
Abstract
A-to-I RNA editing operated by ADAR enzymes is extremely common in mammals. Several editing events in coding regions have pivotal physiological roles and affect protein sequence (recoding events) or function. We analyzed the evolutionary history of the 3 ADAR family genes and of their coding targets. Evolutionary analysis indicated that ADAR evolved adaptively in primates, with the strongest selection in the unique N-terminal domain of the interferon-inducible isoform. Positively selected residues in the human lineage were also detected in the ADAR deaminase domain and in the RNA binding domains of ADARB1 and ADARB2. During the recent history of human populations distinct variants in the 3 genes increased in frequency as a result of local selective pressures. Most selected variants are located within regulatory regions and some are in linkage disequilibrium with eQTLs in monocytes. Finally, analysis of conservation scores of coding editing sites indicated that editing events are counter-selected within regions that are poorly tolerant to change. Nevertheless, a minority of recoding events occurs at highly conserved positions and possibly represents the functional fraction. These events are enriched in pathways related to HIV-1 infection and to epidermis/hair development. Thus, both ADAR genes and their targets evolved under variable selective regimes, including purifying and positive selection. Pressures related to immune response likely represented major drivers of evolution for ADAR genes. As for their coding targets, we suggest that most editing events are slightly deleterious, although a minority may be beneficial and contribute to antiviral response and skin homeostasis.
Abbreviations
| 1000G | = | 1000 Genomes Pilot Project |
| AGS | = | Aicardi-Goutières Syndrome |
| A to I | = | adenosine to inosine |
| BEB | = | Bayes Empirical Bayes |
| BS-REL | = | branch site-random effects likelihood |
| CEU | = | Europeans |
| CHBJPT | = | Chinese plus Japanese |
| DAF | = | derived allele frequency |
| DIND | = | Derived Intra-allelic Nucleotide Diversity |
| DSH | = | dyschromatosis symmetrica hereditaria |
| eQTL | = | Expression quantitative trait loci |
| FDR | = | false discovery rate |
| GARD | = | Genetic Algorithm Recombination Detection |
| GERP | = | Genomic Evolutionary Rate Profiling |
| IFN | = | Interferon |
| iHS | = | Integrated Haplotype Score |
| LD | = | linkage disequilibrium |
| LRT | = | likelihood ratio test |
| MAF | = | minor allele frequency |
| MEME | = | Mixed Effects Model of Evolution |
| RBD | = | dsRNA binding domain |
| SLAC | = | single-likelihood ancestor counting |
| YRI | = | Yoruba |
Introduction
RNA editing, defined as the post-transcriptional modification of RNA molecules not including splicing, capping, and polyadenylation, is a widespread phenomenon in several living organisms. In metazoans, the most common RNA editing event is the adenosine to inosine (A-to-I) conversion operated by ADAR (adenosine deaminases acting on RNA) enzymes mainly on dsRNA substrates.Citation1 Recent estimates suggest that ∼1.6 million editing sites exist in the human genome.Citation2
Mammalian genomes encode 3 ADAR genes: the catalytically active ADAR and ADARB1, plus ADARB2, thought to be inactive and to serve a regulatory role.Citation3 ADARB2 expression is brain-specific, whereas the other 2 ADAR genes are transcribed in many tissues.Citation1 A unique feature of ADAR is the presence of an interferon (IFN)-inducible promoter that drives expression of a full-lenght ADARp150 protein; the constitutive, non IFN-responsive promoter determines the synthesis of a shorter N-terminally truncated ADARp110 product. In line with its IFN-inducible properties, ADAR was shown play a role in antiviral responses.Citation4 Mutations in ADAR are responsible for 2 different genetic diseases: Aicardi-Goutières Syndrome (AGS) and dyschromatosis symmetrica hereditaria (DHS).Citation5 This latter is a pigmentary skin disease, whereas AGS is an autoinflammatory condition mainly affecting the brain and the skin. AGS patients carrying ADAR mutations display up-regulation of IFN stimulated genes, suggesting that the gene acts as a suppressor of IFN responses.Citation6
A-to-I RNA editing is thought to be involved in several physiological and pathological processes. Inosine is recognized as guanosine by the translation and splicing machineries. Therefore, editing events can result in a wide range of effects and may affect protein sequence (recoding events) and function when they occur in coding regions. Compared to other mammals, primates exhibit much higher levels of transcriptome editing, mainly as a result of their genome being rich in Alu sequences, which represent preferential editing sites by virtue of their propensity to form double-stranded RNA structures.Citation7 Thus, the overwhelming majority of editing events occurs in non-coding repetitive sequences. Nonetheless, several A-to-I conversions in coding regions have a pivotal physiological role. Among these, the best studied examples include brain-specific ion channels and neurotransmitter receptors. For instance, editing at a single site (known as the Q/R site) in the ionotropic glutamate receptor subunit GLUR2 alters Ca2+ permeability and is essential for normal brain development. Indeed, Adarb1-/- mice suffer from epileptic seizures and die several weeks after birth. The phenotype is rescued by introduction of a transgene that allows expression of the Glur2 edited form.Citation8 Whereas the GLUR2 Q/R editing event is shared by humans and rodents, conserved mammalian editing sites are a small minority.Citation9
In humans and other primates most editing events occur in the brain and, in analogy to GLUR2, involve neuronal genes. Paz-Yaacov and coworkers also indicated that human-specific editable Alu insertions are enriched in genes related to neuronal functions or implicated in neurological diseases.Citation10 This observation, together with the higher editing levels in the brain of humans compared to chimpanzees and macaques, led some authors to suggest that A-to-I RNA editing contributed to the development of higher brain functions.Citation8,10
From an evolutionary perspective, RNA editing is an extremely interesting phenomenon. In analogy to alternative splicing, editing can provide transcriptome variability and, as noted, it might allow variation at sites that would otherwise be inaccessible to mutation, which instead imposes high fitness costs.Citation11 Nevertheless, recent data have indicated that editing of coding sequences is generally nonadaptive in humans, although the presence of few beneficial recoding events was postulated.Citation12 On this basis, we set out to perform an evolutionary analysis of ADAR family genes and of their coding targets.
Results
ADAR evolved adaptively in primates
To analyze the evolutionary history of ADAR genes (ADAR, ADARB1, and ADARB2) in primates, we obtained coding sequences for available species in public databases; the tree shrew sequence was also included (Table S1). The three DNA alignments were generated using RevTrans and screened for the presence of recombination breakpoints using GARD (genetic algorithm recombination detection).Citation13,14 No breakpoint was detected for any gene.
We next calculated the average nonsynonymous substitution/synonymous substitution rate ratio (dN/dS, also referred to as ω) using the single-likelihood ancestor counting (SLAC) method.Citation15 In all cases dN/dS was lower than 1 (), indicating purifying selection as the major driving force in shaping diversity at ADAR genes in primates. This is not unusual, as most mammalian genes display variable levels of purifying selection at their coding regions.Citation16 A major effect of negative selection is not incompatible with positive selection acting on specific sites or domains. To assess whether positive selection acted on ADAR family members, we applied likelihood ratio tests (LRT) implemented in the codeml program.Citation17,18 Codeml compares models of gene evolution that allow (NSsite models M2a and M8, positive selection models) or disallow (NSsite models M1a and M7, null models) a class of codons to evolve with dN/dS > 1. In the case of ADAR, but not of ADARB1 and ADARB2, both neutral models were rejected in favor of the positive selection models; these results were confirmed using different models of codon frequency ().
Table 1. Genomic position and average dN/dS for ADAR family genes.
Table 2. Likelihood ratio test (LRT) statistics for models of variable selective pressure among sites and branches.
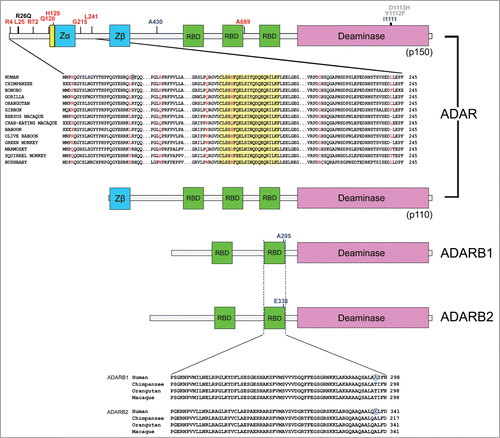
In order to identify specific ADAR sites targeted by positive selection, we applied the Bayes Empirical Bayes (BEB) analysis and the Mixed Effects Model of Evolution (MEME).Citation19-21 To be conservative, only sites detected using both methods were considered. We identified a total of 8 positively selected sites. Interestingly, 7 of these are located in the additional amino-terminal portion of the interferon-induced isoform of ADAR (p150) (). One of the positively selected sites (H129) maps to a nuclear exporting sequence (NES) (). The positively selected L25 residue immediately flanks a missense mutation identified in patients with DSH ().Citation5
Figure 1. Adaptive evolution at ADAR genes in primates. Schematic representation of the domain structure of ADAR family members. Domains are color-coded: nuclear export signal (NES), yellow; Z-DNA binding domains, cyan; RNA binding motifs (RBD), green; deaminase domain, pink. The position of positively selected sites is shown together with sequence alignments for a few representative primates. Positively selected sites in primates and in the human lineage are shown in red and blue, respectively. Some missense mutations associated with AGS and DSH are shown in gray and black, respectively.Citation5
We next extended our analysis to explore possible variations in selective pressure among primate lineages at ADAR. To this aim, we tested whether models that allow dN/dS to vary along branches had significant better fit than models that assume one same dN/dS across the entire phylogeny.Citation22 Because this hypothesis was verified, we used the branch site-random effects likelihood (BS-REL) method to analyze selection along specific lineages.Citation23 BS-REL identified 3 branches: green monkey, bonobo and tree shrew (Fig. S1). These were cross-validated using codeml (branch-site LRT models), with application of false discovery rate (FDR) correction, as suggested.Citation24,25 This analysis confirmed the bonobo branch only (Table S3), but detected no lineage-specific positively selected sites. We note that this is not unusual, as the simultaneous inference of both the site and the branch subject to diversifying selection is difficult;Citation21,24 thus, BEB analysis is accurate but has low power in this context.Citation24
Positive selection of ADAR family genes in the human lineage
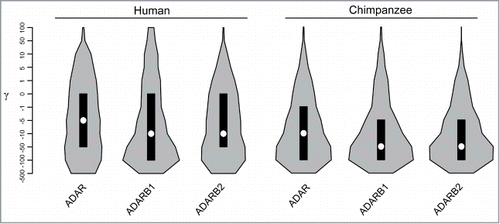
The combined analysis of intra-species polymorphism and between-species divergence may allow increased power to detect sites targeted by positive selection in one species. Moreover, this approach provides information on the distribution of selective effects along gene regions. We thus used gammaMap, a recently developed program that models intragenic variation in selection coefficients (γ), to study the evolution of ADAR family members in the human and chimpanzee lineages. For humans, we exploited data from the 1000 Genomes Pilot Project (1000G) for Europeans (CEU), Yoruba (YRI), and Chinese plus Japanese (CHBJPT).Citation26 For chimpanzees, we used phased SNP information of 10 Pan troglodytes verus.Citation27 Ancestral sequences were reconstructed by parsimony from the human, chimpanzee, orangutan and macaque sequences. In line with the results obtained above, we observed a general preponderance of codons evolving under negative selection (γ < 0) for all genes. In particular, in both species ADAR was found to be less constrained than ADARB1 and ADARB2 ().
Figure 2. Analysis of selective pressure in the human and chimpanzee lineages. Violin plot of selection coefficients (median, white dot; interquartile range, black bar). Selection coefficients (γ) are classified as strongly beneficial (100, 50), moderately beneficial (10, 5), weakly beneficial (1), neutral (0), weakly deleterious (−1), moderately deleterious (−5, −10), strongly deleterious (−50, −100), and inviable (−500).
We next used gammaMap to identify specific codons evolving under positive selection in humans and chimpanzees. To be conservative, we declared a codon to be targeted by positive selection when the cumulative posterior probability of γ ≥ 1 was > 0.75, as suggested.Citation28
No positively selected codon was identified for P. troglodytes. For humans, 2 positively selected sites were identified in ADAR; one of these (I1111) is located within the deaminase domain and is immediately adjacent to 2 mutations (Y1112F and D1113H) responsible for AGS ( and Table S2).Citation5 One selected site was identified in both ADARB1 and ADARB2 (Table S2); the selected sites are in the second dsRNA binding (RBD) domain: alignment of ADARB1 and ADARB2 indicated that the corresponding position is targeted by selection ().
Non-coding regulatory variants represent targets of positive selection in human populations
We next investigated whether natural selection acted on ADAR family members during the recent evolutionary history of human populations. The 1000G data for YRI, CEU, and CHBJPT were used to this purpose.
Integration of different tests can improve the power to detect selective sweeps and, importantly, allows identification of the causal adaptive variant(s).Citation29-31 We applied the DIND (Derived Intra-allelic Nucleotide Diversity) test, which is powerful in most derived allele frequency (DAF) ranges and less sensitive than iHS (Integrated Haplotype Score) to low genotype quality or low coverage (i.e. it is well suited for the 1000G data).Citation32,33 DIND results were combined with pairwise FST analyses, whereas DH was calculated in sliding-windows to account for local events and, for this reason, used as an a posteriori validation.Citation34 Statistical significance (in terms of percentile rank) for all tests was obtained by deriving empirical distributions from a control set of ∼1000 genes (see Materials and Methods). We declared a variant to be selected if it displayed both a DIND and an FST percentile rank >0.95. DH was used to validate high-frequency sweeps, in line with the power profile of this test.Citation34
We detected distinct selection signals in ADAR. In CEU and CHBJPT, the same variant (rs884618) had unusually high FST (YRI/CEU and YRI/CHBJPT comparisons) and represented a DIND outlier (). The variant is located ∼700 bp upstream the ADAR transcription start site, in a region where transcription factor binding sites have been annotated together with histone marks associated with regulatory elements and DNase hypersensitivity peaks (). In CEU and CHBJPT, rs884618 is in full linkage disequilibrium (LD) (r2=1 in both populations) with an eQTL SNP (rs9427108) in naive CD14+ monocytes;Citation35 the selected variant also shows LD (r2= 0.78 and 1 in CEU and CHBJPT, respectively) with rs9427114, which acts as an interferon-dependent eQTL in monocytes ( and ).Citation35 As for YRI, several linked variants (r2>0.85) were found to be outliers in the DIND test and FST distributions (, ). One of these is located close to rs884618 and falls within regulatory elements ().
Table 3. Candidate targets of positive selection in human populations.
Figure 3. Location of the most likely selection targets in human populations. Candidate targets are shown for ADAR (A), ADARB1 (B), ADARB2 (C) within the UCSC Genome Browser view. Relevant annotation tracks are shown. For ADARB2 a sliding-window analysis of DH is also shown in green (YRI) and blue (CEU). The horizontal dashed line represents the 5th percentile of DH. Variants in blue, red and green represent selection targets in CEU, CHBJPT, and YRI, respectively. Additional color codes are as follows: yellow highlight indicates SNPs mapping to regulatory elements; cyan indicates eQTL.
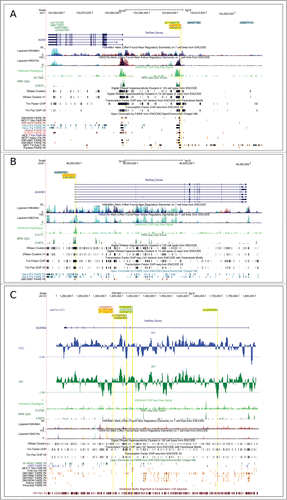
Similarly to ADAR, we detected one variant in ADARB1 which represents the likely selection target in CEU and CHBJPT. Indeed, rs4819027 represented a DIND outlier in both populations and had very high FST in comparisons with YRI (). The SNP is located in the 5' portion of the first intron, where ENCODE data indicate the presence of regulatory elements (). Interestingly, analysis of brain methylation and histone modification patterns showed that rs4819027 also maps to a region where unmethylated CpGs and H3K4me3 marks are located; these signals are associated with active transcription. Finally, rs4819027 is in moderate LD (r2= 0.73 and 0.80 in CEU and CHBJPT, respectively) with rs2838763, an IFN-induced eQTL for ADARB1 in monocytes.Citation35
As for ADARB2, 5 different selection signals were detected. In CEU, 2 high-frequency sweeps were detected at rs60741147 and rs10794743/rs4880500 (). rs60741147 and rs10794743/rs4880500 are in no LD (r2 = 0) and in all cases variants mapped to DH valleys in CEU. rs60741147 is located within the 3' UTR, in a region extremely conserved in mammals (). Likewise, rs10903528 was in a DH valley for YRI (). In this population, a second lower frequency event was detected, which involved variants rs2820600/rs2820599/rs2805512 (in no LD with rs10903528). Finally, in CHBJPT 3 nearby variants were found to represent DIND and FST outliers ( and ). In most instances selected variants were found to map within ENCODE regulatory elements ().
A minority of editing events occurs at highly conserved nonsynonymous sites
We next analyzed the evolutionary history of ADAR/ADARB1 editing sites located in human coding regions. To this aim, we retrieved A-to-I editing sites from the RADAR database (http://rnaedit.com/).Citation36 We limited our analysis to sites within coding regions and located outside of repetitive elements; editing sites corresponding to SNP positions (in the dbSNP137 database) were removed. The remaining sites were divided based on the change the editing causes (i.e., synonymous or nonsynonymous substitution). We also distinguished editing events based on their conservation in mammals. In particular, we designated as "shared" editing events that occur in at least one other species (among chimpanzee, macaque, and mouse; n shared sites = 69); we refer to events that have been described in humans only as "non-shared" (n non-shared = 664). Due to the small sample size (n shared = 17), editing sites that entail synonymous substitutions were not separated based on their being shared or not.
The Genomic Evolutionary Rate Profiling (GERP) score, which measures the base-wise conservation across mammals, was next used to evaluate sequence conservation at the editing site and at flanking synonymous and nonsynonymous positions (4 codon extension at both sides). For non-shared events, editing sites at nonsynonymous positions were found to be significantly less conserved compared to flanking positions (); the same occurred for editing sites at synonymous positions (), but not at shared nonsynonymous editing sites (, p values not shown), possibly due to the small sample size (n = 52). As a further comparison, 1000 positions (reference sites), with flanking synonymous and nonsynonymous sites were randomly selected from the set of genes harboring the editing events. Analysis of GERP scores indicated that for both the synonymous and nonsynonymous editing events, the regions surrounding editing sites were significantly less conserved than the reference sites; this was observed at both shared and non-shared editing sites for nonsynonymous changes (Wilcoxon rank sum test, 2-tailed, p values = 0.0064 and 2.2×10–16, respectively; ) and at editing sites that cause synonymous changes (Wilcoxon rank sum test, 2-tailed, p value = 0.0024, ).
Figure 4. Conservation at ADAR editing sites. (A) Box plot representation of GERP conservation scores for A-to-I editing events that cause nonsynonymous substitutions. Red and orange denote “non-shared” editing sites and their flanking sites, respectively; blue and cyan indicate “shared” editing sites and their flanking sites; dark gray indicates control nonsynonymous positions with their flanking codons in light gray (see text). Wilcoxon rank sum test (2-tailed) p values are also reported. (B) Box plot representation of GERP conservation scores for A-to-I editing events that cause synonymous substitutions. Dark and light green indicate editing sites and their flanking regions, respectively; dark gray indicates control synonymous positions, with light gray indicating their flanking sites. Wilcoxon rank sum test (2-tailed) p values are reported. (C) Distributions of GERP scores at editing-sites are reported for "non-shared" and "shared" nonsynonymous editing sites, as well as for synonymous editing sites. Color codes are as in the previous panels, with 100 random control distributions in gray. Flanking sites are represented with dashed lines.
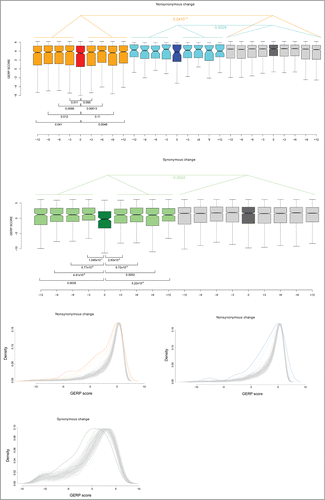
In order to gain further insight, we compared GERP score distributions at editing sites to those deriving from 100 random samples of the same number of synonymous and nonsynonymous sites. In particular, site samples were randomly drawn from the same set of genes as those where the editing events occur (see Materials and Methods). Results confirmed a general shift of editing sites toward lower conservation scores (). Nonetheless, for both shared and non-shared editing events that cause nonsynonymous substitutions, the distribution was significantly wider than that of random samples, with a fraction of sites (and flanking positions) showing very high GERP scores (). This was not observed for editing sites (and flanks) that cause synonymous changes.
To summarize, the editing site and its flanking positions are significantly less conserved than the average, both when the event causes a synonymous and a nonsynonymous substitution. Nonetheless, a fraction of editing events determine nonsynonymous substitution at highly conserved positions; this is not the case for events that cause synonymous changes and is not depended upon sharing of editing events among mammals.
We next used the WebGestalt tool to assess whether nonsynonymous edited sites showing low and high conservation scores impinge on specific pathways or biological processes.Citation37 Two related pathways, host interactions of HIV factors/HIV infection, were significantly enriched for genes that carry highly conserved editing sites (in the top 10% of GERP scores) compared to the overall set of edited genes (i.e. genes that carry at least one nonsynonymous editing site) (). In this same set, GO terms related to hair cycle and hair follicle/epidermis development were also enriched. No significant difference was observed for genes edited at poorly conserved positions.
Table 4. Pathway and GO term enrichment analysis for genes carrying recoding events at highly conserved positions.
Discussion
Recent technological advances are providing an increasingly detailed picture of the extent, regulation and location of A-to-I editing events in the genome of humans and other species. More than one million editing sites have currently been described in humans and RNA editing has been implicated in a number of processes and diseases.Citation2,5
Results indicated that over diverse time periods, diversity at the 3 genes has been mainly shaped by purifying selection. We detected stronger constraint at ADARB1 and ADARB2 compared to ADAR, although this observation most probably reflects our using the long IFN-induced isoform of ADAR (encoding ADARp150) in both the SLAC and gammaMap analyses. Indeed, we show that ADAR evolved adaptively in primates and that most positively selected sites are located in the Zα domain-containing N-terminal portion specific to ADARp150. The Zα domain is functionally active (whereas Zβ is not) and can bind both dsDNA and dsRNA in a Z conformation.Citation38 Similar Z-DNA binding domains are generally found in proteins that participate in the interferon response pathway.Citation39 In line with its INF-responsiveness, ADAR has been suggested to play a role during viral infection, although both antiviral and proviral effects have been described.Citation4
The N-terminal portion of ADARp150 also carries a NES that overlaps with the Zα domain and drives ADAR shuffling from the nucleus to the cytoplasm.Citation40,41 Thus, the N-terminal protein region widens the activity range of ADAR in terms of substrate recognition and cellular localization. One of the positively selected sites we identified is located within the NES, suggesting that it modulates the level or timing of nuclear-cytoplasmic transport. In turn, the cytoplasmic localization of ADARp150 might be relevant to viral detection and binding, as well as to stress response, as the protein localizes to stress granules. These structures form during viral infection or, more generally, during cell stress conditions, and also contain other editing enzymes such as APOBEC family members.Citation42 Thus, the variation pattern at ADAR in primates suggests that the selective pressures acting on the gene are related to its roles in immune or stress responses.
Conversely, the selection signal identified for the human lineage was located in the ADAR deaminase domain and positively selected sites were also detected for ADARB1 and ADARB2. We note that, although gammaMap detected positively selected sites in humans but non in chimpanzees, the much larger sample size of human chromosomes compared to P. troglodytes might partially account for the different pattern in the 2 species.
The positively selected site in ADAR (I1111) is immediately adjacent to 2 positions that were found to be mutated in AGS patients (Y1112F and D1113H) ().Citation6 These residues lie along the dsRNA interaction surface and the AGS mutations did not alter the editing of a known ADAR substrate in an in vitro assay.Citation6 Thus, the pathogenic substitutions were hypothesized to act in a substrate- or cell type-specific manner. Clearly, this also represents an attractive possibility for the positively selected site we detected in humans, as it might modulate editing at human-specific sites.
Interestingly, we found the corresponding residue to be targeted by selection in human ADARB1 and ADARB2. The sites are located at the C-terminus of the highly conserved α2 helix structure of the second dsRNA binding domain (RBD2). Analysis of homologous domains indicated that C-terminal extensions of this helix can affect the RNA binding capability of the entire RBD domain.Citation43 Furthermore RBD2 is the homologous of the third ADAR RBD, which was demonstrated to contribute to the corrected combination of a nuclear localization signal formed by 2 flexible fragments flanking the folded domain.Citation44 An interesting possibility is that the corresponding sites in the 2 proteins evolved in our species to modulate binding to a common interactor.
Population genetics analysis in humans revealed that the 3 ADAR genes were targeted by selection during the most recent history of human populations, as well. The approach we applied to detect selection is based on the integration of 2 tests, DIND and FST, which rely on distinct signatures left by selective sweeps, namely haplotype homozygosity and population genetic differentiation, respectively. As mentioned above, the combined use of distinct tests is expected to afford higher resolution in detecting the causal variant underlying the adaptive phenotype and to reduce the rate of false positive signals.Citation29–31 As for DH, which has more power than the original Fay and Wu's H statistic, it was used as a confirmatory test.Citation34 This choice was motivated by the difficulty of assessing statistical significance in sliding-window analyses, as multiple non-independent tests are performed. DH has very good power for high-frequency sweeps:indeed, the 3 ADARB2 selected alleles at frequency >0.75 (rs60741147 and rs10794743 in CEU, and rs10903528 in YRI) were all located within DH valleys ().Citation34 DH reaches values lower than the 5th percentile in few relatively small regions along ADARB2 (4 and 5 valleys in CEU and YRI, respectively) () and is based on a feature partially independent from population genetic differentiation and haplotype homozygosity. Thus, the DH results do provide support to the strategy we applied to detect selective events. Additional confirmation comes from the observation that most selected variants are located within regions with regulatory function, as assessed by ENCODE annotations.Citation45 In the case of ADAR and ADARB1 the selection targets were also found to be in partial or full LD with previously described eQTLs. Overall, these data are in agreement with recent analyses indicating that regulation of gene expression is a major determinant of phenotypic variation in our species, as well as a common target of natural selection.Citation46,47
In particular, the positively selected variants in ADAR and ADARB1, both shared by CEU and CHBJPT, are in linkage with eQTLs described in CD14+ monocytes, suggesting that a major selective pressure underlying adaptive evolution at these genes is accounted for by infectious agents. Interestingly, the positively selected ADARB1 variant also maps to a region which likely regulates transcription in the brain. Given the central role of this enzyme in the editing of brain-specific genes, these data warrant further analysis of the modulatory effects of the 2 SNP alleles. ADARB2 is also (and preferentially) expressed in the brain, but its biological role and regulation are poorly understood. We detected multiple sweep events at this gene, with diverse variants targeted in the same and in distinct populations, suggesting strong selective pressure.
Overall, data herein indicate that ADAR family genes were targeted by positive selection in primates, in the human lineage, and in the recent history of human populations. These findings and a wealth of previous data suggest a central role for these enzymes in physiological and pathological processes, a role at least partially mediated by the specific editing of coding sites with well-known effects.Citation3,5,48 These include the already cited GLUR2 Q/R site, several nonsynonymous editing sites in HTR2C (serotonin receptor), an S/G recoding event in AZIN1 which predisposes to hepatocellular carcinoma, and an editing event in NEIL1 that alters the enzyme's specificity.Citation49,50 Beside these and a few more examples, though, the scenario of A-to-I editing in coding regions and its overall significance have remained elusive. We reasoned that further insight into this issue might be gained through an evolutionary analysis of ADAR/ADARB1 coding targets.
Recently, a study in macaques indicated that purifying selection is the major force acting at editing sites and at their flanking positions.Citation49 In partial agreement, a previous analysis had suggested that editing sites are less conserved across primates than their flanks, but that the overall region carrying the editing site is more conserved than control sequences.Citation51 Herein we used a score of conservation across mammals and we focused on editing sites located in coding regions by separately analyzing editing events that originate synonymous and nonsynonymous substitution, to account for underlying variation in sequence conservation in coding sequences. In contrast to previous reports, our findings indicate that the editing site is less conserved than its flanks which, in turn, are more variable than control positions randomly drawn from the edited coding regions. This effect is observed at shared and non-shared nonsynonymous sites, as well as at positions that entail synonymous substitutions when edited. The possible reasons for these discrepancies are manifold. With respect to Bahn and coworkers' data, the overwhelming majority of editing sites they analyzed was accounted for by non-coding sites. As for the macaque editome data, the authors analyzed fewer than 30 editing sites in coding regions shared among macaques, chimpanzees and humans; sequence conservation was measured in terms of human-macaque percentage identity or dS.Citation49 Also, the authors did not consider the effect of editing on the protein sequence (whether or not recoding occurs) and did not use a comparison with control sequences.
As suggested, the lower conservation we observed at the editing site might reflect fixation of the edited form in some mammals or correction of G-to-A mutations through editing.Citation51 Nonetheless, these possibilities do not explain why flanking positions are less conserved than control sites. We suggest that most editing events are slightly deleterious and are therefore counter-selected within regions that are poorly tolerant to change and thus, by definition, highly conserved. This might be the case for both synonymous and nonsynonymous editing events, with the former excluded from regulatory regions (e.g. in splicing regulatory elements) and the latter counter-selected in constrained protein regions.
These conclusions are in agreement with a recent work showing that in humans the frequency and level of editing is lower at nonsynonymous than at synonymous sites, and that recoding events are rarer in essential genes or in genes subject to strong functional constraint (measured as dN/dS).Citation12 Based on these and other observations the authors proposed that most recoding events are deleterious byproducts, although few events might be functionally relevant and beneficial.Citation12 In fact, analysis of GERP score distributions revealed that a minority of recoding events does occur at highly conserved positions. These might represent the functional fraction. This hypothesis is supported by the identification of enriched pathways and process for genes that harbor recoding events at highly conserved positions. We note that the functionally relevant recoding events we mentioned above (in GLUR2, HTR2C, AZIN1, and NEIL1) all occur at highly conserved positions (GERP score > 4.5), but are not included in the enrichment analysis because they remain below the 90th percentile threshold (GERP score = 5.7) we set.
Interestingly, the significant pathways and processes we detected are related to ADAR/ADARB1 known functions. In HIV-1 infection both antiviral and proviral effects have been described for ADAR.Citation52-55 The 3 genes that contribute to the pathway (PMSC4, ANAPC7, and NUPL2) represent host factors for HIV-1 replication. Unlike restriction factors, which are specifically devoted to antiviral response and often fast-evolving, host factors carry out central physiological functions and are exploited by the virus for infection.Citation56 Therefore, genes coding for host factors usually evolve under purifying selection. In this respect editing might represent an advantage on the host side to allow some level of variability that may affect the viral replication process with a low overall fitness cost. It will be interesting to evaluate whether editing at these genes can affect HIV-1 infection or replication efficiency.
Finally, genes harboring recoding events are involved in epidermis development and hair cycle/hair follicle development. As mentioned above, mutations in ADAR are responsible for dyschromatosis symmetrica hereditaria (DSH), a pigmentary skin disease. More recently, hair anomalies have also been described in patients with DHS.Citation57 Consistently, a conditional mouse model that lacks ADAR expression in the epidermis shows fur loss and skin pathology. In particular, epidermal necrosis and abnormal hair follicles were evident in these animals.Citation58 The pathogenic mechanism underlying DSH is poorly understood, but is thought to result from loss or decreased editing at specific target genes. Those we identified herein represent promising candidates.
In summary, our data indicate that both ADAR family genes and their targets evolved under variable selective regimes, including purifying and positive selection. We suggest that pressures related to immune response were major drivers of evolution for ADAR genes and, possibly, some of their targets. These analyses do not support nor dismiss the previous suggestion whereby A-to-I RNA editing contributed to the development of higher brain functions in humans.Citation8,10 Further analyses will be necessary to clarify this issue, although result herein do not reveal exceptionally fast evolution at ADAR genes in humans compared to other primates.
Materials and Methods
Evolutionary analysis in primates
Primate sequences for ADAR, ADARB1 and ADARB2 were retrieved from the Ensembl and NCBI databases (http://www.ensembl.org/index.html; http://www.ncbi.nlm.nih.gov/). All primate genes represented 1-to-1 orthologs of the human genes, as reported in the EnsemblCompara GeneTrees database (Tab. S1).Citation59 This information was not available for Papio hamadryas, Macaca fascicularis, and Saimiri boliviensis. BLAT search of the 3 ADAR gene coding sequences against the genome of these species (genome assemblies: Pham_1.0, MacFas_5.0, and SaiBol1.0) was performed using the Ensembl BLAST/BLAT utility; in all cases hits were consistent with the presence of a single ortholog, with no evidence of gene duplication. A phylogenetic tree of the 3 ADAR family proteins was constructed using phyML with the best-fitting model (JTT plus gamma-distributed rates) generated by ProtTest 3.Citation60,61 The list of species and the phylogenetic tree are reported in Table S1 and in Figure S2, respectively.For the analysis of positive selection, DNA alignments were performed using the RevTrans 2.0 utility, which uses the protein sequence alignment as a scaffold to construct the corresponding DNA multiple alignment.Citation13 This latter was checked and edited by hand to remove alignment uncertainties. Alignments were first screened for the presence of recombination breakpoints using GARD (Genetic Algorithm Recombination Detection);Citation14 the average nonsynonymous substitution/synonymous substitution rate ratio (dN/dS, also referred to as ω) was calculated using the single-likelihood ancestor counting (SLAC) method.Citation15
The site models implemented in PAML have been developed to detect positive selection affecting only a few aminoacid residues in a protein. To detect selection, site models that allow (M2a, M8) or disallow (M1a, M7) a class of sites to evolve with ω >1 were fitted to the data using the F3×4 and the F61 codon frequency models. For these analyses we used trees generated by maximum-likelihood using the program PhyML.Citation18,60 Positively selected sites were identified using the Bayes Empirical Bayes (BEB) analysis (with a cut-off of 0.90), which calculates the posterior probability that each codon is from the site class with ω >1 (under model M8).Citation19 A second method, the Mixed Effects Model of Evolution (MEME) (with the default cutoff of 0.1), which allows the distribution of ω to vary from site to site and from branch to branch at a site, was applied.21To explore possible variations in selective pressure among different lineages, we applied the free-ratio models implemented in the PAML package: the M0 model assumes all branches to have the same ω, whereas M1 allows each branch to have its own ω.Citation17 The models are compared through likelihood-ratio tests (degree of freedom= total number of branches -1). In order to identify specific branches with a proportion of sites evolving with ω>1, we used BS-REL with the PhyML-generated tree.Citation23 Branches identified using this approach were cross-validated with the branch-site likelihood ratio tests from PAML (the so-called modified model A and model MA1, "test 2").Citation24 A false discovery rate (FDR) correction was applied to account for multiple hypothesis testing (i.e., we corrected for the number of tested lineages), as suggested.Citation25 BEB analysis from MA (with a cut-off of 0.90) was used to identify sites that evolved under positive selection on specific lineages.Citation24
GARD, SLAC, MEME and BS-REL analyses were performed through the DataMonkey server (http://www.datamonkey.org) or run locally (through HyPhy).Citation14,15,21,62
Population genetics-phylogenetics analysis
For gammaMap analysis,Citation63 we assumed θ (neutral mutation rate per site), k (transitions/transversions ratio), and T (branch length) to vary among genes following log-normal distributions. For each gene we set the neutral frequencies of non-STOP codons (1/61) and the probability that adjacent codons share the same selection coefficient (p = 0.02). For selection coefficients we considered a uniform Dirichlet distribution with the same prior weight for each selection class. For each gene we run 10,000 iterations with thinning interval of 10 iterations.
Population genetics analyses
Genotype data from the Pilot 1 phase of the 1000 Genomes Project were retrieved from the dedicated website, organized in a MySQL database, and analyzed according to selected regions/populations; these analyses were performed using the GeCo++ and the libsequence libraries.Citation26,64,65
The pairwise FST and the DIND (Derived Intra-allelic Nucleotide Diversity) test were calculated for all SNPs mapping to ADAR, ADARB1 and ADARB2, as well as for SNPs mapping to a control set of ∼1,000 genes; these latter were used as a reference, as previously described.Citation30-32,66
FST values are not independent from allele frequencies, so we binned variants based on their minor allele frequency (MAF, 50 classes) and calculated FST empirical distributions for each MAF class. The same procedure was applied for the DIND test; thus, we calculated statistical significance by obtaining an empirical distribution of DIND values for variants located within control genes; in particular, the DIND test was calculated using a constant number of 20 upstream and downstream flanking variants, as previously described.Citation30,31 DIND values for YRI, CEU and AS were binned in derived allele frequency (DAF) intervals (100 classes) and for each class the distributions were calculated. As suggested, for values of iπD=0 we set the DIND value to the maximum obtained over the whole dataset plus 20. Only SNPs with both FST and DIND with a percentile rank >0.95 were considered as selection targets.Citation32
DH was calculated in 5kb sliding windows moving with a step of 500 bp.Citation34,67 Sliding window analyses have an inherent multiple testing problem that is difficult to correct because of the non-independence of windows. In order to partially account for this limitation, we calculated DH also for the control gene set, and the distribution of the statistic was obtained for the corresponding windows. This allowed calculation of the 5th percentile and the identification of regions below this threshold.
LD was calculated through the SNAP utility (http://www.broadinstitute.org/mpg/snap/) Citation68
ADAR editing sites analysis
We retrieved A-to-I editing sites from the RADAR database (http://rnaedit.com/), limiting our search to sites within coding regions and located outside of repetitive elements.Citation36 Information concerning the presence of the same editing event in other species (chimpanzee, macaque, and mouse) was also based on RADAR annotations.
The Genomic Evolutionary Rate Profiling (GERP) scorewas obtained from UCSC tables (table name: GERP Scores for Mammalian Alignments) and used to evaluate conservation: positive scores represent a deficit in substitutions and indicate evolutionary constraint.Citation69 To generate 100 comparison distributions, nonsynonymous and synonymous positions were randomly drawn from the same genes harboring the editing events. In particular, distributions were generated by 100 resamplings of the same number of positions as the number of non-shared and shared editing events for nonsynonymous changes (n = 443 and n = 52, respectively), and for all editing sites for synonymous changes (n = 238).
GO term and pathway enrichment
To evaluate whether low and high conserved nonsynonymous editing sites are involved in specific pathways or biological processes we used the WEB-based GEne SeT AnaLysis Toolkit.Citation37 Specifically, unique gene lists that carry highly or poorly conserved editing sites (in the 10% tails of GERP score distributions, Table S4) were used as queries; the background list was accounted for by the total set of genes carrying recoding events. The 87% of recoding events occurred in distinct genes (one event/gene). When the same gene carried more than one recoding event, it was counted only once in the relative list (for instance, if the same gene presented 2 recoding events at highly conserved positions, it was included only once in the highly conserved list). The minimum number of genes for a category was set to 3, and we applied a Benjamini and Hochberg correction for multiple testing. We queried for enrichment in GO categories, KEGG pathways and in the Pathway Commons database.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary_Material.pdf
Download PDF (338 KB)Funding
CP and DF are supported by fellowships of the Doctorate School of Molecular and Translational Medicine, University of Milan.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Nishikura K. Functions and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem 2010; 79:321-349; PMID:20192758; http://dx.doi.org/10.1146/annurev-biochem-060208-105251
- Bazak L, Haviv A, Barak M, Jacob-Hirsch J, Deng P, Zhang R, Isaacs FJ, Rechavi G, Li JB, Eisenberg E, et al. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res 2014; 24:365-376; PMID:24347612; http://dx.doi.org/10.1101/gr.164749.113
- Savva YA, Rieder LE, Reenan RA. The ADAR protein family. Genome Biol 2012; 13:252; PMID:23273215; http://dx.doi.org/10.1186/gb-2012-13-12-252
- Samuel CE. ADARs: Viruses and innate immunity. Curr Top Microbiol Immunol 2012; 353:163-195; PMID:21809195
- Slotkin W, Nishikura K. Adenosine-to-inosine RNA editing and human disease. Genome Med 2013; 5:105; PMID:24289319; http://dx.doi.org/10.1186/gm508
- Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, et al. Mutations in ADAR1 cause aicardi-goutieres syndrome associated with a type I interferon signature. Nat Genet 2012; 44:1243-1248; PMID:23001123; http://dx.doi.org/10.1038/ng.2414
- Daniel C, Silberberg G, Behm M, Ohman M. Alu elements shape the primate transcriptome by cis-regulation of RNA editing. Genome Biol 2014; 15:R28; PMID:24485196; http://dx.doi.org/10.1186/gb-2014-15-2-r28
- Li JB, Church GM. Deciphering the functions and regulation of brain-enriched A-to-I RNA editing. Nat Neurosci 2013; 16:1518-1522; PMID:24165678; http://dx.doi.org/10.1038/nn.3539
- Pinto Y, Cohen HY, Levanon EY. Mammalian conserved ADAR targets comprise only a small fragment of the human editosome. Genome Biol 2014; 15:R5; PMID:24393560; http://dx.doi.org/10.1186/gb-2014-15-1-r5
- Paz-Yaacov N, Levanon EY, Nevo E, Kinar Y, Harmelin A, Jacob-Hirsch J, Amariglio N, Eisenberg E, Rechavi G. Adenosine-to-inosine RNA editing shapes transcriptome diversity in primates. Proc Natl Acad Sci U S A 2010; 107:12174-12179; PMID:20566853; http://dx.doi.org/10.1073/pnas.1006183107
- Gommans WM, Mullen SP, Maas S. RNA editing: A driving force for adaptive evolution? Bioessays 2009; 31:1137-1145; PMID:19708020; http://dx.doi.org/10.1002/bies.200900045
- Xu G, Zhang J. Human coding RNA editing is generally nonadaptive. Proc Natl Acad Sci U S A 2014; 111:3769-3774; PMID:24567376; http://dx.doi.org/10.1073/pnas.1321745111
- Wernersson R, Pedersen AG. RevTrans: Multiple alignment of coding DNA from aligned amino acid sequences. Nucleic Acids Res 2003; 31:3537-3539; PMID:12824361; http://dx.doi.org/10.1093/nar/gkg609
- Kosakovsky Pond SL, Posada D, Gravenor MB, Woelk CH, Frost SD. Automated phylogenetic detection of recombination using a genetic algorithm. Mol Biol Evol 2006; 23:1891-1901; PMID:16818476; http://dx.doi.org/10.1093/molbev/msl051
- Kosakovsky Pond SL, Frost SD. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol Biol Evol 2005; 22:1208-1222; PMID:15703242; http://dx.doi.org/10.1093/molbev/msi105
- Lindblad-Toh K, Garber M, Zuk O, Lin MF, Parker BJ, Washietl S, Kheradpour P, Ernst J, Jordan G, Mauceli E, et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature 2011; 478:476-482; PMID:21993624; http://dx.doi.org/10.1038/nature10530
- Yang Z. PAML: A program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci 1997; 13:555-556; PMID:9367129
- Yang Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol Biol Evol 2007; 24:1586-1591; PMID:17483113; http://dx.doi.org/10.1093/molbev/msm088
- Anisimova M, Bielawski JP, Yang Z. Accuracy and power of bayes prediction of amino acid sites under positive selection. Mol Biol Evol 2002; 19:950-958; PMID:12032251; http://dx.doi.org/10.1093/oxfordjournals.molbev.a004152
- Yang Z, Wong WS, Nielsen R. Bayes empirical bayes inference of amino acid sites under positive selection. Mol Biol Evol 2005; 22:1107-1118; PMID:15689528; http://dx.doi.org/10.1093/molbev/msi097
- Murrell B, Wertheim JO, Moola S, Weighill T, Scheffler K, Kosakovsky Pond SL. Detecting individual sites subject to episodic diversifying selection. PLoS Genet 2012; 8:e1002764; PMID:22807683; http://dx.doi.org/10.1371/journal.pgen.1002764
- Yang Z, Nielsen R. Synonymous and nonsynonymous rate variation in nuclear genes of mammals. J Mol Evol 1998; 46:409-418; PMID:9541535; http://dx.doi.org/10.1007/PL00006320
- Kosakovsky Pond SL, Murrell B, Fourment M, Frost SD, Delport W, Scheffler K. A random effects branch-site model for detecting episodic diversifying selection. Mol Biol Evol 2011; 28:3033-3043; PMID:21670087; http://dx.doi.org/10.1093/molbev/msr125
- Zhang J, Nielsen R, Yang Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol Biol Evol 2005; 22:2472-2479; PMID:16107592; http://dx.doi.org/10.1093/molbev/msi237
- Anisimova M, Yang Z. Multiple hypothesis testing to detect lineages under positive selection that affects only a few sites. Mol Biol Evol 2007; 24:1219-1228; PMID:17339634; http://dx.doi.org/10.1093/molbev/msm042
- 1000 Genomes Project Consortium, Durbin RM, Abecasis GR, Altshuler DL, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA. A map of human genome variation from population-scale sequencing. Nature 2010; 467:1061-1073; PMID:20981092; http://dx.doi.org/10.1038/nature09534
- Auton A, Fledel-Alon A, Pfeifer S, Venn O, Segurel L, Street T, Leffler EM, Bowden R, Aneas I, Broxholme J, et al. A fine-scale chimpanzee genetic map from population sequencing. Science 2012; 336:193-198; PMID:22422862; http://dx.doi.org/10.1126/science.1216872
- Quach H, Wilson D, Laval G, Patin E, Manry J, Guibert J, Barreiro LB, Nerrienet E, Verschoor E, Gessain A, et al. Different selective pressures shape the evolution of toll-like receptors in human and african great ape populations. Hum Mol Genet 2013; 22:4829-4840; PMID:23851028; http://dx.doi.org/10.1093/hmg/ddt335
- Grossman SR, Andersen KG, Shlyakhter I, Tabrizi S, Winnicki S, Yen A, Park DJ, Griesemer D, Karlsson EK, Wong SH, et al. Identifying recent adaptations in large-scale genomic data. Cell 2013; 152:703-713; PMID:23415221; http://dx.doi.org/10.1016/j.cell.2013.01.035
- Forni D, Cagliani R, Tresoldi C, Pozzoli U, De Gioia L, Filippi G, Riva S, Menozzi G, Colleoni M, Biasin M, et al. An evolutionary analysis of antigen processing and presentation across different timescales reveals pervasive selection. PLoS Genet 2014; 10:e1004189; PMID:24675550; http://dx.doi.org/10.1371/journal.pgen.1004189
- Forni D, Cagliani R, Pozzoli U, Colleoni M, Riva S, Biasin M, Filippi G, De Gioia L, Gnudi F, Comi GP, et al. A 175 million year history of T cell regulatory molecules reveals widespread selection, with adaptive evolution of disease alleles. Immunity 2013; 38:1129-1141; PMID:23707475; http://dx.doi.org/10.1016/j.immuni.2013.04.008
- Barreiro LB, Ben-Ali M, Quach H, Laval G, Patin E, Pickrell JK, Bouchier C, Tichit M, Neyrolles O, Gicquel B, et al. Evolutionary dynamics of human toll-like receptors and their different contributions to host defense. PLoS Genet 2009; 5:e1000562; PMID:19609346; http://dx.doi.org/10.1371/journal.pgen.1000562
- Fagny M, Patin E, Enard D, Barreiro LB, Quintana-Murci L, Laval G. Exploring the occurrence of classic selective sweeps in humans using whole-genome sequencing data sets. Mol Biol Evol 2014; 31:1850-68; PMID:24694833; http://dx.doi.org\10.1093/molbev/msu118
- Zeng K, Fu YX, Shi S, Wu CI. Statistical tests for detecting positive selection by utilizing high-frequency variants. Genetics 2006; 174:1431-1439; PMID:16951063; http://dx.doi.org/10.1534/genetics.106.061432
- Fairfax BP, Humburg P, Makino S, Naranbhai V, Wong D, Lau E, Jostins L, Plant K, Andrews R, McGee C, et al. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science 2014; 343:1246949; PMID:24604202; http://dx.doi.org/10.1126/science.1246949
- Ramaswami G, Li JB. RADAR: A rigorously annotated database of A-to-I RNA editing. Nucleic Acids Res 2014; 42:D109-13; PMID:24163250; http://dx.doi.org/10.1093/nar/gkt996
- Wang J, Duncan D, Shi Z, Zhang B. WEB-based GEne SeT AnaLysis toolkit (WebGestalt): Update 2013. Nucleic Acids Res 2013; 41:W77-83; PMID:23703215; http://dx.doi.org/10.1093/nar/gkt439
- Athanasiadis A, Placido D, Maas S, Brown BA,2nd, Lowenhaupt K, Rich A. The crystal structure of the zbeta domain of the RNA-editing enzyme ADAR1 reveals distinct conserved surfaces among Z-domains. J Mol Biol 2005; 351:496-507; PMID:16023667; http://dx.doi.org/10.1016/j.jmb.2005.06.028
- Athanasiadis A. Zalpha-domains: At the intersection between RNA editing and innate immunity. Semin Cell Dev Biol 2012; 23:275-280; PMID:22085847; http://dx.doi.org/10.1016/j.semcdb.2011.11.001
- Poulsen H, Nilsson J, Damgaard CK, Egebjerg J, Kjems J. CRM1 mediates the export of ADAR1 through a nuclear export signal within the Z-DNA binding domain. Mol Cell Biol 2001; 21:7862-7871; PMID:11604520; http://dx.doi.org/10.1128/MCB.21.22.7862-7871.2001
- Strehblow A, Hallegger M, Jantsch MF. Nucleocytoplasmic distribution of human RNA-editing enzyme ADAR1 is modulated by double-stranded RNA-binding domains, a leucine-rich export signal, and a putative dimerization domain. Mol Biol Cell 2002; 13:3822-3835; PMID:12429827; http://dx.doi.org/10.1091/mbc.E02-03-0161
- Anderson P, Kedersha N. RNA granules: Post-transcriptional and epigenetic modulators of gene expression. Nat Rev Mol Cell Biol 2009; 10:430-436; PMID:19461665; http://dx.doi.org/10.1038/nrm2694
- Masliah G, Barraud P, Allain FH. RNA recognition by double-stranded RNA binding domains: A matter of shape and sequence. Cell Mol Life Sci 2013; 70:1875-1895; PMID:22918483
- Barraud P, Banerjee S, Mohamed WI, Jantsch MF, Allain FH. A bimodular nuclear localization signal assembled via an extended double-stranded RNA-binding domain acts as an RNA-sensing signal for transportin 1. Proc Natl Acad Sci U S A 2014; 111:E1852-61; PMID:24753571; http://dx.doi.org/10.1073/pnas.1323698111
- ENCODE Project Consortium, Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature 2012; 489:57-74; PMID:22955616; http://dx.doi.org/10.1038/nature11247
- Enard D, Messer PW, Petrov DA. Genome-wide signals of positive selection in human evolution. Genome Res 2014; 24:885-95; PMID:24619126; http://dx.doi.org\10.1101/gr.164822.113
- Fraser HB. Gene expression drives local adaptation in humans. Genome Res 2013; 23:1089-1096; PMID:23539138; http://dx.doi.org/10.1101/gr.152710.112
- Tomaselli S, Locatelli F, Gallo A. The RNA editing enzymes ADARs: Mechanism of action and human disease. Cell Tissue Res 2014; 356:527-532; PMID:24770896; http://dx.doi.org/10.1007/s00441-014-1863-3
- Chen JY, Peng Z, Zhang R, Yang XZ, Tan BC, Fang H, Liu CJ, Shi M, Ye ZQ, Zhang YE, et al. RNA editome in rhesus macaque shaped by purifying selection. PLoS Genet 2014; 10:e1004274; PMID:24722121; http://dx.doi.org/10.1371/journal.pgen.1004274
- Yeo J, Goodman RA, Schirle NT, David SS, Beal PA. RNA editing changes the lesion specificity for the DNA repair enzyme NEIL1. Proc Natl Acad Sci U S A 2010; 107:20715-20719; PMID:21068368; http://dx.doi.org/10.1073/pnas.1009231107
- Bahn JH, Lee JH, Li G, Greer C, Peng G, Xiao X. Accurate identification of A-to-I RNA editing in human by transcriptome sequencing. Genome Res 2012; 22:142-150; PMID:21960545; http://dx.doi.org/10.1101/gr.124107.111
- Doria M, Neri F, Gallo A, Farace MG, Michienzi A. Editing of HIV-1 RNA by the double-stranded RNA deaminase ADAR1 stimulates viral infection. Nucleic Acids Res 2009; 37:5848-5858; PMID:19651874; http://dx.doi.org/10.1093/nar/gkp604
- Doria M, Tomaselli S, Neri F, Ciafre SA, Farace MG, Michienzi A, Gallo A. ADAR2 editing enzyme is a novel human immunodeficiency virus-1 proviral factor. J Gen Virol 2011; 92:1228-1232; PMID:21289159; http://dx.doi.org/10.1099/vir.0.028043-0
- Phuphuakrat A, Kraiwong R, Boonarkart C, Lauhakirti D, Lee TH, Auewarakul P. Double-stranded RNA adenosine deaminases enhance expression of human immunodeficiency virus type 1 proteins. J Virol 2008; 82:10864-10872; PMID:18753201; http://dx.doi.org/10.1128/JVI.00238-08
- Biswas N, Wang T, Ding M, Tumne A, Chen Y, Wang Q, Gupta P. ADAR1 is a novel multi targeted anti-HIV-1 cellular protein. Virology 2012; 422:265-277; PMID:22104209; http://dx.doi.org/10.1016/j.virol.2011.10.024
- Sawyer SL, Elde NC. A cross-species view on viruses. Curr Opin Virol 2012; 2:561-568; PMID:22835485; http://dx.doi.org/10.1016/j.coviro.2012.07.003
- Kantaputra PN, Chinadet W, Ohazama A, Kono M. Dyschromatosis symmetrica hereditaria with long hair on the forearms, hypo/hyperpigmented hair, and dental anomalies: Report of a novel ADAR1 mutation. Am J Med Genet A 2012; 158A:2258-2265; PMID:22821605; http://dx.doi.org/10.1002/ajmg.a.35488
- Sharma R, Wang Y, Zhou P, Steinman RA, Wang Q. An essential role of RNA editing enzyme ADAR1 in mouse skin. J Dermatol Sci 2011; 64:70-72; PMID:21788117; http://dx.doi.org/10.1016/j.jdermsci.2011.06.013
- Vilella AJ, Severin J, Ureta-Vidal A, Heng L, Durbin R, Birney E. EnsemblCompara GeneTrees: Complete, duplication-aware phylogenetic trees in vertebrates. Genome Res 2009; 19:327-335; PMID:19029536; http://dx.doi.org/10.1101/gr.073585.107
- Guindon S, Delsuc F, Dufayard JF, Gascuel O. Estimating maximum likelihood phylogenies with PhyML. Methods Mol Biol 2009; 537:113-137; PMID:19378142; http://dx.doi.org/10.1007/978-1-59745-251-9_6
- Darriba D, Taboada GL, Doallo R, Posada D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011; 27:1164-1165; PMID:21335321; http://dx.doi.org/10.1093/bioinformatics/btr088
- Delport W, Poon AF, Frost SD, Kosakovsky Pond SL. Datamonkey 2010: A suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 2010; 26:2455-2457; PMID:20671151; http://dx.doi.org/10.1093/bioinformatics/btq429
- Wilson DJ, Hernandez RD, Andolfatto P, Przeworski M. A population genetics-phylogenetics approach to inferring natural selection in coding sequences. PLoS Genet 2011; 7:e1002395; PMID:22144911; http://dx.doi.org/10.1371/journal.pgen.1002395
- Cereda M, Sironi M, Cavalleri M, Pozzoli U. GeCo++: A C++ library for genomic features computation and annotation in the presence of variants. Bioinformatics 2011; 27:1313-1315; PMID:21398667; http://dx.doi.org/10.1093/bioinformatics/btr123
- Thornton K. Libsequence: A C++ class library for evolutionary genetic analysis. Bioinformatics 2003; 19:2325-2327; PMID:14630667; http://dx.doi.org/10.1093/bioinformatics/btg316
- Wright S. Genetical structure of populations. Nature 1950;166:247-249; PMID:15439261; http://dx.doi.org/10.1038/166247a0
- Fay JC, Wu CI. Hitchhiking under positive darwinian selection. Genetics 2000; 155:1405-1413; PMID:10880498
- Johnson AD, Handsaker RE, Pulit SL, Nizzari MM, O'Donnell CJ, de Bakker PI. SNAP: A web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics 2008; 24:2938-2939; PMID:18974171; http://dx.doi.org/10.1093/bioinformatics/btn564
- Cooper GM, Stone EA, Asimenos G, NISC Comparative Sequencing Program, Green ED, Batzoglou S, Sidow A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res 2005; 15:901-913; PMID:15965027; http://dx.doi.org/10.1101/gr.3577405