown of annexin VII enhances nasopharyngeal carcinoma cell radiosensitivity in vivo and in vitro. Cancer Biomark. 2020;28:129–139.
- Donato L, Scimone C, Alibrandi S, et al. Transcriptome analyses of lncRNAs in A2E-stressed retinal epithelial cells unveil advanced links between metabolic impairments related to oxidative stress and retinitis pigmentosa. Antioxidants. 2020;9:318.
- Donato L, Scimone C, Alibrandi S, et al. Discovery of GLO1 new related genes and pathways by RNA-Seq on A2E-stressed retinal epithelial cells could improve knowledge on retinitis pigmentosa. Antioxidants. 2020;9:416.
- Rinaldi C, Bramanti P, Scimone C, et al. Relevance of CCM gene polymorphisms for clinical management of sporadic cerebral cavernous malformations. J Neurol Sci. 2017;380:31–37.
- Donato L, Bramanti P, Scimone C, et al. miRNA expression profile of retinal pigment epithelial cells under oxidative stress conditions. Wiley-Blackwell Online Open. 2018;8:219–233.
- Zhou J, Luo Y-H, Wang J-R, et al. Gambogenic acid induction of apoptosis in a breast cancer cell line. Asian Pac J Cancer Prev Apjcp. 2013;14(12):7601–7605.
- Reed JC. Double identity for proteins of the Bcl-2 family. Nature. 1997;387:773–776.
- Francesca M, Condelli V, Valentina DS, et al. TRAP1 enhances Warburg metabolism through modulation of PFK1 expression/activity and favors resistance to EGFR inhibitors in human colorectal carcinomas. Mol Oncol. 2020 Oct 6. Online ahead of print. DOI:10.1002/1878-0261.12814.