
ABSTRACT
SR1 is a dual-function sRNA from B. subtilis that acts as a base-pairing regulatory RNA and as a peptide-encoding mRNA. Both functions of SR1 are highly conserved. Previously, we uncovered that the SR1 encoded peptide SR1P binds the glycolytic enzyme GapA resulting in stabilization of gapA mRNA. Here, we demonstrate that GapA interacts with RNases Y and J1, and this interaction was RNA-independent. About 1% of GapA molecules purified from B. subtilis carry RNase J1 and about 2% RNase Y. In contrast to the GapA/RNase Y interaction, the GapA/RNaseJ1 interaction was stronger in the presence of SR1P. GapA/SR1P-J1/Y displayed in vitro RNase activity on known RNase J1 substrates. Moreover, the RNase J1 substrate SR5 has altered half-lives in a ΔgapA strain and a Δsr1 strain, suggesting in vivo functions of the GapA/SR1P/J1 interaction. Our results demonstrate that the metabolic enzyme GapA moonlights in recruiting RNases while GapA bound SR1P promotes binding of RNase J1 and enhances its activity.
Abbreviations
| aa | = | amino acid |
| BSA | = | bovine serum albumin |
| DRaCALA | = | Differential Radial Capillary Action of Ligand Assay |
Introduction
Small regulatory RNAs (sRNAs) are the most important posttranscriptional regulators in all 3 kingdoms of life (rev. inCitation1-3). During the past years, computer-based searches combined with RNAseq identified on average 100–200 sRNAs/genome in a multitude of Gram-negative and Gram-positive species. These sRNAs can be divided into 2 major groups, base-pairing and protein-binding sRNAs. Regulatory mechanisms employed by all base-pairing sRNAs comprise inhibition or activation of translation and promotion of RNA decay or stability. Additionally, inhibition of primer maturation, transcriptional attenuation and transcriptional interference has been reported for some cis-encoded sRNAs (rev. inCitation4). Dual-function sRNAs are a subgroup of trans-encoded sRNAs. On the one hand they act as base-pairing sRNAs, but on the other hand as peptide encoding mRNAs. The first example was S. aureus RNAIII encoding δ-haemolysin (26 aa) and using a base-pairing mechanism to activate translation of hla mRNACitation5 or repress translation of rot, spa, coa and lytM (e.g.Citation6,7; rev. inCitation8). Subsequently, the streptolysin SLS-ORF of Streptococcus Pel RNACitation9 and the 43-codon-SgrT ORF on E. coli SgrSCitation10 were identified. SgrS and SgrT downregulate PtsG glucose transporter activity and have a physiologically redundant, but mechanistically distinct function in inhibition.Citation10 Recently, S. aureus sRNA Psm-mec was discovered that encodes the 22 aa secreted cytolytic toxin PSMα and inhibits translation of agrA mRNA by base-pairing with the coding sequence.Citation11 By contrast, no functions have been uncovered to date for the hyp7 ORF on Clostridium perfringens VR,Citation12 the 32-codon RivX-ORF,Citation13 the RSs0019-ORF of Rhodobacter sphaeroidesCitation14 and the 37-codon PhrS-ORF of Pseudomonas aeruginosa.Citation15
The 205 nt long sRNA SR1 from the B. subtilis genome was found by a combination of computer predictions and Northern blotting.Citation16 Using 7 complementary regions, SR1 base-pairs with its primary target, ahrC mRNA, the transcriptional activator of the rocABC and rocDEF arginine catabolic operons.Citation17 Base-pairing induces structural changes downstream of the ahrC ribosome-binding site that inhibit translation initiation.Citation18 SR1 is expressed under gluconeogenic and repressed under glycolytic conditions by CcpN binding to 2 operator sites upstream of and overlapping psr1Citation19 and, to a minor extent, CcpA binding to a cre-site at −139.Citation16 CcpN requires both ATP and a slightly acidic pH Citation20 to prevent promoter escape via direct contacts with the α-subunit of the B. subtilis RNA polymerase.Citation21 In 2010 we demonstrated that the small ORF on SR1 is translated into a 39 aa peptide SR1P (synonym YkzW) that binds GapA (glyceraldehyde-3-P-dehydrogenase A) resulting in stabilization of gapA mRNA.Citation22 SR1 is the first dual-function sRNA in B. subtilis. A computer-based search identified 23 SR1 homologues in Bacillus, Geobacillus, Anoxybacillus and Brevibacillus species.Citation23 All homologues share a high structural similarity with B. subtilis SR1, and the encoded SR1P peptides are highly similar. In the Bacillus cereus group, the sr1p region is present in triplicate or duplicate resulting in longer SR1 species. Both SR1 functions – the base-pairing and the mRNA function – have been conserved over ≈one billion years of evolution.Citation23
Bacterial RNA degradation occurs in a multiprotein complex, the so-called degradosome, which displays a high degree of evolutionary divergence. Whereas the E. coli degradosome is assembled around the C-terminus of the main endoribonuclease RNase E and contains 3′-5′ exoribonuclease PnpA, a helicase and the glycolytic enzyme enolase,Citation24 the B. subtilis degradosome was proposed to comprise the major endoribonuclease RNase Y, 3′-5′ exoribonuclease PnpA, RNases J1, J2, helicase CshA and the glycolytic enzymes enolase (Eno) and phosphofructokinase (PfkA).Citation25 RNases J1 and J2 were first described to be endonucleasesCitation26 and J1 was thought to be the main endonuclease in B. subtilis, but later, J1 and J2 were found to have in addition so far unprecedented 5′-3′ exoribonuclease activities (rev. in 27; 28). Instead, RNase Y was discovered in 2009,Citation25 later investigated in more detailCitation29 and confirmed to be the main endoribonuclease in B. subtilis that fulfils the same role as RNase E in E. coli.
Here, we report on the biological function of the GapA/SR1P interaction. We demonstrate that the glycolytic enzyme GapA moonlights in RNA degradation by recruiting RNases. Far Western blots show that GapA can bind the RNases J1 and Y and SR1P promotes the GapA-J1 interaction. Furthermore, it enhances the RNase activity of GapA-bound RNase J1. A certain percentage of GapA and GapA/SR1P isolated from B. subtilis contain RNase J1 and RNase Y. GapA/SR1P isolated from a wild-type, but not from a ΔrnjA strain, is able to degrade a known RNase J1 substrate in vitro. This finding could be substantiated by an increased half-life of the J1 substrate in a ΔgapA strain. The gapA RNA itself that is stabilized in the presence of SR1P,Citation22 is not only – as shown previously – substrate of RNase Y, which makes a stabilizing cut upstream of the gapA ORF shown before,Citation25,30 but also for RNase J2 and, to a much lesser extent, RNase J1. We present a working model on the function of GapA/SR1P in RNA degradation in B. subtilis and speculate how these interactions lead to the observed stabilization of gapA mRNA. Taken together our data suggest a broader function for GapA/SR1P in RNA turnover.
Results
B. subtilis GapA cannot bind gapA operon mRNA
Previously, we discovered that SR1P interacts with GapA and that this interaction results in stabilization of the gapA mRNA.Citation22 Based on these results, the following scenarios are conceivable: In the absence of SR1P, GapA could i) act as RNase and degrade its own RNA or ii) recruit an RNase that degrades gapA mRNA. Thereby, SR1P might induce a conformational change in GapA that either inhibits intrinsic RNase activity or prevents binding of an RNase. Alternatively, if stabilization of gapA RNA in the presence of SR1P is due to stabilizing cuts by one or several RNases, SR1P might induce a conformational change in GapA that either promotes an internal RNase activity of GapA or facilitates binding of an RNase responsible for such a cut(s).
To investigate whether or not GapA can bind its own RNA, we employed DRaCALA (Differential Radial Capillary Action of Ligand Assay, ref. 31) with internally labeled 2.5 kb cggR-gapA-pgk’ mRNA and GapA or GapA/SR1P purified from B. subtilis as ‘ligands’ along with control proteins of the same concentration. BSA and buffer served as negative, Hfq and RNase A as positive controls. reveals that Hfq known to have a strong RNA binding capacity but no RNase activity bound the substrate RNA tightly, indicated by a dark small dot on the filter. RNase A that simultaneously binds and cleaves RNA yielded a larger, less dark spot. By contrast, GapA and GapA/SR1P from B. subtilis behaved like the negative controls, i.e. were not able to retain the labeled RNA. Therefore, we conclude that GapA does neither bind its own RNA nor act as RNase on gapA operon RNA. Instead, it might be involved in a more complex protein-protein interaction that affects the stability of gapA RNA, and SR1P might interfere with this interaction.
Figure 1. GapA does not bind its own mRNA. DRaCALA with 60 fmol in vitro transcribed, [α-32P]UTP-labeled gapA operon mRNA was mixed with indicated amounts of GapA purified from a B. subtilis wild-type or Δsr1 strain as described in Materials and Methods.
![Figure 1. GapA does not bind its own mRNA. DRaCALA with 60 fmol in vitro transcribed, [α-32P]UTP-labeled gapA operon mRNA was mixed with indicated amounts of GapA purified from a B. subtilis wild-type or Δsr1 strain as described in Materials and Methods.](/cms/asset/351f7e09-7420-42c5-8e63-9bedc72cef53/krnb_a_1208894_f0001_b.gif)
Localization of regions within the gapA operon mRNA required for stabilization of gapA RNA
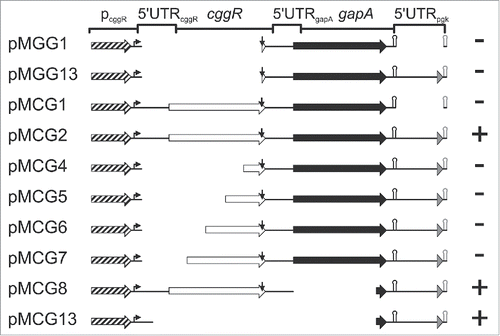
To narrow down the regions of the gapA operon mRNA required for stabilization by GapA/SR1P, we constructed a series of plasmids comprising wild-type and truncated regions of the gapA operon (). These plasmids were integrated into the amyE locus of a ΔgapA/Δsr1 knockout strain and either an empty vector or multicopy-plasmid pWSR1 providing SR1 in trans Citation22 were transferred into these strains. Cells were grown in complex TY medium until onset of stationary phase (OD560 = 4.5), sr1 transcription induced by anhydro-tetracycline, total RNA isolated and subjected to Northern blotting (Fig. S1, summarized in ). Surprisingly, neither the gapA ORF itself (pMGG1) nor the gapA ORF with the 5′ part of the downstream pgk region (pMGG13) or the bicistronic cggR-gapA region lacking the downstream pgk part (pMCG1) needed SR1P for stabilization of gapA mRNA, as they were already stable in its absence. Moreover, gapA with the downstream pgk region comprising in addition progressively longer 3′ portions of cggR (pCG4 to pCG7), but lacking the 5′ UTR and RBS of cggR, were also stable in the absence of SR1P. We only observed a stabilizing effect of SR1P with full-length cggR-gapA-pgk’ (pMCG2), a mutant lacking the gapA sequence, but containing entire cggR with the 5′ UTR of gapA as well as the pgk downstream region (pMCG8), and a mutant containing solely pcggR with the 5′ UTR of cggR and the downstream part of gapA with pgk’ (pMCG13). Apparently, neither the gapA nor the cggR coding sequence were needed for SR1P-dependent stabilization, but regions upstream and downstream of them. These results suggest destabilizing cuts within these 2 regions in the absence of SR1P or stabilizing cuts in the presence of SR1P.
Figure 2. Localization of gapA operon RNA regions dependent on stabilization by GapA/SR1P. Schematic representation of the analyzed gapA operon mutants. Mutants were integrated into the amyE locus of B. subtilis MG1P (Δsr1::phleo; ΔgapA::ery). For pMCG8 and pMCG13 that do not encode a functional gapA gene, strain MG2P (Δsr1::phleo; ΔgapA::ery, ΔthrC::gapA) was used. Strains were transformed with either inducible sr1 overexpression plasmid pWSR1 or empty vector pWH353. Cells were grown as described in Materials and Methods and SR1P dependent stabilization of mutated gapA operon RNA analyzed by Northern blotting (Fig. S1). Genes: boxed arrows (cggR: white; gapA: black; pgk’: gray); pcggR with 200 nt upstream region: hatched arrow; transcription start site: bent arrow; RNaseY processing site: vertical arrow; alternative gapA terminator: black hairpin; artificial bsrF terminator: gray hairpin. +, SR1P required for stabilization; -, RNAs stable in the absence of SR1P (concluded from 3 independent experiments).
B. subtilis GapA interacts with RNase J1 and RNase Y, and SR1P promotes the GapA/J1 interaction
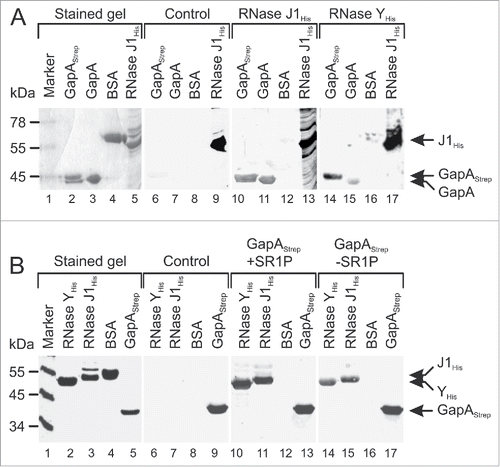
In our previous co-elution assays, Strep-tagged or His-tagged SR1P was bound to an affinity column and crude protein extracts from B. subtilis Δsr1 strains were applied.Citation22,23 In the elution fractions, we did not observe any band in addition to GapA. However, since we used cytosolic extracts, a hypothetical third interaction partner localized in the membrane might have escaped our attention. Therefore, we repeated the co-elution with crude extracts containing cytosolic and membrane proteins, and used a larger scale to prepare highly concentrated extracts. These were applied onto a streptactin column with already bound GapA/SR1P. After elution with desthiobiotin we observed in the protein gel besides GapA additional weak bands corresponding to proteins of ≈55–60 kDa (Fig. S2). Unfortunately, it was not possible to identify these proteins unequivocally by mass spectrometry, as each of the bands contained at least 10–20 proteins, and all scored below the threshold of the validation program. However, among these proteins were also RNases J1 and Y. This prompted us to employ Far Western blotting to study a potential direct interaction between GapA and these RNases. First, we used GapAStrep and GapA as targets and RNase J1His and RNase YHis as bait (). Detection was performed with anti-His-tag antibodies, which proved to be specific (lanes 6–9). RNases J1 and Y bound specifically both GapA preparations (lanes 10, 11, 14, 15), indicating that the Strep-tag does not interfere with binding. In the second Far Western blot (), we used RNases J1His and YHis as targets and GapAStrep with or without SR1P as bait, and detection was with anti-Strep-tag antibodies, which were also specific (lanes 6–9). As expected, GapAStrep and GapAStrep/SR1P bound to RNases J1 and Y.
Figure 3. GapAStrep and GapA interact with RNases J1 and RNase Y. Far-Western blotting. A representative blot of 3 independently performed experiments is shown. Proteins were separated on 10% SDS-PAA gels and either stained with Coomassie (lanes 1–5) or blotted on PVDF membrane (lanes 6–17) as described in Materials and Methods. His-tagged RNases J1 and Y were purified from E. coli, GapAStrep was purified from B. subtilis DB104 (ΔamyE::gapAStrep) and untagged GapA was co-purified with Strep-tagged SR1P from B. subtilis Δsr1::cat (pWSR1/M25). (A) After blocking all blots were incubated with PBST gelatine (control, lanes 6–9) or PBST-gelatine containing either 170 µg RNase J1His (lanes 10–13) or 170 µg RNase YHis (lanes 14–17 RNase binding was detected with mouse anti-His-tag antibodies. Both RNases were able to bind GapAStrep and GapA. (B) Far Western Blot as in A) except that blots were incubated with 100 µg GapAStrep purified from either B. subtilis DB104 (ΔamyE::gapAStrep) (lanes 10–13) or B. subtilis DB104 (Δsr1::phleo, ΔamyE::gapAStrep) (lanes 14–17). GapAStrep binding was detected with mouse anti-Strep-tag antibodies.
GapA isolated from B. subtilis wild-type strains contains RNases J1 and Y
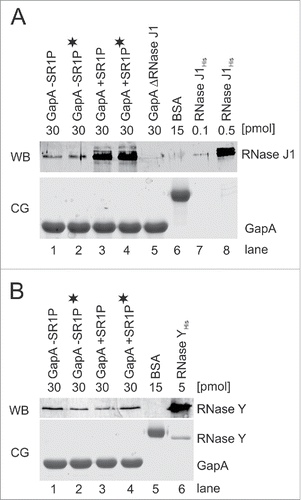
To corroborate the results from the Far Western blotting with an independent method, we used co-elution assays followed by Western blotting with antibodies against RNase J1 and RNase Y (). To investigate if RNA might be needed to bridge the interaction between GapA and RNase J1 or Y, we treated an aliquot of the protein crude extract with RNase A prior to loading onto the streptactin column. shows the Western blot with RNase J antibodies and that with RNase Y antibodies. In both cases, GapA without SR1P and GapA/SR1P copurified the RNase, and this was independent of the presence of RNA (, lanes 1 vs. 2 and 3 vs. 4). Whereas co-elution of RNase Y with GapA was SR1P-independent, GapA/SR1P compared to GapA alone bound approximately 3-fold higher amounts of RNase J1. To substantiate that RNA was neither needed to visualize the GapA/RNase J1 nor the GapA/RNase Y interaction in Far Western blots, we used the same RNase A-treated aliquot to repeat the Far Western blot (Fig. S3). No RNase J was detectable when GapA was prepared from the ΔrnjA strain indicating that GapA co-purifies exclusively RNase J1 and not RNase J2 (). Based on band intensities in , we calculated a stoichiometric GapA/SR1P-RNase J1 ratio of 80:1, i.e. 1.25% of GapA molecules from the wild-type strain carried RNase J1. In the case of GapA lacking SR1P, the ratio was about 200:1 corresponding to 0.5%. About 2% of both the GapA and the GapA/SR1P preparations contained RNase Y as calculated from . However, due to the purification procedure that did not involve cross-linking, we cannot exclude that in vivo a considerably larger fraction of GapA might be bound to RNase J1 or Y.
Figure 4. RNases J1 and Y co-purify with GapA. Co-elution assays Citation22 followed by Western blotting as described in Materials and Methods. A) Top: Western blot (WB) for detection of co-eluted RNase J1 within GapA preparations. GapAStrep was either purified from B. subtilis DB104 (ΔamyE::gapAStrep), DB104 (Δsr1::phleo; ΔamyE::gapAStrep) or DB104 (ΔrnjA::spec; ΔamyE::gapAStrep). To exclude that RNA bridges a possible interaction, an aliquot of the protein crude extracts was incubated with RNase A prior to protein purification (indicated by black asterisk). BSA and RNase J1His purified from E. coli serve as controls. RNase J specific antibodies were used. Bottom: Coomassie stained gel (CG) of the Western blot (loading control). The amount of protein loaded onto the gels is indicated. (B) Top: Western Blot (WB) as in (A) for detection of co-eluted RNase Y within GapA preparations. RNase Y specific antibodies were used. Bottom: CG of Western blot as loading control. The amount of protein loaded onto the gels is indicated.
We conclude that GapA and GapA/SR1P interact directly with RNase J1 and RNase Y, and this interaction is RNA-independent. Whereas SR1P promotes the GapA/RNase J1 interaction, the GapA/RNase Y interaction is not influenced by SR1P.
GapA/SR1P preparations from B. subtilis can cleave a known RNase J1 substrate in vitro
As our GapA and GapA/SR1P preparations from B. subtilis contain sub-stoichiometric concentrations of RNase J1, these preparations should exhibit RNase J1 activity. In and S1 we have shown that for the stabilizing effect of SR1 a gapA mRNA of at least 2.5 kb is required. Degradation of an RNA of this length cannot be analyzed in a PAA gel, and agarose gels cannot be dried to visualize sharp bands. Therefore, we used to test our assumption as first substrate the small (162 nt) internally labeled sRNA SR5, the antitoxin of a type I toxin-antitoxin system investigated in our group. SR5 has a 2-fold longer half-life in a ΔrnjA strain, whereas its half-life was not affected by RNase Y.Citation32 In an in vitro degradation assay (), purified RNase J1 digested SR5 similarly to RNase A used in parallel. By contrast, the same amount of BSA or RNase Y added in the same buffer had no cleavage activity. Therefore, SR5 seemed to be a suitable substrate for the analysis of RNase J1-dependent degradation. To analyze if GapA-J1/Y (GapA with co-purified RNases J1 and Y) or GapA/SR1P-J1/Y can degrade SR5, we tested GapA purified from either B. subtilis wild-type or a Δsr1 strain. Whereas SR5 was completely degraded after addition of 10 pmol GapA/SR1P-J1/Y, the same amount of SR1P-free GapA-J1/Y was not able to cleave SR5 (). This indicates that only the GapA/SR1P-J1/Y complex displays RNase activity. Moreover, the 0.125 pmol J1 (calculated from ) that are present in 10 pmol GapA/SR1P-J1/Y could degrade the substrate (), whereas 0.1 pmol of purified RNase J1 could not (). Therefore, we conclude that not only binding, but also the activity of RNase J1 is enhanced by the presence of SR1P in the purified GapA-J1/Y complex. In contrast to GapA/SR1P-J1/Y purified from the wild-type, GapA/SR1P-Y purified from an isogenic ΔrnjA strain (lane 6) was not able to degrade SR5 () confirming that the RNase activity of the GapA/SR1P preparation observed in lane 5 was indeed due to co-purified RNase J1. presents a time-course for the degradation of SR5 with 10 pmol RNase J1 purified from E. coli (left) and 10 pmol GapA/SR1P-J1/Y purified from B. subtilis (right). In both cases, a similar degradation pattern was observed.
Figure 5. GapA/SR1P is involved in RNA degradation. RNA degradation assays with internally [α-32P]-UTP-labeled sRNA SR5. BSA and protein-free buffer were used as negative controls and 10 pmol RNase A as positive control. [γ-32P]-ATP-labeled pBR322xMspI served as size marker. Incubation was for 30 min at 37°C, followed by denaturation and separation on 8% denaturing polyacrylamide (PAA) gels, if not stated otherwise. (A) SR5 was incubated with RNase Y and RNase J1. The amount of protein used, full-length SR5 and degradation products (DP) are indicated. (B) Degradation assay as in A) with GapAStrep/SR1P purified from B. subtilis DB104 (ΔamyE::gapAStrep), GapAStrep purified from B. subtilis DB104 (Δsr1::phleo, ΔamyE::gapAStrep) and GapAStrep/SR1P purified from the ΔrnjA strain. (C) Time-course RNA degradation assay. SR5 was incubated with 10 pmol GapAStrep purified from Bacillus subtilis DB104 (ΔamyE::gapAStrep) or 10 pmol RNase J1His purified from E. coli. Incubation times and degradation products (DP) are indicated. (D) Degradation of the known RNase J1 target threonyl tRNA with purified RNases J1 and Y and GapAStrep +/−-SR1P. A schematic representation of the RNase J1 cleavage pattern of threonyl tRNA based on Citation33 is shown in Fig. S3. (E) Assay as in A) with GapAStrep purified from B. subtilis cells harvested from log- or stationary phase TY cultures. 10pmol of GapAStrep with or without co-purified SR1P were used. To exclude that the presence of SR1P increases an intrinsic RNase activity of GapA rather than that of co-purified RNase J1, SR1PHis was purified separately and added to the degradation assays. Neither SR1PHis alone (lane 8) nor addition of SR1PHis to SR1P-free GapAStrep (lanes 2 and 5) increased the RNase activity. SR1P, peptide co-purified with GapAStrep; SR1PHis; 50 pmol peptide purified separately and added later to purified GapAStrep.
![Figure 5. GapA/SR1P is involved in RNA degradation. RNA degradation assays with internally [α-32P]-UTP-labeled sRNA SR5. BSA and protein-free buffer were used as negative controls and 10 pmol RNase A as positive control. [γ-32P]-ATP-labeled pBR322xMspI served as size marker. Incubation was for 30 min at 37°C, followed by denaturation and separation on 8% denaturing polyacrylamide (PAA) gels, if not stated otherwise. (A) SR5 was incubated with RNase Y and RNase J1. The amount of protein used, full-length SR5 and degradation products (DP) are indicated. (B) Degradation assay as in A) with GapAStrep/SR1P purified from B. subtilis DB104 (ΔamyE::gapAStrep), GapAStrep purified from B. subtilis DB104 (Δsr1::phleo, ΔamyE::gapAStrep) and GapAStrep/SR1P purified from the ΔrnjA strain. (C) Time-course RNA degradation assay. SR5 was incubated with 10 pmol GapAStrep purified from Bacillus subtilis DB104 (ΔamyE::gapAStrep) or 10 pmol RNase J1His purified from E. coli. Incubation times and degradation products (DP) are indicated. (D) Degradation of the known RNase J1 target threonyl tRNA with purified RNases J1 and Y and GapAStrep +/−-SR1P. A schematic representation of the RNase J1 cleavage pattern of threonyl tRNA based on Citation33 is shown in Fig. S3. (E) Assay as in A) with GapAStrep purified from B. subtilis cells harvested from log- or stationary phase TY cultures. 10pmol of GapAStrep with or without co-purified SR1P were used. To exclude that the presence of SR1P increases an intrinsic RNase activity of GapA rather than that of co-purified RNase J1, SR1PHis was purified separately and added to the degradation assays. Neither SR1PHis alone (lane 8) nor addition of SR1PHis to SR1P-free GapAStrep (lanes 2 and 5) increased the RNase activity. SR1P, peptide co-purified with GapAStrep; SR1PHis; 50 pmol peptide purified separately and added later to purified GapAStrep.](/cms/asset/b4722e0c-e11c-4f2a-bdb5-402b18d2ec51/krnb_a_1208894_f0005_b.gif)
To demonstrate that GapA/SR1P-J1/Y has a specific RNase J1 activity, we employed a degradation assay with a known RNase J1 target, B. subtilis threonyl tRNA.Citation33 As shown in and S4, a similar degradation pattern of threonyl tRNA was observed for RNase J1 and GapA/SR1P-J1/Y from B. subtilis, but not for GapA-J1/Y alone. This confirms that degradation of SR5 is not due to an unspecific co-purified RNase activity, but to the co-purified RNase J1. In addition, to analyze if RNase J1 acts as endo- or as 5′-3′ exoribonuclease, we performed a degradation assay with 5′ labeled SR5 and purified RNase J1 (Fig. S5). The degradation pattern shows both small degradation products that run with the migration front and internal cleavage products. Therefore, we conclude that RNase J1 does not only act as an endonuclease, but most probably also as 5′-3′ exoribonuclease on SR5.
Based on these results we conclude that only GapA purified from a B. subtilis strain expressing both SR1P and RNase J1 has RNase activity on a known J1 substrate, and this activity is due to the presence of sub-stoichiometric amounts of RNase J1 bound to and co-purified with GapA/SR1P.
The ability of the GapA/SR1P-J1 complex to degrade RNA is growth-phase dependent
Formerly, we demonstrated that SR1 is ≈20-fold repressed by CcpN under glycolytic conditions (e.g., complex TY medium, log phase), but expressed under gluconeogenic conditions (e.g. TY medium, stationary phase).Citation16 When we assayed GapA/SR1P-J1/Y preparations from log phase cultures (, lanes 6 and 7), we observed no RNase activity on our substrate RNA SR5, whereas those from stationary phase cultures used before () displayed RNase activity. These data confirm that SR1P that is much more abundant in cells from stationary phase TY cultures, does indeed modulate the GapA-RNase J1-complex.
Neither His-tagged SR1P (SR1PHis6) purified separately by affinity chromatography () nor SR1PHis6 added afterwards to a GapA preparation from the Δsr1 strain from log or stationary phase () were active in degrading SR5 ruling out an intrinsic RNase activity of SR1P or the GapA/SR1P complex. Apparently, subsequent binding of SR1P did not increase the amount of RNase J1 already bound to GapA before. Thus, we conclude that SR1P triggers the RNase activity only when it is co-expressed with GapA.
Whereas RNase J1 binds SR5, GapA does not, confirming that it is not an RNA binding protein
In the DRaCALA () we had only tested if GapA can bind its own (gapA operon) mRNA, which could be excluded. However, as SR5 proved to be a suitable substrate for degradation by RNase J1 and by GapA/SR1P containing sub-stoichiometric J1 amounts, we used labeled SR5 to perform an electrophoretic mobility shift assay (EMSA) with increasing concentrations of RNase J1 and in parallel with the same concentrations of GapA/SR1P purified from B. subtilis. As shown in Fig. S6, already 6 pmol of J1 completely bound SR5 whereas 54 pmol of GapA containing SR1P did not suffice to produce a complex with even a small percentage of labeled SR5. The amount of RNase J1 present in the 54 pmol of the GapA/SR1P preparation would not result in a visible shift as it is still below the lowest amount of RNase J1His that was used in parallel. This result confirms that GapA is not an RNA binding protein, as it can neither bind its own RNA nor a small RNA that is degraded by GapA/SR1P-J1/Y.
The half-life of the RNase J1 substrate SR5 is altered in a gapA knockout strain
SR5 is 2-fold more stable in the absence of RNase J1.Citation32 demonstrate that GapA can interact with RNase J1. Moreover, GapA/SR1P preparations from B. subtilis wild-type strains contain sub-stoichiometric amounts of RNase J1 and are active in degrading SR5 in vitro (). This prompted us to assay if GapA affects the stability of SR5 in vivo. To this end, we determined the half-life of SR5 in a ΔgapA strain and a Δsr1 strain compared to the isogenic wild-type strain. As shown in , the half-life of SR5 was 4.6 min in the wild-type strain 168 and with 11.5 min more than 2-fold higher in the ΔgapA strain, supporting that GapA is indeed involved in the degradation of SR5 by RNase J1. The determined half-life in the absence of GapA (11.5 min) is in perfect agreement with the 2-fold higher half-life in the absence of RNase J1 (12.2 min, ref. 32). In the Δsr1 strain, the half-life of SR5 was with 7.2 min 1.5-fold higher compared to the wild-type strain, confirming a smaller, but also detectable contribution of SR1P to the degradation of SR5 in vivo. Nevertheless the observed increased half-life of SR5 in the Δsr1 strain is in line with the effect observed in the ΔgapA and the ΔrnjA strains and corroborates the participation of both GapA and SR1P in SR5 degradation.
Figure 6. Determination of SR5 half-lives. B. subtilis strains 168, 168 (Δsr1::cat) and 168 (ΔgapA::ery) were grown in complex TY medium until onset of stationary growth phase, samples taken at the indicated times after rifampicin addition, total RNA prepared and separated on 6% denaturing PAA gels as described.Citation16 [α-32P]UTP-labeled riboprobes were used. Reprobing was performed with [γ-32P]ATP-labeled SB767 specific for 5S rRNA. Autoradiograms of Northern blots are shown. Half-lives presented under the gels are averaged from 3 independent determinations.
![Figure 6. Determination of SR5 half-lives. B. subtilis strains 168, 168 (Δsr1::cat) and 168 (ΔgapA::ery) were grown in complex TY medium until onset of stationary growth phase, samples taken at the indicated times after rifampicin addition, total RNA prepared and separated on 6% denaturing PAA gels as described.Citation16 [α-32P]UTP-labeled riboprobes were used. Reprobing was performed with [γ-32P]ATP-labeled SB767 specific for 5S rRNA. Autoradiograms of Northern blots are shown. Half-lives presented under the gels are averaged from 3 independent determinations.](/cms/asset/32bd5554-2e07-466e-8e5f-3ce0a6645fc0/krnb_a_1208894_f0006_b.gif)
Taken together, this data indicates a role of GapA and SR1P in the modulation of RNA degradation in vivo.
B. subtilis gapA mRNA is not only a substrate for cleavage by RNase Y, but also by RNases J2 and J1
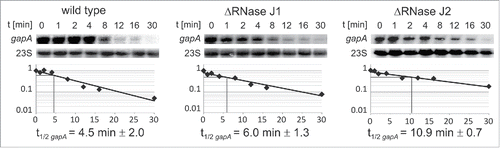
Formerly, it had been shown by Stülke and colleagues that the main endoribonuclease RNase Y makes a stabilizing cut upstream of the gapA ORF within the 3′ end of the cggR ORF yielding a 1.2 kb gapA RNA species that is more stable than the bicistronic 2.2 kb cggR-gapA RNA.Citation25,30 On the other hand, we had demonstrated that the monocistronic 1.2 kb gapA mRNA is more stable in the presence of SR1P.Citation22 As shown above, GapA/SR1P interacts with RNases J1 and Y. This prompted us to investigate if the stability of gapA RNA is also dependent on RNase J1 or its in vivo interaction partner RNase J2. To this end, we determined the half-life of the 1.2 kb gapA RNA in isogenic wild-type, ΔrnjA and ΔrnjB strains. Whereas the half-life of gapA mRNA in the wild-type 168 strain was 4.5 min, it was with 10.9 min 2- to 3-fold longer in the ΔrnjB strain lacking RNase J2 and with ≈6 min 1.5-fold increased in the ΔrnjA strain lacking RNase J1 (). Our data ( and S1) suggest that regions upstream and downstream of the cggR-gapA coding sequence are substrate for destabilizing cuts in the absence of SR1P. The results in demonstrate that RNase J2 is involved in degradation of gapA RNA. RNases J1 and J2 form a complex that is likely the predominant form in vivo.Citation34 RNase J2 is a very weak 5′-3′ exoribonuclease, whereas J1 and the J1/J2 complex have robust 5′-3′ exoribonuclease activity in vitro. However, J2 might, in the complex with J1, be also an endoribonuclease.Citation34 Most likely, RNase J2 is, in complex with RNase J1, responsible for degradation of the gapA operon RNA.
Figure 7. Determination of gapA mRNA half-life. B. subtilis strains 168, 168 (ΔrnjA::spec); 168 (ΔrnjB::ery) were grown in complex TY medium until onset of stationary growth phase, samples taken at the indicated times after rifampicin addition, total RNA prepared and separated on 1.5% agarose gels, probed and reprobed as described.Citation22 Autoradiograms of representative Northern blots are shown. Half-lives are averaged from at least 2 independent determinations.
Discussion
To date, the biological function of the encoded peptides has been unravelled for only a handful of dual-function sRNAs (see also Introduction). In all these cases, the peptide function is connected to the base-pairing function of the RNAs: Both in S. aureus RNAIII and Psm-mec RNA as well as in Streptococcus pyogenes Pel RNA, the encoded peptides – δ-haemolysin, PSMα and streptolysin SLS – and the base-pairing functions of the sRNAs play a role in virulence.Citation8,11,9 Likewise, the base-pairing function of E. coli SgrS is important for glucose and mannose metabolism, and the SgrS-encoded peptide SgrT down-regulates PtsG glucose transporter activity.Citation10,35,36
By contrast, we report that the 2 functions of B. subtilis SR1 are relevant in different pathways: Whereas SR1 acts as a base-pairing sRNA in arginine catabolism,Citation17,18 SR1P plays a broader role, as it is involved in modulating RNA degradation: SR1P binds GapA,Citation22 promotes GapA binding of RNase J1 and this binding enhances the activity of GapA-bound RNase J1 on its corresponding substrates. On the one hand, we demonstrated that GapA can bind RNase J1 in vitro, and this binding is promoted by SR1P. On the other hand, we showed that the GapA/SR1P complex isolated from the B. subtilis wild-type, but not from the RNase J1 knockout strain, contains sub-stoichiometric amounts of RNase J1, which enable it to degrade 2 known in vivo substrates of RNase J1, the sRNA SR5, and threonyl tRNA, in vitro. Furthermore, the presence or absence of GapA and SR1P does also affect SR5 degradation in vivo supporting that the GapA/SR1P-RNase J1 interaction has a biological function. The smaller effect of the sr1 deletion compared to the gapA deletion might be attributed to the fact that GapA is the moonlighting protein whereas SR1P is only the mediator of this moonlighting activity.
As GapA binding to RNases J1 and Y was independent of the presence of RNA, we can exclude a bridging function of RNA in these interactions.
Our findings indicate that GapA might affect the proposed B. subtilis degradosome, which was reported to comprise the main endoribonuclease RNase Y, the main 3′-5′-exoribonuclease PnpA, RNases J1 and J2, helicase CshA, and the glycolytic enzymes Eno and PfkA.Citation25 However, in contrast to the E. coli degradosome, the B. subtilis degradosome cannot be isolated in the absence of cross-linking reagents indicating that it has a more dynamic structure.Citation37 Using a bacterial 2-hybrid system, it was shown that RNase J1 interacts with RNase Y, PnpA and PfkA. RNase Y interacts in addition with PnpA, CshA, Eno and PfkA, whereas PnpA interacts with RNase J1, RNase Y and PfkA.Citation25 However, the authors did not report interactions of GapA with itself (GapA is a tetramer) or with one of the proposed degradosome components. On the one hand, this might be due to masked GapA interactions sites in the constructed chimera. On the other hand, it might also be attributed to the use of a 2-hybrid system from E. coli that does not encode SR1P in its genome.Citation23 Later, some of the initially found interactions were confirmed biochemically with purified proteins, among them Eno-PfkA, Eno-RNase Y, PNPase-RNase J1 and PNPase-RNase Y, whereas the RNase Y-RNase J1 interaction could not be corroborated.Citation38 Based on our data we speculate about a possible involvement of GapA/SR1P in the dynamic B. subtilis degradosome: i) GapA/SR1P could provide additional interactions with RNase J1 and RNase Y, ii) GapA/SR1P could replace PfkA or enolase as scaffolding component, or iii) GapA/SR1P could displace RNase J2 from its interaction with RNase J1 ().
Figure 8. Working model on the potential function(s) of GapA/SR1P in the B. subtilis degradosome. Illustration of the possible effects of GapA/SR1P on the integrity of the B. subtilis degradosome. The interaction of GapA/SR1P might lead to 1) the replacement of the metabolic enzymes PfkA or enolase; 2) the displacement of RNase J2 from its interaction with RNase J1, or 3) additional interactions with RNase J1 and Y. In all cases the presence of GapA/SR1P within the degradosome might result in modulation of the RNase activity or specificity of the RNA degradation machinery. Gray arrows: interactions between the components of the RNA degradosome as observed by Commichau et al., 2009 Citation25 and Lehnik-Habrink et al., 2010,Citation48 black arrows and black crosses: possible effects of GapA/SR1P; gray ovals: proteins.
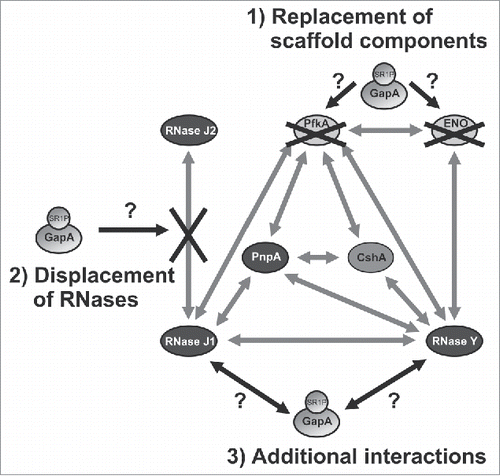
How can we explain our previous finding of SR1P-dependent stabilization of gapA operon mRNA Citation22? Our current data suggest that, on the one hand, regions upstream and downstream of cggR-gapA are necessary for degradation in the absence of SR1P (), most likely because proper folding of the full-length mRNA is important for recognition by RNases. On the other hand, the stable 1.2 kb gapA RNA is generated by two independent processing events: one stabilizing endonucleolytic cut by RNase Y Citation25 and prevention of a destabilizing endonucleolytic cleavage by RNase J2. We speculate that the stabilizing effect of SR1P might be due to one of the above mentioned alterations in the B. subtilis degradosome. As RNase J2 seems to be important for gapA mRNA degradation () an SR1P-mediated displacement of RNase J2 from its interaction with RNase J1 could be a possible scenario for the observed stabilization of gapA mRNA (Fig. S7).
In 2012 Newman et al. observed that citrate modulates B. subtilis enolase activity. They argued that glycolytic enzymes like Eno and PfkA may act as sensors of nutritional stress and may coordinate this with the RNA degrading machinery to decrease the rate of global mRNA turnover under energy-limiting conditions.Citation38 Interestingly, already in 2011 it was reported that citrate alters the activity of E. coli PNPase by direct binding to the enzyme Citation39 suggesting that metabolism and RNA degradation may be linked in all organisms. In our case, CcpN-mediated ≈20-fold repression of sr1 transcription during glycolysis prevents the synthesis of high amounts of SR1P under these conditions whereas under gluconeogenic conditions, the amount of SR1 increases to at least 200–250 molcules/cell.Citation16,18 It is difficult to estimate the amount of SR1P expressed from these SR1 molecules, as an additional regulation of sr1p translation cannot be entirely excluded. Nevertheless, the different RNase activities of logarithmic and stationary phase GapA/SR1P-J1/Y preparations () support that SR1P might, by promoting the GapA-RNase J1 interaction, also act as a sensor to modulate RNA degradation in response to the nutritional state of the cell. In addition, B. subtilis has two Gap enzymes, GapA active in glycolysis and GapB active in gluconeogenesis.Citation40 Thus, GapA is not required in metabolism under gluconeogenic conditions when high amounts of SR1P are available. Consequently, under these conditions, it can moonlight in RNA degradation by recruiting RNase Y or RNase J1. SR1P improves GapA-RNase J1 binding. In addition, it promotes in GapA-J1 complexes the degradation of substrates of this RNase, as shown for SR5.
As we did not detect – in addition to GapA – RNase J1 or RNase Y in our routine co-elution experiments with tagged SR1P (e.g., Citation22), we suggest that the interactions between GapA/SR1P or GapA with RNase J1 or RNase Y are weak, so that only a few molecules of GapA retain the bound RNases. Weak interactions would also facilitate a rapid exchange of transient degradosome components. Furthermore, in the case of J1, different populations might be present in B. subtilis, some associated with the proposed degradosome, others free in the cytosol. Both assumptions would also explain why Newman et al. Citation38 could not confirm biochemically an interaction of RNase J1 with RNase Y, which provides – bound to the membrane – the scaffold for the degradosome. The existence of a larger amount of J1 outside of the degradosome would also suggest that not all J1 targets might depend on GapA or GapA/SR1P. However, this does not seem to apply to the J1 target SR5, for which the 2-fold increased half-life in the ΔgapA strain is in perfect agreement with the 2-fold increased half-life in the ΔrnjA strain compared to the isogenic wild-type strain.Citation32
In 2002, Hackenberg et al. reported that dehydrogenases from rabbit muscle, 2 bacterial species (E. coli and B. stearothermophilus) and the hyperthermophilic archeon Sulfolobus solfataricus cleave RNA.Citation41 In 2012, we discovered in Bacillales, but neither in other Gram-positive nor in Gram-negative bacteria, 23 homologues of SR1, whose base-pairing and peptide-encoding functions are highly conserved over one billion years of evolution.Citation23 Therefore, we assume that SR1P might play a comparable role in the other Bacillales. Furthermore, as all GAPDHs are highly similar, it cannot be excluded that other bacterial GAPDHs might also recruit and modulate the activity of RNases, perhaps in some cases depending on small, so far unknown, peptides, and that the role of GapA in RNA degradation is not confined to Bacillales like B. subtilis. In the light of our present data it is tempting to speculate that the RNase activities observed for the bacterial GAPDH's by Hackenberg et al. might be also modulated by so far unknown co-purified peptides or, alternatively, the result of substoichiometric amounts of a co-purified RNase rather than GAPDH itself.
Interestingly, in E. coli, an alteration of RNase E activity by binding of small proteins has been discovered some years ago: The 17.4 kDa protein RraA Citation42 or the 15.6 kDa RraB Citation43 bind weakly – with a KD value in micromolar range – to and inhibit RNase E. RraA masks the RNA binding domains in the C-terminus of RNase E and additionally binds to the helicase RhlB Citation44 leading to global changes in RNA turnover. It was argued that remodelling of the degradosome by such proteins allows for differential regulation of RNA cleavages in E. coli. RraA is widely distributed among Gram-negative bacteria and also plants,Citation42 whereas RraB is confined to γ-proteobacteria. Gao et al. proposed that RraA and RraB might also exert indirect effects on the amount of free, not degradosome bound PNPase or RhlB.Citation43 These data suggest that modulation of RNase activity involving weak interactions with peptides or small proteins might be a more common principle in the regulation of RNA turnover.
Future research will focus on the effect of the GapA-RNase Y interaction on specific targets, the detailed analysis of a genome-wide impact of GapA on RNA degradation, the molecular mechanism by which SR1P binding to GapA activates bound RNase J1 and the identification of functional homologs of SR1P in other Gram-positive bacteria.
Materials and methods
Strains, growth conditions, enzymes
E. coli strain TG1 was used for cloning and B. subtilis strain DB104 (ΔamyE::pMGG19) or DB104 (Δsr1::cat; ΔamyE::pMGG19) for GapA purification with or without SR1P. E. coli strains CCE093 and SSC420 were used for purification of RNase J1His or RNase YHis (all strains are listed in Table S1). TY medium Citation16 and TFB medium (12 g bacto tryptone, 24 g yeast extract, 4 ml glycerol, 2.31 g KH2PO4, 12.54 g K2HPO4 x 3 H2O/l) served as complex medium for B. subtilis and E. coli, respectively. Taq-polymerases were purchased from Roche or Solis Biodyne (Estonia) and T7 RNA polymerase from New England Biolabs.
Protein purification and co-elution assay
Strep-tagged GapA carrying or lacking SR1P was purified from B. subtilis strain DB104 (ΔamyE::gapAStrep) or DB104 (Δsr1::cat; ΔamyE::gapAStrep) as described Citation22 except that 1.5% Triton-X100 was added to the lysis buffer. For purification of His-tagged RNases J1 and Y, 1 l TFB medium was inoculated with an overnight culture to OD560 of 0.1 and grown for 1.5 hr. RNase expression was induced with 2 mM IPTG for 3 hours, cells harvested by centrifugation and pellets frozen overnight. Protein extracts were prepared by sonication and supernatants applied to 1 ml Ni-NTA-columns. Washing and elution were performed as described.Citation45 Six 500 µl elution fractions were collected for each column, stored at 4°C and, prior to use, concentrated using Amicon(R) Ultra 10k centrifugal filters. Proteins were centrifuged at 4°C with 16.000 g until no further concentration was possible, washed twice with 100 µl storage buffer (300 mM NaCl, 50 mM Na phosphate pH7.5, 25% glycerol) and again centrifuged as before. Proteins were collected by reverse spin with 50 µl storage buffer for 2 min at 1000 g. For purification of SR1PHis, 2 l TY medium were inoculated with an overnight culture of B. subtilis strain MG1P (pWSR1/M31) to an OD560 of 0.2, grown for 7 hr, and sr1 expression induced for 20 min by addition of 0.5 µg/ml anhydro-tetracycline. Cells were harvested by centrifugation, protein crude extracts prepared as described,Citation22 and applied to 1 ml Ni-NTA columns as above. Prior to use, SR1PHis was separated from elution fractions by Amicon(R) Ultra 10k centrifugal filters, and the filtrate concentrated with Amicon(R) Ultra 3k centrifugal filters. Concentration, washing with storage buffer and protein collection was performed as above. The co-elution assay was performed as described.Citation22
Western blotting and Far Western blotting
Western blotting was performed as described,Citation22 but a polyclonal antiserum raised against RNase J2 that recognizes both RNase J1 and RNase J2 (H. Putzer, personal communication) (1:5000) was used. For Far-Western blotting, proteins were separated on SDS/PAA gels and transferred onto PVDF membranes that were blocked for 2 hr. Blots were incubated overnight with 10 ml PBST-gelatine or PBST-gelatine supplemented with either 100 µg GapAStrep purified from B. subtilis or 170 µg His-tagged RNase purified from E. coli. Binding of GapAStrep and His-tagged RNase was detected by incubation with mouse-anti-Strep-tag antibody (1:1000; IBA Göttingen) or mouse-anti-His-tag antibody (1:2000; IBA Göttingen), respectively, and subsequently horseradish peroxidase coupled anti-mouse antibody (1:2000; IBA Göttingen).
DRaCALA (Differential Radial Capillary Action of Ligand Assay)
In vitro transcribed [α-32P]-UTP labeled RNA was diluted to a final concentration of 24 nM. 2.5 µl RNA were mixed with 2.5 µl purified protein and incubated for 10 min at 25°C. Subsequently, the reaction mixture was spotted onto nitrocellulose membrane NC45 (pore size 0.45 µm, Serva Heidelberg). Spots were allowed to dry for 30 min at room temperature and signals detected after overnight exposure to a phosphorImager plate using Aida Image Analyzer v.4.50.
RNA degradation assay
One µl in vitro transcribed, [α-32P]-UTP labeled RNA (10.000 cpm) was incubated with 2 µl 5x reaction buffer (100 mM Tris-HCl pH8.0; 40 mM MgCl2, 500 mM NH4Cl; 0,5 mM DTT) and 7 µl diluted protein for 30 min at 37°C. The reaction was stopped by addition of 10 µl formamide loading dye Citation16 and 5 min at 95°C. Samples were separated on 6% to 12.5% denaturing PAA gels. Dried gels were analyzed by phosphorImaging using Aida Image Analyzer v.4.50.
RNA preparation, Northern blotting, in vitro transcription and EMSA
Strains were cultivated until onset of stationary phase, 0.5 ml samples flash-frozen in liquid nitrogen and stored at -20°C. Preparation of total RNA, separation on 6% denaturing PAA gels, Northern blotting and determination of RNA half-lives were carried out as described previouslyCitation16,17 except that Aida Image Analyzer v.4.50 was used. [α-32P]-UTP labeled riboprobes (SR5) or [α-32P]-dATP labeled DNA fragments (gapA, SR1) were used for detection and reprobing performed as described.Citation16,22 In vitro transcription for the generation of [α-32P]-UTP labeled RNAs was carried out as described.Citation46 EMSA was carried out as described.Citation18
Plasmid constructions
For construction of plasmid pMGG13, a PCR was performed on chromosomal DNA from B. subtilis DB104 with primer pair SB1818/SB1993, the resulting fragment digested with BsrGI and BamHI and inserted into vector pMGG1Citation47 (primers are listed in Table S2, plasmids in Table S3). For expression of the bicistronic cggR-gapA mRNA, a fragment was generated by PCR with primer pair SB1217/SB1083 as above, digested with EcoRI/BsrGI and inserted into pMGG1 yielding pMCG1. Plasmid pMCG2 was constructed by ligation of the 1.2 kb BsrGI/BamHI fragment from pMGG13 into pMCG1. Plasmids that allow expression of 5′ truncated cggR-gapA mRNA were constructed as follows: The 1.7 kb EcoRI/BamHI pcggR-gapA fragment from pMGG13 was ligated into pMG7 yielding plasmid pMGG14 that was later used as a host for the ‘cggR-gapA fragments. Next, ‘cggR-gapA fragments were generated by PCR on pMCG2 as template using primer SB1083 and either primer SB2142, SB2249, SB2250 or SB2251. The obtained fragments were inserted into the pMGG14 PstI vector yielding plasmids pMCG4-7. Plasmid pMCG8 that lacks most of the gapA ORF was constructed using 2 independent PCRs on pMCG2 as template and primer pairs SB1217/SB2291 and SB2292/SB1993, respectively, the resulting fragments digested with EcoRI/Acc65I or Acc65I/BamHI and jointly inserted into pUC19. The resulting 1.8 kb EcoRI/BamHI fragment was cloned into pMG6 yielding pMCG8. Further truncation of the cggR portion from the 3′-end was achieved by integration of a fragment generated by PCR with primer pair SB1217/SB2387 using pMCG2 as template. The resulting fragment was digested with EcoRI and Acc65I and ligated into pMCG8 yielding pMCG13. Plasmid pMGG19 for the expression of gapAStrep from the amyE locus was generated by insertion of a fragment obtained by 2 subsequent PCRs, the first with primer pair SB1818/SB2437, and the second with primer pair SB1818/SB2438, into the HindIII/BamHI vector of pMG7.Citation47 All inserts were confirmed by sequencing.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary_Data.pdf
Download PDF (2.7 MB)Acknowledgments
The authors thank Ciaran Condon (Paris) for E. coli strain CCE093 for overexpression and purification of His-tagged RNase J1, B. subtilis strain CCG434 (ΔrnjA) and antibodies against RNase Y, and Harald Putzer (Paris) for E. coli strain SSC420 for overexpression and purification of His-tagged RNase Y and antibodies against RNases J1/J2. Furthermore, we thank Bernhard Schlott, FLI Jena, for performing the mass spectrometry analysis on the bands shown in Fig. S2. This work was supported by Deutsche Forschungsgemeinschaft BR1552/8–1 (to S. B.)
Related Research Data
References
- Brantl S. Bacterial chromosome-encoded small regulatory RNAs. Future Microbiol 2009; 4:85-103; PMID:19207102; http://dx.doi.org/10.2217/17460913.4.1.85
- Brantl S. Acting antisense: plasmid- and chromosome-encoded sRNAs from Gram-positive bacteria. Future Microbiol 2012; 7:853-71; PMID:22827307; http://dx.doi.org/10.2217/fmb.12.59
- Brantl S, Brückner R. Small regulatory RNAs from low-GC Gram-positive bacteria. RNA Biol 2014; 11:443-56; PMID:24576839; http://dx.doi.org/10.4161/rna.28036
- Brantl S. Regulatory mechanisms employed by cis-encoded antisense RNAs. Curr Opin Microbiol 2007; 10:102-9; PMID:17387036; http://dx.doi.org/10.1016/j.mib.2007.03.012
- Morfeldt E, Taylor D, von Gabain A, Arvidson S. Activation of alpha-toxin translation in Staphylococcus aureus by the trans-encoded antisense RNA, RNAIII. EMBO J 1995; 14:4569-77; PMID:7556100
- Boisset S, Geissmann T, Huntzinger E, Fechter P, Bendridi N, Possedko M, Chevalier C, Helfer AC, Benito Y, Jacquier A, et al. Staphylococcus aureus RNAIII coordinately represses synthesis of virulence factors and the transcription regulator Rot by an antisense mechanism. Genes Dev 2007; 21:1353-66; PMID:17545468; http://dx.doi.org/10.1101/gad.423507
- Chevalier C, Boisset S, Romilly C, Masquida B, Fechter P, Geissmann T, Vandenesch F, Romby P. Staphylococcus aureus RNAIII binds to two distant regions of coa RNA to arrest translation and promote RNA degradation. PLoS Pathog 2010; 6:1000809; PMID:20300607; http://dx.doi.org/10.1371/journal.ppat.1000809
- Fechter P, Caldelari I, Lioliou E, Romby P. Novel aspects of RNA regulation in Staphylococcus aureus. FEBS Lett 2014; 588:2523-9; PMID:24873876; http://dx.doi.org/10.1016/j.febslet.2014.05.037
- Mangold M, Siller M, Roppenser B, Vlamincks BJ, Penfound TA, Klein R, Novak R, Novick RP, Charpentier E. Synthesis of group A streptococcal virulence factors is controlled by a regulatory RNA molecule. Mol. Microbiol 2004; 53:1515-27; PMID:15387826
- Wadler CS, Vanderpool CK. A dual function for a bacterial small RNA: SgrS performs base-pairing dependent regulation and encodes a functional polypeptide. Proc Natl Acad Sci USA 2007; 104:20454-9; PMID:18042713; http://dx.doi.org/10.1073/pnas.0708102104
- Kaito C, Saito Y, Ikuo M, Omae Y, Mao H, Nagano G, Fujiyuki T, Numata S, Han X, Obata K, et al. Mobile genetic element SCCmec-encoded psm-mec RNA suppresses translation of agrA and attenuates MRSA virulence. PLoS Pathog 2013; 9:e1003269; PMID:23592990; http://dx.doi.org/10.1371/journal.ppat.1003269
- Shimizu T, Yaguchi H, Ohtani K, Banu S, Hayasi H. Clostridial VirR/VirS regulon involves a regulatory RNA molecule for expression of toxins. Mol. Microbiol 2002; 43:257-65
- Roberts SA, Scott JR. RivR and the small RNA RivX: the missing links between the CovR regulatory cascade and the Mga regulon. Mol. Microbiol 2007; 66:1506-22; PMID:18005100
- Berghoff B, Glaeser J, Sharma CM, Vogel J, Klug G. Photooxidative stress-induced and abundant small RNAs in Rhodobacter sphaeroides. Mol. Microbiol 2009; 74:1497-512
- Sonnleitner E, Gonzalez N, Sorger-Domenigg T, Heeb S, Richter AS, Backofen R, Williams P, Hüttenhofer A, Haas D, Bläsi U. The small RNA PhrS stimulates synthesis of the Pseudomonas aeruginosa quinolone signal. Mol. Microbiol 2011; 80:868-85; PMID:21375594; http://dx.doi.org/10.1111/j.1365-2958.2011.07620.x
- Licht A, Preis S, Brantl S. Implication of CcpN in the regulation of a novel untranslated RNA (SR1) in B. subtilis. Mol. Microbiol 2005; 58:189-206; PMID:16164558
- Heidrich N, Chinali A, Gerth U, Brantl S. The small untranslated RNA SR1 from the B. subtilis genome is involved in the regulation of arginine catabolism. Mol. Microbiol 2006; 62:520-36; PMID:17020585
- Heidrich N, Moll I, Brantl S. In vitro analysis of the interaction between the small RNA SR1 and its primary target ahrC mRNA. Nucleic Acids Res 2007; 35:4331-46; PMID:17576690; http://dx.doi.org/10.1093/nar/gkm439
- Licht A, Brantl S. (2006) Transcriptional repressor CcpN from Bacillus subtilis compensates asymmetric contact distribution by cooperative binding. J Mol Biol 2006; 364:434-48; PMID:17011578; http://dx.doi.org/10.1016/j.jmb.2006.09.021
- Licht A, Golbik R, Brantl S. Identification of ligands affecting the activity of the transcriptional repressor CcpN from Bacillus subtilis. J Mol Biol 2008; 380:17-30; PMID:18511073; http://dx.doi.org/10.1016/j.jmb.2008.05.002
- Licht A, Brantl S. The transcriptional repressor CcpN from Bacillus subtilis uses different repression mechanism at different promoters. J Biol Chem 2009; 284:30032-8; PMID:19726675; http://dx.doi.org/10.1074/jbc.M109.033076
- Gimpel M, Heidrich N, Mäder U, Krügel H, Brantl S. A dual-function sRNA from B. subtilis: SR1 acts as a peptide encoding mRNA on the gapA operon. Mol Microbiol 2010; 76:990-1009; PMID:20444087; http://dx.doi.org/10.1111/j.1365-2958.2010.07158.x
- Gimpel M, Preis H, Barth E, Gramzow L, Brantl S. SR1 – a small RNA with two remarkably conserved functions. Nucleic Acids Res 2012; 40:11659-72; PMID:23034808; http://dx.doi.org/10.1093/nar/gks895
- Evguenieva-Hackenberg E, Klug G. New aspects of RNA processing in prokaryotes. Curr Opin Microbiol 2011; 14:587-92; PMID:21945217; http://dx.doi.org/10.1016/j.mib.2011.07.025
- Commichau, FM, Rothe FM, Herzberg C, Wagner E, Hellwig D, Lehnik-Habrink M, Hammer E, Völker U, Stülke J. Novel activities of glycolytic enzymes in Bacillus subtilis. Mol Cell Proteomics 2009; 8:1350-60; PMID:19193632; http://dx.doi.org/10.1074/mcp.M800546-MCP200
- Even S, Pellegrini O, Zig L, Labas V, Vinh J, Bréchemmier-Baey D, Putzer H. Ribonucleases J1 and J2: two novel endoribonucleases in B. subtilis with functional homology to E. coli RNase E. Nucleic Acids Res 2005; 33:2141-52; PMID:15831787; http://dx.doi.org/10.1093/nar/gki505
- Bechhofer DH. B. subtilis mRNA decay: new parts in the toolkit. Wiley Interdiscip Rev RNA 2011; 2:387-94; PMID:21957024; http://dx.doi.org/10.1002/wrna.66
- Laalami S, Zig L, Putzer H. Initiation of mRNA decay in bacteria. Cell Mol Life Sci 71:1799-828; PMID:24064983; http://dx.doi.org/10.1007/s00018-013-1472-4
- Shahbabian K, Jamalli A, Zig L, Putzer H. RNase Y, a novel endoribonuclease, initiates riboswitch turnover in Bacillus subtilis. EMBO J 2009; 28:3523-33; PMID:19779461; http://dx.doi.org/10.1038/emboj.2009.283
- Lehnik-Habrink M, Schaffer M, Mäder U, Diethmaier C, Herzberg C, Stülke J. RNA processing in Bacillus subtilis: identification of targets of the essential RNase Y. Mol Microbiol 2011; 81:1459-73; PMID:21815947; http://dx.doi.org/10.1111/j.1365-2958.2011.07777.x
- Roelofs KG, Wang J, Sintim HO, Lee VT. Differential radial capillary action of ligand assay for high-throughput detection of protein-metabolite interactions. Proc Natl Acad Sci USA 2011; 108:15528-33; PMID:21876132; http://dx.doi.org/10.1073/pnas.1018949108
- Müller P, Jahn N, Ring C, Maiwald C, Neubert R, Meißner C, Brantl S. A multistress responsive type I toxin-antitoxin system: bsrE/SR5 from Bacillus subtilis. RNA Biol 2016; in press; PMID:26940229; http://dx.doi.org/10.1080/15476286.2016.1156288
- Daou-Chabo R, Condon C. RNase J1 endonuclease activity as a probe of RNA secondary structure. RNA 2009; 15:1417-25; PMID:19458035; http://dx.doi.org/10.1261/rna.1574309
- Mathy N, Hébert A, Mervelet P, Bénard L, Dorléans A, Li de la Sierra-Gallay I, Noirot P, Putzer H, Condon C. Bacillus subtilis ribonucleases J1 and J2 form a complex with altered enzyme behaviour. Mol Microbiol 2010; 75:489-98; PMID:20025672; http://dx.doi.org/10.1111/j.1365-2958.2009.07004.x
- Rice JB, Vanderpool CK. The small RNA SgrS controls sugar-phosphate accumulation by regulating multiple PTS genes. Nucleic Acids Res 2011; 39:3806-19; PMID:21245045; http://dx.doi.org/10.1093/nar/gkq1219
- Balasubramanian D, Vanderpool CK. Deciphering the interplay between two independent functions of the small RNA regulator SgrS in Salmonella. J Bacteriol 2013; 195:2620-30; PMID:23935052; http://dx.doi.org/10.1128/JB.00586-13
- Lehnik-Habrink M, Lewis RJ, Mäder U, Stülke J. RNA degradation in Bacillus subtilis: an interplay of essential endo- and exoribonucleases. Mol Microbiol 2012; 84:1005-17; PMID:22568516; http://dx.doi.org/10.1111/j.1365-2958.2012.08072.x
- Newman JA, Hewitt L, Rodrigues C, Solovyova AS, Harwood CR, Lewis RJ. Dissection of the network of interactions that links RNA processing with glycolysis in the Bacillus subtilis degradosome. J Mol Biol 2012; 416:121-36; PMID:22198292; http://dx.doi.org/10.1016/j.jmb.2011.12.024
- Nurmohamed S, Vincent HA, Titman CM, Chandran V, Pears MR, Du D, Griffin JHL, Callaghan AJ, Luisi BF. Polynucleotide phosphorylase activity may be modulated by metabolites in Escherichia coli. J Biol Chem 2011; 286:14315-23; PMID:21324911; http://dx.doi.org/10.1074/jbc.M110.200741
- Fillinger S, Boschi-Muller S, Azza S, Ervyn E, Branlant G, Aymerich S. Two glyceraldehyde-3-phosphate dehydrogenases with opposite physiological roles in a nonphotosynthetic bacterium. J Biol Chem 2000; 275:14031-7; PMID:10799476; http://dx.doi.org/10.1074/jbc.275.19.14031
- Evguenieva-Hackenberg E, Schiltz E, Klug G. Dehydrogenases from all three domains of life cleave RNA. J Biol Chem 2002; 277:46145-50; PMID:12359717; http://dx.doi.org/10.1074/jbc.M208717200
- Lee K, Zhan X, Gao J, Qiu J, Feng Y, Meganathan R, Cohen SN, Georgiou G. RraA, a protein inhibitor of RNase E activity that globally modulates RNA abundance in E. coli. Cell 2003; 114:623-34; PMID:13678585; http://dx.doi.org/10.1016/j.cell.2003.08.003
- Gao J, Lee K, Zhao M, Qiu J, Zhan X, Xaxena A, Moore CJ, Cohen SN, Georgiou G. Differential modulation of E. coli mRNA abundance by inhibitory proteins that alter the composition of the degradosome. Mol Microbiol 2006; 61:394-406; PMID:16771842; http://dx.doi.org/10.1111/j.1365-2958.2006.05246.x
- Gorna MW, Pietras Z, Tsai YC, Callaghan AJ, Hernández H, Robinson CV, Luisi BF. The regulatory protein RraA modulates RNA-binding and helicase activities of the E. coli degradosome. RNA 2010; 16:553-62; PMID:20106955; http://dx.doi.org/10.1261/rna.1858010
- Steinmetzer K, Brantl S. Plasmid pIP501 encoded transcriptional repressor CopR binds asymmetrically at two consecutive major grooves of the DNA. J Mol Biol 1997; 269:648-93; PMID:9223633; http://dx.doi.org/10.1006/jmbi.1997.1083
- Heidrich N, Brantl S. Antisense-RNA mediated transcriptional attenuation: importance of a U-turn loop structure in the target RNA of plasmid pIP501 for efficient inhibition by the antisense RNA. J Mol Biol 2003; 333:917-29; PMID:14583190; http://dx.doi.org/10.1016/j.jmb.2003.09.020
- Gimpel M, Brantl S. Construction of a modular plasmid family for chromosomal integration in Bacillus subtilis. J Microbiol Methods 2012; 91:312-7; PMID:22982324; http://dx.doi.org/10.1016/j.mimet.2012.09.003
- Lehnik-Habrink M, Pförtner H, Rempeters L, Pietack N, Herzberg C, Stülke J. The RNA degradosome in Bacilus subtilis: identification of CshA as the major RNA helicase in the multiprotein complex. Mol Microbiol 2010; 77:958-71; PMID:20572937; http://dx.doi.org/10.1111/j.1365-2958.2010.07264.x