
Abstract
Cigarette smoke is highly toxic and is a major risk factor for airway inflammation, oxidative stress, and decline in lung function—the starting points for chronic obstructive pulmonary disease. Quercetin is a potent dietary antioxidant that displays anti-inflammatory activities. The goal of this study was to evaluate the effects of quercetin on reducing the redox imbalance and inflammation induced by short-term cigarette smoke exposure. In vitro, 25 and 50 μM quercetin attenuated the effects of cigarette smoke extract (increased generation of reactive oxygen species and nitric oxide) on J774A.1 cells (macrophages). We further examined the effects of quercetin in vivo. Male C57Bl/6 mice that received 10 mg/kg/day of quercetin via orogastric gavage before exposure to five days of cigarette smoke demonstrated reduced levels of leukocyte, oxidative stress, histological pattern changes of pulmonary parenchyma, and lung function alterations compared to the group that did not receive quercetin. These results suggest that quercetin may be an effective adjuvant for treating the effects of cigarette smoke exposure.
Introduction
In the last century, approximately 100 million people were killed by smoking, and more than 8 million people a year will die from diseases related to cigarette smoke [Citation1,Citation2]. Cigarette smoke is highly toxic, containing a variety of particulate matter, chemicals, and reactive oxygen species (ROS) that are capable of inducing inflammation throughout the pulmonary epithelium [Citation3,Citation4]. Chronic smoking is a major risk factor for the development of chronic obstructive pulmonary disease [Citation5–9]. Although pulmonary changes caused by short-term cigarette smoke exposure do not show all the features of chronic obstructive pulmonary disease (COPD) compared to long-term use, both models exhibit similar pathophysiological mechanisms, including inflammation and redox imbalance [Citation10–12].
The airway epithelium acts as a protective barrier between the lungs and environmental toxins; thus, the changes caused by the inhalation of cigarette smoke can be first observed here [Citation13]. The initial cellular activation—epithelial, endothelial, and resident macrophages—caused by ROS and other toxins present in cigarette smoke induces cytokine and chemokine secretion and subsequent recruitment of immune cells, mainly macrophages and neutrophils [Citation4, Citation14]. These processes stimulate further production of cytokines, as well as proteases and ROS, which amplify the inflammatory response and redox imbalance. These processes collectively cause tissue destruction and is considered the starting point for COPD pathogenesis [Citation15,Citation16].
Redox imbalance occurs whenever ROS levels exceed the body’s antioxidant system, either transiently or persistently. High ROS levels can interfere with cellular homeostasis and redox-sensitive signaling pathways, causing oxidative damage that can culminate in cell death [Citation17].ROS are produced by living organisms during normal cellular metabolism or by activated inflammatory cells. They are also present in environmental pollutants. Due to this constant contact with ROS, organisms have gradually developed antioxidant defense systems, including antioxidant enzymes and non-enzymatic substances with antioxidant effects, to prevent damage to proteins, lipids and DNA [Citation18]. The enzymatic system is primarily dependent on the action of three key players: superoxide dismutase, which converts superoxide anions to hydrogen peroxide; glutathione peroxidase, which reduces peroxides; and catalase, which converts hydrogen peroxide into water and molecular oxygen [Citation17]. Non-enzymatic ROS scavengers include minerals, phenolic compounds, and vitamins. Of the phenolic compounds, flavonoids have rich antioxidant properties, with quercetin (3,5,7,3′,4′-pentahydroxyl flavone) being the most abundant flavanol and present in a variety of foodstuffs. Moreover, it has strong antioxidant capacity due to its ability to remove O2-, OH-, singlet oxygen [Citation19–22]. It interacts with a broad spectrum of cell targets reducing the expression of inflammatory cytokines and growth factors, reducing secretion of mucin proteins and metalloproteinases activities [Citation23–25].Thus, quercetin has been explored as a treatment option for pulmonary diseases, specifically, its ability to modulate inflammation [Citation22] and redox imbalance [Citation26]. However, the ability of quercetin to counteract the deleterious effects of cigarette smoke have not been elucidated. The goal of this study was to determine whether treatment with quercetin can correct the redox imbalance induced by cigarette smoke exposure using cellular (alveolar macrophages) and in vivo approaches (C57BL/6 mice).
Materials and methods
In vitro experiments
Murine J774A.1 macrophages were cultured in 75 cm2 growth flask (Sardesdt AG& Co., Germany) at 37 °C in a 5% CO2 atmosphere and grown in Dulbecco’s Modified Eagle’s Medium (DMEM, Sigma) supplemented with 10% (v/v) fetal bovine serum (FBS) (Invitrogen Co. Ltd., Carlsbad, CA, USA), 1% (v/v) penicillin, 3.7% (v/v) sodium bicarbonate, 1% (v/v) glutamine (200 mM), and 2.5 mL of HEPES (1 M) (final concentration 200 g/mL). When the cells reached 80% confluency, they were collected with a cell scraper and used for experiments.
Preparation of cigarette smoke extract (CSE)
Before experimentation, CSE was prepared from filtered Marlboro Red cigarettes (10 mg of tar, 0.9 mg of nicotine, and 10 mg of carbon monoxide per cigarette) as previously described [Citation27]. The cigarette smoke was bubbled through 10 mL of supplemented culture medium in a modified syringe-driven apparatus. The resulting suspension was titrated to pH 7.4 and sterilized with a 0.22 μm Millipore filter. This solution was considered to be 100% CSE. For experiments, the CSE was diluted with culture medium to 0.5% CSE. Control medium was prepared by bubbling air through 10 mL of culture medium in the same conditions [Citation11].
Cytotoxicity of cigarette smoke extract and quercetin
The toxicity of cigarette smoke and quercetin (Sigma Aldrich) was determined using a colorimetric assay based on the reduction of resazurin, basically in the presence of viable cells, resazurin suffers a colorimetric change from blue to red [Citation28,Citation29]. J774A.1 cells (1 × 106 cells/mL in 200 µL of culture medium) were seeded in 96-well plates and exposed to decreasing concentrations of CSE: 20; 10; 5; 2.5; 1.25; 0.6; 0.3; 0.15% in culture medium or quercetin (QC): 100; 50;25; 12.5; 6.25; 3.12; 1.6; 0.8 µM in culture medium for 24 h. Next, 20 µL of resazurin (1 mM) was added to each well, and the absorbances were measured with a Microplate reader (λex = 570 nm; λem = 600 nm) to determine cell viability [Citation30]. These data were used to calculate the 50% cytotoxic concentrations (CC50) for quercetin and CSE.
Reactive oxygen species (ROS) production in J774A.1 cells
Intracellular ROS levels were detected using the 5-(and 6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (carboxy-H2DCFDA) assay as previously described [Citation11]. J774A.1 cells (2.5 × 104 cells/well) were seeded in 96-well plates containing DMEM supplemented with 10% fetal bovine serum and cultured for 45 min at 37 °C in humidified air supplemented with 5% CO2. The supernatant was then removed and the cells were resuspended in DMEM containing 10, 25, and 50 μM of quercetin for 6 h (37 °C and 5% CO2 in the dark). Next, the cells were washed with Hanks’ balanced salt solution (HBSS) and exposed to medium containing 0.5% CSE and incubated in the dark with carboxy-H2DCFDA for 24 h at 37 °C in humidified air supplemented with 5% CO2. Cells were washed with HBSS, medium without red phenol was added, and fluorescence intensity was determined using an automated plate reader model (λex = 485 nm; λem = 535 nm; VICTOR X3 equipped with the PerkinElmer 2030 workstation and Workout 2.5). Mean fluorescence intensity was reported as the percent (%) increase over control (culture medium without quercetin or CSE.
Nitric oxide (NO) production in J774A.1 cell
NO production by J774.A1 cells was determined based on the accumulation of nitrite in cell supernatant using the Griess method [Citation31]. Cells (1 × 106 cells/well of RPMI supplemented with 10% of fetal bovine serum) were added to 48-well microplates and maintained for 2 h at 37 °C in humidified air supplemented with 5% CO2 to allow for cell adhesion. The wells were washed with 200 μL of sterile PBS to remove non-adhered cells, and 200 μL of RPMI supplemented with 10% of fetal bovine serum was added. Next, 200 μL of RPMI with 10% of SFB containing quercetin (10, 25, and 50 μM) or quercetin + 0.5% CSE was added to the wells. After 24 h, 50 μL of the supernatant was collected and mixed with 100 μL of Griess reagent (a 1:1 mixture of 1% sulfanilamide in 2.5% phosphoric acid and 0.1% dihydrochloride N-[1-naftil]-ethylenediamine in 2.5% phosphoric acid). The plate was incubated in the dark for 10 min at 37 °C. The absorbance reading was measured using an ELISA reader (λ = 570 nm). The nitrite concentration was calculated by linear regression using a standard curve of 1 mM sodium nitrite in 10% RPMI of fetal bovine serum (R2> 0.99).
In vivo experiment
All in vivo experiments were approved by the ethics committee of Federal University of Ouro Preto (2015/20). Thirty Male C57BL/6 mice (10-12 weeks old) obtained from the Animal Science Center of the Federal University of Ouro Preto (CCA–UFOP) were housed under controlled conditions (12 h light/dark cycle, 21 °C ± 2 °C, 50% ± 10% humidity) and provided food and water ad libitum. Mice were divided into 5 groups (n = 5 per group): a control group exposed to ambient air (CG); a group that received 200 µL of vehicle solution containing 50% propylene glycol (Sigma Aldrich) and 50% saline (VG) by orogastric gavage; a group that received 10 mg/kg/day of quercetin diluted in propylene glycol solution (QG); a group exposed to cigarette smoke (CSG); a group treated with quercetin and exposed to cigarette smoke (QCSG). The administration of quercetin was performed via orogastric gavage 1 h before exposure to cigarette smoke.
In vivo cigarette smoke exposure protocol
In an inhalation chamber, mice were exposed to 12 commercial full-flavor filtered Virginia cigarettes (tar: 10 mg; nicotine: 0.9 mg and carbon monoxide: 10 mg) per day divided into three times daily (morning, afternoon, and night) for 5 consecutive days as previously described [Citation11, Citation32]. The cigarette smoke (CS) and QCSG animals were initially placed in an inhalation chamber (40 cm × 30 cm × 25 cm) inside an exhaustion chapel. Each cigarette was coupled to a 60-mL plastic syringe through which the smoke was injected inside the inhalation chamber. The cigarette smoke and QCSG groups were maintained in the smoke atmosphere for 6 min; after, the cover was removed from the inhalation chamber for 1 min to allow the air to completely exhaust. The procedure was then repeated with the remaining cigarettes. The control group, VG, and QG groups were subjected to the same conditions except without the cigarette exposure.
Lung function
Lung function analysis was performed 24 h after the last exposure. The mice were individually anesthetized via intraperitoneal administration with ketamine (100 mg/kg) and xylazine (20 mg/kg). A small neck incision was made, and the trachea was cannulated with an 18 G catheter coupled to a 2-cm long tube, which connected the trachea to the flow head (MLT1L). Breathing was monitored using a differential pressure transducer (Spirometer, ADInstruments, Bella Vista, NSW, Australia). The lung function of each mouse was recorded for 60 s with three repetitions each; this signal was amplified (Octal Bridge Amp model ML228; Powerlab 4/35 model ML870; ADInstruments) and recorded with LabChart v8.1.11 using a standard desktop PC. The evaluated parameters were tidal volume, respiratory rate, and minute ventilation.
Blood collection
After recording lung function, the animals received an overdose of ketamine (130 mg/kg) and xylazine (0.3 mg/kg). Their blood was collected by cardiac puncture and stored in polypropylene tubes (2 mL) with 15 μL of heparin. Erythrocyte, hematocrit, and hemoglobin counts were evaluated using an electronic counting device (Mindray® Bio-Medical Electronics Co. Ltda, Shenzhen, China).
Assessment and analysis of the bronchoalveolar lavage fluid (BALF)
Immediately after blood collection, the chest of each animal was opened to collect the bronchoalveolar lavage fluid (BALF). Briefly, the thorax of each animal was opened, the left main bronchus was clamped, the trachea was cannulated, and the right lung was washed with 1.5 mL of saline solution (3 × 500 µL). The samples were kept on ice to avoid cell lysis. A Neubauer chamber was used to determine the total leukocyte counts. For the differential cell count in the BALF, 250 μL of each sample was centrifuged in a cytocentrifuge (INBRAS, São Paulo, BR). Cells were stained with a fast panoptic coloration kit (Laborclin, Pinhais, Parana), and 100 cells were counted per slide [Citation11, Citation33].
Homogenized tissue
After BALF collection, the right ventricle of each animal was perfused with saline to remove blood from the lungs. The right lung was removed and homogenized in 1 mL of phosphate buffer (pH 7.4) and centrifuged at 15,521 g for 10 min. The supernatant was collected and stored at -80 °C for biochemical analyses.
Antioxidant defense and oxidative stress biomarkers in lung homogenates
Lung homogenates were used to measure antioxidants enzyme activity and oxidative damage. Superoxide dismutase (SOD) activity was measured using ELISA (λ = 570 nm) as previously described [Citation34], which is based on the enzyme’s ability to inhibit the autoxidation of pyrogallol. Catalase activity (conversion of hydrogen peroxide (H2O2) to water and molecular O2 per minute)was measured spectrophotometrically (λ = 240 nm) as previously described [Citation35]. Glutathione analysis was determined using an assay adapted from a commercial kit (CS0260, Sigma, St. Louis, MO, USA); the method is based on the ability to reduce the 5.5′-Dithio Acid -bis- (2-nitrobenzoic) to thio-2-nitrobenzoic acid according to the Griffith assay [Citation36]. To calculate the concentration of total glutathione (GSH + GSSG) and oxidized glutathione (GSSG) a standard serial dilution curve is prepared, and the concentration of reduced form (GSH) is calculated from the subtraction of the total glutathione value by the value of oxidized glutathione. Oxidative damage was determined based on lipid peroxidation of thiobarbituric acid reactive substances as previously described [Citation37] and by measuring the levels of oxidatively modified proteins via the carbonyl protein assay [Citation38]. Total protein was measured according to the Bradford method [Citation39].
Stereological analyses
The left lung was fixed, processed, and stained with hematoxylin and eosin. Twenty random images obtained from the histological slides of the lungs were scanned with a 40× objective lens. The volume density analyses of the alveolar septum (Vv[sa]) and the alveolar spaces (Vv[a]) were performed in a test system composed of sixteen points and a test area delineated by the prohibited line to avoid overestimation of the number of structures. The test system was connected to a monitor attached to a microscope. The number of points (PP) that reached the alveolar septa was determined according to the total number of test points (PT) in the system using the equation VV = PP/PT. To ensure uniformity and proportionality in the pulmonary samples, we analyzed 18 random fields in a cycloid test system attached to a monitor. The reference volume was estimated by point counting using the test point system. A total area of 1.94 mm2 was analyzed to determine Vv[sa] and Vv[a] [Citation40].
Statistical analysis
The normal distribution of each variable was assessed using the Kolmogorov-Smirnov test; data are presented as the mean and standard error of the mean (SEM). For comparison among groups, a one-way ANOVA followed by Tukey’s post-test was used. For non-parametric data, we used Kruskal Wallis test followed by Dunn’s post-test, and the data are expressed as the median, minimum, and maximum values. In both cases, the difference was considered significant when the P value was < 0.05. The statistical analyses were performed using Prism v.5 (Windows 7, GraphPad Software, San Diego, CA USA).
Results
Cytotoxicity of cigarette smoke extract and quercetin
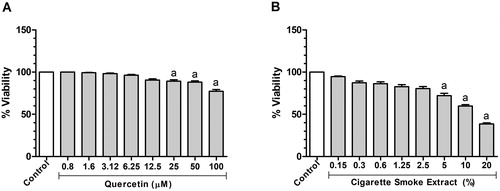
To determine the nontoxic working concentrations of CSE and quercetin, their cytotoxicity was measured using J774.A1 cells by the resazurin assay. Both CSE (Kruskal-Wallis, p < 0.0001) and quercetin (Kruskal-Wallis, p < 0.0001) decreased the number of viable cells in dose-dependent manners (). Basically, a decrease in cell viability was observed after 24 h exposure to 25 (p < 0.05), 50 (p < 0.05) and 100 μM (p = 0.001) quercetin when compared to that in the CG (untreated cells). In cells exposed to CSE, cell viability decreased after 24 h exposure to 5% (p = 0.01), 10% (p = 0.001) and 20% (p = 0.001) when compared to control. To obtain assurance that CSE and quercetin were not toxic to the cell, the half cytotoxic concentrations (CC50) were determined, specifically 14.8% for CSE and 221.5 µM for quercetin at 24 h. Thus, 10, 25, and 50 μM quercetin [Citation41] and 0.5% CSE [Citation11] were used for the subsequent experiments.
Figure 1. Effect of quercetin and cigarette smoke extract on J774A.1 cell viability within 24 h. (A) Analysis of quercetin administration (0.8, 1.6, 3.12, 6.25, 12.5, 25, 50 and 100 μM) on cell viability of J774. (B) Analysis of cigarette smoke extract administration (0.15, 0.3, 0.6, 1.25, 2.5, 5, 10 and 20%) on cell viability of J774. Data were expressed in median, minimum and maximum value and were analyzed by Kruskal-Wallis followed by Dunn’s post-test (p < 0.05). The letter (a) represents a significant difference when compared to control. This test was performed in sextuplicate from two independent experiments.
Effect of CSE on ROS and nitric oxide production in J774A.1 cell
ROS production increased significantly when the cells were exposed to CSE for 24 h (ANOVA, p < 0.0001) in relation to control (p = 0.01). When the cells were pretreated with quercetin at 25 (p = 0.001) and 50 μM (p = 0.0001) and then exposed to CSE for 24 h, we observed a reduction in ROS production compared to CSE treatment alone ().
Figure 2. Effect of cigarette smoke extract and quercetin on ROS and NO production in J774A.1 cell. (A) Quantitative analysis of ROS production in J774 cells administered with Quercetin (10, 25 and 50 μM) and/or CSE (0.5%). (B) NO production measured as nitrite content J774 cells administered with Quercetin (10, 25 and 50 μM) and/or CSE (0.5%). Data are expressed as mean ± standard error of the mean and were analyzed by one-way ANOVA followed by Tukey’s post-test (n = 5-8, p < 0.05). The letter (a) represents a significant difference when compared to control. The letter (b) represents a significant difference when compared to Q10. The letter (c) represents a significant difference when compared to Q25. The letter (d) represents a significant difference when compared to Q50. The letter (e) represents a significant difference when compared to CSE.
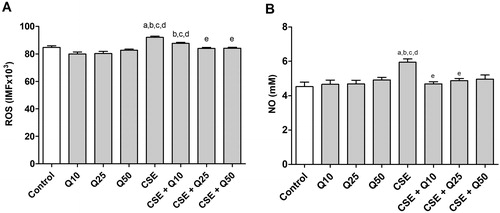
We next measured NO production, as it is an important inflammatory mediator. Relative to the control condition (no CSE), NO concentration increased significantly (ANOVA, p = 0.0011) when the cells were exposed to CSE for 24 h in relation to control (p = 0.001). However, pretreatment with quercetin at all of the tested concentrations 10 (p = 0.01) and 25 μM (p < 0.05) prevented this increase in relation to cells exposed to cigarette smoke extract ().
Lung function in C57BL/6 mice
We found that cigarette smoke (CSG) induced changes in breathing patterns, including respiratory rate (ANOVA, p = 0.0002) and the tidal volume (ANOVA, p < 0.0001) compared to the control group (CG) exposed to ambient air (p = 0.001). Surprisingly, quercetin (QCSG) prevented these variations in tidal volume (p = 0.001) compared to CSG ().
Table 1. Assessment of lung function in mice.
Action of quercetin on hematological parameters in C57BL/6 mice
The exposure to cigarette smoke (CSG) promoted increases in erythrocytes (ANOVA, p < 0.0001), hemoglobin (ANOVA, p < 0.0001) and hematocrit (ANOVA, p < 0.0001) when compared to the control group exposed to ambient air (p = 0.001). However, pretreatment with quercetin (QCSG) did not promote changes in these parameters compared to CSG ().
Table 2. Hematological parameters of experimental groups.
Recruitment of total and differential cell count in BALF
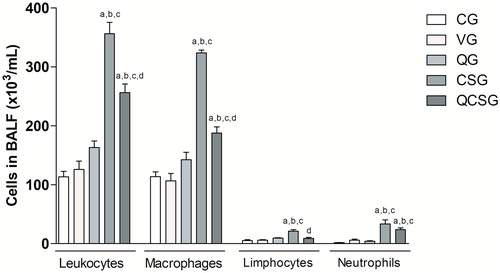
Exposure to cigarette smoke for five days promoted an increase in influx of total leukocytes (ANOVA, p < 0.0001), macrophages (ANOVA, p < 0.0001), lymphocytes (ANOVA, p < 0.0001), and neutrophils (ANOVA, p < 0.0001) into the BALF compared to CG (p = 0.001)(). Interestingly, pretreatment with quercetin before cigarette smoke exposure resulted in decreased numbers of total leukocytes (p = 0.001), macrophages (p = 0.001) and lymphocytes (p = 0.001) when compared to exposed group (CSG).
Figure 3. Total and differential count of bronchoalveolar lavage fluid. CG: control group; VG: vehicle group; QG: quercetin group; CSG: cigarette smoke group; QCSG: quercetin and cigarette smoke group. The letter (a) represents a significant difference between groups when compared to CG. The letter (b) represents a significant difference between groups when compared to VG. The letter (c) represents a significant difference between groups when compared to QG. The letter (d) represents a significant difference between groups when compared to CSG. Data are expressed as mean ± standard error of the mean and were analyzed by one-way ANOVA followed by Tukey’s post-test, n = 5 animals per group, p < 0.05.
Antioxidant defense and oxidative stress biomarkers in lung homogenates
Cigarette smoke exposure promoted a decrease of SOD (ANOVA, p = 0.0002) and catalase activity (ANOVA, p = 0.0025) in the CSG compared to the control group (CG) (p < 0.05), which was attenuated by quercetin pretreatment (p = 0.01) (). The GSH/GSSG ratio was lower (Kruskal-Wallis, p = 0.0027) in the CSG than to the control group (p < 0.05), VG and QG, and pretreatment with quercetin had no effect on this reduction. In contrast, the damage to lung parenchyma was evaluated based on levels of carbonylated proteins (ANOVA, p = 0.0028) and TBARS (ANOVA, p = 0.0001), which both increased in the CSG compared to the control group (ambient air) (p = 0.01). In addition, the QCSG showed a reduction in carbonylated proteins (p < 0.05) and TBARS (p = 0.001) concentration compared to the CSG.
Table 3. Antioxidant defense and oxidative stress biomarkers in lung homogenates.
Stereological analyses of the lung tissue
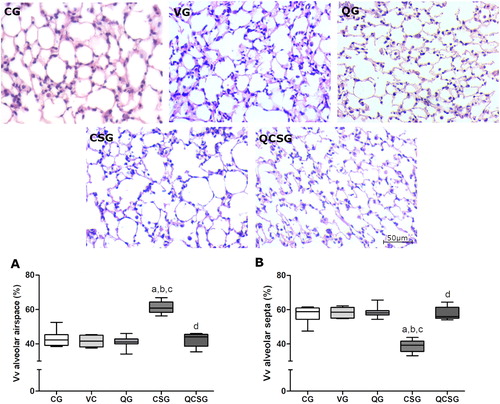
In the stereological analyses (), the volume densities of the alveolar air space (Vv[a]) were increased in the CSG (Kruskal-Wallis, p = 0.001) compared to the control group (p < 0.05) (), and the volume densities of alveolar septa (Vv[sa]) were decrease in CSG (Kruskal-Wallis, p = 0.0011) compared to control group (p = 0.01) (). However, quercetin administration promotes a decrease of Vv[a] (p < 0.05) and an increase of Vv[sa] (p < 0.05) compared to the CSG.
Figure 4. Morphometric evaluations of the lung parenchyma. Photomicrographs of lung sections stained with hematoxylin and eosin. Bar = 50 µm, 400x magnification. CG: control group; VG: vehicle group; QG: quercetin group; CSG: cigarette smoke group; QCSG: quercetin and cigarette smoke group. (A) Volume density of alveolar septa. (B) Volume density of alveolar airspace. The letter (a) represents a significant difference between CSG when compared to CG. The letter (b) represents a significant difference between CSG when compared to VG. The letter (c) represents a significant difference between CSG when compared to QG. The letter (d) represents a significant difference between QCSG when compared to CSG. For A and B, data were expressed in median, minimum and maximum value and were analyzed by Kruskal-Wallis followed by Dunn’s post-test, n = 5 animals per group, p < 0.05.
Discussion
The initial changes caused by cigarette smoke exposure in relation to the pathogenesis of COPD and other lung diseases typically involve inflammation and redox imbalance. Thus, studies of natural products with antioxidant and anti-inflammatory proprieties that could be used to counteract the deleterious effects of cigarette smoke have been encouraged. In this study, we showed that the antioxidant effect of quercetin was effective against the oxidative stress and changes in lung morphology and function induced by cigarette smoke. To our knowledge, this is the first study that examines the initial changes in lung function caused by exposure to cigarette smoke.
First, we evaluated the effects of quercetin on J774A.1 alveolar macrophage exposed to cigarette smoke extract. Alveolar macrophages reside in the pulmonary alveoli and act as the first line of defense in the lungs by orchestrating airway inflammatory events in response to the toxins present in cigarette smoke. These cells secrete multiple inflammatory mediators and ROS [Citation42–46]. In the present study, ROS and NO production were measured in J774.A1 alveolar macrophages exposed to cigarette smoke extract with or without quercetin pretreatment. We found that exposure to cigarette smoke extract increased ROS and NO production, which contributes to an increase in oxidative stress. This result is consistent with the fact that NO is an important modulator of macrophage activity. Pretreatment of quercetin at all doses reduced ROS and NO to baseline levels. This dramatic result may be explained by the ability of quercetin to diffuse at high rates through cell membranes and on the surfaces of lipid bilayers and also in the aqueous phase [Citation47].
After demonstrating the antioxidant effects of quercetin in vitro, we next tested the in vivo effects using a short-term cigarette smoke exposure model. We observed significant hematological alterations in response to cigarette smoke exposure. Specifically, erythrocyte, hemoglobin, and hematocrit levels were all increased. This result is unsurprising given that the gas phase of the cigarette smoke is rich in carbon monoxide (CO). After inhalation, CO reaches the bloodstream and combines with hemoglobin to form carboxyhemoglobin (HbCO), which blocks oxygen binding. Because HbCO circulates faster than oxyhemoglobin, the serum O2 concentration decreases dramatically, causing hypoxia [Citation48,Citation49]. Hypoxia then activates the xanthine oxidase (XO) pathway, resulting in increased conversion of ATP to ADP, AMP, iosin, and hypoxanthine (the substrate for XO), whose metabolism favors the generation of ROS [Citation50,Citation51]. Because quercetin does not interact with CO, it was unsurprising that quercetin pretreatment did not prevent the hematological alterations. Thus, our results suggest that quercetin acts by neutralizing the ROS generated by XO.
Exposure to cigarette smoke promoted increased influx of inflammatory cells into the airways, corroborating our previous studies [Citation11, Citation33, Citation52]. Specifically, we showed that oxidants present in cigarette smoke can stimulate alveolar macrophages to release ROS and chemotactic factors that attract neutrophils and other inflammatory cells. Activated macrophages and neutrophils also generate ROS via nicotinamide adenine dinucleotide phosphate (NADPH) oxidases [Citation53,Citation54]. In our study, cigarette smoke exposure led to a greater recruitment of macrophages and neutrophils in CSG compared to the control group, suggesting a greater generation of ROS in CSG. We also found that pretreatment with quercetin reduced the influx of inflammatory cells, particularly macrophages, to the BALF in animals exposed to cigarette smoke. A similar result was shown by Ganesan and colleagues (2010) using a model of lung injury induced by elastase/LPS that was pretreated with quercetin at 10 mg/kg/day for 10 days [Citation24].
The oxidant load in the lungs due to cigarette smoke exposure leads to a redox imbalance that culminates in oxidative damage to macromolecules, such as lipids and proteins. Previous data showed that changes in antioxidant enzyme activity and oxidative damage occur from the third day of CS exposure [Citation40]. Here, we evaluated the redox imbalance by measuring oxidative damage. Our results showed that contact with cigarette smoke significantly decreased the activity of the antioxidants, enzymes SOD and catalase. Pretreatment with quercetin restored their activity to control levels. Quercetin—one of the most potent antioxidants in the flavonoid family—has been shown to be effective against redox imbalance in several models and is a strong superoxide scavenger [Citation55]. Antioxidant enzymes, in particular SOD, lose part of their activity when there is an excess of their substrate. Thus, the O2- present in the cigarette smoke as well as the increase in O2- generation by NADPH oxidases resulting from cigarette smoke exposure likely contributed to the loss of SOD activity observed herein [Citation56]. Because the product of O2- dismutation by SOD is H2O2, which is the substrate for catalase, a reduction in SOD activity would also result in loss of catalase activity. Thus, it is likely that the effect of quercetin in restoring SOD and catalase activity is due, at least in part, to its effect on reducing O2- levels. Another marker of redox imbalance is the ratio GSH/GSSH. Several authors have correlated the decrease in GSH/GSSH ratio to oxidative stress and the emergence of various diseases [Citation57–59]. Because the lung contains high levels of glutathione [Citation60] and COPD is a disease related to oxidative stress, the progression of COPD is substantially affected by the status of glutathione. In this study cigarette smoke exposure caused a decrease in the GSH/GSSH ratio and pretreatment with quercetin had no effect. These results suggest that pretreatment with quercetin before cigarette smoke exposure promotes the endogenous antioxidant defense mechanisms—i.e. by increasing the activity of antioxidant enzymes, such as SOD and catalase. The decreased SOD and catalase activities and decreased GSH/GSSG ratio reflect the susceptibility of the lung to cigarette smoke-induced oxidative stress. These results are corroborated by our experiments showing higher levels of lipid peroxidation and protein carbonylation in the lung parenchyma of mice exposed to cigarette smoke. Interestingly, quercetin supplementation was prevented this damage. Despite the low administered dose of quercetin, its antioxidant effects were clearly demonstrated in our experimental model. This may be due to the ability of quercetin to sit between lipid bilayers; the increase in its local concentration would allow quercetin to exert its antioxidant activity more efficiently [Citation19, Citation61].
Redox imbalance causes structural damage to essential lung components and is responsible for irreversible damage to the lung parenchyma [Citation62,Citation63]. Our results demonstrate that exposure to cigarette smoke for a short time caused alterations in the alveolar structure as shown by an increase in alveolar volume density and a decrease in alveolar septa volume density. Pretreatment with quercetin was able to prevent these alterations. These results corroborate our previous studies that showed similar results using other antioxidants [Citation11, Citation52]. The stereological changes in the pulmonary parenchyma caused by short-term exposure to cigarette smoke are well-established in our group [Citation33, Citation40, Citation52, Citation64], but this is the first time that we have related these changes to lung function parameters.
Changes in pulmonary architecture caused by short-term exposure to cigarette smoke reflected a change in the breath control balance. The group exposed to cigarette smoke presented a higher level of tidal volume compared to the other groups, and pretreatment with quercetin attenuated this effect. The CSG also presented a lower respiratory rate index when compared to CG. We believe that these changes occurred due to as a physiological response to short exposure to cigarette smoke in the attempt to reestablish respiratory balance, as there was no significant change in minute volume between groups.
Quercetin has low bioavailability, mainly due to its low solubility and to undergo extensive metabolism before reaching the bloodstream [Citation65,Citation66]. Li and colleagues (2016) suggest an absorption rate of the aglycone form of approximately 2% [Citation25]. A strength of our study is the low dose of quercetin that was used (10 mg/kg/day), which is equivalent to a dose of 48.65 mg/day for a human of 60 kg [Citation67]. This amount can be easily achieved by increasing the intake of foods rich in this flavonoid, such as capers (180.77 mg/100g), purple onions (19.93 mg/100g) and apple with the peel (4.4 mg/100g) [Citation68] without the need for supplementation.
Conclusions
Quercetin is a highly potent antioxidant both in alveolar macrophages and in the pulmonary parenchyma of mice. Importantly, it significantly prevented the oxidative damage and consequently the morphological and functional pulmonary alterations produced by short-term exposure to cigarette smoke, in this study. Thus, we propose that an increase in consumption of this flavonoid would be an effective adjuvant in the treatment of pulmonary lesions induced by cigarette smoke and can act to prevent or even decrease the rate of lesion progression.
Declaration of interest statement
The author(s) declared no potential conflicts of interest with respect to research, authorship, and/or publication of this article.
Acknowledgments
N.A.M and F.S.B. would like to express their gratitude to CAPES for Post-Doctoral fellowship (N.A.M.: PNPD/CAPES-Process # 88882.316933/2019-01 in Laboratory of Experimental Pathophysiology (LAFEx), Center of Research in Biological Sciences (NUPEB), UFOP, Brazil; F.S.B.: CAPES-PVEX-Process # 88881.172437/2018-01 in the Interdepartmental Division of Critical Care Medicine, St. Michael’s Hospital, University of Toronto, Toronto, ON, Canada). The authors would like to express their gratitude to Keila Karine Duarte Campos (in memorian) and Maria Terezinha Bahia for your technical assistance during this study. FSB is in credit with the CNPq for the fellowship of research productivity.
Additional information
Funding
References
- Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3(11):e442. doi:10.1371/journal.pmed.0030442.
- Asma S, Song Y, Cohen J, et al. CDC grand rounds: global tobacco control. Morb Mortal Wkly Rep. 2014;63(13):277–280.
- Crotty Alexander LE, Shin S, Hwang JH. Inflammatory diseases of the lung induced by conventional cigarette smoke: a review. Chest. 2015;148(5):1307–1322. doi:10.1378/chest.15-0409.
- Kumar A, Cherian SV, Vassallo R, et al. Current concepts in pathogenesis, diagnosis, and management of smoking-related interstitial lung diseases. Chest. 2018;154(2):394–408. doi:10.1016/j.chest.2017.11.023.
- Dalrymple A, Ordonez P, Thorne D, et al. An improved method for the isolation of rat alveolar type II lung cells: Use in the Comet assay to determine DNA damage induced by cigarette smoke. Regul Toxicol Pharmacol. 2015;72(1):141–149. doi:10.1016/j.yrtph.2015.03.013.
- Austin V, Crack PJ, Bozinovski S, et al. COPD and stroke: are systemic inflammation and oxidative stress the missing links? Clin Sci. 2016;130(13):1039–1050. doi:10.1042/CS20160043.
- Zinser JW. Tobacco use and lung cancer. Salud Publica Mex. 2019;61(3, may-jun):303–307. doi:10.21149/10088.
- Aghapour M, Raee P, Moghaddam SJ, et al. Airway epithelial barrier dysfunction in chronic obstructive pulmonary disease: role of cigarette smoke exposure. Am J Respir Cell Mol Biol. 2018;58(2):157–169. doi:10.1165/rcmb.2017-0200TR.
- Durham AL, Adcock IM. The relationship between COPD and lung cancer. Lung Cancer. 2015;90(2):121–127. doi:10.1016/j.lungcan.2015.08.017.
- Seifart C, Plagens A. Genetics of chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2007;2(4):541–550.
- Campos KKD, Araujo GR, Martins TL, et al. The antioxidant and anti-inflammatory properties of lycopene in mice lungs exposed to cigarette smoke. J Nutr Biochem. 2017;48:9–20. doi:10.1016/j.jnutbio.2017.06.004.
- Valenca SS, Pimenta WA, Rueff-Barroso CR, et al. Involvement of nitric oxide in acute lung inflammation induced by cigarette smoke in the mouse. Nitric Oxide. 2009;20(3):175–181. doi:10.1016/j.niox.2008.11.003.
- Dye JA, Adler KB. Effects of cigarette smoke on epithelial cells of the respiratory tract. Thorax. 1994;49(8):825–834. doi:10.1136/thx.49.8.825.
- Billatos E, Faiz A, Gesthalter Y, et al. Impact of acute exposure to cigarette smoke on airway gene expression. Physiol Genomics. 2018;50(9):705–713. doi:10.1152/physiolgenomics.00092.2017.
- Ponce-Gallegos MA, Ramírez-Venegas A, Falfán-Valencia R. Th17 profile in COPD exacerbations. COPD. 2017;12:1857–1865. doi:10.2147/COPD.S136592.
- Zinellu E, Zinellu A, Fois AG, et al. Circulating biomarkers of oxidative stress in chronic obstructive pulmonary disease: a systematic review. Respir Res. 2016;17(1):17. doi:10.1186/s12931-016-0471-z.
- Lushchak VI. Free radicals, reactive oxygen species, oxidative stresses and their classifications. UkrBiochemJ. 2015;87(6):11–18. doi:10.15407/ubj87.06.011.
- Lee IT, Yang CM. Role of NADPH oxidase/ROS in pro-inflammatory mediators-induced airway and pulmonary diseases. Biochem Pharmacol. 2012;84(5):581–590. doi:10.1016/j.bcp.2012.05.005.
- Chiş IC, Mureşan A, Oros A, et al. Protective effects of Quercetin and chronic moderate exercise (training) against oxidative stress in the liver tissue of streptozotocin-induced diabetic rats. Physiol Int. 2016;103(1):49–64. doi:10.1556/036.103.2016.1.5.
- Birben E, Sahiner UM, Sackesen C, et al. Oxidative stress and antioxidant defense. World Allergy Organ J. 2012;5(1):9–19.
- Kashyap D, Garg VK, Tuli HS, et al. Fisetin and quercetin: promising flavonoids with chemopreventive potential. Biomolecules. 2019;9(5):174. doi:10.3390/biom9050174.
- Farazuddin M, Mishra R, Jing Y, et al. Quercetin prevents rhinovirus-induced progression of lung disease in mice with COPD phenotype. PLoS One. 2018;13(7):e0199612. doi:10.1371/journal.pone.0199612.
- Li N, Li Q, Zhou XD, et al. The effect of quercetin on human neutrophil elastase-induced mucin5AC expression in human airway epithelial cells. Int Immunopharmacol. 2012;14(2):195–201. doi:10.1016/j.intimp.2012.07.008.
- Ganesan S, Faris AN, Comstock AT, et al. Quercetin prevents progression of disease in elastase/LPS-exposed mice by negatively regulating MMP expression. Respir Res. 2010;11(1):131. doi:10.1186/1465-9921-11-131.
- Li Y, Yao J, Han C, et al. Quercetin, inflammation and immunity. Nutrients. 2016;8(3):167. doi:10.3390/nu8030167.
- Veith C, Drent M, Bast A, et al. The disturbed redox-balance in pulmonary fibrosis is modulated by the plant flavonoid quercetin. Toxicol Appl Pharmacol. 2017;336:40–48. doi:10.1016/j.taap.2017.10.001.
- Palozza P, Simone R, Catalano A, et al. Lycopene prevention of oxysterol-induced proinflammatory cytokine cascade in human macrophages: inhibition of NF-kappaB nuclear binding and increase in PPARgamma expression. J Nutr Biochem. 2011;22(3):259–268. doi:10.1016/j.jnutbio.2010.02.003.
- Ahmed SA, Gogal RM, Jr., Walsh JE. A new rapid and simple non-radioactive assay to monitor and determine the proliferation of lymphocytes: an alternative to [3H]thymidine incorporation assay. J Immunol Methods. 1994;170(2):211–224. doi:10.1016/0022-1759(94)90396-4.
- Xiao J, Zhang Y, Wang J, et al. Monitoring of cell viability and proliferation in hydrogel-encapsulated system by resazurin assay. Appl Biochem Biotechnol. 2010;162(7):1996–2007. doi:10.1007/s12010-010-8975-3.
- Wang S, Holyoak GR, Panter KE, et al. Resazurin reduction assay for ram sperm metabolic activity measured by spectrophotometry. Proc Soc Exp Biol Med. 1998;217(2):197–202. doi:10.3181/00379727-217-44223.
- Green LC, Wagner DA, Glogowski J, et al. Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal Biochem. 1982;126(1):131–138. doi:10.1016/0003-2697(82)90118-X.
- Valenca SS, da Hora K, Castro P, et al. Emphysema and metalloelastase expression in mouse lung induced by cigarette smoke. Toxicol Pathol. 2004;32(3):351–356. doi:10.1080/01926230490431466.
- Pena K, Ramos C, Soares N, et al. The administration of a high refined carbohydrate diet promoted an increase in pulmonary inflammation and oxidative stress in mice exposed to cigarette smoke. COPD. 2016;11:3207–3217. doi:10.2147/COPD.S119485.
- Marklund S, Marklund G. Involvement of the superoxide anion radical in the autoxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur J Biochem. 1974;47(3):469–474. doi:10.1111/j.1432-1033.1974.tb03714.x.
- Aebi H. Catalase in vitro. Methods Enzymol. 1984;105:121–126.
- Griffith OW. Determination of glutathione and glutathione disulfide using glutathione reductase and 2-vinylpyridine. Anal Biochem. 1980;106(1):207–212. doi:10.1016/0003-2697(80)90139-6.
- Buege JA, Aust SD. Microsomal lipid peroxidation. Methods Enzymol. 1978;52:302–310.
- Levine RL, Williams JA, Stadtman ER, et al. Carbonyl assays for determination of oxidatively modified proteins. Methods Enzymol. 1994;233:346–357.
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72(1-2):248–254. doi:10.1016/0003-2697(76)90527-3.
- Campos KK, Manso RG, Goncalves EG, et al. Temporal analysis of oxidative effects on the pulmonary inflammatory response in mice exposed to cigarette smoke. Cell Immunol. 2013;284(1-2):29–36. doi:10.1016/j.cellimm.2013.07.002.
- de Araújo GR, Rabelo ACS, Meira JS, et al. Baccharis trimera inhibits reactive oxygen species production through PKC and down-regulation p47phox phosphorylation of NADPH oxidase in SK Hep-1 cells. Exp Biol Med (Maywood)). 2017;242(3):333–343. doi:10.1177/1535370216672749.
- Schuliga M. NF-kappaB signaling in chronic inflammatory airway disease. Biomolecules. 2015;5(3):1266–1283. doi:10.3390/biom5031266.
- Phipps JC, Aronoff DM, Curtis JL, et al. Cigarette smoke exposure impairs pulmonary bacterial clearance and alveolar macrophage complement-mediated phagocytosis of streptococcus pneumoniae▿. Infect Immun. 2010;78(3):1214–1220. doi:10.1128/IAI.00963-09.
- Murugan V, Peck MJ. Signal transduction pathways linking the activation of alveolar macrophages with the recruitment of neutrophils to lungs in chronic obstructive pulmonary disease. Exp Lung Res. 2009;35(6):439–485. doi:10.1080/01902140902759290.
- Barnes PJ. Alveolar macrophages in chronic obstructive pulmonary disease (COPD). Cell Mol Biol (Noisy-le-Grand). 2004;50.
- Thomassen MJ, Kavuru MS. Human alveolar macrophages and monocytes as a source and target for nitric oxide. Int Immunopharmacol. 2001;1(8):1479–1490. doi:10.1016/S1567-5769(01)00092-3.
- Kasmi S, Bkhairia I, Harrabi B, et al. Modulatory effects of quercetin on liver histopathological, biochemical, hematological, oxidative stress and DNA alterations in rats exposed to graded doses of score 250. Toxicol Mech Methods. 2017;25:1–11.
- Gavrilovska-Brzanov A, Shosholcheva M, Kuzmanovska B, et al. The influence of smoking on the variations in carboxyhemoglobin and methemoglobin during urologic surgery. Med Arch. 2017;71(3):178–182. doi:10.5455/medarh.2017.71.178-182.
- Zeng Q, Shen LJ, Li S, et al. The effects of hemoglobin levels and their interactions with cigarette smoking on survival in nasopharyngeal carcinoma patients. Cancer Med. 2016;5(5):816–826. doi:10.1002/cam4.647.
- Heunks LM, Vina J, van Herwaarden CL, et al. Xanthine oxidase is involved in exercise-induced oxidative stress in chronic obstructive pulmonary disease. Am J Physiol. 1999;277(6):R1697–704. doi:10.1152/ajpregu.1999.277.6.R1697.
- Kayyali US, Budhiraja R, Pennella CM, et al. Upregulation of xanthine oxidase by tobacco smoke condensate in pulmonary endothelial cells. Toxicol Appl Pharmacol. 2003;188(1):59–68. doi:10.1016/S0041-008X(02)00076-5.
- Ramos CO, Campos KKD, Costa GP, et al. Taurine treatment decreases inflammation and oxidative stress in lungs of adult mice exposed to cigarette smoke. Regul Toxicol Pharmacol. 2018;98:50–57. doi:10.1016/j.yrtph.2018.07.008.
- Rahman I, Adcock IM. Oxidative stress and redox regulation of lung inflammation in COPD. Eur Respir J. 2006;28(1):219–242. doi:10.1183/09031936.06.00053805.
- Lee SU, Ryu HW, Lee S, et al. Lignans Isolated From Flower Buds of Magnolia fargesii Attenuate Airway Inflammation Induced by Cigarette Smoke in vitro and in vivo. Front Pharmacol. 2018;9:970.
- D’Andrea G. Quercetin: a flavonol with multifaceted therapeutic applications? Fitoterapia. 2015;106:256–271.
- Chang KH, Park JM, Lee CH, et al. NADPH oxidase (NOX) 1 mediates cigarette smoke-induced superoxide generation in rat vascular smooth muscle cells. Toxicol in Vitro. 2017;38:49–58. doi:10.1016/j.tiv.2016.10.013.
- Jiang X, Chen C, Liu Y, et al. Critical role of cellular glutathione homeostasis for trivalent inorganic arsenite-induced oxidative damage in human bronchial epithelial cells. Mutat Res Genet Toxicol Environ Mutagen. 2014;770:35–45. doi:10.1016/j.mrgentox.2014.04.016.
- Monostori P, Wittmann G, Karg E, et al. Determination of glutathione and glutathione disulfide in biological samples: an in-depth review. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877(28):3331–3346. doi:10.1016/j.jchromb.2009.06.016.
- Ballatori N, Krance SM, Notenboom S, et al. Glutathione dysregulation and the etiology and progression of human diseases. Biol Chem. 2009;390(3):191–214.
- Cantin AM, North SL, Hubbard RC, et al. Normal alveolar epithelial lining fluid contains high levels of glutathione. J Appl Physiol. 1987;63(1):152–157. doi:10.1152/jappl.1987.63.1.152.
- Kosinova P, Berka K, Wykes M, et al. Positioning of antioxidant quercetin and its metabolites in lipid bilayer membranes: implication for their lipid-peroxidation inhibition. J Phys Chem B. 2012;16(4):1309–1318. doi:10.1021/jp208731g.
- Dekhuijzen P, van Beurden W. The role for N-acetylcysteine in the management of COPD. Int J Chron Obstruct Pulmon Dis. 2006;1(2):99–106. doi:10.2147/copd.2006.1.2.99.
- Posso SV, Quesnot N, Moraes JA, et al. AT-RVD1 repairs mouse lung after cigarette smoke-induced emphysema via downregulation of oxidative stress by NRF2/KEAP1 pathway. Int Immunopharmacol. 2018;56:330–338. doi:10.1016/j.intimp.2018.01.045.
- Machado DF, Campos KKD, da Silva NP, et al. The administration of surfactant decreased oxidative stress in lungs of mice exposed to cigarette smoke. Int Immunopharmacol. 2018;54:275–279. doi:10.1016/j.intimp.2017.11.023.
- Cai X, Fang Z, Dou J, et al. Bioavailability of quercetin: problems and promises. CMC. 2013;20(20):2572–2582. doi:10.2174/09298673113209990120.
- Guo Y, Bruno RS. Endogenous and exogenous mediators of quercetin bioavailability. J Nutr Biochem. 2015;26(3):201–210. doi:10.1016/j.jnutbio.2014.10.008.
- Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008;22(3):659–661. doi:10.1096/fj.07-9574LSF.
- Guc M, Schroeder G. Application of molecularly imprinted polymers (MIP) and magnetic molecularly imprinted polymers (mag-MIP) to selective analysis of quercetin in flowing atmospheric-pressure afterglow mass spectrometry (FAPA-MS) and in electrospray ionization mass spectrometry (ESI-MS). Molecules. 2019;24(13):2364. doi:10.3390/molecules24132364.