
Abstract
Chronic obstructive pulmonary disease (COPD) is a slowly progressive and poorly reversible airway obstruction disease. It is caused either alone or in combination of emphysema, chronic bronchitis (CB), and small airways disease. COPD is thought to be a multi-factorial disorder in which genetic susceptibility, environmental factors and tobacco exposure could be doubly or simultaneously implicated. Available medicines against COPD include anti-inflammatory drugs, such as β2-agonists and anticholinergics, which efficiently reduce airflow limitation but are unable to avert disease progression and mortality. Advanced glycation end products (AGE) and their receptors i.e. receptor for advanced glycation end products (RAGE) are some molecules that have been implicated in the complication of COPD. Several RAGE single nucleotide polymorphic (SNP) variants are produced by the mammalian cells. Based on the ethnicity some SNPs aggravate the COPD severity. Mammalian cells produce several alternative RAGE splice variants including a soluble RAGE (sRAGE) and an endogenous soluble RAGE (esRAGE). Both of these act as decoy receptor and thus may help to arrest the COPD complications. Several lines of evidences indicate a decreased level of sRAGE in the COPD subjects. One of the new strategies to reduce COPD complication may be sRAGE therapeutic administration to the COPD subjects. This comprehensive discussion sheds light on the role of RAGE and its polymorphic variants in the COPD complication along with sRAGE therapeutic significance in the COPD prevention.
Introduction
Chronic obstructive pulmonary disease (COPD) is characterized by the functional abnormality of airway obstruction caused either alone or in combination of emphysema, chronic bronchitis (CB) and small airways disease. COPD is a slowly progressive and poorly reversible disease with phenotypic heterogeneity [Citation1]. Based on the high-resolution computed tomography (HRCT) of the chest, emphysema is characterized as permanent expansion of air spaces distal to the lung terminal bronchioles and with time, sustained inflammation may lead to parenchymal destruction in affected regions [Citation2, Citation3]. However, bronchitis is characterized by inflammation of the bronchial tubes (the bronchi), the air passages that extend from trachea into the small airways and lung alveoli. CB is characterized by excessive tracheobronchial mucus generation which aggravates cough with expectoration for 3 or more months, continuing for minimal 2 years in succession. In CB, lung alveolar epithelium is the inflammation initiator as well as target [Citation4]. In clinical practice for COPD diagnosis and staging, the forced expiratory volume per second (FEV1) is measured but Vestbo and colleagues claimed its inadequacy for a wholesome characterization of established COPD sufferers [Citation5].
Chronic tobacco or cigarette smoking (CS) and environmental pollutant exposure along with specific genetic predisposition are the prominent COPD pathogenic initiators [Citation6–8]. CS induced aggravated oxidative and inflammatory stress result in macromolecular and structural alteration of lung tissues leading to tissue damage and inevitable destruction [Citation9–11]. However, in a large, well characterized and controlled COPD cohort (ECLIPSE), Agusti and associates claimed COPD clinical manifestations as highly variable with the relative extent of airflow limitation being rendered unable to incarcerate disease heterogeneity [Citation12]. While anti-COPD therapy undoubtedly reduces the complication, there is no established specific drug that completely normalizes a COPD patient. Thus, perhaps Mathers and colleagues rightfully predicted that by 2030, CS-related diseases would be accounting for ∼8.3 million global human deaths [Citation13].
A number of studies revealed that in COPD subjects, the level of advanced glycation end products (AGEs) and its receptor i.e. receptor for advanced glycation end products (RAGE) is much higher in lung tissues compared to smokers without airflow limitation. This expression extent was subsequently observed instrumental in deteriorating lung functioning characterized by altered lung parenchymal variations [Citation14–17]. Of note, RAGE is a cell surface pattern recognition receptor of immunoglobulin superfamily molecule [Citation18, Citation19]. In normal physiological conditions, RAGE is maximally expressed within the lung tissues, particularly basolateral face of alveolar type I (AT-1) epithelial cells, the squamous cells involved in alveoli and blood gaseous exchange wherein RAGE plays a key role in lung development [Citation20–24]. Incidentally, CS inhalation elevates the RAGE expression in lung epithelial cells [Citation25]. The other major ligands of multiligand RAGE molecule are high mobility group box protein 1 (HMGB1) and S100/calgranulins, the molecules involved in inflammatory reactions [Citation26, Citation27]. Further, separate studies by Repapi and Hancock concluded that a locus on chromosome 6p21, housing RAGE (AGER) gene is associated with lung function (forced expository volume 1 or FEV1) [Citation28, Citation29]. However, this association was noticed in nonsmokers. Interactions of AGE with RAGE lead to enhanced level of oxidative stress and inflammation which complicate the manifold diseased conditions including COPD [Citation30–41]. Genetic polymorphism of RAGE molecule performs a decisive role in RAGE dependent pathological alterations in COPD and several other disease conditions including COPD [Citation42].
RAGE is produced not only as a membrane bound protein but also in the soluble secretory form [Citation43–45]. Soluble RAGE is generated either as a splice variant of RAGE (AGER) gene [Citation45] referred as endogenous soluble RAGE (esRAGE) or via proteolysis of the membrane bound RAGE, called as sRAGE [Citation46, Citation47]. Interestingly, in a study Smith and coworkers screened COPD patients and 42 healthy control subjects, demonstrating a significant sRAGE and FEVA1 correlation. Based on their experimental findings, it was concluded that, lower the sRAGE plasma concentration, the greater is the extent of airflow obstruction [Citation48]. A number of other reports also confirmed that sRAGE levels are significantly lower in COPD sufferers than control subjects [Citation49–51]. Sukur and colleagues further confirmed that neutrophilic airway inflammation in asthma and COPD exhibit an inverse correlation with sRAGE expression [Citation52]. Additionally, sRAGE acts as a decoy receptor that binds to RAGE ligands and inhibits the signaling of cell surface or membrane receptor i.e. mRAGE [Citation53]. Thus, sRAGE may be used as a therapeutic molecule against various diseases including COPD, which are complicated by RAGE. The present review article describes the role of AGE and RAGE including various RAGE single nucleotide polymporhic variants (SNPs) in the COPD complication with the sRAGE anti-COPD efficacy.
Role of AGE and RAGE in COPD complication
Experimentally, when proteins (such as bovine serum albumin) or lipids are incubated at 37 °C with aldose sugars for a long duration (∼ 3-4 weeks), a non-enzymatic and irreversible glycation proceeds (through Maillard reaction), and generating molecules known as advanced glycation end products (AGE). Thus, in the mammalian body AGE levels increase on increasing blood glucose concentration [Citation54]. Physiologically blood is the most ideal location of a mammalian body for AGE generation [Citation55, Citation56]. Alternatively, AGEs can also form through a variety of other reactions, including oxidation of sugars, lipids, and amino acids, producing reactive aldehydes that eventually form AGEs. The most common of the multiple AGEs forms is N(6)-carboxymethyl lysine (AGE-CML) [Citation55]. After production AGE accumulates in various tissues including skin.
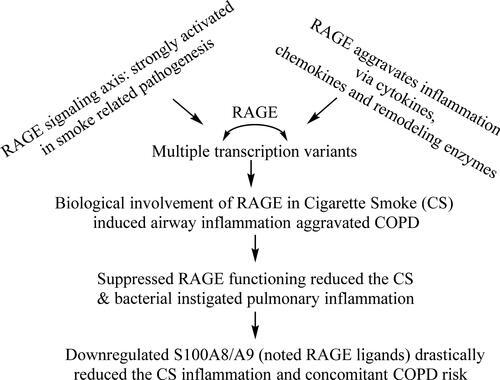
depicts the RAGE involvement in COPD pathogenesis on CS exposure and aggravated inflammation activities, wherein the ligands play contributory roles in aggravating the inflammation, even though all of these are not associated with enhanced expressions for a COPD manifestation. The RAGE involvement in pulsating pulmonary inflammation (PI) is supported by a PI reduction following RAGE knockdown. Several different AGEs receptors have been discovered, one but most prominent of which is the RAGE. This receptor was initially discovered from bovine pulmonary tissues by Schmidt and her associates [Citation18]. In normal physiological conditions, mammalian lung tissues express an elevated RAGE extent compared to others [Citation57] and physiologically RAGE may be involved in the lung development [Citation20, Citation24, Citation58]. Using experimental animal models such as RAGE knockout mice models, Wolf and associates have shown that RAGE regulates the alveolar epithelial cell differentiation, impacting the development and maintenance of lung tissue structure [Citation59]. However, in pathopysiological conditions supraphysiological RAGE expression in the lung tissues is involved in the complication of various diseased conditions including COPD [Citation23, Citation60].
Figure 1. Ligand signaling and inflammation mediated RAGE involvement in COPD pathogenesis.
A number of studies examined the AGE-RAGE axis implication in the COPD complication. In one such study Wu and colleagues determined the level of AGE and RAGE in the alveolar wall and small airways (lung parenchyma). In their experimental design, human subjects with COPD and controls (healthy individuals) were similar in age and smoking history. However, FEV1% prediction was lower for COPD subjects. Results of this study indicated that intensity of staining for AGEs was greater in the airways (pZ0.025) and alveolar walls (pZ0.004) in COPD subjects and FEV1% predicted was correlated with the intensity of staining for AGEs in the airways and alveoli. Additionally, intensity of staining for RAGE was also significantly increased in alveolar walls (pZ0.03) but not in the airways. Based on the increased staining of AGEs (ligand) and RAGE (receptor) in the COPD subjects, Wu and accomplices concluded that AGE-RAGE interaction may have a role in the COPD pathogenesis [Citation15]. In another study, Hoonhorst and associates detected enhanced AGE accumulation in the skin of COPD patients compared to healthy smoking and never-smoking controls [Citation61]. Further studies by the same group showed that in COPD, AGEs accumulate differentially in various body compartments, i.e. they accumulate in the skin, but not in plasma, sputum or bronchial biopsies [Citation16]. Additionally, Gopal and associates also detected an enhanced level of AGE precursor, i.e. N(6)-carboxymethyllysine (CML) levels in the plasma of COPD subjects compared to non-COPD controls [Citation62]. However, the above studies failed to establish whether AGE accumulation in various tissues of a COPD subject could be a COPD cause or consequence.
Besides AGE accumulation, a number of studies assessed the RAGE expression with smoking history and COPD. In one such attempt using animal model, Reynolds and associates showed an enhanced RAGE expression in pulmonary epithelium on being exposed to tobacco smoke. This study also demonstrates the significance of Egr-1, a transcription factor regulating RAGE expression during embryonic development. Additionally this study also predicted the Egr-1 and RAGE positive feedback involvement in cigarette smoke (CS)-related disease [Citation25]. Additional studies by Ferhani and associates revealed an enhanced RAGE and another of its ligands, HMGB1 expression in the bronchoalveolar lavage (BAL) of the COPD subjects with RAGE-HMGB1 interaction sustaining an inflammatory reaction [Citation26]. Furthermore, it has been shown that AGE-RAGE immuno-staining is enhanced in bronchial biopsies and lung parenchyma of COPD patients [Citation15]. In transgenic mouse models, RAGE was identified as essential in the alveolar morphogenesis during embryonic lung development with a conditional overexpression being implicated in the emphysema-like phenotype in adult mice [Citation58]. A number of similar studies on murine models with conditional overexpressed RAGE revealed aggravated emphysema with RAGE–/– mice being guarded against airspace enlargement (emphysema) subsequent to prolonged CS exposure [Citation59, Citation63, Citation64]. Based on these results on animal model experiments it can be concluded that increased RAGE is detrimental to lung physiology via promoting emphysema like conditions.
In this context, in a significant study Guerassimov and colleagues used NZWLac/J, C57BL6/J, A/J, SJ/L and AKR/J mice strains and exposed them to CS for 6 months. Analysis revealed only AKR mice as being sensitive toward CS induced inflammatory reactions needed for emphysemal manifestation. Based on the results, Guerassimov and accomplices further concluded that Lm, elastance and inflammation are the signatures needed for emphysemal phenotype in mice. Importantly, treatment of emphysema-susceptible AKR mice with the RAGE inhibitor, FPS-ZM125 also inhibited the airspace enlargement following CS exposure [Citation65], re-confirming the RAGE involvement in the development of emphysema like conditions. Since only AKR mice (and not other strains) developed emphysema like conditions following CS exposure, the findings infer certain strain specific genetic factors as responsible for emphysemal complications. Additionally, Niu and coworkers demonstrated RAGE gene as a promising candidate in Han Chinese population for genetically heterogeneous COPD and asthma [Citation66]. Thus, gaining a genetic understanding of RAGE aggravated COPD complication is highly essential.
Noted RAGE genetic polymorphism with COPD
COPD is a multifaceted disorder in which genetic susceptibility, environmental factors, and tobacco exposure may all be implicated [Citation67]. The RAGE gene prevails in chromosome 6 (6p21.3) and comprising 11 exons interlaced with 10 introns [Citation68]. Additionally, RAGE gene along with Pre-B-cell leukemia transcription factor 2 (PBX2) homeobox gene and a notch homolog are positioned near the human leukocyte antigen (HLA) locus [Citation69]. The polymorphic nature of RAGE gene is now well-characterized. Thus, several alternatively spliced RAGE variants have been discovered with protein coding and non-coding traits. While ensemble described 15 distinct RAGE transcript variants [Citation70], some other studies have reported up to 19 transcripts [Citation45, Citation71, Citation72]. The four major transcripts are well characterized by (NM_001136), comprising of 11 exons besides encoding a 404 amino acid protein, with 55 kDa molecular weight (cDNA: 1492 bp, DNA sequence: 4557 bp) [Citation45]. The (NM_001206929) as the longest isoform has 11 exons along with encoding a 420-amino acid protein. The next transcript is N terminus truncated RAGE (N-RAGE) having an initiation codon at exon 3 besides an in-frame stop codon in the intron sequence. So, it expresses a short protein of merely 303 amino acids (42 kDa) without the V-type immunoglobulin domain. This isoform though has an ability to be transported and localized in the plasma membrane, cannot bind any of the RAGE ligands and its biological functions are not well-understood [Citation45, Citation73]. The 3rd transcript is C-truncated, lacking the transmembrane and cytoplasmic domains of the full-length RAGE. This transcript is generally referred as endogenous soluble RAGE or esRAGE. The 4th transcript being encoded is known as soluble RAGE (sRAGE), formed by proteolytic cleavage of full-length RAGE, principally ADAM10 (a disintegrin and metalloproteinase domain-containing protein 10). Thus, sRAGE and esRAGE are two soluble C-truncated RAGE proteins, circulating in the blood and other biological fluids such as broncho-alveolar lavage (BAL). These proteins i.e. both esRAGE and sRAGE can bind AGEs as well as other RAGE ligands [Citation43, Citation44, Citation74] and act as endogenous competitive RAGE inhibitors. Devoid ligand engagement with the membrane bound RAGE (mRAGE) curtailed an initiation of various mRAGE driven metabolic pathways. Thus, esRAGE (46 kDa) and sRAGE (50 kDa) are decoy receptors [Citation53]. Of note, esRAGE serum levels are two- to five folds lower than sRAGE of healthy subjects, wherein sRAGE and esRAGE may not have same biomarker expressions [Citation43].
Besides multiple splice variants, several SNPs of RAGE gene have been identified. presents the major aspects of select RAGE gene polymorphisms, with most implications being ethnicity driven. However, rs2070600 (Gly82Ser) and rs2071288 (-6 G > A) are known for their comparatively significant involvement in COPD pathogenesis [Citation42]. As it is known, a SNP is characterized by single nucleotide alteration in various regions of RAGE gene. So, it is highly essential to understand a specific RAGE gene splice variant or SNP(s) being involved in the COPD development or complication. Of note, two separate genome-wide association studies in healthy individuals of European ancestry reported a significant association between RAGE gene rs2070600 and spirometry measures of airflow obstruction [Citation28, Citation75]. Apart from this, epidemiological studies too, point out toward the RAGE genetic variation being associated with the COPD risk. For instance, Li Y and colleague’s analysis of Chinese population found G82S SNP in the RAGE gene being associated with an increased COPD risk (). More specifically, smokers possessing G82S genotype of RAGE gene exhibit a comparatively greater risk for COPD development [Citation76].
Figure 2. Summary of -374 T/A, -429 T/C and G82S RAGE gene SNPs possible involvement in inflammation and COPD manifestation. The representations pinpoint the findings of 2014 study by Li Y and colleagues, wherein the depicted RAGE gene SNPs feature in top five of most noted ones.
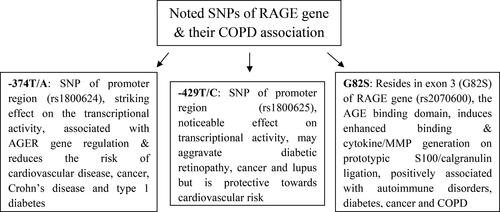
Table 1. Noted polymorphisms of RAGE gene (AGER) with their implicit SNP prospects, molecular significance and characteristic involvement in COPD pathogenesis.
In another recent attempt using Japanese population, the G82S polymorphism in the RAGE gene was found associated with combined pulmonary fibrosis and emphysema (CPFE) [Citation77]. Of note, this G82S SNP on exon 3 prevails as the ligand-binding site of the RAGE gene and operates as the N-linked glycosylation site [Citation42, Citation68]. This genetic variant also determines sRAGE extent and may act as a emphysema and COPD biomarker [Citation78, Citation79]. Inspection of UK natives also found the G82S RAGE variant to affect the serum sRAGE and lung functions in smokers [Citation80]. In a recent comprehensive review article, Dancer and colleagues have zeroed in on the various contradictory results pertaining to the RAGE involvement in the complication of various disorders that could vary as per the ethnicity dependent distinct RAGE gene SNPs frequencies [Citation42].
Mechanism of RAGE dependent COPD complication
RAGE complicates various diseased conditions including COPD through aggravating inflammation and oxidative stress. As early as in 2001, Schmidt and colleagues claimed that interactions of RAGE with its various ligands such as AGE has no causative association vis-à-vis diseased complications and rather, it intensifies the inflammatory responses mitigated in the activation of cellular events such as cellular dysfunction and tissue destruction [Citation34]. Stogsdill and team have shown that the embryonic overexpression of RAGE results in severe lung hypoplasia associated with apoptosis of alveolar epithelium, leading to an embryonic lethal phenotype [Citation81]. In a consecutive study, the same group has further shown that conditional overexpression of RAGE in the adult murine lung results in airspace enlargement and inflammation induction [Citation81, Citation82].
Likewise, a study by Waseda and colleagues on RAGE knockout (RAGE-/-) versus RAGE sufficient (RAGE+/+) mice observed RAGE as being critically involved in elastase-induced PI and emphysema. The investigators also noted that RAGE expressed on radio-resistant structural cells such as bronchial type 1 and type II epithelial cells and fibroblasts is essential for these PI outcomes [Citation63]. Several studies examined the effects of multiligand-RAGE binding with its various ligands such as S100/calgranulin family (S100A12 and S100B), amyloid β-fibrils, high-mobility group box 1 (HMGB1) and Mac-1. All attempts concluded that binding of RAGE with its various ligands stimulates the inflammatory stress [Citation83–85]. Further, Ferhani and associates showed that HMGB-1 forms complexes with IL-1β [Citation26], a proinflammatory cytokine involved in amplifying and prolonging the inflammatory and remodeling responses in the airways of smokers and COPD patients [Citation86]. On the same lines, Smith and colleagues assessed amyloid A (amyloid β-peptides) and S100A12 (S100/calgranulin) proteins in COPD sufferers. Results showed enhanced level of plasma amyloid A in patients with stable COPD compared to healthy control subjects, whereas plasma S100A12 extents in COPD patients and healthy controls were similar. In a similar study on CS administered mice models by Chen and associates, S100A8/A9 was found as a causative factor of RAGE induced airway inflammation. The investigators reached on this conclusion through RAGE knockdown driven amelioration of S100A8/A9 dependent airway inflammation [Citation41]. In a comprehensive review article, Provinciali and colleagues described various proinflammatory genes involved in the COPD complication [Citation87]. Thus, interaction of RAGE with its various ligands including AGE, HMGB1, S100 and amyloid β-peptides etc. exaggerates the inflammatory stress. Subsequent paragraphs discuss the various immunological cells whose activation by CS or other stimuli leads to increased RAGE expression which subsequently enhances immunological reactions through a vicious self-propagation cycle.
As early as in 1974 Niewoehner and associates observed pigmented alveolar macrophages (AMs) in the respiratory bronchioles of young smokers amidst their early phase of smoking. The accumulated AMs in these subjects persist through the manifested pulmonary disease [Citation88]. Subsequently, in two separate studies Shapiro and Tetley claimed that AMs play a decisive role in the COPD genesis [Citation89, Citation90]. AMs are tasked with defending the alveolar compartment from incursions by the external environment molecules such as pathogens or CS lung damaging constituents. However, a large number of AMs accumulations within these alveolar compartments are anticipated as both initiator and perpetrator of lung injury in response to CS. The number of accumulated AMs is inversely correlated with alveolar parenchymal tissue density [Citation91]. Erstwhile of this, and the number of occupied airways by the innate (e.g. AMs, neutrophils etc) and acquired immunological cells (B cells, CD4 + T cells and CD8+ T cells) elevates with advancing COPD progression [Citation92]. In a study on mice models, Robinson and associates noticed that smoking increases RAGE expression by AMs leading to aggravated lung inflammation [Citation36].
In their study on adult mice models, Stogsdill and colleagues noticed that conditional RAGE overexpression in alveolar epithelium (lungs) caused airspace enlargement associated with intensified apoptosis of alveolar epithelial cells along with AMs and neutrophils accumulation [Citation82]. The investigators showed that mice overexpressing RAGE in the lung tissues exhibited increased expression of macrophage inflammatory protein-2 (MIP-2) and interferon gamma (IFN-γ) in the BAL, besides increased matrix metalloproteinase-9 (MMP-9) expression. Their results further claimed that RAGE overexpression spontaneously induces PI and alveolar destabilization independent of CS exposure. In another similar experiment, Sanders and associates exposed wild type and RAGE-/- mice with or without tobacco-smoke either for 7 days or 4 months. Following smoking exposure, the isolated AMs from these mice were utilized to monitor the expression pattern of genes involved in oxidative stress, endoplasmic reticulum (ER) stress and cytokine actions. The results of this study claimed that RAGE expressed by AMs is a vital mediator of tobacco-smoke dependent oxidative stress. The authors further claimed that their findings provided a novel insight to the central importance of RAGE, the source of oxidative stress, AMs activation and the lung diseases pathogenesis due to tobacco-smoke exposure, suggesting manifold therapeutic options herein [Citation35]. summarizes the involvement of RAGE gene expression in COPD, vis-à-vis early smokers, adult mice models through a concurrent involvement of neutrophilic recruitment and functioning.
Figure 3. Implication of RAGE in COPD pathogenesis pinpointed distinctly in young smokers, adult mice and mitigated via neutrophilic inflammation.
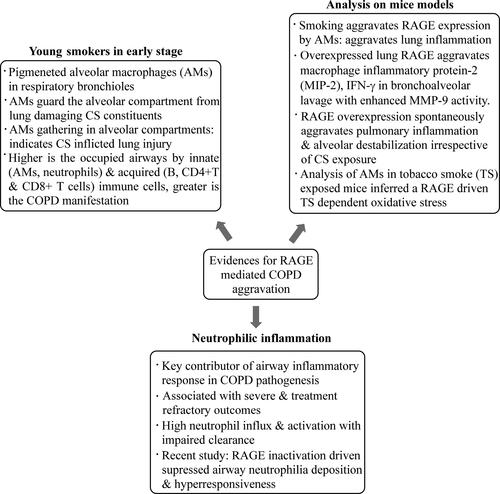
In addition, studies using mouse models have demonstrated a role for RAGE in several pathological processes associated with COPD, particularly airway neutrophilia. Neutrophilic inflammation is an important component of the airway inflammatory response amidst COPD pathogenesis [Citation49] wherein, it is implicated in severe and treatment-refractory outcomes. The mechanisms underlying airway neutrophilia are not well understood but involve persistent neutrophil influx and activation, together with an impaired neutrophil clearance [Citation39]. In a very recent study, it has been shown that inhibition of RAGE but not toll like receptor 4 (TLR4) (both are pattern recognition receptors) signaling may protect against airway neutrophilia and hyperresponsiveness during COPD progression [Citation93].
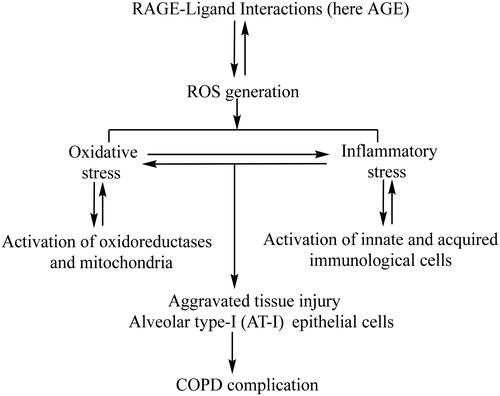
Regarding the mechanism of RAGE dependent complication of various diseases, one study by our group has shown that the proinflammatory cytokine TNF-α not only enhances the RAGE expression through increased generation of reactive oxygen species (ROS) and subsequent oxidative stress. It was noticed that once expressed, RAGE further stimulates the aggravated ROS generation by NADPH oxidase, a redox sensitive oxidoreductase and mitochondria (a cell organelle). So, RAGE and ROS cross-talk with each other through a vicious self-propagation cycle [Citation33]. In another study by our group, the AGE-RAGE interaction was observed to aggravate the inflammatory stress through activating NF-ĸB, a proinflammatory transcription factor [Citation94]. pinpoints the AGE-RAGE interaction mediated enhanced oxidative and inflammatory stress as the factors leading to COPD occurrence.
Figure 4. Alveolar type I (AT-I) epithelial cells are predominant RAGE expressing in the lungs. The interaction of RAGE with AGE/other ligands generates reactive oxygen species from oxidoreductases and mitochondria which accumulates the troubling oxidative and inflammatory stress. Enhanced oxidative and inflammatory stress causes tissue injury (AT-I cells) and COPD complications.
Besides NF-κB, Egr-1, a zinc-finger containing transcription factor, reportedly gets activated subsequent to tobacco smoke-induced RAGE expression [Citation25]. Erstwhile of the above, the Ras-raf-MAPK pathway assumes a decisive role for cell-cycle regulation [Citation95, Citation96]. Further, in a similar study on smoking induced mice models, Reynolds and associates showed that RAGE-Ras-NF-κB axis likely contributes to inflammation associated with several smoking-related lung diseases [Citation97]. Besides the transcription factors NF-ĸB and Egr-1, another transcription factor Nrf2 is also involved in guarding RAGE mediated lung epithelial cells integrity. Inhibition of RAGE attenuates CS-induced lung epithelial cell damage via RAGE-mediated Nrf2/DAMP signaling [Citation98]. In this reference, previously through a separate publication Schmidt and associates have shown that RAGE induces the expression of vascular cell adhesion molecule 1 (VCAM-1), a cell surface proinflammatory and prooxidative adhesion molecule, prevailing on endothelial cell surface and thereby causing vasculopathy [Citation99]. Further, in trauma-induced leukocyte adhesion observed in cremaster muscle venules, the ligand-RAGE axis, the sRAGE and intercellular adhesion molecule 1 (ICAM-1), another cell surface proinflammatory and prooxidative adhesion molecule affects the neutrophilic inflammation [Citation38, Citation100]. Henceforth, Sukkar and associates confirmed that neutrophilic airway inflammation in asthma and COPD is associated with reduced sRAGE expression [Citation52]. In this context, it is essential to understand the sRAGE significance vis-à-vis COPD complication.
Role of soluble RAGE in COPD complication
Importantly, as mentioned in the involvement of RAGE genetic polymorphic variants in COPD complication, that besides membrane bound RAGE (mRAGE), C-(carboxy) terminus truncated RAGE prevails in the circulating blood and other body fluids such as BAL. The circulating RAGEs are designated either as soluble RAGE (sRAGE, 50 kDa) or endogenous soluble RAGE (esRAGE, 46 kDa). While, alternative splicing of the RAGE (AGER) gene leads to esRAGE formation [Citation45], sRAGE is generated via proteolytic cleavage of RAGE extracellular domain [Citation46, Citation47]. The process is regulated by metalloproteinase driven mRAGE electrodomain shedding, wherein activities of MMP-9 and A disintegrin and metalloprotease (ADAM) 10 are prominent [Citation47, Citation101]. However, the extent to which proteolysis contributes to the circulating sRAGE pool is unclear [Citation47, Citation102]. In this reference, Hudson and colleagues found approximately 80% of lung RAGE prevailing as membrane-associated receptor or mRAGE, while about 7% occurs as esRAGE [Citation45] and the rest (∼13%) existing as sRAGE. Thus, in healthy subjects, esRAGE serum levels are two-to-five folds lower than sRAGE, resulting in distinct biomarker values for sRAGE and esRAGE [Citation43].
A prevailing hypothesis is that when sRAGE levels are sufficiently adequate to out-compete proinflammatory membrane bound RAGE for AGE binding, the possibility of inflammatory outcomes including COPD decreases. This hypothesis is based on multiple investigations including a crucial one by Smith and accomplices who used 61 COPD patients, 42 healthy control subjects to demonstrate that plasma sRAGE is reduced in patients with stable COPD relative to healthy controls. Levels of plasma sRAGE correlated inversely with the airflow obstruction severity, as measured by FEV1. The study further concluded that the inverse relation of mRAGE (high) and sRAGE (low) expressions in the circulation is important with respect to understanding the RAGE-COPD severity association [Citation48]. sRAGE seems useful for identifying frequent exacerbating COPD phenotypes. This parameter may also be used in the estimation of time needed for exacerbation in COPD (ECOPD) [Citation103]. Another study by Miniati and associates claimed significantly lower sRAGE expression in COPD patients than similar aged and sex-matched individuals not exhibiting any airflow obstruction. In this study, emphysema and chronic pulmonary diseases have been claimed as independent predictors of reduced sRAGE expression in COPD subjects [Citation51]. Another attempt by Gopal P and colleagues claimed an association of plasma sRAGE but not esRAGE with lung function impairment in COPD [Citation62]. In continuation of a similar line of studies, a number of investigations confirmed that sRAGE is a strong emphysema marker. Thus, based on the results of these investigations it is conclusively claimed that sRAGE can act as a decoy receptor via neutralization and clearance of circulating AGEs as well as by preventing AGEs to mRAGE ligation. This possible ‘protective’ mechanism may be reduced in COPD, as sRAGE levels are comparatively lower in COPD patients than in non-COPD controls [Citation50, Citation78, Citation104].
Monitoring an age dependent COPD pathogenesis controlled by RAGE and sRAGE expressions, Hoonhorst and accomplices analyzed young (18-40), old (40-75) smokers and never-smokers along with mild-to very severe COPD patients and measured the AGE, RAGE and sRAGE accumulations in various compartments (tissues) of their body. Based on the findings, the investigators concluded that the protective mechanism of sRAGE as a decoy-receptor is impaired in COPD subjects due to low sRAGE level in bronchial samples of these patients [Citation16]. Further cross-sectional studies revealed a consistent association between sRAGE and other aspects of COPD. For example, separate studies led by Cockayne and Iwamoto tracked the systemic sRAGE levels with smoking and/or COPD disease status and observed that plasma sRAGE levels in COPD as well as overlapping COPD and asthma patients were lower than asthmatic patients and healthy controls. Likewise, in all participants, lower sRAGE levels were associated with predicted FEV1/FVC decline, monitored over 4 years [Citation49, Citation104]. It is known that certain SNPs of RAGE gene are associated with lower levels of plasma sRAGE in both healthy and Alzheimer’s disease states [Citation105]. In light of the evidence that RAGE is a genetic determinant of pulmonary function (FEV1/FVC) [Citation28], it is important to ascertain whether genetic factors are predictors of sRAGE deficiency and neutrophil-dominated inflammation in asthma and COPD. In this context, Cheng and colleagues examined the relationship between RAGE SNPs and specific clinical COPD characteristics but could not come-up with any consensus [Citation78]. However, in 2016, another study by Miller and colleagues elucidated that T allele of rs2070600 (codes for the RAGE-Ser82 variant), is associated with increased lung function in a cohort of UK smokers. Further, the relative sRAGE extents were lower in the serum of individual smokers carrying this variant. These observations provide a novel insight toward the RAGE SNP involvement in lung biology besides supporting a functional role for rs2070600 in determining sRAGE levels, which may in turn explain the association between this variant and the related functions thereof [Citation80]. More experiments on COPD subjects with or without smoking history as well as this RAGE SNP are necessary to completely understand this SNP role in COPD complications and also whether sRAGE therapeutic administration can reduce the COPD complications including emphysema, bronchitis or CB and small airway diseases.
Conclusion and future perspectives
Available medicaments against COPD such as (anti-inflammatory drugs, β2-agonists and anticholinergics) efficiently reduce airflow limitation, but are unable to prevent disease progression and mortality [Citation106]. The COPD implications are compounded by the absence of novel drug targets in the area of pulmonary medicine, substantially resulting from inadequate knowledge pertaining to disease progression, particularly from slow to the advanced stage [Citation107, Citation108]. Although it is observed that CS induced lung complications in humans are similar to those in mice models [Citation109], the disease modeling has proven difficult since no single in vivo COPD model entirely captures the breadth of symptoms experienced by patients, particularly in the later stages [Citation110–113]. In this scenario it is important to survey the current COPD research landscape to promote successful models for understanding the disease pathology and developing more effective treatment options. So, exploring other advanced therapeutic options have become a present day necessity. In a recent study by Harrell CR and accomplices, the effects of “Exosome-derived Multiple Allogeneic Protein Paracrine Signaling (Exo-d-MAPPS)” in the attenuation of CS induced chronic airway inflammation in animal model as well as human COPD subjects were analyzed. Based on results, the scientists associated have claimed that Exo-d-MAPPS could be considered as a potentially new therapeutic agent for treating chronic inflammatory lung diseases including COPD though its efficacy needs validation via large clinical trials [Citation114].
One of the new strategies to reduce the COPD complications may be to use sRAGE as therapeutic agent. This review article is aimed at providing a comprehensive discussion for a possible implication of AGE, RAGE and their polymorphic variants in the COPD complication alongwith exploring sRAGE therapeutic evaluation against COPD complication. It is now well understood that RAGE expression aggravates in CS induced respiratory complications including COPD. Thus, RAGE could be a promising risk prediction and prognostic source in many pathological conditions including COPD. It is also well understood that sRAGE which acts as a decoy receptor, is diminished in COPD subjects and its therapeutic administration could guard against COPD complications. At present, patient specific survey vis-à-vis specific RAGE polymorphic variants is highly essential and whether a particular group of COPD patients or an individual patient having a characteristic RAGE polymorphic variant is sensitive to sRAGE therapy or not. Further, based on the individualized data, large scale sRAGE clinical trial against smoking and never smoking platforms, using various age and gender groups, both with and without COPD complications is highly essential to assess the real impact of sRAGE therapy in reducing the COPD complications.
Declaration of interest
No potential conflict of interest was reported by the authors.
Funding
The author(s) reported there is no funding associated with the work featured in this article.
References
- Pauwels RA, Buist AS, Calverley PM, et al. GOLD scientffic committee, global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO global initiative for chronic obstructive lung disease (GOLD) Workshop summary. Am J Respir Crit Care Med. 2001;163(5):1256–1276. DOI:https://doi.org/10.1164/ajrccm.163.5.2101039
- Jelic TM. Emphysema [Online First], IntechOpen, 2019. DOI:https://doi.org/10.5772/intechopen.83273 Available from: https://www.intechopen.com/online-first/emphysema.
- Saetta M, Turato G, Maestrelli P, et al. Cellular and structural bases of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2001;163(6):1304–1309. DOI:https://doi.org/10.1164/ajrccm.163.6.2009116
- Wilson R, Rayner CF. Bronchitis. Curr Opin Pulm Med. 1995;1(3):177–182.
- Vestbo J, Anderson W, Coxson HO, ECLIPSE investigators, et al. on behalf of the ECLIPSE investigators: Evaluation of COPD longitudinally to identify predictive surrogate end-points (ECLIPSE). Eur Respir J. 2008;31(4):869–873. DOI:https://doi.org/10.1183/09031936.00111707
- Saetta M, Di Stefano A, Turato G, et al. CD8+ T-lymphocytes in peripheral airways of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998;157(3 Pt 1):822–826. DOI:https://doi.org/10.1164/ajrccm.157.3.9709027
- Barnes PJ, Shapiro SD, Pauwels RA. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J. 2003;22(4):672–688.
- Mayer AS, Newman LS. Genetic and environmental modulation of chronic obstructive pulmonary disease. Respir Physiol. 2001;128(1):3–11. DOI:https://doi.org/10.1016/s0034-5687(01)00258-4
- Vestbo J, Prescott E, Almdal T, et al. Body mass, fat-free body mass, and prognosis in patients with chronic obstructive pulmonary disease from a random population sample: findings from the Copenhagen City Heart Study. Am J Respir Crit Care Med. 2006;173(1):79–83. DOI:https://doi.org/10.1164/rccm.200506-969OC
- Tuder RM, Petrache I. Pathogenesis of chronic obstructive pulmonary disease. J Clin Invest. 2012;122(8):2749–2755. DOI:https://doi.org/10.1172/JCI60324
- Agusti AG, Sauleda J, Miralles C, et al. Skeletal muscle apopotosis and wight loss in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2002;166(4):485–489. DOI:https://doi.org/10.1164/rccm.2108013
- Agusti AG, Calverley P, Celli B, et al. Characterization of COPD heterogeneity in the ECLIPSE cohort. Respir Res. 2010;11:122.
- Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3(11):e442. DOI:https://doi.org/10.1371/journal.pmed.0030442
- Cerami C, Founds H, Nicholl I, et al. Tobacco smoke is a source of toxic reactive glycation products. Proc Natl Acad Sci U S A. 1997;94(25):13915–13920. DOI:https://doi.org/10.1073/pnas.94.25.13915
- Wu L, Ma L, Nicholson LFB, et al. Advanced glycation end products and its receptor (RAGE) are increased in patients with COPD. Respir Med. 2011;105(3):329–336. DOI:https://doi.org/10.1016/j.rmed.2010.11.001
- Hoonhorst SJM, Pouwels SD, Faiz A, et al. Advanced glycation end products and their receptor in different body compartments in COPD. Resp Res. 2016;17(1):46.
- Zaigham S, Persson M, Jujic A, et al. Measures of lung function and their relationship with advanced glycation end-products. ERJ Open Res. 2020;6(2):00356–2019. DOI:https://doi.org/10.1183/23120541.00356-2019
- Schmidt AM, Vianna M, Gerlach M, et al. Isolation and characterization of two binding proteins for advanced glycosylation end products from bovine lung which are present on the endothelial cell surface. J Biol Chem. 1992;267(21):14987–14997.
- Neeper M, Schmidt A, Brett J, et al. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J Biol Chem. 1992;267(21):14998–15004.
- Demling N, Ehrhardt C, Kasper M, et al. Promotion of cell adherence and spreading: a novel function of RAGE, the highly selective differentiation marker of human alveolar epithelial type I cells. Cell Tissue Res. 2006;323(3):475–488. DOI:https://doi.org/10.1007/s00441-005-0069-0
- Shirasawa M, Fujiwara N, Hirabayashi S, et al. Receptor for advanced glycation end-products is a marker of type I lung alveolar cells. Genes Cells. 2004;9(2):165–174. DOI:https://doi.org/10.1111/j.1356-9597.2004.00712.x
- Fehrenbach H, Kasper M, Tsherinig T, et al. Receptor for advanced glycation end products (RAGE) exhibits highly differential cellular and Sub-cellular localisation in rat and human lung. Cell Mol Biol (Noisy-le-Grand). 1998;44(7):1147–1157.
- Mukherjee TK, Mukhopadhyay S, Hoidal JR. Implication of receptor for advanced glycation end product (RAGE) in pulmonary health and pathophysiology. Respir Physiol Neurobiol. 2008;162(3):210–215. DOI:https://doi.org/10.1016/j.resp.2008.07.001
- Al-Robaiy S, Weber B, Simm A, et al. The receptor for advanced glycation end-products supports lung tissue biomechanics. Am J Physiol Lung Cell Mol Physiol. 2013;305(7):L491–L500. DOI:https://doi.org/10.1152/ajplung.00090.2013
- Reynolds PR, Kasteler SD, Cosio MG, et al. RAGE: developmental expression and positive feedback regulation by Egr-1 during cigarette smoke exposure in pulmonary epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2008;294(6):L1094–L1101. DOI:https://doi.org/10.1152/ajplung.00318.2007
- Ferhani N, Letuve S, Kozhich A, et al. Expression of high-mobility group box 1 and of receptor for advanced glycation end products in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;181(9):917–927. DOI:https://doi.org/10.1164/rccm.200903-0340OC
- Schmidt AM, Yan SD, Yan SF, et al. The biology of the receptor for advanced glycation end products and its ligands. Biochim Biophys Acta. 2000;1498(2–3):99–111. DOI:https://doi.org/10.1016/S0167-4889(00)00087-2
- Repapi E, Sayers I, Wain LV, Wellcome Trust Case Control Consortium, and NSHD Respiratory Study Team, et al. Genome-wide association study identifies five loci associated with lung function. Nat Genet. 2010;42(1):36–44. DOI:https://doi.org/10.1038/ng.501
- Hancock DB, Eijgelsheim M, Wilk JB, et al. Meta-analyses of genome-wide association studies identify multiple loci associated with pulmonary function. Nat Genet. 2010;42(1):45–52. DOI:https://doi.org/10.1038/ng.500
- Ahmad S, Khan MY, Z. Rafi Z, et al. Oxidation, glycation and glycoxidation-The vicious cycle and lung cancer . Semin Cancer Biol. 2018;49:29–36. DOI:https://doi.org/10.1016/j.semcancer.2017.10.005
- Reynaert NL, Gopal P, Rutten EPA, et al. Advanced glycation end products and their receptor in age-related, non-communicable chronic inflammatory diseases; Overview of clinical evidence and potential contributions to disease . Int J Biochem Cell Biol. 2016;81(Pt B):403–418. DOI:https://doi.org/10.1016/j.biocel.2016.06.016
- Wautier MP, Chappey O, Corda S, et al. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. The am. Am J Physiol Endocrinol Metab. 2001;280(5):E685–E694. DOI:https://doi.org/10.1152/ajpendo.2001.280.5.E685
- Mukherjee TK, Mukhopadhyay S, Hoidal JR. The role of reactive oxygen species in TNFalpha-dependent expression of the receptor for advanced glycation end products in human umbilical vein endothelial cells. Biochim Biophys Acta. 2005;1744(2):213–223. DOI:https://doi.org/10.1016/j.bbamcr.2005.03.007
- Schmidt AM, Yan SD, Yan SF, et al. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest. 2001;108(7):949–955. DOI:https://doi.org/10.1172/JCI14002
- Sanders KA, Delker DA, Huecksteadt T, et al. RAGE is a critical mediator of pulmonary oxidative stress, alveolar macrophage activation and emphysema in response to cigarette smoke. Sci Rep. 2019;9(1):231. DOI:https://doi.org/10.1038/s41598-018-36163-z
- Robinson AB, Johnson KD, Bennion BG, et al. RAGE signaling by alveolar macrophages influences tobacco smoke-induced inflammation. Am J Physiol Lung Cell Mol Physiol. 2012;302(11):L1192–L1199. DOI:https://doi.org/10.1152/ajplung.00099.2012
- Iwashima Y, Eto M, Hata A, et al. Advanced glycation end products-induced gene expression of scavenger receptors in cultured human monocyte-derived macrophages. Biochem Biophys Res Commun. 2000;277(2):368–380.
- Frommhold D, Kamphues A, Hepper I, et al. RAGE and ICAM-1 cooperate in mediating leukocyte recruitment during acute inflammation in vivo. Blood. 2010;116(5):841–849. DOI:https://doi.org/10.1182/blood-2009-09-244293
- Simpson JL, S. Phipps S, P.G. Gibson PG. Inflammatory mechanisms and treatment of obstructive airway diseases with neutrophilic bronchitis. Pharmacol. & Therap. 2009;124(1):86–95. DOI:https://doi.org/10.1016/j.pharmthera.2009.06.004
- Hori O, Brett J, Slattery T, et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of RAGE and amphoterin in developing nervous system. J Biol Chem. 1995;270(43):25752–25761.
- Chen M, Wang T, Shen Y, et al. Knockout of RAGE ameliorates mainstream cigarette smoke-induced airway inflammation in mice. Int Immunopharmacol. 2017;50:230–235. DOI:https://doi.org/10.1016/j.intimp.2017.06.018
- Serveaux-Dancer M, Jabaudon M, Creveaux I, et al. Pathological implications of receptor for advanced glycation end-product (AGER) gene polymorphism. Dis Markers. 2019; 2019:2067353. DOI:https://doi.org/10.1155/2019/2067353
- Prasad K. Low levels of serum soluble receptors for advanced glycation end products, biomarkers for disease state: myth or reality. Int J Angiol. 2014;23(1):11–16. DOI:https://doi.org/10.1055/s-0033-1363423
- Tam XHL, Shiu SWM, Leng L, et al. Enhanced expression of receptor for advanced glycation end-products is associated with low circulating soluble isoforms of the receptor in type 2 diabetes. Clin Sci. 2011;120(2):81–89. DOI:https://doi.org/10.1042/CS20100256
- Hudson BI, Carter AM, Harja E, et al. Identification, classification, and expression of RAGE gene splice variants. Faseb J. 2008;22(5):1572–1580. DOI:https://doi.org/10.1096/fj.07-9909com
- Zhang L, Bukulin M, Kojro E, et al. Receptor for advanced glycation end products is subjected to protein ectodomain shedding by metalloproteinases. J Biol Chem. 2008;283(51):35507–35516. DOI:https://doi.org/10.1074/jbc.M806948200
- Raucci A, Cugusi S, Antonelli A, et al. A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10). Faseb J. 2008;22(10):3716–3727. DOI:https://doi.org/10.1096/fj.08-109033
- Smith DJ, Yerkovich ST, Towers MA, et al. Reduced soluble receptor for advanced glycation end-products in COPD. Eur Respir J. 2011;37(3):516–522. DOI:https://doi.org/10.1183/09031936.00029310
- Cockayne DA, Cheng DT, Waschki B, et al. Fine, systemic biomarkers of neutrophilic inflammation, tissue injury and repair in COPD patients with differing levels of disease severity. PLoS ONE. 2012;7(6):e38629. DOI:https://doi.org/10.1371/journal.pone.0038629
- Gopal P, Rutten EPA, Dentener MA, et al. Decreased plasma sRAGE levels in COPD: influence of oxygen therapy. Eur J Clin Invest. 2012;42(8):807–814. DOI:https://doi.org/10.1111/j.1365-2362.2012.02646.x
- Miniati M, Monti S, Basta G, et al. Soluble receptor for advanced glycation end products in COPD: relationship with emphysema and chronic cor pulmonale: a case-control study. Respiratory Res. 2011;12:37.
- Sukkar MB, Wood LG, Tooze M, et al. Wark, soluble RAGE is deficient in neutrophilic asthma and COPD. Eur Respir J. 2012;39(3):721–729.
- Vazzana N, Santilli F, Cuccurullo C, et al. Soluble forms of RAGE in internal medicine. Intern Emerg Med. 2009;4(5):389–401. DOI:https://doi.org/10.1007/s11739-009-0300-1
- Yan SF, Ramasamy R, Schmidt AM. Mechanisms of disease: advanced glycation end-products and their receptor in inflammation and diabetes complications. Nat Clin Pract Endocrinol Metab. 2008;4(5):285–293.
- Goldin JA, Beckman JA, Schmidt AM, et al. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation. 2006;114(6):597–605. DOI:https://doi.org/10.1161/CIRCULATIONAHA.106.621854
- Singh R, Barden A, T. Mori T, et al. Advanced glycation end-products: a review. Diabetologia. 2001;44(2):129–146. DOI:https://doi.org/10.1007/s001250051591
- Brett J, Schmidt AM, Yan SD, et al. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol. 1993;143(6):1699–1712.
- Winden DR, Ferguson NT, Bukey BR, et al. Conditional over-expression of RAGE by embryonic alveolar epithelium compromises the respiratory membrane and impairs endothelial cell differentiation. Respir Res. 2013;14:108. DOI:https://doi.org/10.1186/1465-9921-14-108
- Wolf L, Herr C, Niederstraßer J, et al. Receptor for advanced glycation endproducts (RAGE) maintains pulmonary structure and regulates the response to cigarette smoke. PLoS One. 2017;12(7):e0180092. DOI:https://doi.org/10.1371/journal.pone.0180092
- Khaket TP, Kang SC, Mukherjee TK. The potential of receptor for advanced glycation end products (RAGE) as a therapeutic target for lung associated diseases. Curr Drug Targets. 2019;20(6):679–689. DOI:https://doi.org/10.2174/1389450120666181120102159
- Hoonhorst SJM, A.T. Loi LT, Hartman JE, et al. The Hacken NHT. Advanced glycation end products in the skin are enhanced in COPD. Metabolism. 2014;63(9):1149–1156. DOI:https://doi.org/10.1016/j.metabol.2014.06.006
- Gopal P, Reynaert NL, Scheijen JLJM, et al. Plasma advanced glycation end products and skin autofluorescence are increased in COPD. Eur Respir J. 2014;43(2):430–438.
- Waseda K, Miyahara N, Taniguchi A, et al. Emphysema requires the receptor for advanced glycation end products triggering on structural cells. Am J Respir Cell Mol Biol. 2015;52(4):482–491.
- Sambamurthy AS, Leme TD, Oury SD, et al. The receptor for advanced glycation end products (RAGE) contributes to the progression of emphysema in mice. PLoS One. 2015;10(3):e0118979. DOI:https://doi.org/10.1371/journal.pone.0118979
- Guerassimov A, Hoshino Y, Takubo Y, et al. The development of emphysema in cigarette smoke-exposed mice is strain dependent. Am J Respir Crit Care Med. 2004;170(9):974–980. DOI:https://doi.org/10.1164/rccm.200309-1270OC
- Niu H, Niu W, Yu T, et al. Association of RAGE gene multiple variants with the risk for COPD and asthma in Northern Han Chinese. Aging (Albany NY). 2019;11(10):3220–3237. DOI:https://doi.org/10.18632/aging.101975
- Decramer M, Janssens W, Miravitlles M. Chronic obstructive pulmonary disease. Lancet. 2012;379(9823):1341–1351. DOI:https://doi.org/10.1016/S0140-6736(11)60968-9
- Vissing H, Aagaard L, Tommerup N, et al. Localization of the human gene for advanced glycosylation end product-specific receptor (AGER) to chromosome 6p21.3. Genomics. 1994;24(3):606–608. DOI:https://doi.org/10.1006/geno.1994.1676
- Sugaya K, Fukagawa T, Matsumoto K, et al. Three genes in the human MHC class III region near the junction with the class II: gene for receptor of advanced glycosylation end products, PBX2 homeobox gene and a notch homolog, human counterpart of mouse mammary tumor gene int-3. Genomics. 1994;23(2):408–419. DOI:https://doi.org/10.1006/geno.1994.1517
- Ensemble genome browser 91. 2018;948–957. Available from: https://www.ensembl.org/index.html.
- Sterenczak KA, Nolte I, Escobar HM. RAGE splicing variants in mammals. Methods Mol Biol. 2013;963:265–276.
- Sterenczak KA, Willenbrock S, Barann M, et al. Cloning, characterisation, and comparative quantitative expression analyses of receptor for advanced glycation end products (RAGE) transcript forms. Gene. 2009;434(1–2):35–42. DOI:https://doi.org/10.1016/j.gene.2008.10.027
- Yonekura H, Yamamoto Y, Sakurai S, et al. Novel splice variants of the receptor for advanced glycation end-products expressed in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury. Biochemical J. 2003;370(3):1097–1109. DOI:https://doi.org/10.1042/bj20021371
- Koyama H, Yamamoto H, Nishizawa Y. RAGE and soluble RAGE: potential therapeutic targets for cardiovascular diseases. Mol Med. 2007;13(11–12):625–635. DOI:https://doi.org/10.2119/2007-00087.Koyama
- GBD 2015 Chronic Respiratory Disease Collaborators. Chronic respiratory disease collaborators. Global, regional, and national deaths, prevalence, disability-adjusted life years, and years lived with disability for chronic obstructive pulmonary disease and asthma, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Respir Med. 2017;5:691–706.
- Li Y, Yang C, Ma G, et al. Association of polymorphisms of the receptor for advanced glycation end products gene with COPD in the Chinese population. DNA Cell Biol. 2014;33(4):251–258. DOI:https://doi.org/10.1089/dna.2013.2303
- Kinjo T, Kitaguchi Y, Droma Y, et al. T.he Gly82Ser mutation in AGER contributes to pathogenesis of pulmonary fibrosis in combined pulmonary fibrosis and emphysema (CPFE) in Japanese patients. Sci Rep. 2020;10(1):12811. DOI:https://doi.org/10.1038/s41598-020-69184-8
- Cheng DT, Kim DK, Cockayne DA, on behalf of the TESRA and ECLIPSE investigators, et al. Systemic soluble receptor for advanced glycation end products is a biomarker of emphysema and associated with ager genetic variants in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;188(8):948–957. DOI:https://doi.org/10.1164/rccm.201302-0247OC
- Jang Y, Kim JY, Kang SM, et al. Association of the Gly82Ser polymorphism in the receptor for advanced glycation end products (RAGE) gene with circulating levels of soluble RAGE and inflammatory markers in nondiabetic and nonobese Koreans. Metabolism. 2007;56(2):199–205. DOI:https://doi.org/10.1016/j.metabol.2006.09.013
- Miller S, Henry AP, Hodge E, et al. The Ser82 RAGE variant affects lung function and serum RAGE in smokers and sRAGE production in vitro. PLoS One. 2016;11(10):e0164041. DOI:https://doi.org/10.1371/journal.pone.0164041
- Stogsdill JA, Stogsdill MP, Porter JL, et al. Embryonic overexpression of receptors for advanced glycation end-products by alveolar epithelium induces an imbalance between proliferation and apoptosis. Am J Respir Cell Mol Biol. 2012;47(1):60–66.
- Stogsdill MP, Stogsdill JA, Bodine BG, et al. Conditional overexpression of receptors for advanced glycation endproducts in the adult murine lung causes airspace enlargement and induces inflammation. Am J Respir Cell Mol Biol. 2013;49(1):128–134.
- Hudson BI, Lippman ME. Targeting RAGE signaling in inflammatory diseases. Annu Rev Med. 2018;69:349–364.
- Buckley ST, Ehrhardt C. The receptor for advanced glycation end products (RAGE) and the lung. J Biomed Biotechnol. 2010;2010:917108. DOI:https://doi.org/10.1155/2010/917108
- Sasaki M, M. Kashima M, Ito T, et al. Differential regulation of metalloproteinase production, proliferation and chemotaxis of human lung fibroblasts by PDGF, interleukin-1beta and TNF-alpha. Mediators Inflamm. 2000;9(3-4):155–160. DOI:https://doi.org/10.1080/09629350020002895
- Harris HE, Andersson U. Mini-review: the nuclear protein HMGB1 as a proinflammatory mediator. Eur J Immunol. 2004;34(6):1503–1512.
- Provinciali M, Cardelli M, Marchegiani F. Inflammation, chronic obstructive pulmonary disease and aging. Curr Opin Pulm Med. 2011;17:S3–S10. DOI:https://doi.org/10.1097/01.mcp.0000410742.90463.1f
- Niewoehner DE, Kleinerman J, Rice DB. Pathologic changes in the peripheral airways of young cigarette smokers. N Engl J Med. 1974;291(15):755–758.
- Shapiro SD. The macrophage in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1999;160(5 Pt 2):S29–S32. DOI:https://doi.org/10.1164/ajrccm.160.supplement_1.9
- Tetley TD. Macrophages and the pathogenesis of COPD. Chest. 2002;121(5 Suppl):156S–159S. DOI:https://doi.org/10.1378/chest.121.5_suppl.156s
- Finkelstein R, Fraser RS, Ghezzo H, et al. Alveolar inflammation and its relation to emphysema in smokers. Am J Respir Crit Care Med. 1995;152(5):1666–1672. DOI:https://doi.org/10.1164/ajrccm.152.5.7582312
- Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(26):2645–2653. DOI:https://doi.org/10.1056/NEJMoa032158
- Allam VSRR, Faiz A, Lam M, et al. RAGE and TLR4 differentially regulate airway hyperresponsiveness: Implications for COPD. Allergy. 2021;76(4):1123–1135. DOI:https://doi.org/10.1111/all.14563
- Mukhopadhyay S, Mukherjee TK. Bridging advanced glycation end product, receptor for advanced glycation end product and nitric oxide with hormonal replacement/estrogen therapy in healthy versus diabetic postmenopausal women: a perspective. Biochim Biophys Acta. 2005;1745(2):145–155. DOI:https://doi.org/10.1016/j.bbamcr.2005.03.010
- Downward J. Cell cycle: routine role for Ras. Curr Biol. 1997;7(4):R258–R260. DOI:https://doi.org/10.1016/S0960-9822(06)00116-3
- Winston JT, Coats SR, Y.Z. Wang YZ, et al. Regulation of the cell cycle machinery by oncogenic Ras. Oncogene. 1996;12(1):127–134.
- Reynolds PR, Stogsdill JA, Stogsdill MP, et al. Up-regulation of receptors for advanced glycation end-products by alveolar epithelium influences cytodifferentiation and causes severe lung hypoplasia . Am J Respir Cell Mol Biol. 2011;45(6):1195–1202. DOI:https://doi.org/10.1165/rcmb.2011-0170OC
- Lee H, Lee J, Hong SH, et al. Inhibition of RAGE attenuates cigarette Smoke-Induced lung epithelial cell damage via RAGE-Mediated Nrf2/DAMP signaling. Front Pharmacol. 2018;9:684.
- Schmidt AM, Hori O, J.X. Chen JX, et al. Advanced glycation endproducts interacting with their endothelial receptor induce expression of vascular cell adhesion molecule-1 (VCAM-1) in cultured human endothelial cells and in mice. A potential mechanism for the accelerated vasculopathy of diabetes. J Clin Invest. 1995;96(3):1395–1403. DOI:https://doi.org/10.1172/JCI118175
- Wang Y, Wang H, Piper MG, et al. sRAGE induces human monocyte survival and differentiation. J Immunol. 2010;185(3):1822–1835. DOI:https://doi.org/10.4049/jimmunol.0903398
- Yamakawa N, Uchida T, Matthay MA, et al. Proteolytic release of the receptor for advanced glycation end products from in vitro and in situ alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2011;300(4):L516–L525. DOI:https://doi.org/10.1152/ajplung.00118.2010
- Yamamoto H, Watanabe T, Yamamoto Y, et al. RAGE in diabetic nephropathy. Curr Mol Med. 2007;7(8):752–757.
- Miłkowska-Dymanowska J, Białas AJ, Szewczyk K, et al. The usefulness of soluble receptor for advanced glycation end-products in the identification of COPD frequent exacerbator phenotype. Int J Chron Obstruct Pulmon Dis. 2018;13:3879–3884.
- Iwamoto H, Gao J, Pulkkinen V, et al. Soluble receptor for advanced glycation end-products and progression of airway disease. BMC Pulm Med. 2014;14:68.
- Li K, Dai D, Zhao B, et al. Association between the RAGE G82S polymorphism and Alzheimer’s disease. J Neural Transm (Vienna). 2010;117(1):97–104. DOI:https://doi.org/10.1007/s00702-009-0334-6
- Barnes PJ, Stockley RA. COPD: current therapeutic interventions and future approaches. Eur Resp J. 2005;25(6):1084–1106. DOI:https://doi.org/10.1183/09031936.05.00139104
- Gross NJ, Barnes PJ. New therapies for asthma and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2017;195(2):159–166. DOI:https://doi.org/10.1164/rccm.201610-2074PP
- Barnes PJ, Bonini S, Seeger W, et al. Barriers to new drug development in respiratory disease. Eur Respir J. 2015;45(5):1197–1207.
- Wright JL, Churg A. Animal models of cigarette smoke induced chronic obstructive pulmonary disease. Expert Rev Respir Med. 2010;4(6):723–734. DOI:https://doi.org/10.1586/ers.10.68
- Jones B, Donovan C, Liu G, et al. Animal models of COPD: what do they tell us? Respirology. 2017;22(1):21–32. DOI:https://doi.org/10.1111/resp.12908
- Fricker M, Deane A, Hansbro PM. Animal models of chronic obstructive pulmonary disease. Expert Opin Drug Discov. 2014;9(6):629–645. DOI:https://doi.org/10.1517/17460441.2014.909805
- Stevenson CS, Birrell MA. Moving towards a new generation of animal models for asthma and COPD with improved clinical relevance. Pharmacol Ther. 2011;130(2):93–105.
- Wright JL, Cosio M, Churg A. Animal models of chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol. 2008;295(1):L1–L15. DOI:https://doi.org/10.1152/ajplung.90200.2008
- Harrell CR, Miloradovic D, Sadikot R, et al. Molecular and cellular mechanisms responsible for beneficial effects of mesenchymal stem cell-derived product “exo-d-mapps” in attenuation of chronic airway Inflammation. Anal Cell Pathol (Amst). 2020;2020:3153891. Article ID 3153891. DOI:https://doi.org/10.1155/2020/3153891