
ABSTRACT
Many thousand long non-coding (lnc) RNAs are mapped in the human genome. Time consuming studies using reverse genetic approaches by post-transcriptional knock-down or genetic modification of the locus demonstrated diverse biological functions for a few of these transcripts. The Human Gene Trap Mutant Collection in haploid KBM7 cells is a ready-to-use tool for studying protein-coding gene function. As lncRNAs show remarkable differences in RNA biology compared to protein-coding genes, it is unclear if this gene trap collection is useful for functional analysis of lncRNAs. Here we use the uncharacterized LOC100288798 lncRNA as a model to answer this question. Using public RNA-seq data we show that LOC100288798 is ubiquitously expressed, but inefficiently spliced. The minor spliced LOC100288798 isoforms are exported to the cytoplasm, whereas the major unspliced isoform is nuclear localized. This shows that LOC100288798 RNA biology differs markedly from typical mRNAs. De novo assembly from RNA-seq data suggests that LOC100288798 extends 289kb beyond its annotated 3' end and overlaps the downstream SLC38A4 gene. Three cell lines with independent gene trap insertions in LOC100288798 were available from the KBM7 gene trap collection. RT-qPCR and RNA-seq confirmed successful lncRNA truncation and its extended length. Expression analysis from RNA-seq data shows significant deregulation of 41 protein-coding genes upon LOC100288798 truncation. Our data shows that gene trap collections in human haploid cell lines are useful tools to study lncRNAs, and identifies the previously uncharacterized LOC100288798 as a potential gene regulator.
Abbreviations
| lncRNA, Long non-coding RNA | = | mRNAs, mRNA (protein coding) |
| RNA-Seq, RNA-sequencing, high throughput sequencing of cDNA ends | = |
Introduction
Long non-coding (lnc) RNAs can regulate gene expression and are abundant in the genomes of various organisms.Citation1 The human genome has been reported to contain about 60,000 lncRNA genesCitation2 and an increasing number is suggested to play important roles in cancer and other diseases.Citation3,4 Moreover, several lncRNAs were reported to serve as disease biomarkersCitation5,6 and potential drug targets.Citation7-9 LncRNAs display a wide range of functions from nuclear scaffoldingCitation10 to post-transcriptional mRNA regulation by “sponging” regulatory miRNAs,Citation11 transcriptional gene activation or repression by binding and guiding histone modifiers to target genesCitation12,13 and silencing by transcription interferenceCitation14 (reviewed inCitation15). Apart from the basic difference between the functions of lncRNAs and mRNAs, lncRNAs also display a number of RNA biology features that make their identification and functional studies more challenging than that of protein-coding genes.Citation16 These features include: low, tissue-specific expression,Citation17 nuclear localizationCitation18 and inefficient co-transcriptional splicing,Citation19,20 transcription initiation from repeat rich regionsCitation21 and unusually high isoform heterogeneity.Citation22
To date, the majority of functional lncRNA studies have depleted the lncRNA of interest via post-transcriptional knock-down approaches using shRNAs,Citation23 morpholinosCitation24 or modified DNA antisense oligos that target nuclear localized transcripts.Citation25 Based on the atypical RNA biology features described above, these approaches might not be generally suited to study a wide range of lncRNAs. For example, shRNAs are unlikely to target lncRNAs in the nucleus,Citation26 while morpholinos or antisense oligos might be difficult to design for targeting complex lncRNA loci expressing multiple lncRNA isoforms. Importantly, lncRNAs that act solely by their transcription will not be affected by post-transcriptional knockdowns.Citation14 Genetic manipulations might be a more universal approach to interfere with lncRNA function independent of RNA-biology features. These manipulations have become more feasible due to the emergence of fast and simple genome editing technologies such as CRISPR/Cas9.Citation27 One strategy is the genetic deletion of the whole gene body or the promoter of the lncRNA of interest.Citation28-31 While this approach is appealing due to its relative simplicity, there is a risk of simultaneous deletion of potential genomic regulatory elements that could be located in the gene body of the targeted lncRNA, which can make the interpretation of the resulting phenotype problematic.Citation16,32 Therefore genetic insertion of transcriptional terminator sequences, or “gene traps” may be preferable to gene deletions as they are less likely to disrupt regulatory elements.
Gene trap technology is based on the insertion of “truncation cassettes," typically containing polyA signals, shortly after the transcriptional start site (TSS) of the lncRNA to stop RNA Polymerase II transcription and create functional lncRNA “knock-outs”. Gene trap mutagenesis has been used extensively in the mouse to identify and study protein-coding genes.Citation33 Classical gene trap cassettes carry a strong splice acceptor and a reporter protein terminated by a strong polyA signal. This cassette is introduced into the cell line using retroviral vectors that cause random integration into the genome. If the cassette integrates into the gene body of a transcribed gene in the correct transcriptional orientation, transcription will be stopped.Citation34 An analysis of mouse lines carrying gene trap insertions that had the goal to identify key genes expressed during embryonic development, led to the isolation of the lncRNA called gene trap locus 2 (Gtl2) gene.Citation35 It is also known as maternally expressed 3 (Meg3), since it is exclusively expressed from the maternally inherited allele, a phenomenon known as genomic imprinting.Citation36 Gtl2/Meg3 was shown to be functional in mouse developmentCitation37,38 and human disease.Citation39 Subsequently a targeted approach was used to introduce polyA signals from rabbit β globin or simian virus 40 to truncate the imprinted Airn, Kcnq1ot1 and Ube3a-as lncRNAs in mice, as occurs in gene trap truncations. These approaches successfully stopped lncRNA transcription and identified these lncRNAs as transcriptional regulators of developmentally important protein-coding genes.Citation40-43 The advent of genome editing tools such as zinc finger nucleases opened the possibility to use similar approaches also for human cells. In this way polyA containing truncation cassettes were targeted at the abundantly expressed MALAT1 lncRNA causing efficient truncation in a number of human cell lines.Citation44
Insertion of a truncation cassette may interrupt cis-acting genetic elements, and although this is notably less likely than with gene body deletions, it should be controlled for. Such controls include insertion of the truncation cassette at different sites, creating lncRNA truncations of different lengths, or the use of non-functional truncation cassette insertions.Citation32 An important advantage of the gene trap approach is the possibility to restore lncRNA transcription by removing the stop cassette.Citation45 However, restoration of lncRNA function will only be possible if continuous expression is required for function.Citation32,46 Taken together, this indicates that the truncation of lncRNAs is a useful tool to study their function in both mouse and human, and in particular gene trap insertion is a well-controlled high-throughput method to achieve this.
While tools to perform genetic manipulations in mouse and human systems are becoming faster and simpler, the creation of a human cell line carrying a lncRNA truncation may still require optimization and thus is time consuming and resource intensive. Therefore it would be beneficial to use existing lncRNA knockout resources to rapidly investigate a lncRNA of interest. Such a resource was reported for protein-coding genes as the “Human Gene Trap Mutant Collection”.Citation45 This library is comprised of a collection of monoclonal cell lines that carry an insertion of a gene trap cassette in the gene body of a large number of genes.Citation45 The cell line used to establish this resource is a nearly haploid (except for chromosome 8) malignant myeloid lineage cell line called KBM7.Citation47 As most chromosomes are present in only one copy, the integration of a gene trap cassette results in a full knock-out in KBM7 cells. Since the creation of this gene trap collection did not select for a particular type of genomic locus, it contains cell lines with gene trap cassettes inserted into protein-coding genes, as well as into transcribed non-coding regions, including various annotated lncRNAs (visit https://opendata.cemm.at/barlowlab/ for the location of all cassettes). Thus, the KBM7 “Human Gene Trap Mutant Collection” could represent a massive ready-to-use collection of lncRNA knockouts that may be useful for rapidly assessing human lncRNA function. Importantly, efficiency of a gene trap depends on splicing from a neighboring exon of the “trapped” gene to the gene trap cassette.Citation34 In the above described case of Gtl2/Meg3 efficient splicing was expected as this lncRNA produces a number of spliced isoforms.Citation48 While “Human Gene Trap Mutant Collection” has been proven to efficiently stop transcription of protein-coding genes, the usefulness of this approach to study lncRNAs is unclear, since it was shown that many of them are inefficiently spliced or completely unspliced.Citation19
In this study we aimed to close this knowledge gap and test if “Human Gene Trap Mutant Collection” can be successfully used for studying lncRNAs, even the inefficiently spliced ones. For this purpose we focused on a lncRNA, that was identified in a tiling array based study to be close to the SLC38A4 protein-coding gene and named “SLC38A4-down”.Citation49 It is noteworthy that mouse Slc38a4 shows imprinted expression in extra-embryonic, embryonic and adult tissuesCitation50 as well as in cell culture cells.Citation51 No lncRNA has been reported to be involved in regulating Slc38a4 imprinted expression which is, to date, considered a solo imprinted gene (http://igc.otago.ac.nz). Although SLC38A4 was not reported to show imprinted expression in human, the identification of SLC38A4-down lncRNA close to the SLC38A4 gene allowed the possibility that this lncRNA might be involved in transcriptional regulation of SLC38A4. SLC38A4-down lncRNA was predicted from its expression profile, that lacked exon peaks, to be mainly unspliced and was also shown to be nuclear-localized.Citation49 These features make it an unsuitable target for a post-transcriptional knock-down approach. Importantly, we identified a number of gene trap insertions in the gene body of this lncRNA in the “Human Gene Trap Mutant Collection” in the correct transcriptional orientation, which allowed us to use this lncRNA as a model in our study. We first identified that SLC38A4-down corresponds to the LOC100288798 lncRNA annotated by NCBI RNA reference sequences collection (RefSeqCitation52). Using publicly available RNA-seq data from various tissues and cellular fractions we found the LOC100288798 lncRNA to be ubiquitously expressed, inefficiently spliced and polyadenylated. Unspliced isoforms are retained in the nucleus, while minor spliced isoforms are exported to the cytoplasm. We also extended the annotation of this lncRNA by showing that it is twice as long as the annotated version, as it is transcribed over 500 kilobases (kb) and overlaps the SLC38A4 protein-coding gene in multiple tissues. Thus we suggest renaming it SLC38A4-AS lncRNA in accordance with recent lncRNA nomenclature guidelines.Citation53 We then obtained three independent KBM7 clones harboring gene trap cassettes in the body of SLC38A4-AS predicted to stop transcription 3kb and 100kb downstream of its transcription start. RNA sequencing (RNA-seq) of control and SLC38A4-AS truncated cell lines showed that SLC38A4-AS was efficiently truncated, which resulted in genome-wide gene expression changes. We applied further stringent filtering to identify a small list of the most plausible SLC38A4-AS targets. Based on this data we conclude that lncRNA truncations available in the “Human Gene Trap Mutant Collection” are useful to study lncRNAs, making this resource a valuable tool for studying lncRNA function in a human system. In order to maximize the usefulness of this data for the scientific community we provide a UCSC genome browser hub to display all the RNA-Seq data as well as the information on gene trap insertion sites presented in this paper (https://opendata.cemm.at/barlowlab/).
Results
LOC100288798 is a ubiquitously expressed, inefficiently processed lncRNA
LOC100288798 lncRNA is annotated by several reference gene databases including RefSeqCitation52 and GENCODE v19 (http://www.gencodegenes.org/releases/19.html,Citation54) as a 269kb lncRNA on human chromosome 12 (A). LOC100288798 lncRNA was also identified by RNA-seq based human lncRNA annotation studies such as Cabili et alCitation17 and MiTranscriptomeCitation2 (A). It is an intergenic lncRNA that initiates from its own CpG island (CpG: 106) and is located between the SLC38A2 and SLC38A4 protein-coding genes (A). Despite the 35 spliced expressed sequence tags (ESTs) mapped to this locus (Human ESTs That Have Been Spliced public track at UCSC Genome Browser), LOC100288798 remains an uncharacterized lncRNA.
Figure 1. RefSeq LOC100288798 is a ubiquitously expressed, inefficiently processed lncRNA (A) Overview of the genomic locus. UCSC Genome Browser screenshot – from top to bottom: CpG island annotation, RefSeq Genes annotation, GENCODE v19 annotation, UCSC Genes annotation, MiTranscriptome lncRNA transcripts,Citation2 Cabili et al lincRNA transcriptsCitation17.(B) LOC100288798 is a ubiquitously expressed lncRNA. Heat map shows expression level of SLC38A2, SLC38A4 and LOC100288798 (marked as “lncRNA” throughout the figure) in multiple tissues and cell types. Letters in brackets after the name of each sample indicate the source and the type of RNA-seq (see Table S1A for details of abbreviations). Expression levels of SLC38A4 and LOC100288798 were calculated as average RPKMs of RefSeq isoforms (SLC38A2 – 1 isoform: NM_018976, SLC38A4 – 2 isoforms: NM_018018 and NM_001143824, LOC100288798 – 5 isoforms: NR_125377, NR_125378, NR_125379, NR_125380, and NR_125381), values are displayed inside each cell. Heat map color legend is displayed on the left. (C) LOC100288798 lncRNA is variably spliced in different tissues. Heat map shows splicing efficiency (Methods) of LOC100288798 and 2 protein-coding genes TPB, SLC38A2 (well-spliced ubiquitously expressed protein coding gene controls) in publicly available total RNA-seq data (Table S1A). Calculated splicing efficiency is displayed inside each cell. Heat map color legend is displayed on the left. (D) Visual inspection of ENCODE HeLa RNA-seq of various cell and RNA fractions suggests that LOC100288798 is an inefficiently processed lncRNA. From top to bottom: Chromosome position; RefSeq annotation; ENCODE HeLa RNA-seq sequencing data. RNA-seq data is displayed using the public ENCODE RNA-seq (CSHL) hub in the UCSC browser (only Replicate 2 from 2 replicates available at ENCODE RNA-seq (CSHL) hub is displayed). From top to bottom: PolyA+ RNA-seq of the whole cell Reverse and Forward strand show absence of SLC38A4 expression from the reverse strand and visible expression from the forward strand corresponding to LOC100288798. Dashed orange lines indicate chromosome positions of RefSeq annotated exons of LOC100288798. Comparison of signal intensities between polyA+ and polyA- indicates LOC100288798 is inefficiently spliced as it appears more abundant in polyA- fraction. Cytoplasm RNA-seq indicates that only spliced and polyadenylated LOC100288798 transcripts can be exported to the cytoplasm (compare peaks in polyA+ and no peaks in polyA-). Nuclear RNA-seq indicates nuclear enrichment of LOC100288798 unspliced form (compare nucleus polyA- to cytoplasm polyA-). RNA-seq tracks are displayed with the default ENCODE RNA-seq (CSHL) hub scale (range - from 0 to 100). (E) PolyA+ enrichment. Bar plot shows PolyA+ enrichment (calculated as the ratio between RPKM in PolyA+ and PolyA- RNA fractions) of the 4 indicated genes in HeLa cells (ENCODE RNA-seq data). RPKMs and consequently PolyA+ enrichment were calculated for spliced isoforms (RPKM over exons, blue bars) and unspliced isoforms (RPKM over whole gene body, purple bars) of the 4 genes. PolyA+ enrichment is a relative value, therefore we indicated the absolute RPKM values of spliced and unspliced isoforms in PolyA- fraction below each respective bar. (F) Nuclear enrichment. Bar plot shows nuclear enrichment (calculated as the ratio between RPKM in nuclear and cytoplasmic fractions) of the 4 indicated genes in HeLa cells (ENCODE RNA-seq data). RPKMs and consequently nuclear enrichment were calculated for spliced isoforms (RPKM over exons, blue bars) and unspliced isoforms (RPKM over whole gene body, purple bars) of the 4 genes in PolyA+ (darker bars) and PolyA- (lighter bars) fractions. Nuclear enrichment is a relative value, therefore we indicated the absolute RPKM values in cytoplasmic fraction below each respective bar.
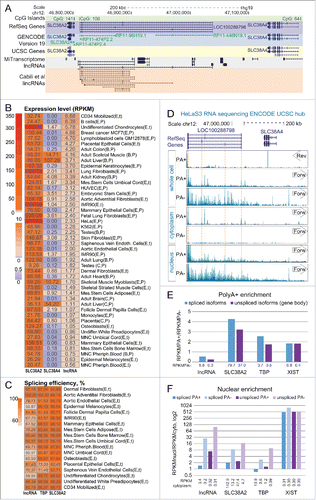
We characterized this lncRNA using publicly available human RNA-seq data. We first asked which tissues and cell types express LOC100288798 lncRNA using polyA+ enriched and total (rRNA depleted) RNA-seq data from 34 healthy primary tissues and cell types as well as 4 normal and 3 malignant cell lines originating from different studies (total of 41 different cell types, 5 of which were replicated twice giving the total of 46 samples, Table S1A, Methods). We downloaded the raw RNA-seq data, aligned it with STARCitation55 and obtained an average of 186 million uniquely mapped reads per sample (ranging from 16 to 371 million reads, Table S1A). We next calculated expression levels of LOC100288798 lncRNA and its neighboring SLC38A2 and SLC38A4 genes by calculating average RPKMs of RefSeq annotated spliced isoforms (Methods). shows the obtained expression profile in the 46 analyzed samples. This shows that SLC38A2 is highly expressed (RPKM>9) in every analyzed sample and its ubiquitous expression is known (http://www.proteinatlas.org/ENSG00000134294-SLC38A2/tissue). In contrast, SLC38A4 is expressed (RPKM > 0.5) in just 18/46 samples (which corresponds to 15/41 different cell/tissue types) with highest expression in liver and skeletal muscle, consistent with previous observations (The Human Protein Atlas: http://www.proteinatlas.org/ENSG00000139209-SLC38A4/tissue, Expression Atlas: http://www.ebi.ac.uk/gxa/genes/ENSG00000139209). Similar to SLC38A2, the LOC100288798 lncRNA is expressed (RPKM>0.5) in all analyzed samples. Notably, the highest LOC100288798 lncRNA expression level, achieved in CD34 cells, is 48 fold lower than the highest expression level of SLC38A2 and 16 fold lower than that of SLC38A4, consistent with previous observations that lncRNAs are generally lower expressed than protein-coding genes.Citation17 We next asked if LOC100288798 lncRNA expression showed any correlation with the 2 nearby genes, since it is known that some lncRNAs can regulate their nearby protein-coding genes.Citation13,40 Although LOC100288798 lncRNA and its closest gene SLC38A2 were both ubiquitously expressed, they did not show correlation in expression level (Pearson correlation = 0.17, 46 samples). This, together with the fact that their transcription start sites are separated by 11kb and located in 2 separate CpG islands, indicates that these 2 genes initiate from independent promoters, and while they seem to belong to the same transcription network, the regulation of their expression level may be independent. LOC100288798 lncRNA and SLC38A4 showed a striking difference in cell type expression profile and no correlation in expression among the tested tissues and cell types (Pearson correlation = 0.07, 46 samples), which indicates independent transcriptional regulation. When we analyzed correlation only in tissues that express both LOC100288798 lncRNA and SLC38A4, correlation between these 2 genes was still negligible (Pearson correlation = 0.11, 18 samples), although the small number of samples may impede the correlation analysis. In summary, we found that LOC100288798 is a ubiquitously, but lowly expressed lncRNA displaying no striking correlation with the expression of its neighboring protein-coding genes.
We next characterized the efficiency of LOC100288798 lncRNA splicing as it was previously reported that lncRNAs show reduced co-transcriptional splicing when compared to mRNAs.Citation19 We used publicly available total RNA-seq data (Table S1A) from 18/41 of the above described different cell types and estimated splicing efficiency for LOC100288798 lncRNA and 2 protein-coding genes TBP and SLC38A2 that were expressed in the same cell types. We calculated the average splicing efficiency of all unique splice sites from all isoforms of the analyzed gene (C) by calculating RPKMs of exonic and intronic 45bp regions surrounding the splice site (Methods). As expected, both protein-coding genes showed high splicing efficiency with an average of 93.0% (TBP) and 96.5% (SLC38A2) among analyzed cell types. Importantly only 2 (for TBP) and one (for SLC38A2) cell types showed splicing efficiencies of less than 90%. The result was different for the LOC100288798 lncRNA. Here average splicing efficiency was 76.0%, with 14/18 cell types showing splicing efficiency of less than 90% and 7 - lower than 70%. It is noteworthy that low splicing efficiencies are not restricted to low expression levels. For example undifferentiated chondrocytes (59% splicing efficiency) and IMR90 cells (68% splicing efficiency) are in the top 25% and top 50% highest expressing tissues for the LOC100288798 lncRNA (B). This indicates that LOC100288798 lncRNA is less well spliced compared to protein-coding genes, and that splicing is variable in different cell types.
It has been reported that lncRNAs tend to be nuclear localized,Citation18,56 and that nuclear export depends on the addition of a 3’ polyA tail, which is connected to splicing.Citation57 To investigate the processing of LOC100288798 lncRNA we used publicly available ENCODE RNA-seq data from nuclear, cytoplasmic, as well as whole cell fractions (Table S1B). Importantly, the RNA from each cell fraction was further divided into polyA enriched (polyA+) and polyA depleted (polyA-), thus providing a source of information about the polyadenylation and cellular localization of LOC100288798 lncRNA spliced/polyadenylated as well as unspliced isoforms. We first visually inspected the RNA-seq signal obtained from HeLa cells in the LOC100288798/SLC38A4 region using the ENCODE (CSHL) RNA-seq hub in the UCSC browser (D). The SLC38A4 protein-coding gene is not expressed in whole cell polyA+ RNA-seq as indicated by the absence of RNA-Seq signal over exons on the reverse strand (D, whole cell, top box, Arrow marked 'Rev'), consistent with our expression calculation (B, RPKM of SLC38A4 = 0.00). In contrast, the forward strand showed abundant RNA-seq signals over LOC100288798 lncRNA exons in polyA+ and over the whole gene body in polyA- RNA-seq data. Interestingly, the signal intensities in polyA+ and polyA- data were comparable confirming inefficient splicing of LOC100288798 lncRNA (D, whole cell, middle and bottom box, Arrow marked 'Forw'). In the cytoplasmic fraction, only spliced and polyadenylated isoforms of LOC100288798 lncRNA were detectable as RNA-seq signal over exons in the polyA+, but not in the polyA- fraction (D, cytoplasm). In the nuclear fraction, stronger RNA-seq signals were detectable over the LOC100288798 lncRNA gene body in polyA- than in the polyA+ faction, and no clear enrichment of exonic signals was visible. This indicated that spliced isoforms of LOC100288798 lncRNA were exported to the cytoplasm, whereas mainly unspliced isoforms were retained in the nucleus.
To quantify this visual analysis we calculated RPKM values for LOC100288798 lncRNA and 2 control protein-coding genes, SLC38A2 and TBP, as well as for the XIST lncRNA, which is known to be polyadenylated, nuclear localized and well spliced.Citation58 We first estimated the efficiency of polyadenylation by calculating the ratio of RNA-seq signal in the PolyA+ fraction over the PolyA- fraction (RPKMPA+/RPKMPA-, E). We observed that all the 3 control genes, which are known to be polyadenylated, show ratios of ∼2-4 for both unspliced (whole gene body, purple bars) and spliced (blue bars) isoforms, indicating efficient polyadenylation of these transcripts. Spliced and unspliced isoforms of LOC100288798 lncRNA showed ratios smaller than 1, indicating inefficient polyadenylation of LOC100288798 lncRNA (E, lncRNA). We next assessed the efficiency of cytoplasmic export by calculating the ratio of RNA-seq signals in the nuclear over the cytoplasmic cell fraction for both PolyA+ and PolyA- RNA-seq datasets (F). As expected, PolyA- fraction showed high ratios for both spliced and unspliced isoforms of the 4 tested genes, indicating nuclear enrichment of unprocessed isoforms (F, light blue and light purple bars). In contrast, the pattern of nuclear enrichment of polyadenylated spliced and unspliced isoforms differed notably between the analyzed genes (F, blue and purple bars). While spliced and polyadenylated XIST isoforms were almost exclusively present in the nucleus (ratio: ∼500), similar processed isoforms of the protein-coding genes SLC38A2 and TBP showed low ratios, indicating no nuclear enrichment (F). Consistent with our conclusions from visual inspection, spliced isoforms of LOC100288798 lncRNA were exported to the cytoplasm and showed low ratios similar to the analyzed protein-coding genes (RPKM of spliced isoforms in the polyadenylated cytoplasmic fraction = 3.4, while RPKM of spliced isoforms in the polyadenylated whole cell fraction = 2.3, B). Interestingly, unspliced isoforms of LOC100288798 lncRNA showed high ratios, indicating nuclear enrichment. Similar profiles were observed for LOC100288798 lncRNA in 4 other analyzed cell lines (Fig. S1, Table S1B). In summary, this analysis showed that LOC100288798 lncRNA is inefficiently polyadenylated in comparison to SLC38A2, TBP and XIST. Whereas the small fraction of polyadenylated LOC100288798 lncRNA isoforms is exported to the cytoplasm, the major fraction consisting of unspliced isoforms is highly enriched in the nucleus. Therefore we show that LOC100288798 lncRNA polyadenylation and nuclear enrichment profiles are distinct from both XIST lncRNA and protein-coding genes.
De novo assembly of LOC100288798 exon structure identifies overlap with SLC38A4
Visual inspection of the RNA-seq data indicated that LOC100288798 transcription extends over the downstream SLC38A4 gene (see continuous RNA-seq signal in D), in spite of RefSeq annotating the 3’ end of LOC100288798 112kb upstream from SLC38A4 ( top). Interestingly, human spliced ESTs annotated continuous spliced transcripts overlapping SLC34A4 (). We next aimed to fully annotate LOC100288798 using publicly available RNA-seq data from multiple cell types. We limited this analysis to reads aligned to a 1 Mega base pairs (Mb) region (chr12:46,500,000-47,500,000) around LOC100288798. We extracted reads from each of the 46 aligned RNA-seq samples used in B (polyA+ as well as ribosomal depleted total RNA-seq) and performed de novo assembly using the Cufflinks software.Citation59 Thus, we obtained 46 assemblies, which we merged using Cuffmerge softwareCitation59 to create an integrative de novo annotation of the investigated region (see for selected isoforms and Table S1Cfor all the isoforms annotated in the region). Importantly, we identified exon models that share exons with LOC100288798 lncRNA and overlap the SLC38A4 protein coding gene, indicating that LOC100288798 is a 558kb long lncRNA (chr12:46777455-47335067, see CUFF.281.86 in and Table S1C). Visual inspection of the LOC100288798 RNA-seq signal in cell types ranging from the highest expressing (CD34 cells, RPKM=6.68) to lowest expressing (MNC Peripheral blood, RPKM=0.56), showed that extended transcription persists independently of expression level (). Therefore LOC100288798 lncRNA is consistently overlapping the SLC38A4 protein-coding gene and should be renamed as SLC38A4-AS according to the recently suggested nomenclature.Citation53 As this nomenclature also appears more intuitive we have used it for the remainder of this study.
Figure 2. LOC100288798 exon structure assembly from various tissues extends its annotation to over 500kb overlapping SLC38A4.UCSC Genome Browser screen shot of the studied locus (chr12:46,772,500-47,422,500). From top to bottom: Chromosome position and the scale; RefSeq gene annotation (all annotated isoforms are displayed), spliced human ESTs (12/35 ESTs displayed), transcriptome assembly of the locus obtained in this study (Results, Methods). Note that only selected transcripts are shown (11/167 de novo isoforms of LOC100288798 and 4/43 de novo isoforms of SLC38A4), and that both EST and transcriptome assembly data reveal extension of LOC100288798 to over 500kb in length. RNA-seq tracks from ENCODE/CSHL UCSC hub with the titles containing cell type name, RNA-seq type and transcriptional orientation are displayed below. Only total whole cell RNA-seq is displayed. Bottom: normalized RNA-seq signal from wild type human haploid KBM7 cell lines (merged data from 2 wild type clones sequenced in this study, Methods). For all RNA-seq tracks: only forward strand (Plus Signal) is displayed.

Gene trap insertion in the haploid human KBM7 efficiently truncates SLC38A4-AS lncRNA
Although visual inspection of RNA-seq and exon model assembly suggested that SLC38A4-AS lncRNA is a single lncRNA gene it is possible that this was an artifact resulting from multiple short overlapping lncRNAs. To address this issue we used the haploid KBM7 cell line for which a collection of gene trap insertion clones was readily available.Citation45 We first confirmed that SLC38A4-AS was expressed in wildtype KBM7 cells and found it well expressed over the predicted length by visual inspection of RNA-Seq data performed in this study ( bottom). Next, we identified 3 cell lines from the publicly available KBM7 gene trap collection where independent insertion events inserted gene trap cassettes in the correct orientation into the gene body of SLC38A4-AS (). Two of these cell lines were predicted to stop SLC38A4-AS transcription at 2,904bp (3kb1 and 3kb2, A), and one cell line at 103,958bp (100kb) downstream of the RefSeq annotated transcription start. To create biological replicates of the single 100kb insertion cell line we recovered 2 batches of this cell line from frozen stocks and cultured them in parallel (100kb1, 100kb2, Methods, A). The production of KBM7 gene trap insertion cell lines is a multi-step procedure including infection of cells with the gene trap cassette, fluorescent activated cell sorting (FACS) and clonal expansion to obtain monoclonal cultures. Also different people may have handled different cell lines. These factors are possible sources of gene expression differences, so we controlled for these factors using multiple control cell lines. First, we obtained 3 different KBM7 cell lines that had not undergone the gene trap insertion procedure but were handled by different people and had different passage numbers (wild type: WT1, WT2, WT3, A). Second, to control for potential effects of the gene trap insertion procedure, we obtained 2 cell lines with gene trap insertions not in SLC38A4-AS, but in the HOTTIP lncRNA gene body of which one was predicted to stop HOTTIP lncRNA and one was not, based on mapping cassette insertion orientation (C1 and C2, , A). To eliminate further batch effects from handling cells and preparing RNA and RNA-Seq libraries, all cell lines were obtained as frozen stocks and recovered, cultured and harvested at the same time by one person. Similarly one person performed RNA extraction and library preparation.
Table 1. Stop cassette insertions overview.
Figure 3. Gene trap technology allows truncation of SLC38A4-AS lncRNA in human haploid KBM7 cell line (A) Overview of the experimental design: SLC38A4-AS truncation and control cell lines used in the study. Top row: Wild type KBM7 cells underwent the gene trap insertion procedure and single clones were selected and expanded to a monoclonal population. Three independently obtained clones with gene trap cassettes mapping within the gene body of SLC38A4-AS lncRNA were available (see ). Two monoclonal cell lines with independent insertion events that integrated a gene trap cassette 3kb downstream of SLC38A4-AS transcription start site (TSS) were available (3kb1 and 3kb2). Only one monoclonal cell line had a gene trap insertion 100kb downstream of the downstream of SLC38A4-AS TSS. Therefore we prepared biological replicates by performing independent thawing and culturing procedures (100kb1 and 100kb2). Left column: We obtained 3 wild type KBM7 control cell lines, which did not undergo any gene trap insertion procedure, were not monoclonal and were cultured by different people at different times prior to culturing for this analysis (WT1, WT2 and WT3). Middle column: To control for changes during gene trap insertion and selection procedure we obtained 2 KBM7 cell lines that did undergo gene trap insertion within the body of HOTTIP lncRNA and were monoclonally expanded (C1 and C2) (see ). (B) Ploidy of KBM7 cell lines assessed by cell size. Bar plot shows peak cell size measured for 9 cultured KBM7 cell lines (Methods). All the cell lines were thawn and processed in one batch by the same person. Cell size was measured at the first splitting (3 days post-thawing, dark gray bars), second splitting (6 days post-thawing, medium gray bars), and prior to harvesting (8 days post-thawing, light gray bars). (C) Ploidy of KBM7 cell lines assessed by total DNA amount. Bar plot shows total DNA mass isolated from 20 million cells. DNA mass in the plot is normalized to WT1 sample (absolute value for WT1 is 109 μg). (D) Confirmation of successful SLC38A4-AS truncation by RT-qPCR. Top: schematic representation of the locus (drawn to scale). Blue bars show RefSeq annotation of LOC100288798 and SLC38A4 genes. Black bar underneath shows the extended annotation of LOC100288798 (SLC38A4-AS) obtained in this study (). White arrows inside the bars indicate transcriptional orientation of the gene. Below the positions of stop cassette insertions () and RT-qPCR probes are displayed (). Bottom: Expression profiling of SLC38A4-AS in the KBM7 cell lines (described in A). Error bars represent standard deviation from 3 RT-qPCR technical replicates. Bars are ordered from left to right as listed (top to bottom) in the legend on the right. For each RT-qPCR probe the expression level in WT1 is set to 100%.
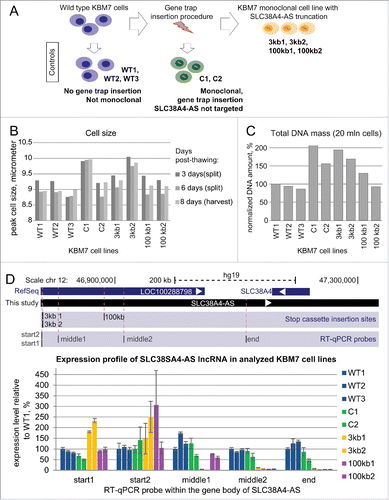
After recovery we cultured the cell lines for 8 days and 2 passages. We measured the cell size prior to splitting and harvesting (Methods) and noticed that the C1 and 3kb2 cell lines showed increased peak cell size (B). It has been reported previously that cell size increases with ploidyCitation60 and therefore this result indicated that these KBM7 cell lines were not haploid. We then harvested the cells using 20 million cells for DNA isolation and 100 million cells for RNA isolation. As a further test for ploidy we measured the DNA amount obtained from the 20 million cells. Consistent with the cell size measurements we found that C1 and 3kb2 cells displayed 2 and 1.5 fold increase in DNA amount compared to wild type controls. Additionally we found that 3kb1 and C2 also showed 2 and 1.5 fold increase in DNA amount (C). As both cell size and DNA content are indirect measures of ploidy we performed karyotyping of selected cell lines (3kb2, 100kb, C1, WT2, Supplemental Figs. 2–5). This confirmed the haploid state of the 100kb and WT2 cell lines and the diploid state of the 3kb2 and C1 cell lines. Also we did not detect large scale chromosomal aberrations in addition to the known t(9;22) translocation.Citation45 This indicated that most cell lines that underwent gene trap insertion and clonal expansion procedure either gained diploidy, or were a mixture of haploid and diploid cells. Note that KBM7 cell ploidy does not interfere with any downstream analyses, as RNA-seq expression analyses are performed on normalized values that correct for increased RNA amount in diploid versus haploid cells. To confirm that both alleles carry the gene trap insertion and to validate the integrity of the genomic locus after the gene trap insertion we performed 2 DNA blotting assays for the 2 3kb truncation cell lines (see Supplemental Figure. 6A-B for maps of restriction enzymes and probes). First, we identified the expected 2.8kb (size of the gene trap cassette) increase in size of a genomic EcoRV fragment including the gene trap insertion site in 3kb1 and 3kb2 cell lines compared to wildtype (Fig. S6C–E). Second, we identified the expected size reduction of a genomic EcoRI/BamHI fragment due to the insertion of a BamHI site with the gene trap cassette (Fig. S6D–F). Importantly, we did not detect any wildtype fragment in the 3kb1 and 3kb2 cell lines indicating that gene trap insertion occurred in sorted haploid cells and that diploidy arose after cassette insertion. Therefore it can be concluded that both chromosomes in diploid cells carry the gene trap.
We next tested if gene trap cassette insertions 3kb and 100kb downstream of the SLC38A4-AS transcription start indeed stopped transcription elongation. We designed 5 RT-qPCR probes inside the body of the SLC38A4-AS gene (, D). We placed 2 probes (start1 and start2) upstream of the 3kb stop cassette insertion site, one probe (middle1) downstream of the 3kb, but upstream of the 100kb stop cassette, and 2 probes (middle2 and end) downstream of the 100kb stop cassette insertion site. Note, that the “end” RT-qPCR probe lies outside of the gene body of RefSeq annotated LOC100288798. We used all these probes to define the profile of SLC38A4-AS transcription in 3 wild type (blue, WT1-3), 2 control (green, C1, C2), 2 3kb (yellow, 3kb1, 3kb2) and 2 100kb (purple, 100kb1, 100kb2) SLC38A4-AS truncation cell lines (D bar plot). Since SLC38A4-AS RNA-Seq signals decreased from 5’ to the 3’ end (see ), we normalized expression levels to WT1 for each RT-qPCR probe. All cell lines displayed transcription of SLC38A4-AS upstream of the 3kb gene trap insertion site, with increased expression in the 2 3kb truncation cell lines (D, start1 and start2). Consistent with expectations, the 2 3kb truncation cell lines displayed dramatic reduction of SLC38A4-AS transcription 28kb downstream of the transcription start (25kb downstream the truncation site, middle 1), while the 100kb truncation cell lines displayed continuous SLC38A4-AS transcription since these cell lines carried the stop cassette downstream of this RT-qPCR probe (D, middle1). Expression levels downstream from the 100kb stop cassette were dramatically reduced in both the 3kb and 100kb truncation cells, but largely unchanged in the wild type and the control cells (D, middle2 and end). Thus, RT-qPCR confirmed that the SLC38A4-AS lncRNA was successfully truncated in KBM7 cells at the gene trap cassette insertion sites. Importantly, lack of transcription at multiple positions downstream of the gene trap cassette insertion sites in all tested cell lines further indicates that the SLC38A4-AS gene generates a single 558kb long transcript.
Table 2. RT-qPCR probes for analyzing expression profile of SLC38A4-AS lncRNA.
RNA-seq of KBM7 cell lines with truncated SLC38A4-AS lncRNA confirms a single transcription unit overlapping SLC38A4
As RT-qPCR only detects transcripts in a very narrow window at the chosen primer position, we performed RNA-seq to obtain a global picture of SLC38A4-AS truncation. We chose 2 cell line replicates per group: wild type (WT2 and WT3), control (C1 and C2), 3kb (3kb1 and 3kb2) and 100kb (100kb1 and 100kb2). 50bp single-end RNA-seq and alignment using STARCitation55 produced an average of 35 million uniquely mapped reads per sample (standard deviation – 1.0 million reads) (Table S1D). Visual inspection showed similar SLC38A4-AS RNA-seq profiles in wild type and control cells with a similar decrease in signal from 5’ to 3’ end as seen before (compare and A wild type). While the 3kb2 cell line showed a clear reduction of RNA-seq signal downstream the 3kb stop cassette insertion site, 3kb1 seemed to have residual transcription and thus truncation might be less efficient. Both the 100kb1 and 100kb2 replicates displayed a similar SLC38A4-AS expression profile with a clear reduction in RNA-seq signal after the gene trap cassette insertion point. We next quantified the RNA-seq signal strength to confirm the conclusions made from visual inspection. To obtain a transcription profile of SLC38A4-AS in each cell line we calculated RPKM of 5 regions (relative to the transcription start): 0-3kb, 3kb-50kb, 50kb-100kb, 100kb-300kb and 300kb-600kb (B). WT, C and 100kb cell lines showed a 3-fold RPKM drop from 0-3kb to 3kb-50kb regions with detectable expression in the 3kb-50kb window (RPKM > 0.2), which is consistent with the reported RNA-seq signal decrease from 5’ to the 3’end for lncRNAs.Citation61 In the 3kb cell lines the gene trap cassette stopped SLC38A4-AS and removed this pattern, and therefore all windows downstream of the gene trap cassette insertion site showed very low expression (RPKM <= 0.05). WT and C cell lines showed a further 1.8- and 1.7-fold signal drop between 50-100kb and 100kb-200kb regions confirming the visual impression that the RNA-Seq signal decreases from 5’ to 3’ end in WT and C cell lines. The 100kb cell lines follow the expression pattern of the WT and C cell lines but the signal drops to very low expression levels (RPKM <= 0.02) after the gene trap insertion site.
Figure 4. RNA-seq confirms truncation and continuity of the SLC38A4-AS lncRNA gene. (A) SLC38A4-AS RNA-seq signal of the 8 clones analyzed in D. Top: schematic representation of the locus (as described for D). Bottom: RNA-seq signal, normalized to sample read number, pink dots indicate RNA-seq signal that exceeds the range presented inside the box. Type of the cell line is indicated on the left, name of the cell line is indicated on the right. Vertical dashed red lines indicate position of the 3kb and 100kb stop cassettes. Low density of RNA-seq signal piles indicate low expression and the smallest size corresponds to 1 read. (B) Expression profile of different regions of SLC38A4-AS lncRNA in the RNA-Seq data shown in (A). Bar plots show RPKM of the regions of SLC38A4-AS indicated on the X axis for 4 types of cell lines (as grouped on A). RPKM value for each clone type is averaged from 2 cell lines, error bars show the RPKM values of the 2 samples. Numbers above the bars show the plotted value. Note that this analysis allows the comparison of regions within one cell line but not between cell lines. (C) Expression profile comparison of SLC38A4-AS between analyzed clones. Bar plot shows RPKM of the regions of SLC38A4-AS indicated on the X axis for each cell line type normalized to the value for “Wild type”. Normalized RPKM values are the average of 2 cell lines of each type, indicated by the error bars.
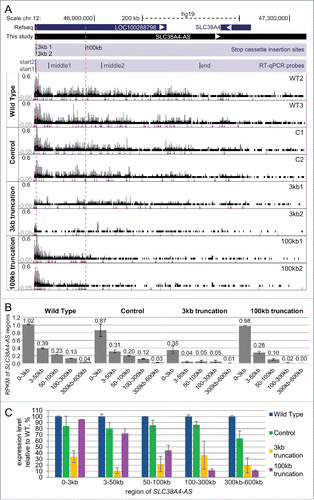
To allow a direct comparison between cell lines we plotted the expression of each window relative to WT (set to 100%, C). The first window (0-3kb) showed similar expression in WT, C and 100kb cell lines but was ∼3-fold lower in 3kb cell lines. The following window (3-50 kb) showed a further ∼3-fold reduction in expression for the 3kb cell lines whereas all other cell lines showed similar expression of SLC38A4-AS. At the 50-100kb window the expression of the 100kb truncation cell lines started to drop ∼2-fold but were still ∼2-fold higher than 3kb truncation cell lines. In the last 2 windows (100-300kb, 300kb-600kb) the 100kb truncation cell lines showed a low residual expression level (∼10-fold less compared to WT, 6-8 fold less than C) whereas 3kb truncation cell lines showed a 2-3 fold higher residual expression likely due to the inefficient truncation of the 3kb1 cell line identified by visual inspection. We observed that while difference between 100kb replicates was low for every analyzed SLC38A4-AS region (maximal difference between 100kb1 and 100kb2 constituted 37% of the mean, at 100-300kb, C), the difference between 3kb1 and 3kb2, which resulted from different integration events, was more notable (maximal difference between 3kb1 and 3kb2 constituted 126% of the mean, at 100-300kb, C). 3kb1 showed 2.5- to 4.4-fold higher expression compared to 3kb2 in the 4 windows downstream the 3kb gene trap insertion (B). In spite of increased RNA-seq signal compared to the 3kb2 and 100kb truncations, the 3kb1 cell line did not reach the wild type and control levels of SLC38A4-AS transcription (C). It was possible that the difference in truncation efficiency between the 3kb1 and the 3kb2 cell lines was due to sequence aberrations in the splice acceptor sequence in the gene trap cassette. Therefore we amplified and sequenced this region of the gene trap cassette and found it to be identical in the 3kb1, 3kb2 and C1 cell lines (Supplemental Fig. 7A–B). In order to discriminate inefficient truncation of SLC38A4-AS from a contamination of the 3kb1 cell line with wildtype cells we performed a PCR assay with primers directly flanking the cassette insertion site. We identified the correct wildtype PCR fragment in all tested cell lines, except for 3kb1 and 3kb2 cell lines, where the cassette insertion separates the primers by 2.8kb, which is not amplified in our settings (Supplemental Fig. 7C). Importantly this indicates that the 3kb1 cell line is not contaminated with wildtype cells to a detectable level. In summary, RNA-seq confirms efficient truncation of SLC38A4-AS in both 100kb truncation cell lines and the 3kb2 cell line. Interestingly, the global transcriptional analysis of 3kb1 truncation revealed reduced truncation efficiency in this cell line.
SLC38A4-AS truncation causes deregulation of several genes in trans
To investigate if SLC38A4-AS truncation had an effect on gene expression in cis or in trans, we calculated expression level of RefSeq annotated protein-coding genes and performed differential gene expression analysis using Cuffdiff software.Citation62 We compared WT2, WT3, C1 and C2 (4 control replicates) with 3kb1, 3kb2, 100kb1 and 100kb2 (4 targeted cell line replicates). This analysis produced a list of 120 significantly differentially expressed genes (excluding chromosomes X and Y, Table S1E) that we further filtered by requiring a 3-fold expression change between the 2 conditions, which resulted in a list of 41 protein-coding genes (Table S1 Elines in bold). This number of genes was 5-fold higher than the average number of genes differentially expressed (3-fold expression change) in 11 mock comparisons (Table S1F). Interestingly, the 41 genes were distributed across almost all chromosomes (Table S1 Elines in bold). One gene (CD163L1) was down-regulated and 3 (CD9, EMP1 and CRY1) were upregulated on chromosome 12, the same chromosome that contains SLC38A4-AS. However, these genes were located 33-61 million bp distant from SLC38A4-AS and therefore their regulation is more likely to arise from trans effects. We then calculated expression levels (FPKM, Methods) of the 41 significantly deregulated genes reported above by Cuffdiff for each of the 8 samples separately to allow unsupervised clustering to be performed (Methods). This analysis correctly grouped the 2 biological replicas of the 3kb truncation, 100kb truncation replicates and wild type replicates (A). Interestingly, C1 and C2, although in the same branch, did not group together, which may relate to the fact that C1 carries a truncated HOTTIP lncRNA (gene trap insertion in sense to HOTTIP, ), while C2 had an antisense insertion in the HOTTIP gene body, and therefore should not truncate ().
Figure 5. Genome-wide differential expression analysis reveals deregulation of protein-coding genes in trans upon SLC38A4-AS lncRNA truncation (A) Expression level of genes differentially expressed between SLC38A4-AS truncation cell lines and the 4 control cell lines allows unsupervised clustering of the cell lines that resembles the different cell groups. Heat map shows expression level (FPKM, Methods) of genes (name indicated on the right) with significant differential expression (p < 0.01, >3 fold expression change, Methods) between 2 conditions: no SLC38A4-AS truncation (WT2, WT3, C1, C2) and genetic truncation of SLC38A4-AS (3kb1, 3kb2, 100kb1, 100kb2). Expression values are normalized to the mean FPKM among all 8 samples. Mean is set to 1. Names of genes that form the filtered stringent list of deregulated genes (, Methods) are displayed in bold blue font. Heat map color legend is displayed on the right. (B) and (C) Examples of up- and downregulated protein coding genes from the stringent list (). CD9 is markedly upregulated (B) and RORB is markedly downregulated (C) upon truncation of SLC38A4-AS. UCSC Genome Browser screen shots show normalized RNA-seq signal. Top to bottom: Chromosome position, RefSeq gene annotation, RNA-seq signal, normalized to sample read number, from eight sequenced cell lines. Each box shows the same range from 0 to 0.6, only forward strand is shown. Pink dots indicate RNA-seq signal that exceeds the range presented inside the box. Name of cell line is indicated on the left.
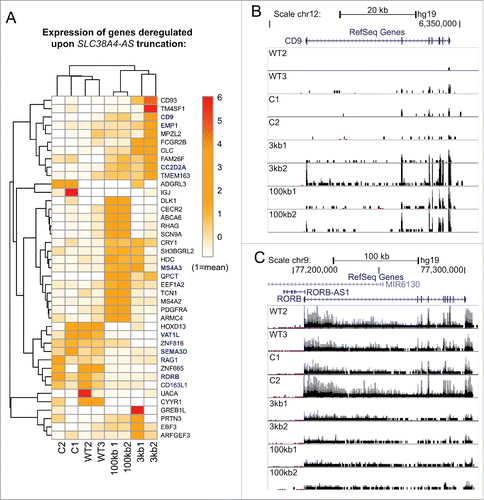
We then performed further filtering to create a small stringent list of the deregulated genes. To increase the stringency of the list of differentially expressed genes we performed 3 filtering steps. First, we filtered out genes that showed significant differential expression between wild type (WT2, WT3) and control (C1, C2) samples and thus might be differentially expressed due to the effect of the gene trap cassette insertion procedure (3/41 genes). Second, we removed the genes that showed differential expression between 3kb and 100kb truncation thus restricting our list to the genes that are regulated by the part of SLC38A4-AS lncRNA downstream of the 100kb cassette insertion site (18/41 genes). Third, we only retained the genes that were differentially expressed in both pairwise comparisons of control to 3kb (3kb1, 3kb2 vs C1, C2, 12 genes) and control to 100kb samples (100kb1, 100kb2 vs C1, C2, 24 genes). These filtering steps resulted in a stringent list of 6 protein-coding genes (). Three of these genes, including CD9 (B) were upregulated upon SLC38A4-AS truncation, and 3, including RORB (C), were downregulated. In summary, these data show that genetic truncation of SLC38A4-AS lncRNA results in genome-wide gene expression changes and provides a stringent list of 6 potential SLC38A4-AS target genes.
Table 3. Stringent list of genes affected by SLC38A4-AS lncRNA truncation.
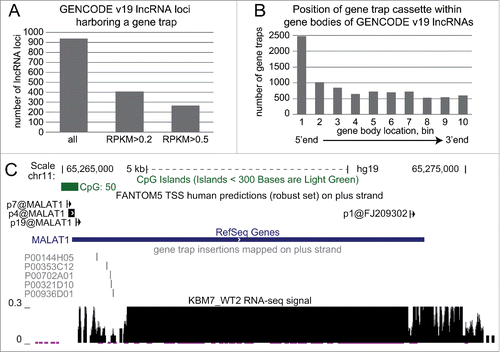
As these results provide clear evidence for the use of the “Human Gene Trap Mutant Collection” to study lncRNAs, we investigated how many lncRNAs can be potentially studied using this collection in its current form. First, we calculated expression for all GENCODE v19 lncRNAs in the 2 wild type cell lines investigated in this study (WT1, WT2) and found 2,307 non-overlapping lncRNA loci to be expressed (i.e. to express at least one lncRNA isoform with RPKM>0.2). Next, we investigated how many GENCODE v19 lncRNAs contained a gene trap insertion on the same strand and found that 938 lncRNAs are likely to be truncated in one of the available cell lines (A left bar). Overlapping these 2 data sets revealed 409 expressed lncRNAs carrying a gene trap insertion in the current collection (A middle bar). If we set a higher expression cut off of RPKM>0.5, we find 266 lncRNAs carrying a gene trap (A right bar). We investigated the position of gene trap insertions relative to the transcriptional start site of lncRNAs and found enrichment at the 5’ end (B). Finally we examined the well-studied lncRNA MALAT1 and identified 5 gene trap insertions close to the 5’ end corresponding to potential knock-out cell lines.()
Figure 6. Haploid gene trap collection represents a rich resource for quick functional assessment of hundreds of lncRNAs. (A) Hundreds of GENCODE v19 lncRNAs expressed in KBM7 cell line are targeted by a gene trap insertion. Bar plot shows number of non-overlapping GENCODE v19 lncRNA loci that contain a gene trap cassette in the same transcriptional orientation in KBM7 clones within the “Human Gene Trap Mutant Collection” (left bar, Methods), and the number of these lncRNA loci that are expressed (middle bar, loci that contain lncRNA transcripts expressed with RPKM > 0.2) and well expressed (right bar, loci that contain lncRNA transcripts expressed with RPKM > 0.5) in wild type KBM7 cells. (B) Gene trap cassettes are preferentially inserted at the 5’ end of lncRNAs. Bar plot shows the number of gene trap cassettes inserted into different regions in the gene bodies of GENCODE v19 lncRNA. Numbers correspond to 10 equally sized, non-overlapping regions investigated for each gene. (C) Five genetic truncations of the well-known lncRNA MALAT1 are available within the “Human Gene Trap Mutant Collection." Shown is the UCSC browser screen shot of the MALAT1 gene region. From top to bottom: chromosome scale, CpG island annotation (UCSC track), FANTOM5 TSS predictions (robust set)Citation82 on the plus strand, RefSeq gene annotation, position of gene trap insertion cassettes available (plus strand), normalized RNA-seq signal from WT2 KBM7 cell line showing wild type expression of MALAT1.
Discussion
Here we report the first use of the “Human Gene Trap Mutant Collection”Citation45 to study the function of a human lncRNA. To demonstrate the utility of this collection we analyzed cell clones that successfully truncated the SLC38A4-AS lncRNA (renamed from LOC10028879) that displays RNA biology features distinct from protein-coding genes, including low expression and inefficient splicing. We also investigated this gene trap collection as a whole for its suitability for the study of lncRNAs, and identified hundreds of lncRNAs with gene trap insertions including the well-studied MALAT1 lncRNA. Therefore we demonstrate here the utility of the “Human Gene Trap Mutant Collection” for studying lncRNAs and also identify SLC38A4-AS as a very long and novel functional regulatory lncRNA.
Prior to analyzing gene trap efficiency we examined the RNA biology of the SLC38A4-AS lncRNA that has not previously been characterized. We showed that SLC38A4-AS, unlike many lncRNAs, does not show tissue-specific expression. While tissue-specificity is often considered as an indication of functionality,Citation63 several ubiquitously expressed lncRNAs have been proven to play important gene regulatory roles.Citation40,64 We used a set of public RNA-seq data to show that SLC38A4-AS lncRNA is inefficiently spliced and that the major unspliced isoform is nuclear localized. Importantly, by comparing SLC38A4-AS to 2 control protein-coding genes, we show that the unspliced isoforms we detect for SLC38A4-AS are not just an intronic signal. We conclude this from the finding that the polyadenylation and localization profiles for unspliced isoforms of the protein-coding genes, which are notably highly expressed, differ dramatically from that of SLC38A4-AS. Minor spliced isoforms of SLC38A4-AS lncRNA are well detectable in the cytoplasm and thus are exported and likely stable. SLC38A4-AS lncRNA is thus a transcript with unusual RNA biology features different from protein-coding genes. We performed de novo transcriptome assembly in the region and were able to show that transcription of SLC38A4-AS extends 289kb downstream the RefSeq annotated 3’ end and overlaps the downstream SLC38A4 gene.
We then obtained KBM7 cells from the “Human Gene Trap Mutant Collection” with gene trap insertions at 2 different locations (3kb and 100kb downstream of the transcription start) in the gene body of SLC38A4-AS lncRNA to test whether the unusual RNA biology features interfered with efficient truncation by the gene trap cassette. By using qRT-PCR as well as RNA-seq we identified one cell line with efficient truncation at both insertion sites. This data not only verifies that gene trap insertions in KBM7 cells efficiently truncate SLC38A4-AS lncRNA, but also confirms our prediction of the extended SLC38A4-AS lncRNA length. Detailed RNA-seq analysis identifies that the 3kb1 cell line shows less efficient truncation compared to 3kb2 cell line despite these cell lines sharing same gene trap insertion site. Differences in the efficiency of truncation between different insertion sites have been documented for one truncation of the Airn lncRNA. In this case a truncation cassette insertion at 3 different genomic loci caused successful truncation of the lncRNA whereas the same cassette was highly inefficient when inserted into a CpG island.Citation14 Also differences in the gene trap efficiency of protein-coding genes were noted for different cassette integration sites.Citation45 However, a difference between similar insertion sites as shown for 3kb1 and 3kb2, was surprising. DNA gel blotting experiments did not detect a large scale rearrangement of the chromosomal locus with the gene trap insertion nor did they identify a contamination of the 3kb1 cell line with wildtype cells. As DNA blotting might not be sensitive enough to detect a low level of wildtype cell contamination we validated these results by a PCR assay. We also validated that the splice acceptor sequence was unchanged in the 3kb1 cell line. Taken this together, an aberration of the genetic sequence in 3kb1 is unlikely to be the cause for the reduced efficiency of transcription termination in this cell line. A connection between chromatin structure and transcription termination has been made in yeastCitation65 and it has been suggested that local chromatin changes influence splicing.Citation66 It is therefore possible that cell line specific local chromatin changes result in differences in truncation efficiency at identical cassette integration points. As global gene-expression analysis showed high similarity between both 3kb truncation cell lines, it is highly likely that the residual level of SLC38A4-AS expression seen in 3kb1 cell line is not sufficient to maintain a wildtype gene expression pattern. We therefore conclude that gene trap approach used for the “Human Gene Trap Mutant Collection” is a useful tool to truncate inefficiently spliced lncRNAs.
We noted that 2 qRT-PCR primers that are close to the 3kb truncation cassette insertion site, showed elevated qRT-PCR signals specifically in 3kb truncation cell lines. Interestingly RNA-seq did not support this elevated transcription on the forward strand, which corresponds to SLC38A4-AS lncRNA, but identified strong transcription from the reverse strand directly at the gene trap insertion site that was absent in the control cell lines. Similar transcription on the reverse strand at the gene trap insertion point was visible albeit at lower levels for the 100kb truncation cell lines (Fig. S8). Thus, we provide evidence that the gene trap cassette used for the “Human Gene Trap Mutant Collection” can drive transcriptional activity, which was suggested earlier.Citation45 Additionally, we also show that this activity can be strong (2-fold higher than SLC38A4-AS) and therefore has to be carefully considered when expression of genes in close proximity is affected, as transactivation of protein-coding genes by the transcriptionally active viral LTRs was reported in gene therapy patients.Citation67
Interestingly, SLC38A4-AS lncRNA shares several unusual RNA biology features with the imprinted mouse lncRNA Airn that also overlaps in antisense orientation and silences the protein-coding Igf2r gene. Although Airn lncRNA is inefficiently spliced, 5% of its nascent transcripts are spliced and give rise to stable lncRNAs that are exported to the cytoplasm.Citation20 These spliced Airn lncRNA isoforms are, however, not connected to the silencing mechanism.Citation14 Interestingly, truncation experiments identified that Airn silences Igf2r due to its transcriptional overlap, a phenomenon called transcriptional interference.Citation14,40 The Airn lncRNA also silences 2 protein-coding genes that it does not overlap in a tissue-specific manner, likely by targeting repressive chromatin to the promoters of these genes.Citation68,69 We tested if the SLC38A4-AS lncRNA silences the SLC38A4 protein-coding gene that it overlaps and/or the SLC38A2, which is located 10kb away in a similar manner. We were surprised to find that neither SLC38A4 nor SLC38A2 protein-coding genes were affected by the truncation of SLC38A4-AS lncRNA. In addition, expression analysis of multiple tissues did not show anti-correlating expression patterns of the 2 protein-coding genes with the lncRNA. In the case of imprinted expression involving a repressor lncRNA, such a pattern would not be expected as one allele expresses the protein-coding gene whereas the other allele expresses the lncRNA. Therefore we conclude that SLC38A4-AS lncRNA most likely does not share functional similarities with the imprinted Airn lncRNA and does not control SLC38A4 or SLC38A2 protein-coding gene expression. This data supports the hypothesis that imprinted expression of Slc38a4 in the mouse, is rodent-specific as it is also absent from the pig and cow.Citation70,71
In order to test the functional importance of SLC38A4-AS lncRNA as a gene regulator in trans, we tested whether the truncation of the lncRNA resulted in gene expression changes in KBM7 cells. In accordance with recent guidelines established for the correct analysis of lncRNA knockout experiments, we included a number of controls in this analysis.Citation32 First, we excluded batch effects from the handling of cells by having all cell lines cultured in parallel by one person. Second, it is possible that the gene trap insertion disrupts an important genetic element that causes gene expression changes of protein coding genes that are not dependent on the lncRNA. Therefore we analyzed 3 independently derived SLC38A4-AS lncRNA truncation cell lines: 3kb1, 3kb2 with an identical insertion site and 100kb. As controls we used 2 batches of wild type KBM7 cell lines. In order to identify genes that are specifically deregulated upon truncation we performed differential gene expression analysis between SLC38A4-AS lncRNA truncation cell lines (3kb1, 3kb2, 100kb1, 100kb2), and all control cell lines (C1, C2 that carried gene traps at unrelated loci, WT1, WT2 that lacked gene traps). This analysis resulted in 120 differentially expressed genes, 41 of which were more that 3-fold up/downregulated in the truncation cell lines. Importantly, none of the differentially expressed genes were located in close proximity to the SLC38A4-AS lncRNA, as reported for well-known cis-regulating lncRNAs, such as Airn or KCNQ1OT1.Citation36 Whereas clustering based on the 41 differentially expressed genes allowed correct grouping of the replicates, performing a similar analysis using the expression of genes in the 10Mbp region around SLC38A4-AS resulted in sporadic clusters. This indicates a lack of consistent changes of these genes between control and truncation cell lines and thus further supports a lack of cis-acting regulatory function of SLC38A4-AS lncRNA (Supplemental Fig. 9). We plotted expression values of the 41 significantly deregulated genes in all the 8 cell lines as a heat map and found that a number of genes seemed to be specifically expressed in one control cell type (C1/C2 or WT1/WT2) or in one of the truncation cell types (3kb1, 3kb2 or 100kb1, 100kb2) rather than in all control vs. all truncation cell types. Therefore, we also performed pairwise comparisons to remove these genes. We do note that this approach limits the part of the lncRNA examined for function to regions downstream of the 100kb truncation cassette (i.e., spanning ∼400kb of the SLC38A4-AS gene body). Additionally we note that the function of the first 3kb of SLC38A4-AS lncRNA (upstream 3kb gene trap cassette position) was not assessed in our study while it is possible that this region may possess a function.
Of the 6 genes that pass the most stringent filters for deregulation in SLC38A4-AS lncRNA truncation cell lines 2 are of special interest. The first is the clusters of differentiation proteins 9 (CD9) that belongs to the superfamily of tetraspanins, integral membrane proteins that play a role in multiple biological processes by interacting with membrane proteins like other tetraspanins, growth factors and cytokine receptors. Clinical data suggests that CD9 is a suppressor of metastasis and modulates tyrosine kinase receptor signaling in cancer.Citation72 CD9 is also a marker for haematopoietic stem cellsCitation73 and was found to be up-regulated upon induction of pluripotent stem cells (iPS) from KBM7 cells,Citation74 although it is not necessary for pluripotency in miceCitation75. The second gene is RAR-related orphan receptor B (RORB or RORβ), which encodes the nuclear receptor subfamily 1, group F, member 2 (NR1F2) protein that binds to DNA and inhibits transcription.Citation76 RORB has not been implicated in cancer,Citation77 but was associated with the mammalian circadian clock,Citation76 and was found to be a member of a gene hub that discriminates human iPS from stem cells.Citation78 Little is known about the importance of RORB in KBM7 cells, however it is unlikely to be essential for this cell line as an unbiased mapping of gene trap insertions in this cell line identified 7 gene trap insertion events in this gene with 4 predicted to stop RORB transcription.Citation79
As mentioned above, gene trap cassette removal could provide a valuable rescue control. Human Haploid Gene Trap Collection contains cell lines with gene trap cassettes flanked by loxP sites that thus can be removed by Cre recombinase expression and the expression of the targeted genes might be restored. Among the analyzed SLC38A4-AS truncation cell lines, 3kb1 and 3kb2 did have loxP sites flanking the gene trap cassette, while 100kb truncation cell lines did not. However, while removal of the truncation cassette by expressing the Cre recombinase and subsequent re-expression of full-length SLC38A4-AS lncRNA could restore its wildtype gene expression pattern, it is possible that the gene expression changes initiated by SLC38A4-AS lncRNA are accompanied by changes in secondary gene expression or in the epigenetic landscape that may not be immediately reversible. Such an example was reported for the Airn lncRNA that silences the Igf2r protein coding gene in early development. After silencing, by Airn transcription, Igf2r acquires repressive epigenetic marks on its promoter and silencing is stably maintained in the absence of Airn lncRNA expression.Citation46 Therefore we conclude that the use of multiple control cell lines may prove a more efficient way to study lncRNA function in comparison to multiple targeted cell lines.
In summary, this report shows that the “Human Gene Trap Mutant Collection” is a useful tool to study lncRNA function. Importantly, we identified 857 GENCODE v19 lncRNAs (http://www.gencodegenes.org/releases/19.html) for which KBM7 gene trap insertions cell lines are available (Methods and https://opendata.cemm.at/barlowlab/). Similar to protein-coding genes, the gene trap cassette preferentially inserts close to the 5’ end of lncRNAs, which is useful for functional studies as the bulk of the lncRNA will not be produced.Citation45 We found that 409 lncRNA loci with a gene trap insertion show an RPKM >0.2 (RPKM of at least one isoform in the locus) and 266 have an RPKM>0.5, which constitutes respectively 44% and 28% of all GENCODE v19 lncRNA gene trap insertion clones. It is to date unclear, which expression cutoff can be used to indicate functional importance, and it is therefore possible that also lncRNAs expressed to a lower level have a functional importance. The “Human Gene Trap Mutant Collection” could be a useful tool to study this question. Also KBM7 cells can be converted to iPS cells and have the potential to be differentiated into different lineages.Citation74 Therefore it is possible that lncRNAs that are lowly expressed in wild-type KBM7 cells are highly expressed in a different lineage, which can also be studied using KBM7 iPS cells. Gene trap KBM7 cells from the “Human Gene Trap Mutant Collection” are simple to obtain and culture and therefore offer a rich resource that allows analysis of lncRNA function in a human system. This is illustrated by the example of the MALAT1 lncRNA. This lncRNA was previously studied using a truncation cassette,Citation44 an experiment that includes (1) cloning of the truncation cassette for homologous recombination (2) optimizing endonuclease to cleave genomic DNA at the desired position (3) selection, screening, expansion and testing of correctly targeted clones.Citation44 This effort linearly increases for the production of cell lines with different truncation cassette insertion sites. In contrast to this time-consuming approach, 5 KBM7 gene trap clones are readily available truncating the MALAT1 lncRNA at different positions close to the 5’ end that are ready to be analyzed.
According to our results, the unusual RNA biology inherent to many lncRNAs does not influence the ability of the gene trap cassette to stop lncRNA transcription, and gene trap truncations are therefore a universal tool for studying a wide range of lncRNAs. The availability of multiple control cell lines is an additional advantage and allows thorough artifact control. Using SLC38A4-AS lncRNA as an example, we also show that gene trap resource together with the already available RNA-seq resources from the ENCODE consortium allow fast characterization of a lncRNA of interest. We anticipate that similar integrated approaches that make efficient use of these publicly available resources will allow the fast functional characterization of the many lncRNAs found in the human genome.
Methods
RPKM calculation
RPKMs were calculated using RPKM_count.py from RSeQC package (https://code.google.com/p/rseqc/) using –skip-multi-hits option.
Estimating expression of LOC100288798 and SLC38A4 in various tissues and cell types
Various public raw RNA-seq datasets (See Table S1A) were downloaded as fastq files and aligned with STAR using the following command: STAR_2.3 –genomeDir hg19genome_no_splice_junction_database_provided –readFilesIn [read1.fastq] [read2.fastq] –outFilterMultimapNmax 10 –outFilterMismatchNmax 10 –outSJfilterOverhangMin 30 16 16 16 –alignSJDBoverhangMin 3 –alignSJoverhangMin 6 –outFilterType BySJout –outSJfilterCountUniqueMin 3 1 1 1 –outSJfilterCountTotalMin 4 2 2 2 –outSAMstrandField intronMotif –outFilterIntronMotifs RemoveNoncanonical –alignIntronMax 300000 –alignMatesGapMax 500000 –outFileNamePrefix [output] –outStd SAM –outSAMmode Full. SAM output was converted to BAM and sorted by position using samtools software. Expression levels (RPKM) were estimated for RefSeq annotated isoforms of SLC38A2, SLC38A4 and LOC100288798 (SLC38A2 – 1 isoform: NM_018976, SLC38A4 – 2 isoforms: NM_018018 and NM_001143824, LOC100288798 – 5 isoforms: NR_125377, NR_125378, NR_125379, NR_125380, and NR_125381). The average RPKM of all isoforms was displayed inside each cell of the heat map (B), which was built in R using the pheatmap function without clustering rows and columns. Rows were sorted according to expression level of LOC100288798. Heat map color scale was skewed toward lower values to highlight non-expressed genes (shades of blue – 0<RPKM<0.5) and display the range of LOC100288798 expression (shades of orange – 0.5<RPKM<10).
Splicing efficiency calculation
Splicing efficiency was calculated using public total (ribosomal depleted) RNA-seq datasets of high depth (135-371 million reads, Table S1A). Splicing efficiency of each RefSeq annotated splice site was estimated by calculating RPKM of exonic and intronic 45bp regions surrounding the splice site starting 5bp away from the precise splice site position to allow for potentially imprecise annotation of the splice site. For each splice site, which passed the coverage cutoff (exonic RPKM > 0.2), “Splicing efficiency” (S), S = 100*(1-RPKMintronic/RPKMexonic), was calculated. Splicing efficiency was within the range from 0 for fully unprocessed splice sites (RPKMintronic>= RPKMexonic, S was set to 0, when it was calculated to be <0) to 100 for perfectly processed splice sites (RPKMintronic=0). We then calculated the average splicing efficiency of all the unique splice sites for each gene and assigned the splicing efficiency of the gene with this value.
Estimation of PolyA+ and nuclear enrichment
Publicly available cellular/PolyA fractionation RNA-seq data for 5 cell lines (HeLa, Lymphoblastoid cell line GM12878, Embryonic stem cells, HUVEC and K562) produced by the ENCODE project were downloaded as raw fastq files, aligned with STAR using default parameters. Expression of spliced products was calculated for: LOC100288798: averaged from NR_125379 NR_125380 NR_125378 NR_125377 NR_125381, SLC32A2: NM_018976, TBP: NM_003194, XIST: NR_001564 (RefSeq identifiers). Expression over the whole gene body was calculated for LOC100288798: over chr12:46777889-47046362 (gene body of NR_125381) and chr12:46777458-47046362 (gene body of NR_125379 NR_125380 NR_125378 NR_125377), SLC38A2: over chr12:46751970-4676664, TBP: over chr6:170863420-170881958, XIST: over chrX:73040485-73072588.
Assembly of SLC38A4-AS exon structure using publicly available RNA-seq data from multiple cell types
Exon structure assembly was performed for each of 46 public RNA-seq data only in the region of interest: samtools view -b [position sorted STAR alignment] chr12:46,500,000-47,500,000 > tissue.1Mb.bam . De novo transcriptome assembly was performed for each one of 1Mb regions in all the samples separately using Cufflinks version 2.2.1 with the following command: cufflinks –multi-read-correct –output-dir [output] -F 0.01 -p 7 –library-type fr-firststrand (if RNA-seq is stranded) –mask-file pseudogenes.gtf tissue.1Mb.bam . Pseudogene annotation was obtained from GENCDOE v19. The resulting transcript assemblies were then merged using Cuffmerge with the following command: cuffmerge -s hg19_fasta –keep-tmp -p 8 –min-isoform-fraction 0 [list of all gtf files from 46 cufflinks assemblies]. Single exon transcripts were discarded.
KBM7 cell culture
All gene trap KBM7 cell lines were obtained frozen from Horizon Genomics GmbH (http://www.horizon-genomics.com/). WT KBM7 cell lines were from Horizon Genomics GmbH or from Sebastian Nijman lab. All cell lines were cultured in filter cap flasks in IMDM (Sigma) medium (with L-Glutamine, supplemented with Penicillin/Streptomycin and 10% Fetal Bovine Serum (PAA Laboratories (GE Healthcare)) at 37°C with 5% CO2. KBM7 are suspension cells. Cell concentration and cell size were measured using Casy cell counter (Schärfe System GmbH).
RNA preparation
RNA was isolated from pelleted KBM7 cells using TRIreagent (Sigma), dissolved in RNA Storage Solution (RSS, Ambion) and stored at −20oC. RNA was DNAse I treated (DNAfree kit, Ambion). Quality control was performed by accessing RNA integrity using Agilent RNA 6000 Nano Kit.
RT-qPCR
RNA was converted to cDNA using RevertAid First Strand cDNA Kit (Fermentas) with –RT (no reverse transcriptase) control reaction for each RNA sample according to manufacturer's protocol. RT-qPCR was performed using MESA GREEN qPCR MasterMix Plus for SYBR Assay I dTTP (Eurogentec). Primers () were designed using Primer3. RT-qPCR was performed using standard curves in 3 technical replicates for each sample and standard deviation between the replicates was used to define the error and plot the error bars.
DNA-blot
DNA extraction, restriction enzyme digestion and DNA gel blots were performed using standard methods. The hybridization probe was amplified by PCR, cloned and gel purified. Membranes were exposed to an imaging plate (FujiFilm) that was scanned (Typhoon TRIO, GE Healthcare). Levels were adjusted on the whole image to increase the visibility of all bands on the image.
Chromosome analysis
Metaphase preparation and FISH were carried out by standard methods. Dividing cells were locked in metaphase by adding colcemid (0.1μg/ml final concentration) (Gibco, ThermoFisher) for 60 minutes. After fixation cells were dropped onto slides, dried at 42°C for 30 minutes and then incubated at 60°C over night. One slide was used for Giemsa-trypsin banding of chromosomes. For FISH analyses a Cy3 labeled probe mix (Kreatech) was used which detects the centromeric regions of chromosomes 1, 5 and 19.
Strand-specific RNA-seq library preparation and RNA sequencing
4 μg of DNase I treated RNA underwent Ribosomal depletion using RiboZero rRNA removal kit Human/Mouse/Rat (Epicentre) following manufacturer's protocol. RNA-seq library was prepared with ribosomal depleted RNA using TruSeq RNA Sample Prep Kit v2 (Illumina) with modifications to preserve strand information as described.Citation80 Quality and size distribution of the prepared libraries was assessed with ExperionTM DNA 1K Analysis Chips, and was used for molarity calculation. 8 RNA-seq libraries were barcoded using TruSeq RNA Sample Prep Kit v2 provided barcodes and pooled in equal molarities. 50bp single-end RNA-sequencing was performed at the Biomedical Sequencing Facility (http://biomedical-sequencing.at/BSF/) using Illumina HiSeq 2000.
KBM7 RNA-seq alignment
Raw RNA-seq data from each sample in fastq format was aligned using STARCitation55 with default parameters: STAR_2.3 –genomeDir hg19genome_no_splice_junction_database_provided –readFilesIn [sample.fastq] –runThreadN 8 –genomeLoad NoSharedMemory –outFileNamePrefix [sample] –outStd SAM –outSAMmode Full. Output was converted to BAM and sorted using samtools software. This resulted in average 35 million of uniquely mapped reads per sample with low standard deviation of 1.0 million reads.(Table S1D).
Differential gene expression analysis
RefSeq annotation downloaded from UCSC table browser on 27th January 2014 was used (filter: “name does match NM*," 36,734 isoforms, RefSeq_NM.gtf). CuffdiffCitation59 (version 2.2.1) was used for expression level (FPKM) estimation and differential expression analysis with the following command: cuffdiff RefSeq_NM.gtf -p 7 [replicates group1] [replicates group2] –labels [label group1], [label group2] –library-type fr-firststrand –mask-file pseudogenes.gtf. The outputted list of significantly differentially expressed genes was additionally filtered and only genes showing at least 3-fold change between non-truncated controls (WT2, WT3, C1, C2, replicate group1) and truncated cell lines (3kb1, 3kb2, 100kb1, 100kb2, replicate group2) were kept resulting in the list of 41 genes. Pairwise comparisons performed for further filtering: WT2, WT3 (replicate group1) versus C1, C2 (replicate group2) and 3kb1, 3kb2 (replicate group1) 100kb1, 100kb2 (replicate group2).
KBM7 cell lines clustering based on differential gene expression
Expression level (FPKM) of RefSeq protein coding genes was calculated in each of 8 samples separately using Cuffdiff (same command as above, no replicates). Expression of 41 significantly differentially expressed genes (A) or was used to perform unsupervised clustering of the samples. Heat map was built in R using pheatmap function with options clustering_distance_cols= "canberra," clustering_distance_rows= "euclidean."
Expression calculation and gene trap insertion analysis
GENCODE v19 lncRNA expression was calculated as RPKM (described above) separately for WT2 and WT3 cell lines. The average RPKM from both calculations was used in the figure. To determine the number of lncRNAs with gene trap insertion sites we downloaded cassette insertion sites from http://kbm7.genomebrowser.cemm.at/ in July 2015. Insertion sites can be updated and gene trap insertion sites used in this publication are available from http://opendata.cemm.at/barlowlab. Overlaps on the same strand with lncRNA annotations from GENCODE v19 were identified and overlapping annotations merged with bedtools software. GENCODE v19 lncRNA annotation was obtained at ftp://ftp.sanger.ac.uk/pub/gencode/Gencode_human/release_19/gencode.v19.long_noncoding_RNAs.gtf.gz. To calculate position of gene trap insertions within the gene body we divided each GENCODE v19 lncRNA into 10 equally sized regions (numbered 1-10 starting at 5’ end). Then we calculated the overlap of mapped gene trap insertion sites with these regions (bedtools) and created a sum of all insertions mapped to similar numbered regions.
Author contributions
A.E.K., D.P.B. and F.M.P. conceived the study and wrote the manuscript. I.V. discovered the SLC38A4-AS lncRNA and performed preliminary experiments characterizing this lncRNA. J.N. performed karyotype analysis and FISH. A.E.K and F.M.P performed DNA blots and PCR analyses. A.E.K. performed bioinformatic analysis, cell culture and RNA-seq.
Data access
Raw RNA-seq data from 8 KBM7 cell lines and the differential expression analysis output of Cuffdiff (Results, A) were deposited in NCBI's Gene Expression Omnibus Citation81 and are accessible through GEO Series accession number GSE71284 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE71284). Full de novo assembly in the 1Mb region around SLC38A4-AS lncRNA, RNA-seq signal in 8 sequenced KBM7 cell lines as well as location of gene trap insertion cassettes used in the study can be viewed in the related UCSC genome browser hub via https://opendata.cemm.at/barlowlab/.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
KRNB_A_1110676_Updated_Supplemental_datas.zip
Download Zip (1,009 KB)Supplementary Files
Download Zip (999.5 KB)Acknowledgments
We thank Horizon Genomics GmbH for providing the KBM7 cell lines, Claudia Kerzendorfer and Sebastian Nijman for handling KBM7 cells and the Biomedical Sequencing Facility (http://biomedical-sequencing.at/) for advice and performing RNA sequencing. We thank Sara Sdelci, Quanah Hudson and Daniel Andergassen for technical assistance and useful discussions. We thank Quanah Hudson and Tilmann Bürckstümmer for advice and reading the manuscript. The study was partially supported by Austrian Science Fund [FWF F43-B09, FWF W1207-B09].
References
- Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell 2013; 154:26–46; PMID:23827673; http://dx.doi.org/10.1016/j.cell.2013.06.020
- Iyer MK, Niknafs YS, Malik R, Singhal U, Sahu A, Hosono Y, Barrette TR, Prensner JR, Evans JR, Zhao S, et al. The landscape of long noncoding RNAs in the human transcriptome. Nat Genet 2015; 47(3):199–208; PMID:25599403; http://dx.doi.org/10.1007/82_2015_444
- Cheetham SW, Gruhl F, Mattick JS, Dinger ME. Long noncoding RNAs and the genetics of cancer. Br J Cancer 2013; 108:2419–25; PMID:23660942; http://dx.doi.org/10.1038/bjc.2013.233
- Batista PJ, Chang HY. Long noncoding RNAs: cellular address codes in development and disease. Cell 2013; 152:1298–307; PMID:23498938; http://dx.doi.org/10.1016/j.cell.2013.02.012
- Prensner JR, Iyer MK, Balbin OA, Dhanasekaran SM, Cao Q, Brenner JC, Laxman B, Asangani IA, Grasso CS, Kominsky HD, et al. Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat Biotechnol 2011; 29:742–9; PMID:21804560; http://dx.doi.org/10.1038/nbt.1914
- Roth A, Diederichs S. Long Noncoding RNAs in Lung Cancer. Curr Top Microbiol Immunol 2015; PMID:26037047
- Meng L, Ward AJ, Chun S, Bennett CF, Beaudet AL, Rigo F. Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature 2015; 518:409–12; PMID:25470045; http://dx.doi.org/10.1038/nature13975
- Wahlestedt C. Targeting long non-coding RNA to therapeutically upregulate gene expression. Nat Rev Drug Discov 2013; 12:433–46; PMID:23722346; http://dx.doi.org/10.1038/nrd4018
- Roberts TC, Wood MJ. Therapeutic targeting of non-coding RNAs. Essays Biochem 2013; 54:127–45; PMID:23829532; http://dx.doi.org/10.1042/bse0540127
- Quinodoz S, Guttman M. Long noncoding RNAs: an emerging link between gene regulation and nuclear organization. Trends Cell Biol 2014; 24:651–63; PMID:25441720; http://dx.doi.org/10.1016/j.tcb.2014.08.009
- Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010; 465:1033–8; PMID:20577206; http://dx.doi.org/10.1038/nature09144
- Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010; 464:1071–6; PMID:20393566; http://dx.doi.org/10.1038/nature08975
- Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, Lajoie BR, Protacio A, Flynn RA, Gupta RA, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 2011; 472:120–4; PMID:21423168; http://dx.doi.org/10.1038/nature09819
- Latos PA, Pauler FM, Koerner MV, Senergin HB, Hudson QJ, Stocsits RR, Allhoff W, Stricker SH, Klement RM, Warczok KE, et al. Airn transcriptional overlap, but not its lncRNA products, induces imprinted Igf2r silencing. Science 2012; 338:1469–72; PMID:23239737; http://dx.doi.org/10.1126/science.1228110
- Kornienko AE, Guenzl PM, Barlow DP, Pauler FM. Gene regulation by the act of long non-coding RNA transcription. BMC Biol 2013; 11:59; PMID:23721193; http://dx.doi.org/10.1186/1741-7007-11-59
- Chu C, Spitale RC, Chang HY. Technologies to probe functions and mechanisms of long noncoding RNAs. Nat Struct Mol Biol 2015; 22:29–35; PMID:25565030; http://dx.doi.org/10.1038/nsmb.2921
- Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 2011; 25:1915–27; PMID:21890647; http://dx.doi.org/10.1101/gad.17446611
- Cabili MN, Dunagin MC, McClanahan PD, Biaesch A, Padovan-Merhar O, Regev A, Rinn JL, Raj A. Localization and abundance analysis of human lncRNAs at single-cell and single-molecule resolution. Genome Biol 2015; 16:20; PMID:25630241; http://dx.doi.org/10.1186/s13059-015-0586-4
- Tilgner H, Knowles DG, Johnson R, Davis CA, Chakrabortty S, Djebali S, Curado J, Snyder M, Gingeras TR, Guigó R. Deep sequencing of subcellular RNA fractions shows splicing to be predominantly co-transcriptional in the human genome but inefficient for lncRNAs. Genome Res 2012; 22:1616–25; PMID:22955974; http://dx.doi.org/10.1101/gr.134445.111
- Seidl CI, Stricker SH, Barlow DP. The imprinted Air ncRNA is an atypical RNAPII transcript that evades splicing and escapes nuclear export. Embo J 2006; 25:3565–75; PMID:16874305; http://dx.doi.org/10.1038/sj.emboj.7601245
- Kelley D, Rinn J. Transposable elements reveal a stem cell-specific class of long noncoding RNAs. Genome Biol 2012; 13:R107; PMID:23181609; http://dx.doi.org/10.1186/gb-2012-13-11-r107
- Hacisuleyman E, Goff LA, Trapnell C, Williams A, Henao-Mejia J, Sun L, McClanahan P, Hendrickson DG, Sauvageau M, Kelley DR, et al. Topological organization of multichromosomal regions by the long intergenic noncoding RNA Firre. Nat Struct Mol Biol 2014; 21:198–206; PMID:24463464; http://dx.doi.org/10.1038/nsmb.2764
- Guttman M, Donaghey J, Carey BW, Garber M, Grenier JK, Munson G, Young G, Lucas AB, Ach R, Bruhn L, et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 2011; 477:295–300; PMID:21874018; http://dx.doi.org/10.1038/nature10398
- Ulitsky I, Shkumatava A, Jan CH, Sive H, Bartel DP. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell 2011; 147:1537–50; PMID:22196729; http://dx.doi.org/10.1016/j.cell.2011.11.055
- Tripathi V, Ellis JD, Shen Z, Song DY, Pan Q, Watt AT, Freier SM, Bennett CF, Sharma A, Bubulya PA, et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell 2010; 39:925–38; PMID:20797886; http://dx.doi.org/10.1016/j.molcel.2010.08.011
- Ohrt T, Muetze J, Svoboda P, Schwille P. Intracellular localization and routing of miRNA and RNAi pathway components. Curr Top Med Chem 2012; 12:79–88; PMID:22196276; http://dx.doi.org/10.2174/156802612798919132
- Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol 2014; 32:347–55; PMID:24584096; http://dx.doi.org/10.1038/nbt.2842
- Sauvageau M, Goff LA, Lodato S, Bonev B, Groff AF, Gerhardinger C, Sanchez-Gomez DB, Hacisuleyman E, Li E, Spence M, et al. Multiple knockout mouse models reveal lincRNAs are required for life and brain development. Elife 2013; 2:e01749; PMID:24381249; http://dx.doi.org/10.7554/eLife.01749
- Yin Y, Yan P, Lu J, Song G, Zhu Y, Li Z, Zhao Y, Shen B, Huang X, Zhu H, et al. Opposing roles for the lncRNA haunt and its genomic locus in regulating hoxa gene activation during embryonic stem cell differentiation. Cell Stem Cell 2015; 16:504–16; PMID:25891907; http://dx.doi.org/10.1016/j.stem.2015.03.007
- Zhang B, Arun G, Mao YS, Lazar Z, Hung G, Bhattacharjee G, Xiao X, Booth CJ, Wu J, Zhang C, et al. The lncRNA Malat1 is dispensable for mouse development but its transcription plays a cis-regulatory role in the adult. Cell Reports 2012; 2:111–23; PMID:22840402; http://dx.doi.org/10.1016/j.celrep.2012.06.003
- Han J, Zhang J, Chen L, Shen B, Zhou J, Hu B, Du Y, Tate PH, Huang X, Zhang W. Efficient in vivo deletion of a large imprinted lncRNA by CRISPR/Cas9. RNA Biol 2014; 11:829–35; PMID:25137067; http://dx.doi.org/10.4161/rna.29624
- Bassett AR, Akhtar A, Barlow DP, Bird AP, Brockdorff N, Duboule D, Ephrussi A, Ferguson-Smith AC, Gingeras TR, Haerty W, et al. Considerations when investigating lncRNA function in vivo. Elife 2014; 3:e03058; PMID:25124674; http://dx.doi.org/10.7554/eLife.03058
- Skarnes WC, von Melchner H, Wurst W, Hicks G, Nord AS, Cox T, Young SG, Ruiz P, Soriano P, Tessier-Lavigne M, et al. A public gene trap resource for mouse functional genomics. Nat Genet 2004; 36:543–4; PMID:15167922; http://dx.doi.org/10.1038/ng0604-543
- Stanford WL, Cohn JB, Cordes SP. Gene-trap mutagenesis: past, present and beyond. Nat Rev Genet 2001; 2:756–68; PMID:11584292; http://dx.doi.org/10.1038/35093548
- Schuster-Gossler K, Simon-Chazottes D, Guenet JL, Zachgo J, Gossler A. Gtl2lacZ, an insertional mutation on mouse chromosome 12 with parental origin-dependent phenotype. Mamm Genome 1996; 7:20–4; PMID:8903723; http://dx.doi.org/10.1007/s003359900006
- Barlow DP, Bartolomei MS. Genomic imprinting in mammals. Cold Spring Harb Perspect Biol 2014; 6:a018382; PMID:24492710; http://dx.doi.org/10.1101/cshperspect.a018382
- Kanduri C. Long noncoding RNAs: Lessons from genomic imprinting. Biochim Biophys Acta 2015; PMID:26004516; http://dx.doi.org/10.1016/j.bbagrm.2015.05.006
- da Rocha ST, Edwards CA, Ito M, Ogata T, Ferguson-Smith AC. Genomic imprinting at the mammalian Dlk1-Dio3 domain. Trends Genet 2008; 24:306–16; PMID:18471925; http://dx.doi.org/10.1016/j.tig.2008.03.011
- Benetatos L, Vartholomatos G, Hatzimichael E. DLK1-DIO3 imprinted cluster in induced pluripotency: landscape in the mist. Cell Mol Life Sci 2014; 71:4421–30; PMID:25098353; http://dx.doi.org/10.1007/s00018-014-1698-9
- Sleutels F, Zwart R, Barlow DP. The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature 2002; 415:810–3; PMID:11845212; http://dx.doi.org/10.1038/415810a
- Mancini-Dinardo D, Steele SJ, Levorse JM, Ingram RS, Tilghman SM. Elongation of the Kcnq1ot1 transcript is required for genomic imprinting of neighboring genes. Genes Dev 2006; 20:1268–82; PMID:16702402; http://dx.doi.org/10.1101/gad.1416906
- Meng L, Person RE, Huang W, Zhu PJ, Costa-Mattioli M, Beaudet AL. Truncation of Ube3a-ATS unsilences paternal Ube3a and ameliorates behavioral defects in the Angelman syndrome mouse model. PLoS Genet 2013; 9:e1004039; PMID:24385930; http://dx.doi.org/10.1371/journal.pgen.1004039
- Shin JY, Fitzpatrick GV, Higgins MJ. Two distinct mechanisms of silencing by the KvDMR1 imprinting control region. Embo J 2008; 27:168–78; PMID:18079696; http://dx.doi.org/10.1038/sj.emboj.7601960
- Gutschner T, Baas M, Diederichs S. Noncoding RNA gene silencing through genomic integration of RNA destabilizing elements using zinc finger nucleases. Genome Res 2011; 21:1944–54; PMID:21844124; http://dx.doi.org/10.1101/gr.122358.111
- Burckstummer T, Banning C, Hainzl P, Schobesberger R, Kerzendorfer C, Pauler FM, Chen D, Them N, Schischlik F, Rebsamen M, et al. A reversible gene trap collection empowers haploid genetics in human cells. Nat Methods 2013; 10:965–71; PMID:24161985; http://dx.doi.org/10.1038/nmeth.2609
- Santoro F, Mayer D, Klement RM, Warczok KE, Stukalov A, Barlow DP, Pauler FM. Imprinted Igf2r silencing depends on continuous Airn lncRNA expression and is not restricted to a developmental window. Development 2013; 140:1184–95; PMID:23444351; http://dx.doi.org/10.1242/dev.088849
- Andersson BS, Beran M, Pathak S, Goodacre A, Barlogie B, McCredie KB. Ph-positive chronic myeloid leukemia with near-haploid conversion in vivo and establishment of a continuously growing cell line with similar cytogenetic pattern. Cancer Genet Cytogenet 1987; 24:335–43; PMID:3466682; http://dx.doi.org/10.1016/0165-4608(87)90116-6
- Schuster-Gossler K, Bilinski P, Sado T, Ferguson-Smith A, Gossler A. The mouse Gtl2 gene is differentially expressed during embryonic development, encodes multiple alternatively spliced transcripts, and may act as an RNA. Dev Dyn 1998; 212:214–28; PMID:9626496; http://dx.doi.org/10.1002/(SICI)1097-0177(199806)212:2%3c214::AID-AJA6%3e3.0.CO;2-K
- Vlatkovic I. PhD thesis: Mapping and characterization of macro non-protein coding RNAs in human imprinted gene regions, University of Vienna; available for download at http://othes.univie.ac.at/12494/1/2010-09-01_0642621.pdf 2010
- Smith RJ, Dean W, Konfortova G, Kelsey G. Identification of novel imprinted genes in a genome-wide screen for maternal methylation. Genome Res 2003; 13:558–69; PMID:12670997; http://dx.doi.org/10.1101/gr.781503
- Andergassen D, Dotter CP, Kulinski TM, Guenzl PM, Bammer PC, Barlow DP, Pauler FM, Hudson QJ. Allelome.PRO, a pipeline to define allele-specific genomic features from high-throughput sequencing data. Nucleic Acids Res 2015; 43(21): e146; PMID:26202974
- Pruitt KD, Brown GR, Hiatt SM, Thibaud-Nissen F, Astashyn A, Ermolaeva O, Farrell CM, Hart J, Landrum MJ, McGarvey KM, et al. RefSeq: an update on mammalian reference sequences. Nucleic Acids Res 2014; 42:D756–63; PMID:24259432; http://dx.doi.org/10.1093/nar/gkt1114
- Mattick JS, Rinn JL. Discovery and annotation of long noncoding RNAs. Nat Struct Mol Biol 2015; 22:5–7; PMID:25565026; http://dx.doi.org/10.1038/nsmb.2942
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res 2012; 22:1760–74; PMID:22955987; http://dx.doi.org/10.1101/gr.135350.111
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013; 29:15–21; PMID:23104886; http://dx.doi.org/10.1093/bioinformatics/bts635
- Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res 2012; 22:1775–89; PMID:22955988; http://dx.doi.org/10.1101/gr.132159.111
- Curinha A, Oliveira Braz S, Pereira-Castro I, Cruz A, Moreira A. Implications of polyadenylation in health and disease. Nucleus 2014; 5:508–19; PMID:25484187; http://dx.doi.org/10.4161/nucl.36360
- Brockdorff N, Ashworth A, Kay GF, McCabe VM, Norris DP, Cooper PJ, Swift S, Rastan S. The product of the mouse Xist gene is a 15 kb inactive X-specific transcript containing no conserved ORF and located in the nucleus. Cell 1992; 71:515–26; PMID:1423610; http://dx.doi.org/10.1016/0092-8674(92)90519-I
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 2012; 7:562–78; PMID:22383036; http://dx.doi.org/10.1038/nprot.2012.016
- Gregory TR. Coincidence, coevolution, or causation? DNA content, cell size, and the C-value enigma. Biol Rev Camb Philos Soc 2001; 76:65–101; PMID:11325054; http://dx.doi.org/10.1017/S1464793100005595
- Huang R, Jaritz M, Guenzl P, Vlatkovic I, Sommer A, Tamir IM, Marks H, Klampfl T, Kralovics R, Stunnenberg HG, et al. An RNA-Seq strategy to detect the complete coding and non-coding transcriptome including full-length imprinted macro ncRNAs. PLoS One 2011; 6:e27288; PMID:22102886; http://dx.doi.org/10.1371/journal.pone.0027288
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol 2013; 31:46–53; PMID:23222703; http://dx.doi.org/10.1038/nbt.2450
- Kowalczyk MS, Higgs DR, Gingeras TR. Molecular biology: RNA discrimination. Nature 2012; 482:310–1; PMID:22337043; http://dx.doi.org/10.1038/482310a
- Wutz A. Gene silencing in X-chromosome inactivation: advances in understanding facultative heterochromatin formation. Nat Rev Genet 2011; 12:542–53; PMID:21765457; http://dx.doi.org/10.1038/nrg3035
- Alen C, Kent NA, Jones HS, O'Sullivan J, Aranda A, Proudfoot NJ. A role for chromatin remodeling in transcriptional termination by RNA polymerase II. Mol Cell 2002; 10:1441–52; PMID:12504018; http://dx.doi.org/10.1016/S1097-2765(02)00778-5
- Luco RF, Misteli T. More than a splicing code: integrating the role of RNA, chromatin and non-coding RNA in alternative splicing regulation. Curr Opin Genet Dev 2011; 21:366–72; PMID:21497503; http://dx.doi.org/10.1016/j.gde.2011.03.004
- Weber EL, Cannon PM. Promoter choice for retroviral vectors: transcriptional strength vs. trans-activation potential. Hum Gene Ther 2007; 18:849–60; PMID:17767401; http://dx.doi.org/10.1089/hum.2007.067
- Zwart R, Sleutels F, Wutz A, Schinkel AH, Barlow DP. Bidirectional action of the Igf2r imprint control element on upstream and downstream imprinted genes. Genes Dev 2001; 15:2361–6; PMID:11562346; http://dx.doi.org/10.1101/gad.206201
- Nagano T, Mitchell JA, Sanz LA, Pauler FM, Ferguson-Smith AC, Feil R, Fraser P. The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science 2008; 322:1717–20; PMID:18988810; http://dx.doi.org/10.1126/science.1163802
- Babak T, DeVeale B, Tsang EK, Zhou Y, Li X, Smith KS, Kukurba KR, Zhang R, Li JB, van der Kooy D, et al. Genetic conflict reflected in tissue-specific maps of genomic imprinting in human and mouse. Nat Genet 2015; 47:544–9; PMID:25848752; http://dx.doi.org/10.1038/ng.3274
- Baran Y, Subramaniam M, Biton A, Tukiainen T, Tsang EK, Rivas MA, Pirinen M, Gutierrez-Arcelus M, Smith KS, Kukurba KR, et al. The landscape of genomic imprinting across diverse adult human tissues. Genome Res 2015; 25:927–36; PMID:25953952; http://dx.doi.org/10.1101/gr.192278.115
- Charrin S, Jouannet S, Boucheix C, Rubinstein E. Tetraspanins at a glance. J Cell Sci 2014; 127:3641–8; PMID:25128561; http://dx.doi.org/10.1242/jcs.154906
- Karlsson G, Rorby E, Pina C, Soneji S, Reckzeh K, Miharada K, Karlsson C, Guo Y, Fugazza C, Gupta R, et al. The tetraspanin CD9 affords high-purity capture of all murine hematopoietic stem cells. Cell Rep 2013; 4:642–8; PMID:23954783; http://dx.doi.org/10.1016/j.celrep.2013.07.020
- Carette JE, Pruszak J, Varadarajan M, Blomen VA, Gokhale S, Camargo FD, Wernig M, Jaenisch R, Brummelkamp TR. Generation of iPSCs from cultured human malignant cells. Blood 2010; 115:4039–42; PMID:20233975; http://dx.doi.org/10.1182/blood-2009-07-231845
- Akutsu H, Miura T, Machida M, Birumachi J, Hamada A, Yamada M, Sullivan S, Miyado K, Umezawa A. Maintenance of pluripotency and self-renewal ability of mouse embryonic stem cells in the absence of tetraspanin CD9. Differentiation 2009; 78:137–42; PMID:19716222; http://dx.doi.org/10.1016/j.diff.2009.08.005
- Kennaway DJ. Clock genes at the heart of depression. J Psychopharmacol 2010; 24:5–14; PMID:20663803; http://dx.doi.org/10.1177/1359786810372980
- Baek SH, Kim KI. Emerging roles of orphan nuclear receptors in cancer. Annu Rev Physiol 2014; 76:177–95; PMID:24215441; http://dx.doi.org/10.1146/annurev-physiol-030212-183758
- Wang A, Huang K, Shen Y, Xue Z, Cai C, Horvath S, Fan G. Functional modules distinguish human induced pluripotent stem cells from embryonic stem cells. Stem Cells Dev 2011; 20:1937–50; PMID:21542696; http://dx.doi.org/10.1089/scd.2010.0574
- Carette JE, Guimaraes CP, Wuethrich I, Blomen VA, Varadarajan M, Sun C, Bell G, Yuan B, Muellner MK, Nijman SM, et al. Global gene disruption in human cells to assign genes to phenotypes by deep sequencing. Nat Biotechnol 2011; 29:542–6; PMID:21623355; http://dx.doi.org/10.1038/nbt.1857
- Sultan M, Dokel S, Amstislavskiy V, Wuttig D, Sultmann H, Lehrach H, Yaspo ML. A simple strand-specific RNA-Seq library preparation protocol combining the Illumina TruSeq RNA and the dUTP methods. Biochem Biophysical Res Commun 2012; 422:643–6; PMID:22609201; http://dx.doi.org/10.1016/j.bbrc.2012.05.043
- Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 2002; 30:207-10; PMID:11752295; http://dx.doi.org/10.1093/nar/30.1.207
- Forrest AR, Kawaji H, Rehli M, Baillie JK, de Hoon MJ, Haberle V, Lassmann T, Kulakovskiy IV, Lizio M, Itoh M, et al. A promoter-level mammalian expression atlas. Nature 2014; 507:462–70; PMID:24670764; http://dx.doi.org/10.1038/nature13182